
Abstract
Recent advances in spatial omics have expanded the spectrum of profiled molecular categories beyond transcriptomics. However, many of these technologies are constrained by limited spatial resolution, hindering our ability to deeply characterize intricate tissue architectures. Existing computational methods primarily focus on the resolution enhancement of transcriptomics data, lacking the adaptability to address the emerging spatial omics technologies that profile various omics types. Here, we introduce soScope, a unified generative framework designed to enhance data quality and spatial resolution for molecular profiles obtained from diverse spatial technologies. soScope aggregates multimodal tissue information from omics, spatial relations and images, and jointly infers omics profiles at enhanced resolutions with omics-specific modeling through distribution priors. With comprehensive evaluations on diverse spatial omics platforms, including Visium, Xenium, spatial-CUT&Tag, and slide-DNA/RNA-seq, soScope improves performances in identifying biologically meaningful intestine and kidney architectures, revealing embryonic heart structure that cannot be resolved at the original resolution and correcting sample and technical biases arising from sequencing and sample processing. Furthermore, soScope extends to spatial multiomics technology spatial-CITE-seq and spatial ATAC-RNA-seq, leveraging cross-omics reference for simultaneous multiomics enhancement. soScope provides a versatile tool to improve the utilization of continually expanding spatial omics technologies and resources.
Similar content being viewed by others

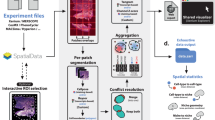

Introduction
Tissues are built upon cells with distinct molecular states and spatial organizations. Extensive dissection of tissue architecture requires the deep characterization of diverse molecular profiles while preserving their spatial contexts. In recent years, significant advances in spatial omics technologies have enabled the spatial profiling of various molecular categories, including transcript (e.g., ST1, Visium2,3,4, Xenium5, Stereo-seq6, seqFISH7,8, slide-seq9,10, STARmap11, and MERFISH12,13), protein (e.g., DBiT-seq14 and spatial-CITE-seq15), epigenetic marker (e.g., spatial ATAC-seq16, spatial ATAC-RNA-seq17, and spatial-CUT&Tag18), and genomic variation (e.g., slide-DNA-seq19 and DNA-seqFISH20). By unveiling the spatial signatures of diverse molecular profiles, spatial omics offers valuable insights across various biological areas, such as cancer21,22, Alzheimer’s disease23,24, and embryo development3,25.
Despite the early success of existing spatial omics approaches, two challenges remain limiting their full potential. Firstly, tissues are often in frozen or formalin-fixed and paraffin-embedded (FFPE) states before sequencing26. The freezing process and formalin-induced deamination can potentially impact the molecule states27,28, leading to reduced sequencing accuracy. Secondly, the majority of spatial technologies use spatial barcodes to mark the positions of targeted molecules and profile omics at the tissue spot resolution. As each spot typically contains tens of cells1,3,29, the multi-cell averaged sequencing readouts reduce the heterogeneity that can be resolved, leading to limited spatial resolution in tissue structure.
Computational techniques hold the promise to reduce data variations and enhance spatial resolutions in spatial omics data, facilitating a more precise characterization of tissue architectures. However, current methods predominantly enhance omics profiles from a single tissue modality, such as spatial positions (e.g., BayesSpace30) or image (e.g., XFuse31 and iStar32), which overlooks the rich, multimodal tissue information available from most spatial platforms. Furthermore, these approaches are primarily tailored for transcriptomics data, where their statistical assumptions are not extendable to other spatial omics categories.
Here, we introduce spatial omics scope (soScope), a fully generative framework that models the generation process of spot-level profiles from diverse spatial omics technologies and aims to enhance their spatial resolution and data quality (Fig. 1a). To achieve this, soScope views each spot as the aggregation of “subspots” at an enhanced spatial resolution, with their omics profiles associated with spatial positions and morphological patterns (Fig. 1b; “Methods” section). Then, soScope integrates spot omics profiles, spatial relations, and high-resolution morphological images using a multimodal deep learning framework and jointly infers omics profiles at the subspot resolution. By selecting omics-specific distributions, soScope allows for accurate modeling and variation reduction of different spatial omics data (Fig. 1c).

a The soScope framework. soScope integrates molecular profiles (\({{{\boldsymbol{X}}}}\)), spatial neighboring relations (\({{{\boldsymbol{A}}}}\)), and morphological image features (\({{{\boldsymbol{Y}}}}\)) from the same tissue using a unified generative model to enhance spatial resolution and refine data quality for diverse spatial omics profiles. b The probabilistic graphical model representation of soScope. Each of the \(N\) spots in the spatial data is considered an aggregation of \(K\) subspots at a higher spatial resolution. The subspot omics profile \(\widehat{{{{\boldsymbol{X}}}}}\) depends on both the latent states \({{{\boldsymbol{Z}}}}\) at the spot level and image features \({{{\boldsymbol{Y}}}}\) at the subspot level. The observed profile \({{{\boldsymbol{X}}}}\) is obtained by summing profiles from its \(K\) subspots. c The neural network structure in soScope. Original spatial profiles (\({{{\boldsymbol{X}}}}\)) and their spatial relations (\({{{\boldsymbol{A}}}}\)) are integrated using a graph encoder to infer the latent states of spots (\({{{\boldsymbol{Z}}}}\)). These latent representations are then combined with subspot-level image features (\({{{\boldsymbol{Y}}}}\)) to jointly learn likelihood parameters for modeling omics profiles at an enhanced resolution. The choice of probabilistic distribution is tailored to the specific omics type to reflect the data characteristics. d An overview of soScope experiments in this study. soScope is evaluated across multiple spatial omics platforms and diverse biological systems. Furthermore, soScope is extended to emerging spatial multiomics technology with simultaneous profiling of multiple molecule types.
soScope offers a unified tool that incorporates multimodal tissue profiles to enhance omics profiles with different molecular categories. With comprehensive benchmarks on diverse spatial omics types (transcripts, epigenetics, DNA, and proteins) from multiple biological systems (Fig. 1d), we demonstrate that soScope can effectively enhance spatial resolutions, reduce unwanted variations, and enable the characterization of complex tissue structure that cannot be detected at the original resolution. Furthermore, soScope extends to the emerging spatial multiomics technology, integrates multimodal, multiomics tissue profiles, and simultaneously enhances their resolutions (Fig. 1d).
Results
soScope architecture
soScope is a unified generative model that integrates molecular profiles (\({{{\boldsymbol{X}}}}\)), spatial neighboring relations (\({{{\boldsymbol{A}}}}\)), and morphological image features (\({{{\boldsymbol{Y}}}}\)) from diverse spatial omics platforms and aims to divide each spot to multiple subspots (\(\widehat{{{{\boldsymbol{X}}}}}\)) (Fig. 1a, b). The model includes three steps (Fig. 1c and Supplementary Fig. 1a; “Methods” section): (1) spot-level representation learning: molecular profiles and their spatial coordinates at the original resolution are embedded into latent representations (\({{{\boldsymbol{Z}}}}\)) with a graph encoder \(q{{{\boldsymbol{(}}}}{{{\boldsymbol{Z|X}}}}{{{\boldsymbol{,}}}}{{{\boldsymbol{A}}}}{{{\boldsymbol{)}}}}\); (2) subspot-level image feature learning: image patches corresponding to subspot regions are segmented from high-resolution tissue images and transformed into subspots’ morphological features; (3) subspot-level omics profile inference: spot representations and subspot image features are combined in an enhancement decoder \(p{{{\boldsymbol{(}}}}\widehat{{{{\boldsymbol{X}}}}}{{{\boldsymbol{|Y}}}}{{{\boldsymbol{,}}}}{{{\boldsymbol{Z}}}}{{{\boldsymbol{)}}}}\) to jointly infer subspot-level profiles that are constrained by the omics-specific distribution and morphological similarities (“Methods” section).
The graph encoder provides high-quality embeddings for spot-level omics profiles with the preservation of spatial neighboring information. The image features are extracted from exact subspot regions and provide information at an enhanced resolution to guide the inference of corresponding omics profiles. The enhancement decoder enables the accurate modeling of diverse subspot omics data with a probabilistic distribution reflecting its statistic variations (“Methods” section; Supplementary Table 1). By optimizing its evidence lower bound (ELBO) (Methods), soScope efficiently generates omics profiles with reduced data noise and enhanced spatial resolutions for a finer characterization of tissue structures.
soScope outperforms enhancement approaches developed for spatial transcriptomics
Existing computational approaches for improving spatial omics resolution predominantly focus on transcriptomics. Therefore, we initially evaluated soScope on spatial transcriptomics (ST) datasets generated from diverse platforms. We compared it with three established methods: iStar32 and XFuse31, which utilize image information to enhance transcript profiles, and BayesSpace30, which employs spatial coordinates to infer high-resolution expression data (“Methods” section). Additionally, we included a variant of soScope that only uses image input, termed image soScope, as a reference (Supplementary Fig. 1b, c and Supplementary Note 5). As there was no high-resolution ground-truth data for evaluation, we proposed simulating “low-resolution” expression profiles by merging transcript data from neighboring spots (“Methods” section). Then, we assessed the performance of resolution enhancement approaches by comparing recovered expressions with original profiles (Fig. 2a; “Methods” section).

a Benchmark setup. Low-resolution spatial profiles are simulated by combining adjacent spot profiles or summing cell profiles within a squared region for spot- and cell-level analysis. Resolution enhancement approaches are then applied to recover the original omics profile. b–e Resolution enhancement analysis for a human intestine tissue profiled by Visuim. b H&E image. c Spatial visualization of example marker gene expressions from the original (first column) and recovered (the rest) profiles in one selected tissue region (results on additional marker genes are in Supplementary Fig. 2b). Pearson correlations are calculated between the recovered and the original expressions. d Performance evaluation using Pearson correlation and mean square error (MSE). Boxplot displays the median (center line), interquartile range (box), and data range (whiskers) based on 9 marker genes. e Evaluation of marker gene enrichments in corresponding tissue regions using Kolmogorov-Smirnov distance (KS distance; “Methods” section). The red dashed line indicates the median KS distance at the original resolution. Percentage indicates the improvement ratio between soScope and the second-best approach. Boxplot is defined in the same format as panel d. f–i Resolution enhancement analysis for a mouse head region profiled by Xenium. f Spatial visualization of an example marker gene expression from the original (first column) and recovered (the rest) profiles. Pearson correlations are calculated between recovered and original expressions. Additional examples are in Supplementary Fig. 3b. g MSE-based performance evaluation. Boxplot is defined in the same format as panel d. h MSE and biological variability analysis. The biological variability quantifies the intrinsic fluctuation in gene expression within the original dataset (“Methods” section). i Spatial visualization of gene correlation in three gene variability groups (“Methods” section). j–l Enhancement analysis at single-cell resolution for a mouse kidney region profiled by Xenium. j H&E image. k Spatial visualization of an example marker gene expression from the original (first), merged (second), and recovered (the rest) profiles. Additional examples are in Supplementary Fig. 3e. l Pearson correlation and MSE between recovered and original expressions (n = 10). Boxplot is the same format as panel d. Source data are provided as a Source Data file.
Firstly, we considered a human intestine dataset generated using Visium platform3, containing 2,649 sequencing spots (“Methods” section) with a high-resolution Hematoxylin and Eosin (H&E) staining image. The layered structure in the intestine sample made it a good example to demonstrate the benefits of resolution enhancement (Fig. 2b, H&E image zoom-in regions). Following our benchmark design, we merged 2,649 spots into 369 “low-resolution” spots with aggregated gene expressions from corresponding neighboring spots. All approaches used the same processed omics data as the input (“Methods” section). For soScope, image features were obtained from a pretrained Inception-v3 model33 on H&E image patches corresponding to subspot regions (“Methods” section), and a negative binomial (NB) distribution was used in the generative modeling of enhanced gene expressions (“Methods” section). We examined three functional regions in the intestine tissue: epithelium, muscularis, and immune regions (Supplementary Fig. 2a), and selected 9 region markers reported in a previous publication34 for resolution enhancement evaluations. Compared with the original expressions, regional “merged” profiles greatly reduced the layered spatial patterns of gene markers (Fig. 2c, second column). From the enhancement analysis (Fig. 2c and Supplementary Fig. 2b), iStar, which utilized image information alone, performed well in the immune region; however, it failed to recover the fine structures of the epithelium and muscularis regions. Another image-based approach, XFuse, performed well in the muscularis region; however, it was misled by strong morphological similarities across regions and did not faithfully retain the tissue structures in the epithelium and immune regions. BayesSpace, which relied on spatial information alone, did not substantially refine the region boundaries in the enhanced profiles. Image soScope failed to recover most patterns due to the lack of information from transcriptomics. As a comparison, soScope achieved reasonable performance in all three regions. From quantitative assessments on enhanced expressions using Pearson correlation and mean square error (MSE) (“Methods” section), soScope consistently demonstrated the highest consistency and the lowest reconstruction error (Fig. 2d). Next, we examined the expression distribution difference for each marker gene within and outside its corresponding tissue region using the Kolmogorov–Smirnov (KS) distance (“Methods” section). soScope and iStar yielded the most pronounced separations for most tested genes (Fig. 2e) and achieved a similar distinguishability as in the original profiles (red dashed line).
Secondly, we evaluated a mouse head dataset obtained from the Xenium platform35 (“Methods” section, Supplementary Fig. 3a). All 379 measured genes were included in the analysis, and we merged 2,619 spots into 291 “low-resolution” spots from original cell-level transcripts for the resolution enhancement analysis (“Methods” section). Corresponding H&E image regions were transformed into deep features, as mentioned earlier, to help the enhancement. From the enhancement results (Fig. 2f, g and Supplementary Fig. 3b), we observed that both iStar and soScope performed well in retaining high-resolution expression profiles. However, by examining genes based on their variability (“Methods” section; Fig. 2h) or abundance (“Methods” section; Supplementary Fig. 3c), we found that soScope exhibited more stable performances across different variations. Furthermore, we investigated whether resolution enhancement would affect gene correlations (“Methods” section). We discovered that most approaches could preserve the correlation patterns across genes after the resolution enhancement, except for XFuse (Fig. 2i), regardless of biological variability differences. Notably, soScope demonstrated the highest performance by MSE or Pearson correlation (Supplementary Fig. 3d).
Thirdly, we assessed the ability of soScope in the enhancement of a single-cell resolution. We used a mouse kidney dataset profiled by Xenium35 (Fig. 2j), which covered 1538 cells. We divided the tissue into small square regions and aggregated the cells’ gene expressions within each region, aiming to recover their cellular expressions (“Methods” section). In the analysis, we used the top 5 abundant and the top 5 marker genes identified from the original study35 (“Methods” section) and deep features of each single cell. As only iStar was previously demonstrated in this task, the comparison was limited between iStar and soScope. From the result, we observed that iStar tended to overestimate the expression levels in regions of high cell density and high expression (Fig. 2k and Supplementary Fig. 3e). Conversely, soScope demonstrated a more consistent expression pattern with the ground truth (Fig. 2l).
Lastly, we investigated how the image input could affect the soScope’s performance by replacing the image with standard Gaussian noise (“Methods” section; Supplementary Note 6). We used a human ductal carcinoma dataset profiled by Xenium5 (Supplementary Fig. 4a) with 1521 spots and 313 genes. From the top 3 principal components (PCs) of deep image features (Supplementary Fig. 4b), tissue subregions could be well separated by the image feature graph (Supplementary Fig. 4c). Based on that, we selected the top 10 highly and the bottom 10 lowly correlated genes with image features (“Methods” section). The similarity graphs constructed using highly correlated genes clearly outlined the boundaries of the tumor regions, while lowly correlated genes failed (Supplementary Fig. 4d). Following the previous simulation setup (“Methods” section), the result of soScope without image input did not reach the accuracy of a standard soScope on highly correlated genes, while for lowly correlated genes, no significant difference was observed between the two models (Supplementary Figs. 4e, f). We further evaluated the impact of graph construction with different distances on soScope’s performance. We tested the model with short-distance (only neighboring spots), medium-distance (connectivity within an 8-spot radius), and long-distance (all spots) for immune cells (Supplementary Fig. 4g) and did not find significant changes in performance (Supplementary Fig. 4h, i).
These findings demonstrated soScope’s capability to enhance spatial resolutions for transcript data across various technological platforms, resolution requirements, and gene statistical characteristics, enabling a detailed characterization of tissue structures.
soScope reduces sequencing noise and uncovers detailed structure on a mouse embryo profiled by spatial-CUT&Tag
Recent advancements in spatial omics technology have expanded our ability to profile molecules in tissues beyond traditional transcriptomics16,36. soScope can be flexibly extended to work with diverse omics types by combining the generative process with omics-specific distributions (“Methods” section). To illustrate this versatility, we applied soScope to a spatial chromatin accessibility dataset generated by spatial-CUT&Tag18. The data were collected on an embryonic day 11 mouse embryo, comprising 1974 sequencing spots arranged in a 50×50 array and an H&E staining image. We selected the top 60 variable peaks from four major organ regions (liver, heart, forebrain, and spinal cord) for the resolution enhancement analysis (“Methods” section; Fig. 3a).

a H&E image of the mouse embryo and four major regions (defined in the original publication18) analyzed in this study. b Evaluation of resolution enhancement performance using Pearson correlation and MSE with simulated low-resolution peak data (“Methods” section). Percentage indicates the improvement/error reduction ratio between soScope and the second-best approach. Boxplot: same format as in Fig. 2d. c Left in each column: spatial visualization of peak counts from four organs using original profiles or enhanced profiles. Right in each column: characterization of high and low expression regions by clustering profiles in the left figure into two groups using \(k\)-means. Results of all comparing methods are provided in Supplementary Fig. 5a; additional peaks are shown in Supplementary Fig. 6. d Detailed investigation of the embryo heart region. Left: high-resolution H&E image reveals two subregions with distinct morphological patterns in the heart region. Right: comparison of marker gene activities from original and enhanced peak expressions. e Spatial visualization of enhanced peak activities from comparing approaches. Results of all comparing methods are provided in Supplementary Fig. 5d. f Validation of the heart structure using a Spatial Transcriptomics dataset from a human embryonic heart section. Left: H&E image with regional annotations. Right: spatial patterns of human orthologous genes. g Quantitative comparison between gene activities in the mouse (left) and gene expressions in the human (right) for heart subregions. Two-sided t-tests were used. n.s.: 0.05 ≤ p; *1E-2 ≤ p < 0.05; **1E-3 ≤ p < 1E-2; ***p < 1E-3 using a two-sided rank-sum test. For Fhl2 in trabecular ventricular myocardium vs. compact ventricular myocardium, Spatial linear: p = 0.020; Image MLP: p = 2.6 × 1E-16; Joint MLP: p = 1.4 × 1E-24; soScope: p = 7.6 × 1E-26; For FHL2, p = 1.1 × 1E-6. For Ldha in trabecular ventricular myocardium vs. compact ventricular myocardium, Spatial linear: p = 0.0012; Image MLP: p = 0.72; Joint MLP: p = 0.60; soScope: p = 1.2 × 1E-10. For LDHA, p = 6.5 × 1E-6. Boxplot: same format as in Fig. 2d. Results of all comparing methods are provided in Supplementary Fig. 5e. Source data are provided as a Source Data file.
As no specific enhancement approach existed for chromatin accessibility data, we adapted six general machine learning models for the enhancement comparison (“Methods” section): (1) spatial-based approaches: estimate resolution-enhanced profiles using spatially nearest neighbors (Spatial linear), or a Gaussian Process mapping (Spatial GP) from spatial coordinates; (2) image-based approaches: predict profiles from image features at subspot resolutions with a linear regressor (Image linear), a Gaussian Process model (Image GP) or a multi-layer perceptron model (Image MLP); and (3) multimodal joint approach: enhance omics from concatenated features of image and spatial positions with a multi-layer perceptron model (Joint MLP). All approaches used the same omics input. In the case where image information was required, the same deep image features were provided, as previously described (“Methods” section). For soScope, we followed the prior work37 and employed a Poisson distribution to model the peak count distribution.
We first evaluated the performance through a simulation study. In detail, we combined every 2 × 2 neighboring spots into a “low-resolution” spot to generate “low-resolution” peak data. Then, we aimed to recover the original profiles using resolution enhancement approaches mentioned above (BayesSpace and XFuse were excluded because their statistical assumptions were tailored to transcriptomics data and not applicable to this context). We quantitatively evaluated the performance by comparing enhanced profiles with original data using Pearson correlation and MSE. soScope achieved the highest correlations and lowest reconstruction errors among benchmarks in retaining the peak counts at the original resolution (Fig. 3b).
Next, we directly enhanced the tissue resolution from the original data by 4 folds (from 1,974 spots to 7,896 enhanced subspots). It is worth noting that the tissue was in a frozen state before sequencing, as mentioned in the original publication18, which can potentially affect chromosome structures and introduce noise to the generated data28. This effect was evident in the original data, where marker peaks associated with specific organs also sporadically appeared in other tissue regions (Fig. 3c, first row), especially when profiles were clustered into highly and lowly expressed groups (second column for each organ; “Methods” section). When applying resolution enhancement to the data, approaches that solely considered spatial positions (Spatial linear and Spatial GP) interpolated inaccurate peak counts based on original noisy data; meanwhile, methods based on predictions from image data (Image linear, Image GP, and Image MLP) were confused by the random noise and failed to capture the expression patterns in the desired regions, particularly in the heart and spinal cord (Fig. 3c and Supplementary Fig. 5a). The direct combination of image and position (Joint MLP) did provide a noticeable improvement. In contrast, soScope, which incorporated both spatial relationships between neighboring spots and image references, correctly identified the expressed regions while effectively suppressing out-of-region expressions (Fig. 3c and Supplementary Fig. 5a). When examining highly and lowly expressed regions (Fig. 3c), highly expressed spots from soScope exhibited a tight colocalization in space, evidenced by their smaller spatial distances in the tissue (Supplementary Fig. 5b).
The enhancement of tissue profiles enabled the study of tissue structure at a finer resolution. As a case study, we focused on the heart region for an in-depth examination. As evident from the zoomed-in H&E image (Fig. 3d, left, separated by a yellow dashed line; Supplementary Fig. 5c), the heart region can be further characterized into two subregions: a trabecular ventricular myocardium and a compact ventricular myocardium. However, when we visualized profiled peaks corresponding to two known marker genes in this region (Fhl2 for trabecular ventricular myocardium, Ldha for compact ventricular myocardium; for comparison, peak intensities were transformed into activity scores; “Methods” section), and their intensities did not form recognizable patterns due to the low resolution (Fig. 3d, right). After resolution enhancement, we visualized spatial peak activities predicted by comparing approaches (Fig. 3e and Supplementary Fig. 5d). As expected, spatial-based approaches did not help improve the structure characterization. Among other approaches, enhanced peak profiles from soScope better reflected the two-layered architecture of the heart based on visual inspection. Specifically, the Fhl2 gene displayed high activity in the trabecular ventricular myocardium, while Ldha showed high activity in the compact ventricular myocardium. To further validate our findings, we made use of a published heart ST dataset from a human embryo25 (Fig. 3f, left; “Methods” section) and cross-checked the spatial expression patterns of these two genes (Fig. 3f, right). Consistently, FHL2 exhibited high expressions in the trabecular ventricular myocardium layer, while LDHA was mainly enriched in the compact ventricular myocardium layer, which aligned with the gene activity scores obtained from the enhanced spatial-CUT&Tag data by soScope. Quantitative analysis further confirmed the similarity of distribution patterns between gene expressions and chromatin accessibility activities (Fig. 3g and Supplementary Fig. 5e). These findings highlight the generative modeling of soScope improved the data quality and provided an in-depth characterization of tissue structure.
soScope achieves consistent performance in tissue structure recovery from extremely low-resolution profiles simulated from slide-DNA/RNA-seq data
In our previous experiments, we showcased the performance of soScope on spatial omics datasets with fixed resolution enhancement ratios. However, it is essential to note that different spatial omics technologies have diverse spatial resolutions. For instance, in the initial ST technology, each sequencing spot may cover ~10–200 cells29, while in the more recent Visium platform, one spot includes 6–10 cells3. These variations in raw data require resolution enhancement approaches to work robustly with tissues profiled under diverse spatial resolutions. Here, we designed a multi-scale stress test to evaluate and calibrate the performance of different methods when enhancing the omics from different original resolutions.
To enable the multi-scale test, we used a mouse liver tumor dataset profiled using slide-seq, which employed barcoded beads to encode spatial information in the tissue and enabled the near-single-cell resolution spatial sequencing9,10. The high resolution in slide-seq raw data allowed us to generate low-resolution data in silico at different scales and test the ability to recover the original profiles from them. The liver tumor tissue19 included two metastatic clone regions and was profiled using both slide-DNA-seq and slide-RNA-seq, comprising 24,679 DNA beads and 31,286 RNA beads (Fig. 4a; “Methods” section). To conduct the test, we divided the entire tissue into small, squared regions at a 60 × 60 resolution and gradually merged neighboring regions at different sizes into one low-resolution spot. Using all tested approaches, we aimed to recover profiles at the original resolution from data at decreased resolutions (Fig. 4b; “Methods” section).

a H&E image of the liver metastasis tissue and its clonal annotations based on spatial DNA principal components (PCs) (“Methods” section). b Illustration of multi-scale resolution enhancement experiment. The entire tissue is divided into 60 × 60 square regions. Neighboring regions at increasing sizes are merged to generate spatial DNA profiles at progressively lower resolutions. Enhancement approaches are required to enhance low-resolution profiles back to their original resolution. c Comparison of enhancement approaches to enhance top 2 DNA PCs from low-resolution profiles at different scales. d Evaluation of recovery accuracy in multi-scale resolution enhancement experiment using Pearson correlation (left) and MSE (right). e Evaluation of tissue structure (\(k\)=3) and substructure (\(k\)=4) identification using DNA PCs enhanced from low-resolution profiles. Subpopulation identification is performed using \(k\)-means clustering. Left: ground truth tissue structures identified from original data. ARI adjusted Rand index. Source data are provided as a Source Data file.
We firstly focused on the enhancement test of the top two DNA principal components (PCs) in slide-DNA-seq, as used in the original work19 (Fig. 4a, right). We employed the Gaussian distribution to model the DNA PCs in the soScope model. In the original data, DNA PC1 exhibited high enrichments in the clone A region (Clone-A PC), while PC2 was enriched in the clone B region (Clone-B PC; Fig. 4a). The DNA regional patterns dramatically degraded with the decrease in resolutions (Fig. 4c, first row). After the resolution enhancement, we found methods only considering spatial relations among spots (Spatial linear and Spatial GP) could capture the correct regions corresponding to two cancer clones; however, these approaches tended to over-smooth the expression patterns, especially when recovering from data with an extremely low resolution (10 × 10). In contrast, image-based or joint predictive approaches (Image linear, Image GP, Image MLP, and Joint MLP) better preserved local expression patterns. However, as mentioned in the original publication19, there was a slight mismatch in spatial coordinates between the H&E image and DNA profiles due to tissue processing. These approaches were misled by the H&E data and reconstructed high-resolution DNA PCs that were more similar to the clone regions defined by the H&E image (Fig. 4a) than the original DNA data (Fig. 4c, left; compared with dashed region outlines). In contrast, soScope successfully preserved the local expression patterns while accurately reconstructing the original clone regions, even from data at extremely low resolutions. This highlights the importance of soScope’s ability to integrate both spatial and cross-modal information using a generative framework to achieve robust resolution enhancement.
From the quantitative evaluation (Fig. 4d), most approaches had expected decreased accuracies as the resolution decreased. soScope consistently showed a stable performance at different resolutions, with a relatively small decrease in accuracy. To further validate whether reconstructed DNA features could reflect tissue structures, we conducted the \(k\)-means clustering on the enhanced profiles. When we set the number of clusters (\(k\)) to 3, all methods successfully identified the three regions that had been previously defined (Fig. 4e, top and Supplementary Fig. 7, left). To explore a more detailed structure, we increased the number of clusters to 4, revealing a subregion within clone A characterized by extremely high expressions in PC1 (Fig. 4e, bottom). Among the various approaches, only the reconstructed DNA PCs by soScope and Spatial GP could consistently capture this substructure across all decreased resolutions (Fig. 4e, bottom and Supplementary Fig. 7, right).
In addition to the DNA analysis, we also conducted the stress test on transcript data obtained from the same tissue using slide-RNA-seq (Supplementary Figs. 8 and 9). Once again, soScope consistently maintained high accuracies in preserving the gene expression patterns for regional gene markers at different resolutions. This highlights the stable performance of soScope in accurately revealing the underlying spatial organization of the tissue across a variety of omics types and spatial resolutions.
Multiomics soScope corrects technical bias by leveraging cross-omics reference on spatial-CITE-seq and spatial ATAC-RNA-seq data
Recent advancements in spatial multiomics have expanded our capabilities for spatial tissue profiling beyond single omics type14,15,17,38. Pioneering technologies include spatial-CITE-seq15 (proteins and genes) and spatial ATAC-RNA-seq17 (chromatin accessibility and genes). The simultaneous collection of multiple molecular features offers the potential to overcome the limitation of single omics and reveal a more complete tissue structure. soScope can be extended to work with spatial multiomics, leverage the advantage from additional molecular types, and simultaneously enhance their spatial resolutions.
We enabled soScope to work with spatial multiomics (termed as multi-soScope) using the following modifications (Fig. 5a): (1) The spatial graph used in the graph encoder was constructed based on similarities from both transcript and protein, providing a combined measurement of spatial heterogeneities (“Methods” section). (2) Features from both transcript and protein modalities were input to the encoder together to generate joint latent representations, enabling the integration of information from both spatial omics profiles. (3) The decoder output distribution parameters of enhanced multiomics profiles simultaneously, allowing the simultaneous modeling of multiomics variations. Specifically, for spatial-CITE-seq applications, we utilized the negative binomial and Poisson distributions to model the enhanced profiles of transcripts and proteins, respectively. In the case of spatial ATAC-RNA-seq, we applied the negative binomial and Gaussian distributions to model the enhanced profiles of transcripts and normalized ATAC peaks (“Methods” section).

a An overview of multiomics soScope framework for a simultaneous enhancement of spatial multiomics profiles. b Spatial visualization of tissue subpopulations identified from original (left) or enhanced (right) transcript profiles. Highlighted regions: i: dermis, ii: pilosebaceous region. Louvain algorithm is used to identify subpopulations as the original publication suggested15. c Spatial visualization of tissue subpopulations identified from original (left) or enhanced (middle, right) protein profiles. d, e Spatial visualization of top 1 differentially expressed genes (d) or proteins (e) from two pilosebaceous subregions and one dermis region before and after enhancement. The Pearson correlations are calculated between expressions from transcript and protein that are identified from the same regions. Source data are provided as a Source Data file.
In the first example, we applied soScope to a spatial-CITE-seq dataset collected from a human skin tissue after the Coronavirus Disease 2019 (COVID-19) mRNA vaccine injection15. The dataset consisted of 1,618 sequencing spots, with each spot encompassing 15,486 genes and 283 proteins. We included all proteins, 114 top variable genes, and a bright-field image in the resolution enhancement analysis (“Methods” section). To provide a reference for tissue structures, we referred to the original study15 and manually annotated two primary regions in this tissue: a dermis layer and a pilosebaceous unit (Fig. 5b, dashed region).
From the original data, subpopulations identified from transcript clusters exhibited high noise levels (Fig. 5b, left). However, tissue regions identified from proteins exhibited stronger spatial colocalization patterns (Supplementary Fig. 10a) - samples belonging to the same subpopulations had smaller distances among each other. This discrepancy could be attributed to transcripts being more susceptible to degradation during sequencing than proteins39. After resolution enhancement using existing transcriptomics approaches, as expected, enhanced transcript profiles were impacted by the quality of original data: both iStar and XFuse exhibited oversimplification of tissue structure around the dermis region (Fig. 5b); BayesSpace discovered more tissue subpopulations; however, it was greatly impacted by the low transcript quality and failed to identify regions with clear spatial coherence (Fig. 5b). In multi-soScope, the resolution enhancement was achieved through joint representations learned from both omics. Therefore, protein data also contributed to the generation of high-resolution transcript profiles. Consequently, gene expressions enhanced by multi-soScope revealed tissue structures with improved spatial coherence in most regions (Fig. 5b), as evidenced by the reduced within-cluster distances compared to clusters identified from the original resolution or from enhanced profiles by comparing approaches (Supplementary Fig. 10b, left). The enhanced protein profiles by soScope with protein-alone input also provided refined tissue boundaries for the pilosebaceous unit and dermis layer separation (Fig. 5c; Supplementary Fig. 10b, right).
Next, we focus on the three major regions within the tissue: two pilosebaceous subregions and one dermis region. We examined the spatial expressions of the top 1 differentially expressed genes in each region. The first two selected genes exhibited varying levels of biases: the expression of FADS1 displayed strong random noise outside the pilosebaceous region, while the expression of RNA18S5 seemed to be distributed along the microfluid array grid used in spatial-CITE-seq (Fig. 5d, first column). When comparing outcomes across various methodologies, both iStar and XFuse performed relatively well in enhancing RNA18S5. However, XFuse failed to generate enhanced expressions for FADS1 and TETM132D, while iStar oversimplified the overexpressed region for the dermis (Fig. 5d). BayesSpace was unable to remove noise for FADS1 or reduce the expression pattern along the microfluid array grid for RNA18S5 (Fig. 5d, fourth column). In contrast, the multiomics soScope method effectively suppressed the expression levels outside the pilosebaceous unit (Fig. 5d, fifth column). Similarly, we explored the protein profiles in the selected regions. The enhanced protein expressions by both approaches accurately represented the expected tissue structures (Fig. 5e). We assessed the spatial consistency of expression patterns between transcripts and proteins identified within the same region (Fig. 5e). Compared to the results obtained at the original resolution, transcripts, and proteins associated with the same region exhibited increased consistency after the enhancement through both methods by Pearson correlation coefficients, with the multi-soScope achieving the best performance (Fig. 5e).
In the second example, we employed multi-soScope on a spatial ATAC-RNA-seq dataset obtained from a mouse embryo at embryonic day 13 (E13). This dataset included 1874 sequencing spots, with 24,017 peaks and 15,748 genes in each spot. For the resolution enhancement analysis, we selected 25 genes with the highest variability along with their corresponding peaks, and a bright-field image (“Methods” section; Supplementary Fig. 11a). After resolution enhancement (“Methods” section), we observed improved spatial enrichment of gene expressions. Additionally, sporadic expressions within the tissue were effectively suppressed, as highlighted by the white frame in Supplementary Fig. 11b. Furthermore, we compared the expression consistency between genes and their ATAC peaks and found that their correlations significantly improved after enhancement (Supplementary Fig. 11c).
In conclusion, using datasets from different molecular categories and generated by various platforms, we demonstrated that multi-soScope could effectively integrate multiomics profiles, compensate the omics with lower quality, and jointly enhance multiomics resolutions.
Discussion
The fast-developing spatial omics technologies have enabled the spatial profiling of different biological molecular features yet suffer from limited spatial resolution and data quality. We propose soScope, a unified generative framework that models the data generation process for diverse spatial omics profiles. By combining omics profiles, spatial neighboring relations, and morphological images from the same tissue, soScope infers omics profiles at an enhanced resolution with omics-specific modeling for data variations.
We extensively evaluate the effectiveness and generalizability of soScope on multiple molecule types profiled by diverse spatial technologies, including Visium, Xenium, spatial-CUT&Tag, slide-DNA-seq, slide-RNA-seq, spatial-CITE-seq, and spatial ATAC-RNA-seq. Across healthy and diseased tissues, we show that soScope refines the tissue domain identifications, improves the distinguishability of known markers, and corrects data and technical bias. Our approach enables the unveiling of a finer tissue structure up to 36-fold of the original resolution. It can be effectively adapted for spatial multiomics data for the simultaneous enhancement of multiomics profiles.
We note that there are several imaging-based spatial omics technologies, such as seqFISH7,8, STARmap11, and MERFISH12,13, which can directly achieve spatial profiling at the single-cell resolution at the cost of lower omics throughputs and smaller tissue regions. While soScope provides enhanced profiles for pre-designated subspot or cell positions, it may not reach subcellular resolutions. To further enhance resolution, soScope can be modified to include paired single-cell omics data from the same tissue to inform subspot inference at a higher resolution. Additionally, soScope incorporates H&E images as inputs, which can be easily annotated by human experts in some clinical studies. We can modify soScope to incorporate human labels and guide posterior inference in a semi-supervised manner for improved latent representation and profile learning. Lastly, for larger datasets with multiple sequential sections from the same organs, soScope can be trained on the part of the data and apply to the rest of the tissue slides to reduce computational costs.
With the continuing expansion of available spatial omics data resources and emerging of new spatial technologies, we believe that soScope holds the potential as a versatile tool to fully leverage spatial omics data and enhance our understanding of complex tissue structures and biological processes.
Methods
soScope framework
A generative model for resolution enhancement of spatial omics
soScope utilizes three modalities from the same tissue: spot-level omics profiles, spatial neighboring relations of spots, and subspot-level morphological features. We aim to divide the observed expression profile \({{{{\bf{x}}}}}^{(n)}\in {{\mathbb{R}}}^{G}\) at the \(n\)th spot \({s}^{(n)}\) into \(K\) subspots \({s}_{1:K}^{(n)}\):
where \({\hat{{{{\bf{x}}}}}}_{1:K}^{(n)}\in {{\mathbb{R}}}^{G}\) are the latent expression to be inferred at \(K\) subspots, \({{{{\mathbf{\epsilon }}}}}^{(n)}\) is drawn from a Gaussian noise with a mean of \({{{\mathbf{0}}}}\) and variance of \({\sigma }^{2}{{{\boldsymbol{I}}}}\), and \(N\) is the total number of spots in a spatial dataset. We formulate the task of spatial omics enhancement into a probabilistic generative model:
where \({{{{\bf{z}}}}}^{(n)}\in {{\mathbb{R}}}^{D}\) is the latent representation of the underlying spatial omics state at spot \({s}^{(n)}\), and \({{{{\bf{y}}}}}_{k}^{(n)}{{{\boldsymbol{\in }}}}{{\mathbb{R}}}^{L}\) is the feature extracted from the corresponding image region at subspot \({s}_{k}^{(n)}\). A neural network \(f(\cdot )\) combines subspot-level image feature \({{{{\bf{y}}}}}_{k}^{(n)}\) and the spot-level latent state \({{{{\bf{z}}}}}^{(n)}\) to learn the parameter \({{{{\mathbf{\omega }}}}}_{k}^{(n)}\) for the probability \(P(\cdot )\) that models expression profiles at the subspot level. The generative probability \(P(\cdot )\) is selected with respect to spatial omics data types to best describe its property. Finally, the profile \({{{{\bf{x}}}}}^{(n)}\) of a spot can be obtained by aggregating profiles at its corresponding subspots jittered by the zero-mean Gaussian noise with \({\sigma }^{2}\) as the variance.
For simplicity, we use matrix notations \({{{\boldsymbol{X}}}}{{{\boldsymbol{=}}}}{\left[{{{{\bf{x}}}}}^{(1)}\,{{\ldots }}\,{{{{\bf{x}}}}}^{(N)}\right]}^{T}{{\in }}{{\mathbb{R}}}^{N\times G}\), \({{{\boldsymbol{Y}}}}{{{\boldsymbol{=}}}}{\left[{{{{\bf{y}}}}}_{1}^{(1)}\,{{\ldots }}\,{{{{\bf{y}}}}}_{K}^{(N)}\right]}^{T}{{\in }}{{\mathbb{R}}}^{{NK}\times L}\), \(\widehat{{{{\boldsymbol{X}}}}}{{=}}{\left[{\widehat{{{{\bf{x}}}}}}_{1}^{(1)}\,{{\ldots }}\,{\widehat{{{{\bf{x}}}}}}_{K}^{(N)}\right]}^{T}{{\in }}{{\mathbb{R}}}^{{NK}\times G}\), \({{{\boldsymbol{Z}}}}{{{\boldsymbol{=}}}}{\left[{{{{\bf{z}}}}}^{(1)}\,{{\ldots }}\,{{{{\bf{z}}}}}^{(N)}\right]}^{T}{{\in }}{{\mathbb{R}}}^{N\times D}\) and obtain the log-likelihood form for the resolution enhancement process in Eq. 2:
Variational inference of soScope
The summation over latent variables makes calculating \(\log p\left({{{\boldsymbol{X}}}} | {{{\boldsymbol{Y}}}}\right)\) intractable. Therefore, we solve the log-likelihood in Eq. 3 via the variational Bayesian inference40. We hypothesize that latent state \({{{\boldsymbol{Z}}}}\) is related to spatial omics profile \({{{\boldsymbol{X}}}}\) and spatial neighboring relations \({{{\boldsymbol{A}}}}\). Then, we estimate its variational distribution in the form of \(q\left({{{\boldsymbol{Z}}}} | {{{\boldsymbol{X}}}},\, {{{\boldsymbol{A}}}}\right)\), where \({{{\boldsymbol{A}}}}\in {[{{\mathrm{0,1}}}]}^{N\times N}\) is the spatial neighboring relation graph41 and is calculated using profiles between neighboring spots in \({{{\boldsymbol{X}}}}\) (Supplementary Note 1). The evidence lower bound (ELBO) is formulated as (Supplementary Note 2):
where \({D}_{{KL}}\left[q\left({{{\boldsymbol{Z}}}} | {{{\boldsymbol{X}}}},\,{{{\boldsymbol{A}}}}\right)\parallel p({{{\boldsymbol{Z}}}})\right]\) is the Kullback-Leibler (KL) divergence between \(q\left({{{\boldsymbol{Z}}}} | {{{\boldsymbol{X}}}},\, {{{\boldsymbol{A}}}}\right)\) and \(p({{{\boldsymbol{Z}}}})\).
Image regularization
To ensure that the enhanced profiles accurately reflect morphological similarities observed in the images, we introduce an additional image regularization term to enforce their consistency. In detail, we calculate the similarity matrices among subspots based on enhanced omics profiles \({{{\boldsymbol{\Lambda }}}}\in {[{{\mathrm{0,1}}}]}^{{NK}\times {NK}}\) and image features \({{{\boldsymbol{W}}}}\in {[{{\mathrm{0,1}}}]}^{{NK}\times {NK}}\), respectively (Supplementary Note 1). Then, we use a soft-label cross entropy42 to evaluate their consistency:
where \({w}^{(i,j)}\) and \({\lambda }^{(i,j)}\) are the entry at the \(i\)th row and the \(j\)th column in \({{{\boldsymbol{W}}}}\) and \({{{\boldsymbol{\Lambda }}}}\), respectively. By minimizing the \({L}_{{{{\rm{Image}}}}}\), enhanced omics profiles are encouraged to reflect the morphological patterns from the images.
soScope objective optimization and inference
We formulate the overall optimization function by combining ELBO and image regularization:
Here, \(\beta\) is a hyperparameter balancing the level of constraint (we set \(\beta=1\) for all experiments). In the implementation of soScope (Fig. 1), we choose \(q\left({{{\boldsymbol{Z}}}} | {{{\boldsymbol{X}}}},\, {{{\boldsymbol{A}}}}\right)\) as a Gaussian distribution, with its mean and covariance matrix given by a graph encoder \(h\left({{{\boldsymbol{X}}}},\, {{{\boldsymbol{A}}}}\right)\). The parameters modeling \(p\left(\widehat{{{{\boldsymbol{X}}}}} | {{{\boldsymbol{Y}}}},\, {{{\boldsymbol{Z}}}}\right)\) are given by an enhancement decoder \(f({{{\boldsymbol{Y}}}},\, {{{\boldsymbol{Z}}}})\) (Supplementary Fig. 1).
The model is optimized with the Adam optimizer43 using a two-step strategy:
(1) Initialize the network \(h\left({{{\boldsymbol{X}}}},\, {{{\boldsymbol{A}}}}\right)\) using a simple variational graph auto-encoder framework44 without resolution enhancement (Supplementary Note 3).
(2) Maximize the overall target function (Eq. 6) to infer subspot profiles.
After optimization, we use the expectation of subspot-level profiles \({\widehat{{{{\boldsymbol{X}}}}}}^{*}\) as the enhanced profiles:
Extension to different omics types
To properly model the generative process, \(P{(\cdot )}\) is determined by the spatial omics type. For transcript data, we use the negative binomial distribution (NB)45; for protein and histone modification, we use the Poisson distribution37,46; for other spatial omics profiles without conclusive probabilistic distribution knowledge (such as principal components (PCs) of DNA, and normalized ATAC peaks), we use the Gaussian distribution (Supplementary Table 1) as a general choice.
Extension to spatial multiomics resolution enhancement
To enable soScope to simultaneously enhance the resolution of multiomics profiles, we modify the original model framework with three major changes: (1) constructing the spatial neighboring relation graph \({{{\boldsymbol{A}}}}\) based on multiomics similarities; (2) mapping latent representations \({{{\boldsymbol{Z}}}}\) using a graph encoder with multiomics inputs; and (3) generating distribution parameters for multiomics subspot profiles simultaneously from the decoder network. The elaborated description of the multiomics soScope framework is provided in Supplementary Note 4.
Datasets
Human intestine dataset from Visium platform
The human intestine tissue section was collected from the colon of a 66-year-old male individual (labeled as A1 in the original data). The tissue was profiled by Visium platform and contained 2,649 sequencing spots. Tissue region annotations and their regional markers were obtained from the previous publication34. For the enhancement analysis, 9 marker genes from 3 regions (epithelium, muscularis, and immune) were included as input. For the image, we took the coordinate information for each subspot and segmented the corresponding H&E image region, with the radius of the region sett to 5 times the spot radius. Then, we resized image patches to 299×299 pixel size and fed them into a pretrained Google Inception-v333 on ImageNet to obtain image features (in 2,048 dimensions).
Mouse head dataset from Xenium platform
The mouse head section was obtained from a one-day-old mouse pup and profiled using the Xenium platform. This region contains 457,781 cells, with each expressing 379 genes. For biological variability analysis, expression counts were normalized against total counts, scaled by median gene expressions, and log1p transformed. These data were then processed using the modelGeneVar function in the scran R package (v1.20.1). Only genes with positive biological variability (n = 194) were selected for gene correlation analysis and categorized into high (top 60 genes), medium (61–120), and low (121–194) variability groups. For the imaging modality, deep features were extracted following the pipeline used in the intestine data.
Mouse kidney dataset from Xenium platform
The mouse kidney dataset is measured from the same mouse pup section using Xenium platform. This tissue region covers 1538 cells and 379 genes. For the enhancement analysis, we selected the top 5 abundant genes and the top 5 marker genes. For the image modality, we segmented the image for each cell from the tissue H&E and extracted deep features using the same method as in our intestinal data analysis. As the cell locations were not arranged in a regular array, we defined the neighborhood of each cell as the five spatially nearest cells using the NearestNeighbors function from the scikit-learn.neighbors Python package (v1.2.0) for the purpose of graph construction.
Human ductal carcinoma dataset from Xenium platform
The dataset is measured from the breast section of a human female using Xenium platform. This tissue region covers 37,894 cells and 313 genes. We identified three distinct gene groups for targeted analyses. For highly correlated genes, we selected the top 10 tumor marker genes as reported in the prior study5. To identify lowly correlated genes, we employed a linear regression model using the LinearRegression function from the scikit-learn.linear_model Python package (v1.2.0), comparing the top 250 abundant genes against the top 50 principal components of image features. We then screened the bottom 10 genes with the lowest \({R}^{2}\), determined via the r2_score function from the scikit-learn.metrics Python package (v1.2.0). For immune cell analysis, we screened 11 marker genes across three cell types (four for T cells, two for B cells, and five for macrophages) based on the previous publication5, and quantified cell density as the summation of log1p-normalized expression of these marker genes following the setting of BayesSpace30. Image modality features were processed using the same pipeline as utilized in the intestinal data analysis, while for soScope without image modality, standard Gaussian noise was employed as a substitute.
Mouse embryo dataset from spatial-CUT&Tag platform
The mouse embryo dataset was collected from a mouse embryo at embryonic day 11. The section was profiled by the spatial-CUT&Tag platform and contained 1,974 sequencing spots. In the original publication18, 11 subpopulations were reported. We selected four organ regions with clear spatial structures (liver, heart, forebrain, and spinal cord) and identified the top 15 variable peaks from each region (getMarkers function in archR v1.0.1 R package) for the resolution enhancement analysis. For the image modality, we followed the established pipeline and learned image features from the H&E image of the embryo. To estimate gene activities from peak counts, we mapped peak regions to genes using bedtools R package (v2.28.0) and performed a negative log transformation on peak data. A higher activity score represents a lower suppression for a gene.
Human embryonic heart dataset from spatial transcriptomics platform
The human heart tissue section was collected from a human embryo at 6.5 post-conception weeks (PCW). The tissue was profiled by the ST platform and contained 186 sequencing spots. Gene expressions were scaled using total transcript counts and log1p-transformed. Regional markers FHL2 and LDHA were identified and reported in the original study25.
Mouse liver dataset from slide-DNA-seq and slide-RNA-seq platforms
The dataset was obtained from a mouse liver metastasis section, encompassing two distinct tumor clone regions labeled as clone A and clone B. The dataset included 24,679 spots for slide-DNA-seq data, and 21,902 spots for slide-RNA-seq data. We used the processed data provided in the original work19. For the resolution enhancement purpose, we divided the tissue slide into 60x60 squared regions and averaged original data in each region into spot-level profiles. Then, we included the top 2 DNA PCs and three marker genes (Hmga2, Tm4sf1, and Aqp5) highlighted in the original work, respectively, in the analysis. For the H&E image data, we extracted deep features from the H&E image for subspots as previously described.
Human skin dataset from spatial-CITE-seq platform
The human skin tissue section was collected from an adult donor who received an early immune activation due to the administration of a Coronavirus Disease 2019 (COVID-19) mRNA vaccine. The tissue contained 1,618 sequencing spots, with each spot including 15,486 genes and 283 proteins. Both omics were normalized using the SCTransform function from R package sctransform (v0.3.5). After that, all proteins and 114 top variable genes (the union of top 20 overexpressed genes from each subpopulation identified using analytical pipeline in the original study15) were included for the enhancement analysis. For the image modality, we extracted deep features from the bright-field image following the same pipeline described above.
Mouse embryo dataset from spatial ATAC-RNA-seq platform
The mouse embryo tissue section was from an E13 (embryonic day 13) mouse embryo. The section included 1,874 sequencing spots, each containing data for 15,748 genes and 24,071 ATAC peaks. We selected the 25 most variable genes (modelGeneVar function in scran v1.20.1 R package) and their corresponding peaks for the enhancement analysis. For the ATAC modality, we performed normalization using the SCTransform function from the R package sctransform (v0.3.5) and subsequently applied min-max normalization. For the image modality, deep features were extracted from the bright-field images using the previously described pipeline.
Experiment setup
In silico “low-resolution” profile simulation
In performance benchmarks, we implemented a simulation strategy wherein we combined neighboring spots to create “low-resolution” spots. Then, we assessed the ability of resolution enhancement techniques to accurately restore the original spot profiles. In the case of Visium datasets, where spots were arranged in a hexagonal pattern, we aggregated the profiles of seven spots within the same hexagon to form a single “low-resolution” spot, with the centroid of the hexagon as its position. For the Xenium datasets, to simulate ST data, we divided the whole tissue regions into the small squared regions (in total 240×240 for mouse head, 50×50 for mouse kidney, and 39×39 for human ductal carcinoma) and integrated the gene expressions of all cells within these square regions to generate regional aggregated expression profiles. For datasets from other spatial platforms, we merged neighboring spots within a square region to produce these “low-resolution” spots. For spatial-CUT&Tag, we merged spots in every 2×2 region. In the multi-scale enhancement experiments with slide-DNA-seq and slide-RNA-seq datasets, we varied the region size from 2×2 to 6×6.
Subpopulation identification
For human skin dataset, we followed the analysis in the original study15 and employed the Louvain algorithm47 (resolution=0.75) to identify subpopulations from the top 10 PCs of normalized proteins or genes.
Comparing methods
All compared methods were benchmarked with the same input features on the same Linux server (Ubuntu 20.04.3 LTS operation system) with Xeon(R) 6226 R CPU and NVIDIA GeForce RTX 3090 GPU (Driver Version: 470.63.01, CUDA Version: 11.4).
Spatial-based enhancement approaches
BayesSpace
(v1.2.1R package)30 is a statistical method for resolution enhancement of ST data. It uses the neighboring expression information to estimate the subspot-level gene expressions. We used the spatialEnhance function from BayesSpace R package for resolution enhancement.
Linear interpolation
(Spatial linear) calculates subspot-level profiles by averaging profiles from the nearest neighboring spots. In the case of Visium data in a hexagonal array, we considered three neighbors within the same triangle. For platforms with a square array configuration, we selected four neighbors from the top, bottom, left, and right directions. This approach was implemented using the interpolate function from the SciPy Python package (v1.4.1).
Spatial gaussian process
(Spatial GP) employs a radial basis function (RBF) to model the covariance matrix of spot coordinates48. This method was trained on spot-level coordinates and profiles and then applied to estimate subspot-level profiles based on their spatial coordinates. We utilized the GaussianProcessRegressor from the SciPy Python package (v1.4.1) for implementation. The choice of the RBF kernel weight was determined by testing across a wide range of values for the best performance. The final parameter settings were provided in Supplementary Table 2a.
Image-based enhancement approaches
iStar32 is a deep regression model designed to enhance the resolution of ST by minimizing the MSEs between the aggregated inferred gene expressions within a spot and the observed gene expression. For our study, the Python implementation of iStar was used, and the model was run using its default configuration settings.
XFuse31 is a deep generative model designed to infer expression maps at enhanced resolution while concurrently reconstructing histological images and spatial gene expressions. We used the Python implementation of XFuse (v0.2.1) and fine-tuned the model under default parameters. Both XFuse and iStar infer profiles at the pixel level. We collected and combined all pixel profiles within the region of a subspot for comparison.
Linear Regression Model from Image (Image linear) employs a linear mapping to predict molecular profiles based on image features. For its implementation, we utilized the LinearRegression module from the scikit-learn Python package (v1.2.0). We trained the model at spot-level data and applied it to the subspot image features to predict subspot profiles.
Image Gaussian Process (Image GP) utilizes image features as the input for the Gaussian Process model to predict molecular profiles. We implemented Image GP using GaussianProcessRegressor from the SciPy Python package (v1.4.1). The weight of the RBF kernel and white noise used for each spatial omics profile was summarized in Supplementary Table 2b.
Image Multilayer Perceptron (Image MLP) models the mapping between image features and molecular profiles through a multilayer perceptron. We implemented the model using MLPRegressor function from scikit-learn Python package (v1.2.0). For each spatial omics dataset, we defined the Image MLP model with the same set of parameters (hidden layer sizes = (128, 32), activation=relu, solver=adam, learning rate init=1E-3, max iter=1E4).
Joint enhancement approach
Joint Multilayer Perceptron (Joint MLP) model has the same framework as Image MLP but takes in the concatenated feature of the image and spatial position for each spot/subspot for omics profile prediction. Implementations and hyperparameters were following the same setting as in Image MLP.
Spatial omics scope (soScope) is implemented in Python with PyTroch (v1.8.0) and PyG (v1.7.2) packages. We provided detailed instructions and a demonstration to run the model on GitHub (details were provided at https://github.com/deng-ai-lab/soScope).
Evaluations
Recovery accuracy evaluation with mean square error
MSE measures the difference between two vectors under \({l}_{2}\) norm. An MSE close to 0 indicates an accurate expression recovery; in the implementation, we first min-max normalized the input vectors and then used the mean_squared_error function from sklearn.metrics Python package (v1.2.0).
Recovery accuracy evaluation with Pearson correlation coefficients
Pearson correlation coefficient is a metric to quantify the consistency between two variables. A coefficient close to 1 indicates a strong positive linear correlation of spatial patterns between reconstructed results and ground truth. In the implementation, we used the pearson function from scipy.stats Python package (v1.10.0).
Differential expression measurement with Kolmogorov–Smirnov distance
The Kolmogorov-Smirnov distance (KS distance) measures the distance between two distributions by calculating the maximum distance between two cumulative distribution functions. A KS distance close to 1 indicates a better separation between two distributions. In the implementation, we used the kstest from scipy.stats Python package (v1.10.0).
Gene variability measurement
We used the modelGeneVar function from the scran R package (v1.20.1) to estimate the biological variability of genes. The biological variability is defined as the difference between the log-normalized expression variance and the technical component. A positive biological variability indicates that the observed variability of a gene exceeds the non-informative variation predicted by the model. Conversely, a negative biological component suggests that the observed variability is less than the non-informative variation expected by the model.
Gene abundance measurement
The abundance of a gene is the total number of expression counts in a tissue. In the implementation, we used the sum function in numpy from the Python package (v1.19.2).
Cluster compactness measurement with average spatial distance
We first min-max normalized the coordinates of each spot. To assess the compactness of clusters, we computed the average distance between every pair of spots within a cluster. A spatial distance of 0 indicates that all spots within the cluster are closely co-localized in space. For implementation, we used the cdist function from the scipy.spatial.distance Python package (v1.10.0).
Software used in this study
The software used for generating images can be accessed via the following link:
Adobe Illustraor: https://www.adobe.com/products/illustrator.html
Software packages used in the study can be accessed via following links:
iStar: https://github.com/daviddaiweizhang/istar
XFuse: https://github.com/ludvb/xfuse
BayesSpace: https://github.com/edward130603/BayesSpace
Seruat: https://satijalab.org/seurat/
ArchR: https://www.archrproject.com/
sctransform: https://github.com/satijalab/sctransform
Linear interpolation: https://docs.scipy.org/doc/scipy/tutorial/interpolate.html
Gaussian Process: https://scikit-learn.org/stable/modules/gaussian_process.html
Linear Regression Model:
https://scikit-learn.org/stable/modules/generated/sklearn.linear_model.LinearRegression.html
Multilayer perceptron: https://scikit.learn.org/stable/modules/neural_networks_supervised.html
Computational efficiency
The detailed runtime and memory consumption are reported in Supplementary Table 3. The experiments were conducted on an NVIDlA GeForce RTX 3090 GPU.
Statistics and reproducibility
The experiments were not randomized. No statistical method was used to predetermine the sample size. For the human intestine dataset, we excluded spots that could not be accommodated within the set of hexagonally arranged “low-resolution” spots. For the mouse liver dataset, we excluded spots located outside the inscribed circular area of the slide-DNA-seq and slide-RNA-seq, as our experimental design involved merging the sampling spots to a lower resolution, and the square region facilitated our experimental procedures. For the mouse head, mouse kidney, human ductal carcinoma, mouse embryo (E11), human heart, human skin, and mouse embryo (E13) datasets, no samples were excluded from the analysis. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Human intestine dataset: the Visium spatial transcriptomics data were downloaded from the Gene Expression Omnibus (GEO) database with the accession number GSE158328, and the H&E image was downloaded from https://data.mendeley.com/datasets/gncg57p5x9/2. Mouse head and kidney dataset: the Xenium data and corresponding H&E image were downloaded from https://www.10xgenomics.com/datasets/mouse-pup-preview-data-xenium-mouse-tissue-atlassing-panel-1-standard. Human ductal carcinoma dataset: the Xenium data and corresponding H&E image were downloaded from https://www.10xgenomics.com/products/xenium-in-situ/preview-dataset-human-breast. Mouse embryo (E11) dataset: the spatial-CUT&Tag data were downloaded from the GEO database with the accession number GSE165217, and the H&E image was shared by authors in the original paper. Human heart dataset: the ST data were downloaded from https://data.mendeley.com/datasets/mbvhhf8m62/2, and the H&E image used was downloaded from https://data.mendeley.com/datasets/dgnysc3zn5/1. Mouse liver dataset: the raw slide-DNA-seq and slide-RNA-seq data were available from the Sequence Read Archive under the accession number PRJNA768453, spatial barcode locations and counts matrices were accessed through the Broad Institute Single Cell Portal under accession number SCP1278, and the authors in the original paper provided the H&E image. Human skin dataset: the spatial-CITE-seq data were downloaded from the GEO database with the accession number GSE213264. The high-resolution bright-field image was downloaded at https://doi.org/10.6084/m9.figshare.20723680. Mouse embryo (E13) dataset: the spatial ATAC-RNA-seq data and the corresponding bright-field image were downloaded at https://brain-spatial-omics.cells.ucsc.edu. Source data are provided with this paper.
Code availability
Source code, demonstrations, and result reproducibility are available on GitHub https://github.com/deng-ai-lab/soScope and on Zenodo49.
References
Moncada, R. et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 38, 333–342 (2020).
Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Fawkner-Corbett, D. et al. Spatiotemporal analysis of human intestinal development at single-cell resolution. Cell 184, 810–826.e823 (2021).
Maynard, K. R. et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci. 24, 425–436 (2021).
Janesick, A. et al. High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis. Nat. Commun. 14, 8353 (2023).
Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777–1792.e1721 (2022).
Shah, S. et al. Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell 174, 363–376.e316 (2018).
Eng, C. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 (2019).
Stickels, R. R. et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 39, 313–319 (2021).
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Wang, X. et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, eaat5691 (2018).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. & Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015).
Moffitt, J. R. et al. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl Acad. Sci. USA 113, 11046–11051 (2016).
Liu, Y. et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 183, 1665–1681.e1618 (2020).
Liu, Y. et al. High-plex protein and whole transcriptome co-mapping at cellular resolution with spatial CITE-seq. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01676-0 (2023).
Llorens-Bobadilla, E. et al. Solid-phase capture and profiling of open chromatin by spatial ATAC. Nat. Biotechnol. 41, 1085–1088 (2023).
Zhang, D. et al. Spatial epigenome-transcriptome co-profiling of mammalian tissues. Nature 616, 113–122 (2023).
Deng, Y. et al. Spatial-CUT&Tag: spatially resolved chromatin modification profiling at the cellular level. Science 375, 681–686 (2022).
Zhao, T. et al. Spatial genomics enables multi-modal study of clonal heterogeneity in tissues. Nature 601, 85–91 (2022).
Takei, Y. et al. Integrated spatial genomics reveals global architecture of single nuclei. Nature 590, 344–350 (2021).
Thrane, K., Eriksson, H., Maaskola, J., Hansson, J. & Lundeberg, J. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res. 78, 5970–5979 (2018).
Berglund, E. et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 9, 2419 (2018).
Chen, W. T. et al. Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell 182, 976–991.e919 (2020).
Zhang, X., Wang, X., Shivashankar, G. V. & Uhler, C. Graph-based autoencoder integrates spatial transcriptomics with chromatin images and identifies joint biomarkers for Alzheimer’s disease. Nat. Commun. 13, 7480 (2022).
Asp, M. et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell 179, 1647–1660.e1619 (2019).
Williams, C. G., Lee, H. J., Asatsuma, T., Vento-Tormo, R. & Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 14, 68 (2022).
Mirzazadeh, R. et al. Spatially resolved transcriptomic profiling of degraded and challenging fresh frozen samples. Nat. Commun. 14, 509 (2023).
Milani, P. et al. Cell freezing protocol suitable for ATAC-Seq on motor neurons derived from human induced pluripotent stem cells. Sci. Rep. 6, 25474 (2016).
Saiselet, M. et al. Transcriptional output, cell-type densities, and normalization in spatial transcriptomics. J. Mol. Cell Biol. 12, 906–908 (2020).
Zhao, E. et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 39, 1375–1384 (2021).
Bergenstrahle, L. et al. Super-resolved spatial transcriptomics by deep data fusion. Nat. Biotechnol. 40, 476–479 (2022).
Zhang, D. et al. Inferring super-resolution tissue architecture by integrating spatial transcriptomics with histology. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-02019-9 (2024).
Szegedy, C., Vanhoucke, V., Ioffe, S., Shlens, J. & Wojna, Z. Rethinking the inception architecture for computer vision. Proc. CVPR IEEE. 2818–2826 (IEEE, 2016).
Bao, F. et al. Integrative spatial analysis of cell morphologies and transcriptional states with MUSE. Nat. Biotechnol. 40, 1200–1209 (2022).
10x Genomics. Whole Mouse Pup Preview Data (Xenium Mouse Tissue Atlassing Panel). https://www.10xgenomics.com/datasets/mouse-pup-preview-data-xenium-mouse-tissue-atlassing-panel-1-standard (2023).
Vandereyken, K., Sifrim, A., Thienpont, B. & Voet, T. Methods and applications for single-cell and spatial multi-omics. Nat. Rev. Genet. 24, 494–515 (2023).
Ji, Z., Zhou, W., Hou, W. & Ji, H. Single-cell ATAC-seq signal extraction and enhancement with SCATE. Genome Biol. 21, 161 (2020).
Vickovic, S. et al. SM-Omics is an automated platform for high-throughput spatial multi-omics. Nat. Commun. 13, 795 (2022).
Shao, W. et al. Comparative analysis of mRNA and protein degradation in prostate tissues indicates high stability of proteins. Nat. Commun. 10, 2524 (2019).
Kingma, D. P. & Welling, M. J. Auto-encoding variational bayes. arXiv https://doi.org/10.48550/arXiv.1312.6114 (2013).
Hu, J. et al. SpaGCN: integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 18, 1342–1351 (2021).
Peterson, J. C., Battleday, R. M., Griffiths, T. L. & Russakovsky, O. Human uncertainty makes classification more robust. arXiv https://doi.org/10.48550/arXiv.1908.07086 (2019).
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. arXiv https://doi.org/10.48550/arXiv.1412.6980 (2014).
Kipf, T. N. & Welling, M. Variational graph auto-encoders. arXiv https://doi.org/10.48550/arXiv.1611.07308 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Gayoso, A. et al. Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nat. Methods 18, 272–282 (2021).
Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, 10008 (2008).
Svensson, V., Teichmann, S. A. & Stegle, O. SpatialDE: identification of spatially variable genes. Nat. Methods 15, 343–346 (2018).
Li, B. & Bao, F. Tissue characterization at an enhanced resolution across spatial omics platforms with deep generative model. deng-ai-lab/soScope: soScope v1.0.0 (v1.0.0). Zenodo https://doi.org/10.5281/zenodo.12740738 (2024).
Acknowledgements
We thank Y. Deng at Yale University for providing H&E data of the mouse embryo; T. Zhao at the Broad Institute of MIT and Harvard for providing H&E data of the mouse liver. Y.D. was supported by the Xinjiang Uygur Autonomous Region Science and Technology Innovation Team (Tianshan Innovation Team) Project (2022TSYCTD0013) and the National Natural Science Foundation of China (No. 62031001 and No. 62325101). Q.D. was supported by the National Natural Science Foundation of China (No. 62088102).
Author information
Authors and Affiliations
Contributions
Q.D. and Y.D. conceived the project. B.L. and F.B. devised the methodology and performed experiments across various spatial omics platforms. F.B., F.L., and Y.H. carried out analytical experiments on biological datasets. H.L. was responsible for the collection of these datasets. B.L., F.B., and Y.D. composed the manuscript. All authors have reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
All authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Can Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, B., Bao, F., Hou, Y. et al. Tissue characterization at an enhanced resolution across spatial omics platforms with deep generative model. Nat Commun 15, 6541 (2024). https://doi.org/10.1038/s41467-024-50837-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-50837-5
This article is cited by
-
Optimization of potential targets for antidepressant Chinese medicines: AI and multi-omics methods
Chinese Medicine (2026)
-
The interpretable multimodal dimension reduction framework SpaHDmap enhances resolution in spatial transcriptomics
Nature Cell Biology (2026)
-
Multisensory Neuromorphic Devices: From Physics to Integration
Nano-Micro Letters (2026)
-
Systematic inference of super-resolution cell spatial profiles from histology images
Nature Communications (2025)
