
Abstract
High-energy exciton emission could allow single-component multi-colour display or white light-emitting diodes. However, the thermal relaxation of high-energy excitons is much faster than the photon emission of them, making them non-emissive. Here, we report quantum dots with light hole-heavy hole splitting exhibiting strong high-energy exciton electroluminescence from high-lying light holes, opening a gate for high-performance multi-colour light sources. The high-energy electroluminescence can reach 44.5% of the band-edge heavy-hole exciton emission at an electron flux density Φe of 0.71 × 1019 s−1 cm−2 − 600 times lower than the photon flux density Φp (4.3 × 1021 s−1 cm−2) required for the similar ratio. Our simulation and experimental results suggest that the oscillator strength of heavy holes reduces more than that of light holes under electric fields. We attribute this as the main reason for strong light-hole electroluminescence. We observe this phenomenon in both CdxZn1-xSe-ZnS and CdSe-CdS core-shell quantum dots exhibiting large light hole-heavy hole splittings.
Similar content being viewed by others

Introduction
Typical inorganic or organic photon emitters exhibit broad absorption spectra, so the carriers can be generated by exciting the band-edge or higher-lying energy states, but those high-energy carriers thermally relax to the band-edge rapidly before they emit photons. As such, we only see a narrow band-edge emission, as generalized by Kasha in 19501. Transition through phonon emission is much faster than that through photon emission, and it happens among the levels that are closely packed, because the quantized phonon energy is rather small compared with that of the photon2,3,4. In addition, the thermal occupation and oscillator strength of high-energy excitons are usually insufficient to observe strong emissions from them. Kasha’s rule holds for the overwhelming majority of emitters, but researchers are interested in finding anti-Kasha light emitters because they allow multi-colour labels and white light emission from a single emitter. Up to now, there are only a few exceptions have been found, such as organic compounds azulene5 or thiophosgene6, which exhibit large energy gaps between the lowest unoccupied molecular orbital (LUMO) and higher-lying energy levels.
Quantum dots (QDs) are nanocrystal semiconductors exhibiting a strong quantum confinement effect, they show much more spaced electron energy levels than that of the bulk counterparts7,8,9,10,11. However, the valence bands of II-VI and III-V semiconductors are still closely packed and the high-energy excitons are still mostly nonradiative. To the best of our literature review, higher-energy exciton emission without ground state filling has been mainly observed in nanocrystals with the intermediate barrier which retards the thermal relaxation of higher-energy excitons12.
Herein, we report that high-energy exciton electroluminescence in light-emitting diodes (LEDs) is much more viable than photoluminescence in QDs. Unlike previous observations of high-energy exciton electroluminescence that relies on filling the ground state with excessive charge carriers13, we can achieve high-energy exciton electroluminescence at low charge concentration by increasing the relative oscillator strength. We observe that the oscillator strength of the ground-state heavy holes (HHs) can be remarkably depressed under electrical fields while that of light holes (LHs) remains intact. We demonstrate strong high-energy exciton electroluminescence in two different weakly confined QDs, both of which feature strong Stark effect and well-split LH and HH levels.
Results
Light hole-heavy hole splitting in polytypic quantum dots
The first type of QDs (CdxZn1-xSe-ZnS based) used in this work were synthesized by modifying a protocol from literature14,15, featuring a graded composition (Fig. 1a) and larger cores (∼15.8 nm). The large core size allows us to observe a strong Stark effect in QD-LED because the wave function can be influenced greatly by the electrical field16,17,18. They were grown by utilizing the reactivity difference between CdSe and ZnSe, and an additional ultrathin ZnS shell (∼0.5 nm) was grown to improve stability (Supplementary Fig. 1, see Methods for details). This radial composition change causes a composition-dependent phase transition: the Zn-rich parts tend to crystallize in a zinc-blende (ZB) structure, while the Cd-rich part tends to crystallize in a wurtzite (WZ) structure, resulting in a sandwich-like polytypic structure, as can be observed in high-resolution transmission electron microscope (HRTEM) images (Fig. 1b, c).

a Schematic of ∼15.8 nm graded CdxZn1-xSe QDs with ∼0.5 nm ZnS shell. Different parts tend to differently crystallize in zinc-blende (ZB) and wurtzite (WZ) structures. b High-resolution transmission electron microscopy (HRTEM) image of polytypic CdxZn1-xSe-ZnS QDs viewing along the [110]WZ and [110]ZB zone axes. A scale bar of 5 nm is also attached (black). c Schematic of ZB/WZ interface of CdxZn1-xSe-ZnS QDs.
According to our previous density functional theory (DFT) computation19, LH and HH degeneracy can be lifted in polytypic QDs, and the splitting between LH and HH can reach 50 meV (Supplementary Fig. 2); however, since quantum confinement of the QD used in this work (∼16.8 nm in diameter) is weak, the energy levels are densely packed8,9, resulting in overlaps of band-edge and higher energy exciton transition. Therefore, in the steady-state absorption spectrum, it is difficult to identify the absorption peaks of different energy levels.
To best distinguish the absorption peaks of LH and HH excitons, we performed femtosecond transient absorption spectroscopy, setting the pumping wavelength in resonance with the energy of the band-edge excitons. By doing so, we can only excite the band-edge electrons without generating high-energy ones. In the transient absorption spectrum of pure wurtzite cores (Fig. 2a, see Methods for synthesis details), only two transition peaks (∼1.95 eV and ∼2.01 eV) can be observed, corresponding to the excitons involving the degenerate band-edge hole state (1S) and higher-order hole state (2S) respectively, as described in the literature4,20,21,22. In contrast, in our polytypic QDs, there are three transition peaks (∼1.92 eV, ∼1.97 eV, and ∼2.03 eV), corresponding to the excitons involving the band-edge heavy-hole state (1Shh), light-hole state (1Slh), and higher-order hole state (2S), respectively. We also observe a decrease in the relative amplitude of the 1Slh transition peak during another non-resonant excitation at ∼2.03 eV (Fig. 2b), indicating a relaxation of LH.

a Transient absorption spectra of pure wurtzite CdxZn1-xSe core (black) and polytypic CdxZn1-xSe-ZnS QDs (purple) under 1.95 eV and 1.92 eV band-edge photoexcitation (at 150 fs). The second derivatives of the bleach spectra (dashed curve) allow us to quantify the splitting energy (∼54 meV). The purple and green arrows indicate the photoexcitation energies. 1Shh: band-edge heavy-hole state; 1Slh: light-hole state; 2S: higher-order hole state. b Transient absorption spectra of polytypic CdxZn1-xSe-ZnS QDs at 200 fs (dash), 250 fs (dot), and 300 fs (solid), pumping at ∼2.03 eV. c Schematic of LH and HH photoexcitation. Under resonant HH photoexcitation (pump 1), band-edge exciton is generated immediately, while under non-resonant LH photoexcitation (pump 2), LH exciton takes some time to relax to the band-edge state (Δt). d Band-edge exciton generation dynamics. The difference in dynamics (shadow) is induced by both LH relaxation and Coulomb correlation, as indicated by arrows. e The differential dynamic of Fig. 2c. The rising edge represents the LH relaxation dynamic, which can be fitted with a single exponential function (orange, dash), and the lifetime of the light holes (τh) can be obtained.
To verify whether phonon bottlenecks exist between LH and HH in our QDs, we performed further femtosecond transient absorption to probe the high-energy exciton relaxation process. When we selectively pump the band-edge (pump condition 1 in Fig. 2a) and higher-lying states (pump condition 2 in Fig. 2a), we can get band-edge exciton either directly or through thermal relaxation of LH (Fig. 2c). We observed that the bleaching signal of band-edge exciton grows slower when pumping the higher-lying LH state (Fig. 2d), indicating a femtosecond time scale relaxation of LH to HH. There is a decrease in the relative change in optical density (ΔA) under band-edge pumping, which is attributed to Coulomb correlation23,24, thus we only focus on the rising edge of bleaching. To quantify the lifetime of the LH relaxation, we subtracted the rising edges of ΔA under pump conditions 1 and 2, fitted the differential with a single exponential growth dynamic (Fig. 2e), and got a lifetime of ∼72 fs. These results suggest that LH and HH will reach a thermal equilibrium rapidly, without any blockage from the phonon bottleneck.
Emission from high-energy light-hole excitons
To characterize the photoluminescence from LH and HH excitons with different exciton populations, we performed pump intensity-dependent photoluminescence spectroscopy on the polytypic QD films. The spectra can be fitted with three Gaussian peaks (Fig. 3a): a band-edge exciton photoluminescence peak (X1) originating from HH excitons, and two higher-order exciton photoluminescence peaks (X2 and X3), originating from LH and 2Sh excitons respectively. The LH photoluminescence is weak at low photoexcitation intensity, which is proportional to the thermal occupation probability and oscillator strength of the LH exciton. It becomes intense when the photoexcited charge population exceeds 2 because of ground state filling. As we further increase the excitation power (photon flux density Φp reaches ∼1.3 × 1022 s−1 cm−2), the proportion of X2 photoluminescence intensity to X1 photoluminescence intensity (X2/X1 intensity ratio) saturates at 45% due to nonradiative Auger recombination (see inset to Fig. 3a).

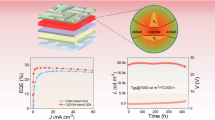
a Pump intensity-dependent photoluminescence spectra of the QDs film. The dashed curves represent the Gaussian fitting of three photoluminescence peaks X1 (purple), X2 (green), and X3 (orange). Inset: the proportion of high-energy exciton emission (X2/X1) versus photon flux density Φp. b Electroluminescence spectra under different voltages. The Stark shift of the electroluminescence peak can reach ∼40 meV as we increase the driving voltage to 9 V. The dashed curves represent Gaussian fitting of three photoluminescence peaks X1 (purple), X2 (green), and X3 (orange). Inset: the proportion of high-energy exciton emission versus electron flux density Φe. c Current density (purple squares) and luminance (green circles) of the CdxZn1-xSe-ZnS QD-LED as a function of voltage. d External quantum efficiency (EQE) of the CdxZn1-xSe-ZnS QD-LED as a function of injected current density.
To confirm that these emission peaks originate from the different states of each single QD rather than from different QDs with different sizes, we compared the photoluminescence spectra before and after size selective gradient centrifugation (Supplementary Fig. 3), and measured the photoluminescence spectra under both the red-edge and higher state excitation (Supplementary Fig. 4, see Methods for details). All these photoluminescence spectra exhibit similar multiple peak emissions, confirming the multiband emission originates from each single QD rather than sub-populations. Furthermore, we also observe that the lifetime of LH photoluminescence X2 (∼4.65 ns) is shorter than that of HH photoluminescence X1 (∼6.95 ns, Supplementary Fig. 5), consistent with the relatively higher oscillator strength of LH excitons.
We then measured the electroluminescence spectra of our QD-LED under different voltages (Supplementary Fig. 6; see Methods for details), which can be fitted with three Gaussian peaks as well (Fig. 3b). However, even under very low electron flux density Φe (∼0.71 × 1019 s−1 cm−2, ∼1.1 A cm−2), the proportion of LH electroluminescence is higher than that under much higher photon flux density Φp (∼4.3 × 1021 s−1 cm−2, ∼2.1 kW cm−2). When the input electron flux density exceeds 1.59 × 1019 s−1 cm−2 (∼2.5 A cm−2), the X2/X1 intensity ratio can reach 80%.
It is worth mentioning that our QD-LEDs show decent luminance and efficiency with a high proportion of hot electroluminescence (Fig. 3c,d). When the X2/X1 intensity ratio approaches 57%, our device has a maximum brightness of ∼80,000 cd m−2 and an external quantum efficiency of 7.5%. Such a high proportion of high-energy exciton electroluminescence with reasonably high luminance and efficiency will potentially enable high-performance multi-colour light sources.
Carrier concentration in quantum dot light-emitting diodes
The observed LH electroluminescence is anomalous. High-energy exciton electroluminescence has been observed in QD-LEDs under high current injection13 (1000 A cm−2). In our photoexcitation experiments, we also need a photon flux density of ∼4.3 × 1021 s−1 cm−2 (equivalent to ∼9 electrons per dot) to let the proportion of LH photoluminescence reach 40%. However, under electrical excitation, we only require a very low electron flux density (∼0.69 × 1019 s−1 cm−2) to generate electroluminescence with a similar LH proportion.
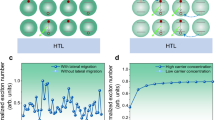
To calculate the average electron population per QD under electrical excitation, we performed a self-developed electrically excited transient absorption (EETA) spectroscopy25,26,27. In EETA, a current pulse is used to excite the sample, and another white light laser pulse probes the change in the absorption after electrical excitation (Fig. 4a and Supplementary Fig. 7). Therefore, the electron behavior in various functional layers can be revealed, such as charge injection dynamics or equilibrated charge concentration. In our polytypic QD-LEDs, as we increase the voltage from 1 V to 10 V, the amplitude of band-edge state bleaching grows continuously, indicating that electron concentration continuously increases (Fig. 4b). Comparing the amplitudes of the bleaching from EETA and nanosecond transient absorption spectra, we can estimate the average electron population in our QDs27, which is less than 0.35 per dot under 10 V voltage (Supplementary Fig. 8). This is far less than the number of carriers required to saturate the ground state, therefore, intense LH electroluminescence observed in our experiments does not originate from ground state filling. Besides, such low injection also excludes strong heat accumulation in QD-LEDs, making the increase in thermal distribution of LH much smaller than the increase in LH electroluminescence (Supplementary Figs. 9, 10). Thus, heat accumulation cannot be the main reason for the strongly enhanced LH electroluminescence.

a Voltage-dependent steady state Electrically excited transient absorption (EETA) spectra of CdxZn1-xSe-ZnS QD-LEDs, averaged within the 10th microsecond. HH: heavy hole; LH: light hole; Pe: higher-order electron state; 2Sh: higher-order hole state. b Amplitude of band-edge state bleaching versus voltage. The increase in amplitude indicates an increasing averaged electron population 〈Ne〉. c Absorbance spectra under different reversed biases. d Normalized absorbance of LH (green, squares) and HH (purple, circles), as a function of bias. e The x-axis cross-section of HH (purple) and LH (green) wavefunctions under an x-direction electric field (500 kV cm−1). The relative directions of the QD and the field are indicated by the schematic. The deformation of potential wells is represented in a dashed line. f Normalized wavefunction overlaps \(\frac{{\left\langle {\psi }_{{{{{\rm{e}}}}}}|{\psi }_{{{{{\rm{h}}}}}}\right\rangle }_{{{{{\bf{E}}}}}}}{{\left\langle {\psi }_{{{{{\rm{e}}}}}}|{\psi }_{{{{{\rm{h}}}}}}\right\rangle }_{0}}\) of LH-electron (green) and HH-electron (purple) as a function of the electric field.
Electrical-field-induced decrease in oscillator strength
We then further investigate whether the oscillator strength of different exciton states changes under different electric fields. In our QDs, the size is large (∼16.8 nm), and quantum confinement is weak, as such, electron and hole wavefunctions can be separated easier than those in QDs with stronger quantum confinement. We measured steady-state absorption spectra of the QD-LEDs under reversed biases (Fig. 4c) and found that the oscillator strength of HH is more prone to reduction than that of LH as a result of the Stark effect (Fig. 4d). We conclude here that the relative decrease in oscillator strength of the HH exciton is the main reason for the strong electroluminescence of LH (Fig. 3b).
We seek to understand this phenomenon by calculating the wavefunctions of a three-dimensional finite sphere well under electric fields28 (Fig. 4e), with effective mass as a variable (Supplementary Table 1, see Methods for details). The potential well deforms and the electron and hole wavefunctions spatially separate under an electric field, resulting in less wavefunction overlap (Fig. 4f), leading to a lower probability of the electron–hole recombination (i.e., a decrease in the exciton oscillator strength). We find that the wavefunction of HH will be more localized, while the wavefunction of LH tends to be more spread. Under an electric field, the wavefunction is more prone to be affected by an electrical field, i.e., easier to shift to one side of the well, resulting in more reduction of normalized wavefunction overlaps \(\frac{{\left\langle {\psi }_{{{{{\rm{e}}}}}}|{\psi }_{{{{{\rm{h}}}}}}\right\rangle }_{{{{{\bf{E}}}}}}}{{\left\langle {\psi }_{{{{{\rm{e}}}}}}|{\psi }_{{{{{\rm{h}}}}}}\right\rangle }_{0}}\) (∼65.9% under 100 kV cm−1) in the HH exciton oscillator strength than that of LH (∼37.7% under 100 kV cm−1). The calculation here is consistent with our experiment observation (Fig. 4c).
We also fabricated QD-LED using biaxially strained CdSe-CdS core-shell QDs29,30, which also features weak electron confinement and well-separated LH and HH levels (Fig. 5a). These devices also exhibit strong LH electroluminescence, therefore, the hot electroluminescence mechanism we discussed is not limited to the specific chemical composition (Fig. 5b).

a Absorbance (purple) and photoluminescence spectra (green) of biaxially strained QDs. A ∼ 48 meV splitting between LH and HH is highlighted (grey, dash). b Electroluminescence spectra of CdSe-CdS QD-LEDs under different voltages. The dashed curves represent the Gaussian fitting of photoluminescence peaks from HH excitons (purple) and LH excitons (green). Inset: the proportion of high-energy exciton emission versus voltage.
In summary, we report strong high-energy exciton electroluminescence from weakly quantum-confined polytypic QDs with the well split LH and HH levels. We perform optically excited transient absorption measurement and find the relaxation of LH to HH is completed within ∼72 fs, excluding the phonon bottleneck as a possible origin. We also conduct electrically excited transient absorption spectroscopy measurements and observe that the maximum charge population is only 0.35 per dot, far less than the amount required to completely fill the ground state, so state filling is also excluded as a possible reason. We measure and calculate the oscillator strength of LH and HH under bias, and found that the oscillator strength of HHs reduces much more than that of LHs under electric fields, leading to enhanced electroluminescence of LHs. We observe this phenomenon in both CdxZn1-xSe-ZnS and CdSe-CdS core–shell quantum dots, both of which exhibit large LH-HH splittings, indicating the finding here is not limited to the specific material composition. This work opens a gate for the development of high-performance white light sources and multi-colour light sources.
Methods
Materials
Cadmium oxide (CdO, 99.99%), zinc oxide (ZnO, 99.9% powder), 1-octadecene (ODE, 90%), oleic acid (OA, 90%), oleylamine (OAm, > 98% primary amine), selenium (Se, 99.99%, powder), 1-octanethiol (OT, 98%), zinc acetate dehydrate (98%) as well as various solvents were purchased from Aldrich and Aladdin.
Preparation of the precursors
Zinc oleate (Zn(OA)2): A 250 mL three-neck flask was used to mix ZnO (60 mmol), OA (60 mL), and ODE (60 mL). The mixture was then degassed at 150 °C for 15 min. Finally, the solution was heated to 310 °C under nitrogen with vigorous stirring to get a colourless clear solution. Cadmium oleate (Cd(OA)2): A 100 mL three-neck flask was used to mix CdO (10 mmol), OA (10 mL), and ODE (10 mL). The mixture was then cooled down to 150 °C to degass for 15 min. Finally, the solution was heated to 240 °C under nitrogen to obtain a colourless clear solution. Se precursor (Se-TOP): Se (10 mmol) was shot in TOP (20 mL) in a glove box under moderate heat overnight until complete dissolution. The S precursor: A 100 mL three-neck flask was used to load a desired amount of Zn(OA)2 and 1-Hexanethiol (1.4 equivalent amount refers to Zn(OA)2). The mixture was then degassed at 120 °C for 15 min under nitrogen with vigorous. To prevent solidification, Cd(OA)2 and Zn(OA)2 were kept at 70 °C and 110 °C respectively.
Synthesis of CdZnSe /ZnS quantum dots
For the preparation of CdZnSe/ZnS core/shell QDs, a 100 mL flask was used for mixing CdO (2.4 mmol), Zn(OAC)2 (4 mmol), and OA (9 mL). Then the mixture was heated to 150 °C for 30 min in flowing high-purity argon. Then, 20 mL of ODE was added to the flask, and the temperature was further heated to 305 °C. 8 mL of Se precursor was injected into the reaction flack at this temperature. The reaction temperature was kept at 305 °C for 30 min for the growth of the cores. To the further growth of gradient CdZnSe cores, Zn(OA)2 (4 mL) and Cd(OA)2 (2 mL) were injected dropwise into the reaction solution, and Se precursor (6 mL) was added with a syringe pump (at a rate of 5 mL h−1) under a temperature of 305 °C. For consecutively overcoating the ZnS shells, Zn(OA)2 (5 mL) and octanethiol (2.8 mmol, 1.4 equivalent amount refers to Zn(OA)2) were injected dropwise into the reaction solution with a syringe pump (at a rate of 5 mL h−1). The temperature was cooled down to room temperature once the reaction was completed.
Synthesis of biaxially strained CdSe/CdS quantum dots
For applying biaxially strain on CdSe cores, a 100 mL flask was used for mixing a hexane solution containing 200 nmol CdSe QDs with ODE (6 mL) and OAm (6 mL). The mixture was heated to 110 °C in a vacuum to evaporate the hexane, oxygen, and water. Then the reaction solution was saturated with nitrogen, and heated to 300 °C for 0.5 h. A desired amount of Cd(OA)2 (diluted in 6 mL ODE) and trioctylphosphine sulfide (diluted in 6 mL ODE) solutions were injected dropwise into the as-prepared CdSe solution with a syringe pump (at a rate of 1.5 mL h−1). Finally, the reaction mixture was naturally cooled to 50 °C, precipitated after 3 min centrifugation at 4323 g and then redispersed in hexane.
Fabrication of quantum dot light-emitting diodes
For the fabrication of QD-LEDs, the patterned ITO-glass substrates were ultrasonically cleaned with detergent, deionized water, acetone, and isopropanol in sequence for 15 min. Then the substrates were dried and treated with ultraviolet ozone for 15 min. PEDOT: PSS was spin-coated on the ITO-glass substrates (4000 rpm for 40 s) and annealed at 150 °C for 15 min. Then, these substrates were transferred into a nitrogen-filled glovebox to further deposit the subsequent layers. TFB (8 mg mL−1 in chlorobenzene, 3000 rpm for 30 s), QDs (15 mg mL−1 in octane, 2000 rpm for 40 s), and ZnO (25 mg mL−1 in ethanol, 2000 rpm for 30 s) were sequentially spin-coated on the substrates under nitrogen. After spin-coating TFB and ZnO, they were annealed at 110 °C and 145 °C respectively for 30 min. Finally, a high vacuum chamber (∼5 × 10−6 mbar) was used to thermally deposit Al cathodes (100 nm).
Device characterization
A semiconductor parameter analyzer with a calibrated Newport silicon diode under ambient conditions was used to analyze the J–V characteristics of the QD-LEDs. The electroluminance was calibrated with a Photo Research spectroradiometer (PR735). An Ocean Optics spectrometer (USB2000) was used to obtain the electroluminescence and time resolution electroluminescence spectra and a source meter was used for the calculations of the EQE.
Photoluminescence measurement
A home-built confocal microscope was used to obtain the photoluminescence (PL) spectra of the film. It was excited by a high-frequency 405 nm pulse laser (Advance Laser System, PIL040X) at a 10 MHz repetition rate. The spectra were collected and analyzed by an Ocean Optics spectrometer (FLMS12313).
Photoluminescence measurement under red-edge excitation
The solution of CdZnSe/ZnS core/shell QDs was excited by a wavelength-tunable pulse laser at a 10 MHz repetition rate. The energy of photons under red-edge excitation is controlled at ∼1.85 eV (670 nm), which is on the red edge of the main peak in PL spectra (∼1.91 eV) but cannot affect the higher order shoulder (∼1.96 eV). Since the required power of excitation is high under red-edge excitation, a spectrum of the blank solvent under red-edge excitation is also collected to filter the signal from the excitation laser. After subtracting the spectrum of the QD solution and blank solvent, the PL spectrum of QDs under red-edge excitation is extracted and compared with the PL spectrum under higher state excitation (∼2.27 eV, 545 nm). Most of the excitation laser can be filtered, but an additional peak can be observed in the red-edge excited PL of QDs because of the disturbances of the incompletely filtered excitation laser.
Femtosecond transient absorption measurement
Transient absorption (TA) spectroscopy measurements are performed on a Time-Tech Spectra (TA100) transient absorption spectrometer. The 1030 nm femtosecond pulse (10 kHz, 190 fs) generated by a Yb: KGW laser (PHAROS, Light Conversion) is split proportionally into a probe beam (10%) and a pump beam (90%). After passing through a delay stage, the probe beam is focused on a YAG crystal to produce a white light continuum in the wavelength range of 500−1000 nm. Then the optical parametric amplifier (ORPHEUS-HE, Light Conversion) converts the pump beam into a laser pulse with a continuously tunable wavelength. The pump beam is incident on the sample at a small angle with the probe beam after passing through a chopper. The transmitted probe is collected and sent to the charge-coupled device and monochromator. The collected signal is the difference in probe intensity without and with the pump beam. According to the absorption spectrum, the probe intensity absorbed by the sample is obtained. Combining with the TA signal, the value of optical gain can be calculated from the original probe intensity and the probe intensity when the pumping beam is present.
Electrical excitation transient absorption measurement
An arbitrary waveform generator was used to apply the electrical pulse (1 kHz, 10 μs) with different voltages on a QD-LED sample. Meanwhile, a supercontinuum white laser (Leukos, Disco) generated a white laser pulse, which was then split proportionally into a probe beam (90%) and a reference beam (10%) by a beam splitter. An electronic time delay module controlled the delay between the probe beam sent to the excitation area and the electrical pulse applied to QD-LEDs. Then the reflected probe beam was collected by a monochromator and charge-coupled device. The reference beam was collected simultaneously to eliminate the effect of optical jitter. The collected signal was the difference in probe intensity without and with the pump electrical pulse.
Absorption measurement under reversed bias
The white light was generated by a halogen lamp (Ideaoptics, HL2000) in the wavelength range of 360–2500 nm. Spectra reflected by the device were collected and the absorbance was calculated by an Ocean Optics spectrometer (FLMS12313). The reversed bias was applied to the device by a programmable linear DC power supply (Rigol, DP832).
Calculation of the wavefunctions
The calculation of the wavefunctions was carried out using COMSOL Multiphysics. The models used were sphere-shaped CdxZn1-xSe-ZnS core-shell QDs, and the diameters were based on our measurements. Herein the overlap between electron and hole wavefunction was represented by the product of the calculated wavefunctions under an electric field \({\left\langle {\psi }_{{{{{\rm{e}}}}}}|{\psi }_{{{{{\rm{h}}}}}}\right\rangle }_{{{{{\bf{E}}}}}}\), and normalized to the overlap without an external electric field as \(\frac{{\left\langle {\psi }_{{{{{\rm{e}}}}}}|{\psi }_{{{{{\rm{h}}}}}}\right\rangle }_{{{{{\bf{E}}}}}}}{{\left\langle {\psi }_{{{{{\rm{e}}}}}}|{\psi }_{{{{{\rm{h}}}}}}\right\rangle }_{0}}\).
Data availability
The datasets that support the findings of this study are available at Figshare (https://doi.org/10.6084/m9.figshare.26133955)31. More detailed data are available from the corresponding authors.
References
Kasha, M. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 9, 14–19 (1950).
Englman, R. & Jortner, J. The energy gap law for radiationless transitions in large molecules. Mol. Phys. 18, 145–164 (1970).
Caspar, J. V. & Meyer, T. J. Application of the energy gap law to nonradiative, excited-state decay. J. Chem. Phys. 87, 952–957 (1983).
Kambhampati, P. Hot exciton relaxation dynamics in semiconductor quantum dots: radiationless transitions on the nanoscale. J. Phys. Chem. C. 115, 22089–22109 (2011).
Beer, M. & Longuethiggins, H. C. Anomalous light emission of azulene. J. Chem. Phys. 23, 1390–1391 (1955).
Levine, S. Z., Knight, A. R. & Steer, R. P. Fluorescence from second excited singlet-state of thiophosgene vapor. Chem. Phys. Lett. 29, 73–76 (1974).
Bawendi, M. G., Steigerwald, M. L. & Brus, L. E. The quantum-Mechanics of Larger Semiconductor Clusters (Quantum Dots). Annu. Rev. Phys. Chem. 41, 477–496 (1990).
Efros, A. L. et al. Band-edge exciton in quantum dots of semiconductors with a degenerate valence band: Dark and bright exciton states. Phys. Rev. B 54, 4843–4856 (1996).
Korkusinski, M., Voznyy, O. & Hawrylak, P. Fine structure and size dependence of exciton and biexciton optical spectra in CdSe nanocrystals. Phys. Rev. B 82, 245304 (2010).
Brus, L. E. Electron-electron and electron-hole interactions in small semiconductor crystallites - the size dependence of the lowest excited electronic state. J. Chem. Phys. 80, 4403–4409 (1984).
Kambhampati, P. Nanoparticles, Nanocrystals, and Quantum Dots: What are the Implications of Size in Colloidal Nanoscale Materials? J. Phys. Chem. Lett. 12, 4769–4779 (2021).
Khan, A. H. et al. CdSe/CdS/CdTe Core/Barrier/Crown nanoplatelets: synthesis, optoelectronic properties, and multiphoton fluorescence upconversion. Acs. Nano. 14, 4206–4215 (2020).
Jung, H. et al. Two-band optical gain and ultrabright electroluminescence from colloidal quantum dots at 1000 A cm−2. Nat. Commun. 13, 3734 (2022).
Lim, J., Park, Y. S. & Klimov, V. I. Optical gain in colloidal quantum dots achieved with direct-current electrical pumping. Nat. Mater. 17, 42 (2018).
Cheng, Y. et al. Continuously graded quantum dots: synthesis, applications in quantum dot light-emitting diodes, and perspectives. J. Phys. Chem. Lett. 12, 5967–5978 (2021).
Empedocles, S. A. & Bawendi, M. G. Quantum-confined stark effect in single CdSe nanocrystallite quantum dots. Science 278, 2114–2117 (1997).
Jacobsson, T. J. & Edvinsson, T. Quantum confined stark effects in ZnO quantum dots investigated with photoelectrochemical methods. J. Phys. Chem. C 118, 12061–12072 (2014).
Zhang, L. et al. Quantum-confined stark effect in the ensemble of phase-pure CdSe/CdS quantum dots. Nanoscale 11, 12619–12625 (2019).
Xu, H. Y. et al. Dipole–dipole-interaction-assisted self-assembly of quantum dots for highly efficient light-emitting diodes. Nat. Photon. 18, 186–191 (2024).
Klimov, V. I. Optical nonlinearities and ultrafast carrier dynamics in semiconductor nanocrystals. J. Phys. Chem. B 104, 6112–6123 (2000).
Sewall, S. L., Cooney, R. R., Dias, E. A., Tyagi, P. & Kambhampati, P. State-resolved observation in real time of the structural dynamics of multiexcitons in semiconductor nanocrystals. Phys. Rev. B 84, 235304 (2011).
Wang, J. H. et al. Spin blockade and phonon bottleneck for hot electron relaxation observed in n-doped colloidal quantum dots. Nat. Commun. 12, 550 (2021).
Sieh, C. et al. Coulomb memory signatures in the excitonic optical Stark effect. Phys. Rev. Lett. 82, 3112–3115 (1999).
Xiang, D. M. et al. Electron and hole spin relaxation in CdSe colloidal nanoplatelets. J. Phys. Chem. Lett. 12, 86–93 (2021).
Li, B. et al. Transient absorption spectrometer using excitation by pulse current. PCT Patent WO/2022/121082 (2022.06.16).
Li, B. et al. Transient absorption spectrometer using excitation by pulse current. CN Patent CN112683797B (2021.12.14).
Gao, Y. et al. Minimizing heat generation in quantum dot light-emitting diodes by increasing quasi-Fermi-level splitting. Nat. Nanotechnol. 18, 1168–1174 (2023).
Niculescu, E. C., Cristea, M. & Spandonide, A. Exciton states in CdSe/ZnS core-shell quantum dots under applied electric fields. Superlattice Microst 63, 1–9 (2013).
Fan, F. J. et al. Continuous-wave lasing in colloidal quantum dot solids enabled by facet-selective epitaxy. Nature 544, 75–79 (2017).
Song, Y. et al. Enhanced emission directivity from asymmetrically strained colloidal quantum dots. Sci. Adv. 8, 8 (2022).
Wang, X. Z. Source data for “Strong high-energy exciton electroluminescence from the light holes of polytypic quantum dots”. Figshare https://doi.org/10.6084/m9.figshare.26133955 (2024).
Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (Grants No. U22A2072 [H.B.S], 62204078 [F.J.F.]), Innovation Program for Quantum Science and Technology (Grants No. 2021ZD0301603 [F.J.F.]), Fundamental Research Funds for the Central Universities (Grants No. WK3540000009 [F.J.F.], WK3540000014 [F.J.F.]), the National Key R&D Program of China (Grant No. 2023YFE0205000 [H.B.S]), and Zhongyuan High Level Talents Special Support Plan (Grant No. 244200510009 [H.B.S]).
Author information
Authors and Affiliations
Contributions
F.J.F., H.B.S and B.L. conceived the concept and designed the experiments. X.Z.W., Y.G., and X.N.L contributed equally to this work. X.Z.W. and B.L. performed photoluminescence spectroscopy, transient absorption spectroscopy, and EETA measurement. Y.G., X.N.L., R.X.L., and J.J.S synthesized the materials, fabricated the devices, and collected the performance data of the QD-LEDs. H.Y.X. contributed to the polytypic quantum dots modeling. X.Z.W., B.L., F.J.F., and H.B.S. wrote the manuscript. All authors contributed to the scientific discussion about this work.
Corresponding authors
Ethics declarations
Competing interests
Patents regarding the design and principle of EETA spectroscopy have been disclosed (CN Patent: CN112683797B [2021.12.14]; PCT Patent: WO/2022/121082 [2022.06.16]). B.L. and F.J.F. are the applicants and inventors of these patents. Measurement of the average electron population is performed based on this spectroscopy. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Gao, Y., Liu, X. et al. Strong high-energy exciton electroluminescence from the light holes of polytypic quantum dots. Nat Commun 15, 6334 (2024). https://doi.org/10.1038/s41467-024-50432-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-50432-8
