
Abstract
Psoriasis is a multifactorial, chronic inflammatory skin disease with unresolved questions on its primary events. Iron overload has been described in the epidermis of psoriasis patients, but its relevance remains unknown. We found that the key iron regulatory hormone hepcidin was highly expressed in the epidermis of psoriasis patients, especially the pustular variants resistant to treatments. In a murine model of acute skin inflammation, keratinocyte-derived hepcidin was required for iron retention in keratinocytes, leading to hyperproliferation of the epidermal layer and neutrophil recruitment, two main features of psoriatic skin lesions. Keratinocytes overexpressing hepcidin were sufficient to elicit these psoriasiform features in a transgenic mouse model. Furthermore, transcriptome analysis of these keratinocytes revealed canonical pathways found in human psoriasis, pointing to a causal role for hepcidin in the pathogenesis of the disease. Altogether, our data suggest that hepcidin could be an actionable target for skin psoriasis treatment, in addition to current therapeutics, or targeted as maintenance therapy during remission to prevent recurrence.
Similar content being viewed by others
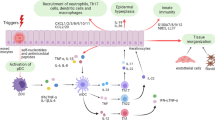


Introduction
Psoriasis affects skin with various phenotypes, and is associated with significant comorbidities1. It is a common disease, affecting 2%–3% of the population worldwide, without distinction of age, sex or ethnicity. Different types of psoriasis exist including plaque psoriasis (or psoriasis vulgaris), the most prevalent and pustular psoriasis (PP), considered as more resistant and particularly difficult to treat. PP, characterized by neutrophil-rich pustules, accounts for only a small fraction of cases but can be life-threatening when extensive2. PP is strongly associated with certain predisposing genetic factors, such as loss-of-function mutations in IL36RN – the interleukin 36 receptor antagonist3, or mutations in CARD14 (caspase recruitment domain-containing protein 14), AP1S3 (adapter protein complex 1 subunit sigma 3) or SERPINA3 (serine protease inhibitor gene serpin family A member 3)4. Despite major advances in our understanding of its pathogenesis and treatment5, psoriasis remains a common chronic disease whose causes have yet to be fully elucidated.
Psoriasis is characterized by abnormal keratinocyte proliferation and immune cell recruitment in the dermis and epidermis. The pathogenesis of psoriasis is complex and involves the interaction of keratinocytes, immune cells, and other skin resident cells. The pathogenic interleukin (IL)−23/IL-17 axis has been identified as playing a key role in psoriasis. The production of TNF-α and IL-23 by dendritic cells (DC) leads to activation of T-helper type 1 (Th1) and type 17 (Th17) cells and subsequent secretion of inflammatory cytokines, including TNF-α, IL-17, and IL-22. These cytokines (especially IL-17) then activate keratinocytes, which produce antimicrobial peptides, cytokines, and chemokines, thereby contributing to the amplification of inflammation. In addition to the TNF-α/IL-23/IL-17/IL-22 pathway, IL-36 and IL36RN are currently the focus of much attention6. Beyond the loss-of-function mutations in IL36RN in PP, IL-36 is also upregulated in plaque psoriasis and the functional activation of IL-36R might contribute to the persistence and perpetuation of psoriatic inflammation together with the TNF-α/IL-23/IL-17/IL-22 axis.
Chemokines, in particular CXCL1, are believed to be secreted in response to IL-17 and recruit neutrophils to the lesional skin. Bridging important pathogenic functions of the innate and adaptive immune systems, neutrophils exhibit complicated interactions with regulatory networks that underlie psoriatic pathophysiology7. Over the last decades, it has been assumed that immune cells drive psoriasis and keratinocytes follow their cues; however, recent evidence shows that keratinocytes can play an important triggering role in such psoriasis immune inflammatory cascades8.
Remarkably, studies going back as far as fifty years have reported increased iron concentrations in psoriatic epidermis compared to controls9,10,11. Hepcidin (encoded by the hamp gene) is a key hormonal peptide, mainly produced by the liver, that maintains systemic iron homeostasis. In conditions of high iron, the production of hepatic hepcidin in the plasma blocks iron absorption from the duodenum and iron release from macrophages. Hepcidin acts on ferroportin (FPN)—the only known cellular iron exporter—to inhibit cellular iron efflux. The efflux of iron out of the cells is controlled by hepcidin binding to ferroportin through two mechanisms: degradation of ferroportin by hepcidin-induced endocytosis12 and occlusion of the open-outward conformation of ferroportin by hepcidin13. Hepatic hepcidin is also induced by select pro-inflammatory cytokines, such as IL-1β or IL-6, and is considered the mediator of anemia of inflammation14. Apart from the liver, many cells and tissues can express hepcidin locally under pathological conditions, such as infection or inflammation, where hepcidin may participate in local iron regulation. To determine the contribution of these hepcidin-producing tissues in body iron homeostasis, we previously developed a mouse model in which the hepcidin gene can be conditionally inactivated15. We recently discovered that hepcidin can be produced by keratinocytes and plays a critical role against bacterial infection by promoting keratinocyte production of CXCL1 and recruitment of neutrophils16. In psoriasis, few studies have explored plasma hepcidin levels within the context of anemia of inflammation17,18,19, but the expression and role of skin hepcidin and iron in the pathogenesis of psoriasis remain to be investigated. Using in vitro approaches, in vivo genetic mouse models, transcriptomics, as well as human 3D organotypic skin model and psoriasis skin biopsies, we found that skin hepcidin has a key role in the initiation of psoriasiform skin inflammation.
Results
Hepcidin expression is increased in the skin of psoriasis patients
Hepcidin expression in biopsies of lesioned skin from patients with psoriasis vulgaris or PP, and in skin specimens from healthy controls, for individuals in different age groups and of either sex, was immunohistochemically assessed (Fig. 1, Supplementary Table 1). Hepcidin expression was highest in keratinocytes, the main cells of the epidermis (Fig. 1a). Hepcidin was mildly expressed in the lesional skin of psoriasis vulgaris patients (Fig. 1a, left) and highly expressed in PP patients (Fig. 1a right). The overexpression in psoriasis patients was correlated with a decrease in FPN expression (Fig. 1a). Interestingly, hepcidin quantified expression was significantly positively correlated to epidermis thickness (Fig. 1b). In addition, in a large cohort of vulgaris psoriasis patients available from a public database20, we also found that hepcidin expression was also increased at the mRNA level in the lesional skin compared to normal skin (Supplementary Fig. 1a). Interestingly, hepcidin expression was the most significantly positively correlated with Il36a and Il23, among different key psoriasis cytokines tested (Supplementary Fig. 1b). We also analyzed mRNA hepcidin levels in lesional skin from PP patients whose mutational status was known for some of them (Supplementary Table 2) and compared them to normal skin. Hepcidin expression was significantly induced in the lesional vs normal skin of PP, with the highest expression in the patients with the IL36RN mutation (Fig. 1c)

a Representative IHC (from n = 14 healthy controls; n = 24 psoriasis vulgaris patients; n = 14 pustular proriasis patients) with or without primary antibody detecting hepcidin, ferroportin (in brown) on sections of cutaneous human biopsies of psoriasis patients and healthy controls. The bar represents 100 μm. Leica DMI3000B microscope; Leica LAS Core software. b Correlation between hepcidin expression (quantified by Image J) and epidermis thickness. Spearman’s r test with 95% confidence intervals. c Hepcidin expression measured by Real-time reverse transcription PCR (qPCR) in the skin of pustular patients (characteristics in supplementary table 2; red dot: IL36RN mutation, blue dots: AP1S3 mutation, green dots: CARD14 mutation, black dots: no mutations identified in IL36RN, CARD14, AP1S3, MPO, SERPINA; n = 3 healthy controls, n = 12 pustular patients (Data are means ± SEM. Two-sided Mann–Whitney test; *p = 0.0176). Source data are provided as a Source Data file.
Increase in hepcidin expression and iron levels in an acute skin inflammation model
Numerous models of genetically engineered mouse models resembling psoriasis have been established, including keratinocyte-specific over-expressing models (IL-23, IL-17A,…), Il36RN or Card14 mutant mice21. Imiquimod (IMQ)-induced skin inflammation in mice has also been extensively used as a model of psoriasiform skin inflammation and has also proved useful for studies of PP pathogenesis22,23. To investigate the role of epidermal hepcidin in psoriasiform skin inflammation, we applied IMQ or Vaseline (Vas) to wild-type (WT) mice (C57BL/6 J) (Fig. 2a). Mice were sacrificed after 3, 6, 24, 48, and 96 h, and the levels of cytokines associated with psoriasis pathogenesis were analyzed. In IMQ-treated mice, Cxcl1, Tnf, Il23, Il6, Il1b, IL22 and Il17 cytokines were induced with various kinetics and intensities (Supplementary Fig. 2a). Hepcidin mRNA levels increased as early as 3 h after treatment (Fig. 2b). At the protein level, hepcidin was significantly increased by a 24 h IMQ treatment and declined from 48 h on, returning to baseline levels (Fig. 2c). The increase in hepcidin was associated with a decrease in FPN expression in keratinocytes, as shown immunohistochemically by comparison of IMQ- and Vas-treated mice (Fig. 2d). As Il36a and Il23 significantly correlated with hepcidin expression in psoriasis and are involved in the development of IMQ-induced skin inflammation22,24, we asked whether intradermal injection of these cytokines could trigger hepcidin expression. We found that hepcidin mRNA levels were highly induced (up to 3500 fold induction) upon intradermal injection of IL-36α, at early time points (Fig.2e and Supplementary Fig. 3).

a Protocol of the IMQ-induced skin inflammation model. b Hepcidin expression measured by qPCR in the skin of IMQ and Vas-treated WT mice (n ≥ 3 mice per group); 24 h vs 6 h:*p = 0.042; 48 h vs 6 h: *p = 0.0324; 96 h vs 6 h: *p = 0.0398 or (c) by ELISA in skin biopsies from IMQ and Vas-treated WT mice (n ≥ 3 mice per group); 24 h vs Vas: **p = 0.0026; 48 h vs Vas: ****p < 0.0001; 24 h vs 3 h: *p = 0.0147; 48 h vs 3 h: ***p = 0.0005; 24 h vs 6 h: *p = 0.0460; 48 h vs 6 h: **p = 0.0014; 96 h vs 24 h: **p = 0.0058; 96 h vs 48 h: ***p = 0.0002. d FPN IHC on the skin of IMQ- or Vas-treated WT mice for 48 h. Scale bar = 100 μm. Representative IHC (n = 5 vas-treated mice and n = 4 IMQ-treated mice). e Hepcidin expression measured by qPCR in the skin of mice with daily intradermal injection of IL-36a (n ≥ 4 mice per group); ****p < 0.0001. f Iron measurement by Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OEP) in the epidermis of IMQ- or Vas-treated WT mice (n ≥ 4 mice per group); 96 h vs Vas: ***p = 0.0001; 96 h vs 3 h: ***p = 0.0001; 96 h vs 6 h: ***p = 0.0002; 96 h vs 24 h: *p = 0.0428; 96 h vs 48 h: *p = 0.0447. g Image of In situ Iron mapping by secondary ion mass spectrometry (SIMS) of skin biopsies from IMQ and Vas-treated WT mice (n = 2 mice per group). Images of CN- ion and PO2- ion allow the identification of cellular str‡uctures. They are used to define the region of interest (ROI). (n = 18 ROI for Vas; n = 24 ROI for IMQ); ****p < 0.0001. Scale bar = 10 μm. FeO- intensity readout for individual nucleus on defined ROIs (yellow circles). Data are means ± SEM. Source data are provided as a Source Data file.
We hypothesized that the hepcidin-FPN axis may contribute to iron retention in keratinocytes. We therefore measured skin iron content by inductively coupled plasma-optical emission spectrometry (ICP-OEP) and showed progressive iron accumulation in IMQ-treated skin, compared to Vas-treated skin (Fig. 2f). Interestingly, we further determined—using nanoscale secondary ion mass spectrometry (NanoSIMS)—that iron was primarily localized in the nuclei of basal keratinocytes of the IMQ-treated epidermis (Fig. 2g). This observation matches the well-known role of iron in cell proliferation, where it acts as a key cofactor of proteins involved in DNA replication and cell cycle control25. Accordingly, we observed that addition of the iron chelator deferoxamine (DFO) decreased keratinocyte proliferation of Normal Human Keratinocytes (NHEKs) (Fig. 3a) and epidermal thickness in organotypic raft cultures (de-epidermalized dermis (DED)) made with human keratinocytes and matched fibroblasts from the same donor (Fig. 3b) or with HaCaT cultures (Supplementary Fig. 4a, b).

a Exponentially growing Normal Human Keratinocytes were treated with (green curve) or without (black curve) 100 μM DFO for 48 h (n = 3 biological replicates); ****p < 0.0001. b Morphological study of 3 DED-Raft made from matched primary human keratinocytes and fibroblasts KCP7 and FCP7, respectively36 treated or not with 100 μM DFO for 72 h following haematoxylin and Eosin staining. Scale bar = 200 μm. c Calcein fluorescence quenching assay in Normal Human Keratinocytes incubated or not (black curve) with Ferric Ammonium Citrate (FAC) (500 μM) ± synthetic hepcidin (3,6 μM) (blue/red curve), for measurement of the labile iron pool. n ≥ 3 biological replicates. ****p < 0.0001; *p = 0.0138. d Left: Representative MTT proliferation assay analysis in NHEK cells, incubated or not (black dots) with FAC ± synthetic hepcidin (blue/red dots). n ≥ 3 biological replicates. ****p = 0.0005; *p = 0.0157. Right: Ratio of KI67-positive cells in NHEK cells incubated with or without (black dots) FAC ± synthetic hepcidin (blue/red dots). Digital images of the immunocyto chemistry-stained slides were acquired using a Widefield Olympus BX63 microscope, and KI-67-positive cells were quantified using the automated software QuPath. Each dot represents the measurement taken from one microscopic slide. (n ≥ 3 biological replicates). ****p = 0.0002; FAC vs -: *p = 0.0305; FAC+hepcidin vs FAC: *p = 0.0430. e MTT proliferation assay analysis in NHEK cells, incubated with DMSO (CTRL: black) with or without Vamifeport ± FAC (resuspended in DMSO) (blue/red/green dots); n = 3 biological replicates. Vamifeport vs CTRL: ***p = 0.0009; FAC vs CTRL: ***p = 0.0002; FAC+Vamifeport vs control: ****p < 0.0001. Data are means ± SEM. Data were analyzed by a one-way Analysis of Variance followed by a Tukey analysis (d), (e), a two-way Analysis of Variance followed by a Tukey analysis (a), (c). Source data are provided as a Source Data file.
Hepcidin controls intracellular iron levels of keratinocytes
Furthermore, we explored the involvement of hepcidin in controlling intracellular iron levels of epidermal cells. In NHEKs (Fig. 3c) and HaCaT (Supplementary Fig. 4c), a calcein fluorescence quenching assay indicated that cellular labile iron pool retention, induced by the addition of ferric ammonium citrate (FAC), was augmented by the addition of hepcidin. The hepcidin-mediated increase in keratinocyte iron retention correlated with an increase in the rate of NHEK cell proliferation, as shown by a MTT proliferation assay and KI-67 staining (Fig. 3d) and HaCat proliferation as shown by real-time analysis using an Incucyte system (Supplementary Fig. 4d). Addition of the FPN inhibitor vamifeport (VIT-2763) was sufficient to induce NHEK cell (Fig. 3e) and HaCat (Supplementary Fig. 4e) proliferation, demonstrating that cellular iron export inhibition contributes to greater cell proliferation. Moreover, FAC increased the rate of proliferation already augmented by VIT-2763 (Fig. 3e). Taken together, these data suggest that hepcidin-mediated FPN degradation is a driver of iron-induced cell proliferation.
Keratinocyte-derived hepcidin contributes to neutrophil recruitment and acanthosis in an acute skin inflammation mouse model
To objectivize the role of hepcidin in psoriasiform skin inflammation, we used our previously generated keratinocyte-specific hepcidin knock-out model16 (Hamp1lox/lox K14-Cre mice = Hamp1∆ker) harboring a deletion efficiency of about 65% as determined by quantitative PCR on mouse genomic DNA (Supplementary Fig. 5). Hamp1∆ker and WT (Hamp1lox/lox) littermates were treated with IMQ on back skin. Iron levels, quantified by ICP-OEP, were reduced (p = 0.06) in the epidermis of IMQ-treated Hamp1∆ker mice compared to that of IMQ-treated Hamp1lox/lox littermates (Fig. 4a). Ki-67 staining of the epidermis revealed significantly fewer Ki-67+ basal keratinocytes (Fig. 4b) in the mutant mice than in controls. Furthermore, the increase in epidermal thickness (acanthosis) was significantly lessened in the knockout mice (Fig. 4c).

a Iron measurement by ICP-OEP in the epidermis of Hamp1lox/lox and Hamp1∆ker mice treated with IMQ for 5 days. n ≥ 7 mice per group; p = 0.06. b Percentage of Ki-67+ cells in epidermal basal cells, quantified in skin sections of Hamp1lox/lox and Hamp1∆ker mice treated with IMQ for 5 days; n ≥ 5 mice per group; **p = 0.0025. c Representative H&E staining and epidermis thickness measurement of Hamp1lox/lox and Hamp1∆ker mice treated with IMQ or Vas for 5 days. n ≥ 4 mice per group; ****p < 0.0001, **p = 0.0106. Scale bar = 100 μm. d Cxcl1 expression measured by qPCR. n ≥ 5 mice per group; ****p < 0.0001, **p = 0.0082. e Representative CXCL1 IHC (n ≥ 4 mice per group) in the skin of Hamp1lox/lox and Hamp1∆ker mice treated with IMQ or Vas for 5 days. Scale bar = 100 μm. Quantification of IHC staining analysis by « ImageJ plugin IHC profiler ». Each dot represents the mean of three measurements taken from one microscopic slide. n ≥ 6 mice per group; **p = 0.0037. f Representative PMN IHC (n ≥ 3 mice per group) on skin biopsies of IMQ-treated Hamp1lox/lox and Hamp1∆ker mice for 5 days and neutrophil count (n ≥ 3 mice per group). ***p = 0.0006; **p = 0.0180. Scale bar = 100 μm (c), (e), (f). Data were analyzed by unpaired two-tailed Student t test (a), (b), (e), a one-way Analysis of Variance followed by a Tukey analysis (c), (d), (f). Hamp1lox/lox are represented by black dots and Hamp1∆ker mice by red dots. Source data are provided as a Source Data file. Pool of three independent experiments. Data are means ± SEM.
We previously demonstrated that hepcidin produced by keratinocytes was critical for neutrophil recruitment in the context of bacterial infection16. The present study similarly revealed that hepcidin contributes to Cxcl1 production and neutrophil infiltration in psoriasiform skin inflammation, as shown by significant lower Cxcl1 expression (Fig. 4d, e) and neutrophil counts in Hamp1∆ker versus Hamp1lox/lox mice after IMQ treatment (Fig. 4f). Levels of IL-17 cytokines previously shown to be involved in epidermal acanthosis and neutrophil recruitment26 were not decreased in the skin of the IMQ-treated Hamp1∆ker mice, compared with control mice (Supplementary Fig. 2b), showing that hepcidin acts independently of these cytokines.
Mice overexpressing hepcidin in the keratinocytes present with neutrophil infiltration and acanthosis
Altogether, the elevated levels of hepcidin expression observed in PP, a resistant form of psoriasis, combined with the finding that hepcidin is expressed in the early phase of a mouse model of acute skin inflammation and contributes to iron-induced hyperproliferation and neutrophil recruitment, suggest that epidermal hepcidin could play a causal role in the initiation / acute phase of psoriasiform skin inflammation. To confirm this, we generated a mouse model overexpressing hepcidin in keratinocytes. The mouse Hamp1 gene sequence was inserted into the ROSA26 locus downstream of a strong CMV promoter and a lox-STOP-lox (LSL) sequence (Supplementary Fig. 6a and methods). After breeding with K14-Cre, the LSL cassette is deleted and Hamp1 is overexpressed in the keratinocytes (Fig. 5a). The newly generated Hamp1 KI-Ker mice displayed an alopecic nude phenotype in the dorsal and ventral regions of the thorax and abdomen (Fig. 5b). Unexpectedly, 50% of the mice died postnatally when approximately 1 month old, for reasons still not understood. Therefore, we sacrificed and analyzed our mice of interest at 20 days of age. We verified that hepcidin overexpression was limited to the skin (Fig. 5c) and not found in other organs (Supplementary Fig. 6b). Hamp1 KI-Ker mice exhibited cutaneous accumulation of iron (Fig. 5d), greater Ki-67 staining of basal keratinocytes than their Hamp1CMV lox-STOP-lox littermates (Fig. 5e), and acanthosis (Fig. 5f), showing that overexpression of skin hepcidin sufficed to induce iron retention and epidermal hyperproliferation. Specific overexpression of hepcidin in keratinocytes was also able to induce epidermal Cxcl1 expression (Fig. 5g, h), and skin neutrophil infiltration (Fig. 5i), but no increase in cutaneous levels of IL-17 (Supplementary Fig. 6c). This transgenic mouse model recapitulates some aspects of psoriasiform skin inflammation, and suggests that hepcidin plays a critical role in its initiation/acute phase.

a Scheme of the knock-in strategy. b Photography of Hamp1 KI-Ker (top) and control littermate. c Hepcidin expression measured by Q-PCR or ELISA in the skin of male and female Hamp1 KI-Ker and control littermates. N ≥ 3 mice per group; ****p < 0.0001; **p = 0.0015. (d) Iron measurement by ICP-OEP in the epidermis of Hamp1 KI-Ker and WT littermates. N≥ 3 mice per group; *p = 0.0361. (e) Percentage of Ki-67+ cells in epidermal basal cells, quantified in skin sections of Hamp1 KI-Ker and control littermates. N ≥ 5 mice per group; *p = 0.0348. f H&E staining (representative of n ≥ 5 mice per group) and epidermis thickness measurement of Hamp1 KI-Ker and control littermates. N ≥ 5 mice per group; ***p = 0.0006. Cxcl1 expression evaluated by Q-PCR; N ≥ 5 mice per group; **p = 0.0011 (g) or IHC (representative of n ≥ 5 mice per group) (h) in the skin of Hamp1 KI-Ker and control littermates. Quantification of IHC staining analysis by « ImageJ plugin IHC profiler » Each dot represents the mean of 3 measurements taken from one microscopic slide. n ≥ 5 mice per group; *p = 0.0415. i Representative PMN IHC on the skin of Hamp1 KI-Ker and control littermate (N ≥ 5 mice per group). Scale bar = 100 μm (f), (h), (i). Data were analyzed by unpaired two-tailed Student’s t test (c–h). Data are means ± SEM. Hamp1CMV lox-STOP-lox are represented by black dots and Hamp1 KI-Ker mice by blue dots. 3-week-old female and male (cf. source data) mice were used. Source data are provided as a Source Data file. Pool of three independent experiments.
GSEA analysis identified enrichment related to neutrophil biology and DNA replication in the skin of Hamp1 KI-Ker mice
We next performed RNA sequencing (RNA-seq) on the skin of Hamp1 KI-Ker mice and control littermates and identified 1938 differentially expressed genes (DEG) (padj<0.01; |fold change|>2). 1172 were upregulated in Hamp1 KI-Ker samples compared to control samples and 766 were downregulated. Gene Set Enrichment Analysis (GSEA) with the Gene Ontology Biological Process (GOBP), REACTOME or HALLMARK gene sets from the Molecular Signatures Database (MSigDB) identified “neutrophil migration”, “cell cycle mitotic”, “inflammatory response” (Fig. 6a) and other enrichments related to neutrophil biology and DNA replication (Supplementary Fig. 7; heat maps in Supplementary Fig. 8) in the skin of Hamp1 KI-Ker mice relative to control mice.

a GSEA plots showing a significant enrichment (p.val = 0.00) of involved genes among upregulated transcripts in Hamp1 KI-Ker samples compared to controls. b Venn diagram comparing differentially expressed genes (DEGs) identified by RNA-seq and those identified involved in psoriasis. Overlap associated with a significant overlap p-value: 6.9E-10 (1.51E-7 after Benjamini-Hochberg adjustment) and a significant z-score: 2.251. c Analysis match between curated IPA analyses and the DEG identified in RNA-seq (overall Z-score). n = 75 analyses (psoriasis vs normal) and n = 45 (post-treated vs pre-treated psoriasis). Data were analyzed by two-sided weighted Kolmogorov–Smirnov test with Benjamini–Hochberg adjustment (a), and One-sided Fisher Exact test with Benjamini–Hochberg adjustment (c). Source data are provided as a Source Data file.
Transcriptome analysis in the skin of Hamp1 KI-Ker mice revealed canonical pathways found in human psoriasis
Among DEG identified in the RNA-seq, 119 genes were known to be involved in psoriasis according to IPA analysis (Fig. 6b). We next performed GSEA analysis and showed that genes overexpressed in two large cohort of psoriasis patients20,27 are enriched among the genes overexpressed in the skin of Hamp1 KI-Ker mice (supplementary file 1). Moreover, by placing our dataset in the context of curated IPA analyses, 97% (72/74) of studies analyzing the transcriptome of human skin psoriasis vs. normal skin had common features (canonical pathways, upstream regulators, causal networks, disease and mechanisms overall Z-score) with our RNA-seq and 72% (55/74) shared more than 30% of these entities (Fig. 6c). Besides, 46% (26/56) of the studies looking at the effect of treatments on skin psoriasis, had at least 30% negatively correlated entities with the features identified in our RNA-seq. These data confirm that hepcidin is positively correlated with the pathogenesis of psoriasis and has a causal role in this disease.
Discussion
We uncovered here the unexpected role of the key iron-regulatory hormone in the pathogenesis of psoriasiform skin inflammation. Our results indicate that keratinocyte-derived hepcidin initiates psoriasiform skin inflammation via iron driven proliferation of the epidermal layer and neutrophil recruitment.
Hepcidin is involved in the initiation of psoriasiform skin inflammation
A combination of results argued for a role of hepcidin in the initiation/acute phase of psoriasiform skin inflammation. First, we found that hepcidin was highly expressed especially in the epidermis of PP patients, the acute form of the disease. Second, in an acute skin inflammation mouse model, hepcidin was expressed in the very early phase highlighting the role of hepcidin during acute inflammation and suggesting its involvement in the initiation of psoriasiform skin inflammation. IL-36 has been not only associated to PP but to various forms of psoriasis and may play a key role in the trigger of hepcidin in early phases. Finally, a transgenic mouse model overexpressing hepcidin in the keratinocytes was sufficient to recapitulate some aspects of psoriasis further substantiating this concept.
Hepcidin expression is downregulated in the late stage of psoriasiform skin inflammation
In the IMQ-induced acute skin inflammation model, the transcriptional increase in hepcidin expression starts as early as 3 h and regains baseline at 96 h, despite daily IMQ application. This decrease may represent an attempt of the keratinocytes to dampen the inflammation cycle fueled by this peptide. This counter-regulation could explain the variability of hepcidin expression observed in the epidermis of vulgar psoriasis patients: we may hypothesize that when the collection of the skin biopsy coincides with the late stage of plaque formation, hepcidin expression may be downregulated.
Hepcidin expression is strongly associated with PP
PP is strongly associated with certain predisposing genetic factors, such as loss-of-function mutations in IL36RN – the interleukin 36 receptor antagonist3 as well as in Serpin A3, the inhibitor of several proteases, especially Cathepsin G expressed by neutrophils and involved in the maturation of IL-36 cytokines28. Our data suggest a strong association between hepcidin and IL-36∝: 1) PP patients with the IL36RN mutation presented the highest hepcidin expression; 2) Intradermal injection of IL-36∝ strongly induced hepcidin expression; 3) Hepcidin expression was positively and significantly correlated with Il36a in two large cohorts of psoriasis patients20,27. In addition, various precipitating factors have also been reported to trigger or flare up acute PP, including corticosteroid use and withdrawal, nonsteroidal anti-inflammatory drugs and acute infections29. Streptococcal Infections are common exacerbating factors during acute GPP (Generalized Pustular Psoriasis)30. We previously showed that hepcidin expression was increased by keratinocytes during this type of infection, triggering the production of CXCL1 and neutrophil recruitment. Therefore, hepcidin may be a contributing factor in the induction of PP.
Nuclear iron accumulation in an acute skin inflammation mouse model
As mentioned before, several studies described an increase in iron concentrations in psoriatic epidermis compared to controls9,11,31. Nanoscale secondary ion mass spectrometry (NanoSIMS) is a unique technology enabling direct identification of chemical elements with high sensitivity/specificity and can be used to visualize elemental distribution in biological samples (chemical mapping). We used here NanoSIMS to visualize iron localization at the subcellular level. We showed here that the iron deposits were located in the nuclei of keratinocytes of the proliferating basal layer in an acute skin inflammation mouse model. This result matches the well-known role of iron in cellular proliferation. Indeed, iron is essential for key enzymes in DNA replication, such a ribonucleotide reductase the rate-limiting enzyme for de novo DNA synthesis25. Iron is also an important cofactor of the G1 phase cyclins E/cdk2, D/cdk5, D1, and the S phase cyclin A/cdk2. Interestingly, Müller et al. found that nuclear iron is responsible for epigenetic modifications of pathways implicated in immune responses and inflammation32, in agreement with the pathogenic role of nuclear iron in the context of psoriasis. Moreover, they described a nuclear action of the iron chelator deferoxamine (DFO). Accordingly, we found that DFO decreased keratinocyte proliferation and epidermal thickness in a human 3D organotypic skin model.
Overexpression of hepcidin in the skin-induced canonical pathways found in psoriasis
Our in vitro and in vivo results convey that during psoriasis the increase of hepcidin leads to FPN degradation and thus iron retention in keratinocytes triggering their hyperproliferation and leading to epidermis thickening, a main feature of psoriasis. Importantly, the only overexpression of hepcidin in the skin, without any other inflammatory stimuli, was sufficient to induce iron accumulation and enrichment in genes involved in neutrophil biology and DNA replication. By comparing our dataset to existing studies analyzing the transcriptome of skin psoriasis vs. normal skin, 97 % had common features with our RNA-seq suggesting that indeed, the overexpression of hepcidin in the skin mirrors psoriasis disease. Besides, 46% of the studies looking at the effect of treatments on skin psoriasis had at least 30% negatively correlated entities with the features identified in our RNA-seq, suggesting that the efficacy of these treatments may be related to the counteracting effect of hepcidin. Interestingly, the uncorrelated studies in this case represent the unresponsiveness to treatment, creating an opportunity for testing hepcidin antagonists.
Inhibiting keratinocyte-derived hepcidin in our acute skin inflammation mouse model did not affect IL-17 expression, a key player in psoriasis pathogenesis. This suggests that hepcidin could be an actionable target for psoriasis treatment, in addition to current therapeutics or targeted as maintenance therapy during remission to prevent recurrence. Adjuvant hepcidin antagonists may benefit PP patients, who suffer from an acute and resistant form of the disease marked by augmented neutrophil recruitment and used as maintenance therapy during remission to prevent recurrence. Finally, IL-36 has been associated to a spectrum of inflammatory skin diseases outside of psoriasis such as hidradenitis suppurativa, atopic dermatitis, and allergic contact dermatitis33; it may be of interest to investigate whether hepcidin play a role in these inflammatory skin conditions.
Methods
Study approval
The animal studies described here were reviewed and approved (Agreements n° APAFIS#9788-2017020314017133 and n° APAFIS#40199-2022102719322238 v8) by the Ministry of Higher Education, Research and Innovation and are in accordance with the principles and guidelines established by the European Convention for the Protection of Laboratory Animals (Council of Europe, ETS 123, 1991).
For human studies, informed written consent to the protocol was obtained for all subjects and was approved by the Ethical Review Committee for publications of the Cochin university Hospital (CLEP), Decision N°: AAA-2022-08049 and by the “Comité de Protection des Personnes” (ID-RCB: 2014-A01017-40 and 2011-A00820-41) at Saint-Louis Hospital. The collection of personal data was approved by the “Commission Nationale de l’Informatique et des Libertés”.
Mice
Mice were housed in specific-pathogen-free animal care facilities at Institut Cochin with 12- hour light-dark cycle, ambient temperature at 22 ± 2 °C, humidity within 30%-70%, and received standard chow ad libitum.
To generate Hamp1∆ker mice, Hamp1lox/lox mice15 were bred with K14-Cre transgenic mice34, in which the Cre recombinase is under the control of the murine K14 promoter. Studies were performed in a C57BL6J background, using 8-12 week old littermates.
To generate Hamp1 KI-Ker mice, we first generated a Rosa26-pCAG-LSL-Hamp mouse line by homologous recombination Knock-in technology into Rosa26 locus. Hamp1 cDNA was inserted downstream the cytomegalovirus (CMV), immediate enhancer/β-actin (CAG) promoter and a loxP-STOP-loxP (LSL) cassette. The vector was transfected into embryonic stem cell (ESC) line and the selection was made in G418. ES cell clones were screened for proper homologous recombination by DNA-PCR before injection into BALB/c blastocysts. Chimeric male mice were crossed with C57BL/6N female mice to obtain the ROSAHamp1 F1 mice. The identification of F1 mice was performed by PCR of tail DNA using primers: sense 5’-TGCTGTCCATTCCTTATTCC-3’ and antisense 5’-TACACCTGTTCAATTCCCCT-3’ that amplify a 310 bp band in case of the mutant allele, and primers: sense 5’-ATACCTTTCTGGGAGTTCTCT-3’ and antisense 5’-TACACCTGTTCAATTCCCCT-3’ that amplify a 192 bp band in the case of the WT Rosa Allele. ROSAHamp1 F1 mice don’t express the Hamp1 cDNA but after breeding with K14-Cre, the LSL cassette was deleted and Hamp1 mRNA was expressed in the keratinocytes. Both male and female Hamp1 KI-Ker and Hamp1 CMV lox-STOP-lox mice were analyzed. Mice were euthanized by cervical dislocation.
Psoriasis biopsies
We had access to lesional skin biopsies taken from 24 patients with vulgaris psoriasis, 26 patients with pustular psoriasis and 11 normal skin specimens from healthy controls matched for age and gender (Supplementary Tables 1 and 2). Patients were recruited at Cochin and Saint-Louis Hospitals. Patients with topical or systemic treatment are indicated in supplementary table 2. The ratio of men to women was 1:1 for pustular psoriasis patients, and the age of patients ranged from 19 to 85 years, with an average of 50 years.
Imiquimod treatment
Female mice were used as 1) the skin of male mice is thicker than that of female mice (making analysis of Imiquimod-induced skin thickening more difficult) and 2) the intensity of response to TLR7 (Imiquimod receptor) is sex-dependent. Three days before the initiation of experiments, female mice were shaved along their backs with electric clippers. Then, mice received a daily topical dose of 62.5 mg of commercially available 5% IMQ cream (Aldara TM 5% cream; MEDA Pharma, Paris, France) on their shaved back for 1 to 5 consecutive days. Control mice were treated similarly with a control vehicle cream (Vaseline, Merck). The kinetic of imiquimod-induced model was established by analyzing mice at different timepoints (3 h, 6 h, 24 h, 48 h and 96 h) of imiquimod application.
Intradermal cytokine injection
Female mice were used to be able to compare the results of intradermal cytokine injection with those of IMQ treatments. Three days before initiation of experiments, female mice were shaved along their backs with electric clippers. Mice were injected intradermally with IL-36∝ (7059-ML, R&D Systems), IL-23 (1887-ML, R&D Systems) or vehicle control (sterile PBS) in two locations on either side of the back for a total of 1 μg protein per mouse using a 29.5-gauge needle. Injections were performed daily until mice were sacrificed. Mice were analyzed at different timepoints (1h30, 3 h, 6 h, 24 h, 48 h, and 96 h).
Immunohistochemistry
Tissues were fixed in 4% formaldehyde and embedded in paraffin. Sections were cut at a thickness of 4 μm. Following dewaxing in xylene baths and rehydratation, sections were subjected to antigen retrieval in a pressure cooker for 15 min at 95 °C with citrate buffer. Human tissues were incubated overnight at 4 °C with a rabbit polyclonal to Hepcidin-25 (1/100, ab30760 Abcam) or FPN (1/200, NBP1-21502 Novus) then stained with the ABC-peroxidase technique. Sections were counterstained with hemalun, dehydrated and mounted.
Sections of mice tissue samples from the dorsal skin were stained with hematoxylin and eosin stain or incubated overnight at 4 °C with a rabbit polyclonal to Ki67 (1/400, ab15580 Abcam, # GR328 5096-1), FPN (1/50, MTP11-A, Alpha Diagnostics, # 116989912.5-L) or CXCL1 (1/500, ab86436 Abcam) then stained with the ABC-peroxidase technique. Sections were counterstained with hemalun, dehydrated and mounted.
A solution of protease K diluted at 1/30 in PBS for 5 min was necessary for unmasking sections before incubating with rat monoclonal anti-PMN primary antibody (1/200, sc-71674 Santa Cruz, # K1620).
ImageJ and ImageJ plugin IHC profiler were applied for quantification of IHC staining analysis as described in ref. 35.
Determination of iron in skin samples by IEC-OP
Sample preparation
The skin samples were weighed in the fresh state then dried in an oven at 70 °C overnight and reweighed in the dry state on a 0.01 mg precision balance (XS205®, Mettler Toledo, Viroflay, France). The dry tissues were put in solution with concentrated nitric acid (HNO3 67-69% PlasmaPure Plus®, SCP Science, Courtaboeuf, France) at 90 °C for one hour in a heating block for digestion (DigiPREP® MS with controller DigiPrep Touch Screen®, SCP Science, Courtaboeuf, France). The minerals were then taken up in ultrapure water with a resistivity greater than 18.2 MΩ (Milli-Q®, Merck-Millipore, Saint-Quentin-en-Yvelines, France) to obtain a final tissue concentration of 10 g/ L in dry weight.
Dosage of iron
Iron was measured by induced coupled plasma optical emission spectrometry (ICP-OES) on an iCAP® 6300 DV spectrometer (Thermo Fisher Scientific, Courtaboeuf, France). The wavelength used is Fe 259.9 nm. Background corrections are made at 259.10 and 259.97 nm. Rhodium (Rh 343.4 nm) is used as an internal standard. A calibration range is carried out between 0 and 5 mg/L. The mineralisates are analyzed purely after the addition of the internal standard. A certified reference sample (SRM 1577b, Bovine Liver, NIST) prepared under the same conditions is analyzed at the start and end of the series. Concentrations are reported by fresh or dry weight (µg/g).
Cell culture
The normal human epidermal keratinocyte (NHEK) cells (PromoCell) were cultured in DermaCult™ Keratinocyte Expansion Medium (Stem cell technologies, #100-0500) supplemented with DermaCult Keratinocyte Expansion Supplement and 1% penicillin/streptomycin (Gibco, #15140122). The human keratinocyte cell line HaCaT was purchased from DKFZ Collection36 (CLS Cell Lines Service, Eppelheim, Germany). HaCaT was cultured in a low calcium medium (DMEM, thermo fisher 21068028). Once confluence was reached, cells were switched in 2.8 mM calcium medium (High Ca2+) to ensure their differentiation.
FCP7 and FEK4 cells are human primary fibroblasts (FCs), whereas KCP7 cells are human primary keratinocytes (KCs). These primary cells were initially procured from the infant foreskin (courtesy of Prof. R.M. Tyrrell’s laboratory, Switzerland to Pourzand’s laboratory)37. The HaCaT cell line, consisting of spontaneously immortalised human skin KCs was similarly sourced from Prof. R.M. Tyrrell’s laboratory for research in Pourzand’s laboratory. HaCaT cells were cultured in a high glucose DMEM supplemented with FCS (10% v/v), 2 mM glutamine and a penicillin/ streptomycin mix (50 IU/ml each). The KCP7 keratinocytes were cultured in an RM+ enriched medium (a blend of DMEM and Ham’s F12 medium at a 3:1 volume ratio) containing 5 µg/ml insulin, 0.4 µg/ml hydrocortisone, 5 µg/ml transferrin, 10 ng/ml epidermal growth factor, 2 × 10−11 M liothyronine, 2 mM l-glutamine, 0.25% w/v sodium bicarbonate, 10% v/v FCS, and a penicillin/ streptomycin mix (50 IU/ml each). Initially, the KCP7 keratinocytes were cultured alongside a Swiss 3T3 feeder layer and later employed in experiments at passage 3. The FCP7 and FEK4 fibroblasts were cultured in EMEM containing 15% v/v FCS, 0.25% w/v sodium bicarbonate, 2 mM l-glutamine and a penicillin/ streptomycin mix (50 IU/ml of each). For the experimental purposes, FCP7 and FEK4 fibroblasts were used at passage 10. The feeder layer Swiss 3T3 (courtesy of Prof I.M. Leigh, Dundee to Pourzand’s laboratory)38 are spontaneously immortalised mouse embryonic fibroblasts which secrete a combination of extracellular matrix proteins and growth factors that facilitate KC adhesion and promote KC proliferation.
Organotypic 3D raft culture using de-epidermalized dermis
The glycerol preserved skin obtained from the Euro Skin Bank in the Netherlands was first rinsed and then stored in PBS for up to ten days at 37oC. To prepare the de-epidermalised dermis (DED), the epidermal layer was mechanically separated from the skin, followed by sectioning the remaining dermis into squares measuring 2 × 2 cm. These squares were then arranged in cell culture plates, papillary dermal side down. Stainless steel rings were positioned atop the dermis to facilitate the seeding of 5 × 105 FCP7 or FEK4 fibroblasts on the reticular dermal surface within the rings. The following day, before repositioning the rings, the DED was flipped to position the papillary dermal side up. Subsequently, KCs (either HaCaT or KCP7) were then seeded at a density of 3 × 105 within the rings on the dermis. The dermis was elevated to the air-liquid interface after 48 h by positioning the composites on stainless-steel grids. The DFO treatment (100 µM for 72 h) was commenced 1-week post-elevation of HaCaT or KCP7 Raft-DEDs. Following a ten-day incubation period without the compounds, the 3D cultures were fixed in a 10% formalin solution and embedded in paraffin. Histological evaluation was performed using Haematoxylin and Eosin staining on sections that had been deparaffinised.
5-bromo-2′-deoxy-uridine assay
The 5-bromo-2′-deoxy-uridine (BrdU) assay was initiated by seeding 8 × 104 cells onto each 10 cm plate. 48 h after seeding DFO treatment was carried out at a concentration of 100 µM over a period of 72 h. To pulse-label the cells with BrdU, 10 µM BrdU was introduced to cell cultures and cells were incubated for an hour at 37oC. A control group was established by omitting BrdU. Post-treatment, the cells underwent trypsinisation followed by centrifugation at 120×g for 8 min at a temperature of 4oC. Subsequently, the medium was discarded. Cells were then permeabilised using ice-cold ethanol at a concentration of 70% and maintained on ice for at least 30 min before proceeding to BrdU labeling. For the BrdU labeling process, the cells were first spun down at 120×g to eliminate ethanol and then rinsed twice with PBS. The DNA within cells was denatured by resuspending them in 2 M HCl for 30 min, intermittently agitating the mixture to ensure the anti-BrdU antibody effectively binds to its epitope within the DNA. Following this, the cells were again centrifuged at 120×g to remove the HCl and washed with PBS-T (PBS containing 0.1% BSA and 0.2% Tween20, pH 7.4). The cells were then incubated with an anti-BrdU primary antibody (sourced from Beckton Dickinson), for 20 min at ambient temperature in a light-protected environment. After a subsequent PBS-T wash, the cells were exposed to a FITC-conjugated secondary antibody from DAKO for another 20 min under similar conditions. A final wash with PBS-T was performed before treating the cells with RNAse (free of DNAse) for 15 min at room temperature to degrade any RNA present. This was followed by the addition of propidium iodide (PI) and a further 30 min incubation in the dark. The RNase treatment is crucial as PI can bind to both DNA and RNA, but our interest lies solely in the DNA-bound PI signal. Flow cytometry was employed for cell analysis, exciting the fluorochromes with a 488 nm laser. The FITC fluorescence was captured within 515 to 545 nm range, while PI fluorescence was detected at wavelengths above 580 nm. We utilized pulse processing of the PI signal to differentiate true G2 phase cells from G1 phase doublets, excluding the latter to accurately gate the G2 population in our experiment. A total of 20,000 events were recorded, ensuring a low flow rate during the collection.
RNA extraction, reverse transcription, and quantitative PCR
Total RNA was isolated with TriReagent (Molecular research center # TR 118), and subsequent cDNA synthesis was performed in Applied Biosystem 2720 thermocycler with the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific # 4368814) according to the manufacturer’s instructions and following this program: hybridization (25 °C − 10 min) - elongation (37 °C − 2 h) – termination (85 °C − 5 min). Real-time PCR was performed in a LightCycler 480 Instrument II (Roche) using the SYBR Green PCR mix (Roche #04887352001) according to the following program: denaturation (95 °C − 10 min) − 45 amplification cycles (95 °C – 10 min followed by 58 °C − 10 min then 72 °C − 5 min) and relative gene expression was determined based on the 2−ΔΔCT method with normalization of the raw data to the human cyclophilin A and murine 36b4. The following primer were used:
Human Hamp (forward: 5’-CTCTGTTTTCCCACAACAGACG-3’; reverse: 5’-CGTCTTGCAGCACATCCCACAC-3’); Human CycloA (forward: 5’- GTCAACCCCACCGTGTTCTT −3’; reverse: 5’-CTGCTGTCTTTGGGACCTTGT-3’); Murine Hamp1(forward: 5’-CCTATCTCCATCAACAGAT-3’; reverse: 5’-TGCAACAGATACCACACTG-3’); Murine Il1bteta (forward: 5’-GCCCATCCTCTGTGACTCAT-3’; reverse: 5’-AGGCCACAGGTATTTTGTCG-3’); Murine Il6 (forward: 5’-ACGATGATGCACTTGCAGAA-3’; reverse: 5’-GTAGCTATGGTACTCCAGAAGAC-3’); Murine Cxcl1 (forward: 5’-GCTGGGATTCACCTCAAGAA-3’; reverse: 5’-TGGGGACACCTTTTAGCATC-3’); Murine Il23p19 (forward: 5’-AGCGGGACATATGAATCTACTAAGAGA-3’; reverse: 5’-GTCCTAGTAGGGAGGTGTGAAGTTG-3’); Murine Tnfa (forward: 5’-AATGGCCTCCCTCTCATCAGTT-3’; reverse: 5’-CGAATTTTGAGAAGATGATCTGAGTGT-3’); Murine Il17A (forward: 5’-TTTTCAGCAAGGAATGTGGA-3’; reverse: 5’-TTCATTGTGGAGGGCAGAC-3’); Murine Il17C (forward: 5′-CCATGGAGATATCGCATCGA-3′; reverse: 5′-CCATGGAGATATCGCATCGA-3′); Murine ll17F (forward: 5’-CAAGAAATCCTGGTCCTTCG-3’; reverse: 5’-GAGCATCTTCTCCAACCTGAA-3’); Murine Il22 (forward: 5’-CATGCAGGAGGTGGTGCCTT-3’; reverse: 5’-CAGACGCAAGCATTTCTCAG-3’); and Murine 36B4 (forward: 5’-ATGTGCAGCTGATAAAGACTGG-3’; reverse: 5’-AGGCCTTGACCTTTTCAGTAAG-3’)
MTT assay
NHEKs cells (5.104 per well) were seeded in 48-well plates and incubated at 37 °C for 24 h. NHEK cells were then treated with FAC (Sigma, # RES20400-A7), hepcidin (Peptide International, PLP-3771-PI), and Vamifeport (FPN inhibitor, VIT-2763; 10 μM). After 72 h of incubation, MTT solution was added at a concentration of 5 mg/ml (Roche, USA, # 11465007001) to each well, and the entire procedure was performed according to the protocol manual. The absorbance was recorded at 550-570 nm using a plate reader (Epoch, Biotek Instruments).
Immunocyto chemistry staining KI67
Sterilized coverslips were placed into 12-well plates and coated with a collagen-coating solution for 2–3 h at 37 °C. After removing the coating solution, the plates with coverslips were dried before seeding of NHEK cells. After incubation for 24 h, the cells were treated with FAC and hepcidin. Following 48 h of further incubation, the cells were fixed in 4% formaldehyde, then a blocking solution of PBS/5% BSA/0.3% Triton™ X-100 was added. After washing, the cells were incubated with a rabbit polyclonal antibody (1/200, Cell signaling technology, D3B5, #9129 S) at 4 °C overnight, followed by staining with Goat anti-Rabbit IgG Secondary Antibody, Alexa Fluor™ 488 conjugate (Invitrogen TM, #A-11008). DAPI solution was added to each well and incubated for 2–5 min at room temperature. Finally, coverslips were removed from the wells, dehydrated, and mounted with an anti-fade mounting medium, with the cells facing towards the microscope slide.
Digital images of the immunocyto chemistry-stained slides were acquired with Widefield Olympus BX63 microscope, and KI-67 positive cells were measured using the automated software QuPath. Only staining intensities exceeding a predefined threshold were considered “positive”.
Incucyte
The Incucyte® S3 Live-Cell Analysis System (Sartorius) is a real-time quantitative live-cell imaging and analysis system that enable visualization and quantification of cell behavior over time, by automatically gathering and analyzing images around the clock within a standard laboratory incubator.
HaCaT cells were seeded into 96-well plates at a density of 16 000 cells/well and treated with or without 500 μM of iron (Ferric Ammomiun Citrate, RES20400-A7, Sigma), and with or without 3.6 µM of synthetic hepcidin (PLP-3771-PI, Peptide International) or 50 nM of FPN inhibitor (VIT-2763). The Red Nuclight Reagent (1/1000, Incucyte® Nuclight Rapid Red Dye for Live-Cell Nuclear Labeling, Sartorius) was added and cells were incubated in the IncuCyte system. Cells were imaged every 2 h for a kinetic of 72 h and the growth rate was measured by acquiring fluorescence images at 20× magnification with the IncuCyte live-cell analysis system.
Calcein fluorescence quenching assay
NHEK cells and HaCaT cells were seeded into 96-well plates and treated overnight with or without 500 μM of iron (Ferric Ammomiun Citrate, RES20400-A7, Sigma). The next day, the medium was removed and the cells were incubated with 0.25 µM of Calcein-AM (InvitrogenTM, # C1430) for 10 min at 37 °C. Afterwards, the wells were washed with PBS and treated with or without 3.6 µM of synthetic hepcidin (PLP-3771-PI, Peptide International) and fluorescence was acquired every 15 min for a kinetic of 180 or 330 min using Clariostar Plus (BMG Labtech).
ELISA
The skin biopsies were added to RIPA lysis buffer, supplemented with Protease Inhibitor Cocktail (Complete Tablets EDTA-free #04693132001, Roche), and homogenized using Tissue Lyser. Skin lysates were then centrifuged at 14000 rpm (18 000×g) for 10 min at 4 °C to remove cell debris. The supernatant was recovered and the quantification of hepcidin, and IL-17 was performed by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions using respectively Mouse hepcidin-25 ELISA (S-1465, BMA Biomedicals) and Mouse IL-17 Immunoassay Quantikine™ ELISA (M1700, R&D Systems).
In situ Iron mapping by secondary ion mass spectrometry
Samples were fixed with 2%-glutaraldéhyde-PFA 4% for 48 h before being dehydrated with sequential EtOH solutions for 10 min each: 50%, 70%, 2 × 90%, 3 × 100%. Samples were then coated with a 1:1 mixture of resin (Electron Microscopy Sciences, dodecenylsuccinic anhydride, 13710, methyl nadic anhydride, 19000, DMP-30, 13600 and LADD research industries: LX112 resin, 21310) and dry EtOH for 1 h. Then, samples were embedded in pure resin for 1 h. Embedding capsules (Electron Microscopy Sciences, 69910-10) were filled with resin, inverted onto the cover slides and placed in an oven at 56 °C for 24 h.
Treated and untreated samples were prepared at the same time. 0.2 μm sections were prepared using a Leica Ultracut UCT microtome. Sample sections of the same thickness were deposited onto a clean silicon chip (Centre de Nanosciences et de Nanotechnologies/CNRS and Université Paris-Saclay) and dried in the air before being introduced into the NanoSIMS-50 ion microprobe (CAMECA, Gennevilliers, France)39,40.
The cellular distribution of iron was imaged by detecting FeO- ion using a Cs+ primary ion. As an absolute quantification is not applicable, the image recording conditions (the primary ion intensity and the field of view) were kept as constant as possible for the same acquisition duration so as to allow reliable comparison between measurements.
For each area, the images of CN- ion, as well as the one for PO2- ion, were recorded in parallel to allow the identification of cellular structure. Further, they were used to define the region of interest (ROI) for FeO- intensity readout using ImageJ.
Additional information on acquisition conditions
The field of view was 60 µm with an image definition of 512 × 512 pixels. Primary Cs+ ion intensity was around 3 pA and multiframe acquisition mode was applied. A total of 300 frames, with a pixel counting time of 0.5 ms, were recorded for each sample area leading to a total acquisition time of about 11 h. Multiframe images were then properly aligned using the TOMOJ plugin41 with CN− images as a reference before a summed image was obtained. Although care was taken to detect specifically the FeO− ion, a weak, not significant level of background signal was counted. Such interference signal was attributed to 35Cl37Cl− cluster ion species (occurring at the same nominal mass as FeO− species) due to the presence of chlorine in the tissue and in the embedding resin.
RNA-Seq analysis
Five different mice per genotype were used for RNA-seq analysis from two independent breedings. Following RNA extraction, RNA concentrations were measured using either a Nanodrop or a Qubit RNA fluorometric assay (Life Technologies, Grand Island, New York, USA). RNA quality, indicated by the RNA integrity number (RIN), was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) according to the manufacturer’s guidelines.
For library construction, 250 ng of high-quality total RNA (RIN > 8) was processed with the Stranded mRNA Prep kit (Illumina), following the manufacturer’s instructions. In brief, poly-A mRNA molecules were purified, fragmented, and reverse-transcribed using random primers. Strand specificity was ensured by substituting dTTP with dUTP during second-strand synthesis. A single A base was added to the cDNA, followed by the ligation of Illumina adapters. Libraries were quantified by Q bit and profiles were assessed using the DNA High Sensitivity LabChip kit on an Agilent Bioanalyzer. Libraries were sequenced on an Illumina Nextseq 500 instrument using 75 base-lengths read V2 chemistry in a paired-end mode.
Post-sequencing, primary analysis was conducted with AOZAN software (ENS, Paris) to demultiplex and quality control the raw data using FastQC modules (version 0.11.5). The resulting fastq files were aligned with the STAR algorithm (version 2.5.2b), and alignment quality was assessed with Picard tools (version 2.8.1). Reads were counted using FeatureCounts (Rsubread version 1.24.1), and statistical analyses on the read counts were performed using the DESeq2 package (version 1.14.1) to determine the proportion of differentially expressed genes between the two conditions.
Ingenuity analysis
Modulated transcripts were selected based on their Benjamini-Hochberg adjusted p-value (p < 0.01) and their fold change (|FC| > 2). DEGs were then analyzed using IPA version 81348237 (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis).
IPA identified significantly enriched pathways, functions, diseases and upstream regulators thanks to a right-tailed Fisher’s Exact Test and their activation status were assessed using z-score calculation. Positive z-score are associated with activation and negative ones with inhibition, the significance threshold is 2 in absolute value.
Enrichments were also performed using GSEA (Gene Set Enrichment Analysis) 4.2.3 software (https://www.gsea-msigdb.org/gsea/) with terms related to Hallmarks, REACTOME pathways and GO biological processes (v2022.1). Settings: tTest, gene_set permutation (n = 1000), collapse.
Heatmapper web server (http://www.heatmapper.ca/) was used for hierarchical clustering and Funrich v3.1.3 (www.funrich.org) for Venn diagrams.
Statistical analysis
Values in the figures are expressed as mean ± SEM. Analysis was performed using GraphPad Prism 6.0. The significance of experimental differences was evaluated by an unpaired (two-tailed) Student’s t-test, when comparing two groups. A one-way or a two-way ANOVA followed by a Tukey analysis was used when more than two groups were compared. Statistical significance is indicated by * symbols (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). ns indicates not significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the findings of this study are available in the article, its Supplementary information, the source data file and from the corresponding author upon request. RNA-sequencing data have been deposited in the Gene Expression Omnibus (GEO) database under accession code GSE220993. Datasets used in Supplementary Fig. 1 are available at GEO, accession number GSE13355. Source data are provided with this paper.
References
Griffiths, C. E. M., Armstrong, A. W., Gudjonsson, J. E. & Barker, J. N. W. N. Psoriasis. Lancet 397, 1301–1315 (2021).
Uppala, R. et al. Autoinflammatory psoriasis’-genetics and biology of pustular psoriasis. Cell. Mol. Immunol. 18, 307–317 (2021).
Onoufriadis, A. et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 89, 432–437 (2011).
Bachelez, H. Pustular psoriasis: the dawn of a new era. Acta Derm. Venereol. 100, adv00034 (2020).
Zhou, X., Chen, Y., Cui, L., Shi, Y. & Guo, C. Advances in the pathogenesis of psoriasis: from keratinocyte perspective. Cell Death Dis. 13, 81 (2022).
Furue, K. et al. Highlighting Interleukin-36 signalling in plaque psoriasis and pustular psoriasis. Acta Derm. Venereol. 98, 5–13 (2018).
Schon, M. P., Broekaert, S. M. & Erpenbeck, L. Sexy again: the renaissance of neutrophils in psoriasis. Exp. Dermatol. 26, 305–311 (2017).
Ni, X. & Lai, Y. Keratinocyte: a trigger or an executor of psoriasis? J. Leukoc. Biol. 108, 485–491 (2020).
Molin, L. & Wester, P. O. Iron content in normal and psoriatic epidermis. Acta Derm. Venereol. 53, 473–476 (1973).
Pinheiro, T. et al. Impact of inflammation on iron stores in involved and non-involved psoriatic skin. Nucl. Instrum. Methods Phys. Res. Sect. B: Beam Interact. Mater. 348, 119–122 (2015).
Forslind, B., Werner-Linde, Y., Lindberg, M. & Pallon, J. Elemental analysis mirrors epidermal differentiation. Acta Derm. Venereol. 79, 12–17 (1999).
Nemeth, E. et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093 (2004).
Aschemeyer, S. et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 131, 899–910 (2018).
Weiss, G., Ganz, T. & Goodnough, L. T. Anemia of inflammation. Blood 133, 40–50 (2019).
Zumerle, S. et al. Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood 123, 3646–3650 (2014).
Malerba, M. et al. Epidermal hepcidin is required for neutrophil response to bacterial infection. J. Clin. Invest. 130, 329–334 (2020).
El-Rifaie, A.-A. A., Sabry, D., Doss, R. W., Kamal, M. A. & Abd El Hassib, D. M. Heme oxygenase and iron status in exosomes of psoriasis patients. Arch. Dermatol. Res. 310, 651–656 (2018).
Ponikowska, M., Tupikowska, M., Kasztura, M., Jankowska, E. A. & Szepietowski, J. C. Deranged iron status in psoriasis: the impact of low body mass. J. Cachexia Sarcopenia Muscle 6, 358–364 (2015).
Dilek, N., Dilek, A. R., Sahin, K., Kaklıkkaya, N. & Saral, Y. Hepcidin expression in psoriasis patients. Indian J. Dermatol. 59, 630 (2014).
Nair, R. P. et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 41, 199–204 (2009).
Gangwar, R. S., Gudjonsson, J. E. & Ward, N. L. Mouse models of psoriasis – a comprehensive review. J. Invest. Dermatol. 142, 884–897 (2022).
Hashiguchi, Y. et al. IL-36α from skin-resident cells plays an important role in the pathogenesis of imiquimod-induced psoriasiform dermatitis by forming a local autoamplification loop. J. Immunol. 201, 167–182 (2018).
Alvarez, P. & Jensen, L. E. Imiquimod treatment causes systemic disease in mice resembling generalized pustular psoriasis in an IL-1 and IL-36 dependent manner. Mediat. Inflamm. 2016, 6756138 (2016).
Chen, W.-C. et al. IL-23/IL-17 immune axis mediates the imiquimod-induced psoriatic inflammation by activating ACT1/TRAF6/TAK1/NF-κB pathway in macrophages and keratinocytes. Kaohsiung J. Med. Sci. 39, 789–800 (2023).
Zhang, C. Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control. Protein Cell 5, 750–760 (2014).
Prinz, I., Sandrock, I. & Mrowietz, U. Interleukin-17 cytokines: effectors and targets in psoriasis—A breakthrough in understanding and treatment. J. Exp. Med. 217, e20191397 (2019).
Foulkes, A. C. et al. A framework for multi-omic prediction of treatment response to biologic therapy for psoriasis. J. Invest. Dermatol. 139, 100–107 (2019).
Frey, S. et al. Rare loss-of-function mutation in SERPINA3 in generalized pustular psoriasis. J. Invest. Dermatol. 140, 1451–1455.e13 (2020).
Benjegerdes, K. E., Hyde, K., Kivelevitch, D. & Mansouri, B. Pustular psoriasis: pathophysiology and current treatment perspectives. Psoriasis (Auckl.) 6, 131–144 (2016).
Choon, S. E. et al. Clinical profile, morbidity, and outcome of adult-onset generalized pustular psoriasis: analysis of 102 cases seen in a tertiary hospital in Johor, Malaysia. Int J. Dermatol. 53, 676–684 (2014).
Pinheiro, T., Alves, Y. M. D., Teixeira, L. C., Ferreira, P. & Filipe, J. Impact of inflammation on iron stores in involved and non-involved psoriatic skin. Nucl. Instrum. Methods Phys. Res. Sect. B: Beam Interact. Mater. 348, 119–122 (2015).
Müller, S. et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat. Chem. 12, 929–938 (2020).
Sachen, K. L., Arnold Greving, C. N. & Towne, J. E. Role of IL-36 cytokines in psoriasis and other inflammatory skin conditions. Cytokine 156, 155897 (2022).
Jonkers, J. et al. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 29, 418–425 (2001).
Varghese, F., Bukhari, A. B., Malhotra, R. & De, A. IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS ONE 9, e96801 (2014).
Boukamp, P. et al. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106, 761–771 (1988).
Zhong, J. L., Yiakouvaki, A., Holley, P., Tyrrell, R. M. & Pourzand, C. Susceptibility of skin cells to UVA-induced necrotic cell death reflects the intracellular level of labile iron. J. Invest. Dermatol. 123, 771–780 (2004).
Proby, C. M. et al. Spontaneous keratinocyte cell lines representing early and advanced stages of malignant transformation of the epidermis. Exp. Dermatol. 9, 104–117 (2000).
Slodzian, G., Daigne, B., Girard, F., Boust, F. & Hillion, F. Scanning secondary ion analytical microscopy with parallel detection. Biol. Cell 74, 43–50 (1992).
Guerquin-Kern, J.-L., Wu, T.-D., Quintana, C. & Croisy, A. Progress in analytical imaging of the cell by dynamic secondary ion mass spectrometry (SIMS microscopy). Biochim. Biophys. Acta (BBA) - Gen. Subj. 1724, 228–238 (2005).
Messaoudii, C., Boudier, T., Sanchez Sorzano, C. O. & Marco, S. TomoJ: tomography software for three-dimensional reconstruction in transmission electron microscopy. BMC Bioinform. 8, 288 (2007).
Acknowledgements
The authors would like to greatly acknowledge the PIME (Alain Schmitt), GenomIC (Benjamin Saintpierre), HistIM and animal facilities of Institut Cochin. The authors thank Jacques Mathieu, Marion Falabregue, Jean-Christophe Deschemin, Sara Guegan, Romain Fontaine, Peter Kanaan and Elio Rivierre for scientific discussions and technical advice or help. This work was supported by fundings from the “Fondation pour la Recherche Médicale” DEQ20160334903, EQ202103012630, PMT202206015641 (C.Peyssonnaux), the Laboratory of Excellence GR-Ex, (ANR-11-LABX-0051), the program “Investissements d’avenir” of the French National Research Agency, reference ANR-11-IDEX-0005-02 (C.Peyssonnaux and S.V.), the IDEX emergence program of the University Paris Cité RM27J21IDXE7-PSOHEPC (C.P.), AREMPH (Association pour les ETudes Medicales et Pharmaceutiques) (S.A. and B.O.), the Swiss National Science Foundation (341 310030_201242) (M.M.) and the LEO Foundation (LF-OC-22-000968) (M.M.).
Author information
Authors and Affiliations
Contributions
C.Peyssonnaux conceived the project; E.A., D.C., N.B., H.D., and M.Malerba performed experiments and analyzed data; T.-D.W. performed and analyzed the Nano-SIMS experiment; M.L.G. performed the Ingenuity analyses; O.R performed the DFO experiment; S.V. and B.O. provided scientific advice; J.P performed and analyzed the ICP-OEP experiment; P.S. performed pathological analyses, and provided scientific advice; S.A., M.Mellet, H.B., F.A, and C.Pourzand. provided essential biopsy material or reagents and scientific advice; Z.K. provided essential mouse models; C.Peyssonnaux. and E.A. designed experiments. C.Peyssonnaux supervised the project and wrote the manuscript with input from E.A., D.C., T-D.W., and J.P. for methods and from all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Abboud, E., Chrayteh, D., Boussetta, N. et al. Skin hepcidin initiates psoriasiform skin inflammation via Fe-driven hyperproliferation and neutrophil recruitment. Nat Commun 15, 6718 (2024). https://doi.org/10.1038/s41467-024-50993-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50993-8
This article is cited by
-
Iron and the immune system
Nature Reviews Immunology (2025)
