
Abstract
The SARS-CoV-2 Omicron variant sparked the largest wave of infections worldwide. Mainland China eased its strict COVID-19 measures in late 2022 and experienced two nationwide Omicron waves in 2023. Here, we investigated lineage distribution and virus evolution in Guangdong, China, 2022-2023 by comparing 5813 local viral genomes with the datasets from other regions of China and worldwide. Additionally, we conducted three large-scale serological surveys involving 1696 participants to measure their immune response to the BA.5 and XBB.1.9 before and after the corresponding waves. Our findings revealed the Omicron variants, mainly the BA.5.2.48 lineage, causing infections in over 90% of individuals across different age groups within a month. This rapid spread led to the establishment of widespread immunity, limiting the virus’s ability to further adaptive mutation and dissemination. While similar immune responses to BA.5 were observed across all age groups after the initial wave, children aged 3 to 11 developed a stronger cross immune response to the XBB.1.9 strain, possibly explaining their lower infection rates in the following XBB.1 wave. Reinfection with Omicron XBB.1 variant triggered a more potent neutralizing immune response among older adults. These findings highlight the impact of age-specific immune responses on viral spread in potential future waves.
Similar content being viewed by others


Introduction
COVID-19 no longer constitutes a public health emergency of international concern1. The emergence of Omicron in late 2021 signified a major turning point in the pandemic. This variant, with exceptional transmissibility and immune evasion, triggered an unprecedented global surge in infections, dwarfing previous waves and eclipsing other Variants of Concern (VOCs)2. After peaking in January 2022, the number of COVID-19 cases dropped steadily in many areas, reaching much lower levels3. The post-Omicron era has presented a complex and dynamic picture of etiological factors and population immunity. Some areas with early outbreaks and high vaccination rates are moving towards a state where the disease is more predictable and less burdensome on health systems, while others still face considerable outbreaks3. The spread of different viral variants and the development of extensive population immunity are key factors driving these differences.
Within the Omicron, a diverse cast of sublineages has emerged. The Omicron wave began with BA.1, which led to an initial surge in global cases in December 20214. BA.2, a more transmissible descendant of BA.1 quickly outpaced its predecessor and became the dominant strain in many regions by February 20225. BA.5 and BF.7 emerged in the middle of 2022, characterized by their ability to evade immunity and high transmission rates, leading to significant case increases in several countries6. XBB.1.5, a recombinant of BA.2.75 and BA.2, was identified in October 2022 and had become the leading global variant by December 20227. A sublineage that should be mentioned is BA.2.86, which was first detected in August 2023 in Israel and Denmark. It possesses over 30 mutations on its spike protein compared to BA.2, including some associated with immune escape and potentially increased transmissibility. The JN.1 variant (BA.2.86.1.1), arising from BA.2.86.1 with the S:L455S substitution, demonstrated enhanced viral fitness, surpassing the previously dominant XBB lineage by early 20248. Subsequent diversification of the JN.1 variant resulted in the emergence of new descendants characterized by spike (S) protein mutations, including S:R346T and S:F456L. Notably, the KP.2 (JN.1.11.1.2) variant, carrying both these substitutions, has demonstrated enhanced immune evasion capabilities9. It has been recognized as a rapidly spreading lineage, affecting multiple regions by April 20243.
Assessing the dynamics of virus prevalence and population immunity is essential for understanding the risks of viral transmission. Reduced public participation in molecular testing for SARS-CoV-2 makes it hard to accurately track how much the Omicron variant is spreading. A serological survey, which can measure the presence of antibodies against specific SARS-CoV-2 variant(s) in a population, has emerged as a crucial tool for understanding population immunity dynamics and the overall infection status of communities. Previous studies showed distinct seroprevalences across different age groups. Individuals who experienced more severe forms of COVID-19 tend to develop higher levels of neutralizing antibodies (nAbs) compared to those with milder cases10, 11. For children between 0 and 19 years, the seroprevalence is generally lower, ranging from 30% to 50% due to factors like less severe illness and limited vaccination12. The highest seroprevalence observed in adults often exceeds 60% due to greater exposure through work, social activities, and vaccination campaigns13. Seroprevalence tends to be lower in older adults aged 60 and above, likely due to reduced exposure chances and possibly faster decline in their immunity14. This figure, however, varies significantly over time and among different demographic groups. The hybrid population immunity attributed to the vaccination and infection history is the key factor.
On 7 December 2022, China implemented a critical shift in its approach to COVID-19, moving away from the rigorous ‘zero-COVID’ policy15. Before lifting of non-pharmaceutical interventions (NPIs), the majority of the population remained infection-naive, with ~70% of the population over 10 years old having received a three-dose vaccination program by December 2021. The consistency of vaccination rates and schedules, coupled with the lack of prior exposure to the virus, helped to minimize confounding factors in studying virus infection dynamics and the development of population immunity. In this study, we investigated the genetic and population immunity landscape across age groups in Guangdong, China, before and after two nationwide Omicron waves. We also explored the influence of population immunity on the spread and mutation of the emerged Omicron variants.
Results
Molecular epidemiology of SARS-CoV-2 before and after the lifting of NPIs
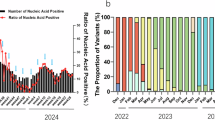
The COVID-19 epidemic in mainland China from late 2022 to July 2023 presented a distinctive opportunity to study the infection, transmission, and evolution of SARS-CoV-2 variants in a population that was largely naive to the virus and had undergone a similar vaccination regimen. To elucidate the molecular epidemiology of SARS-CoV-2 before and after the lifting of NPIs, we sequenced 6183 samples (Supplementary Data 1), from which 5813 high-quality viral genomes were obtained, spanning from the onset of the Omicron outbreak on 22 October 2022, to the end of the epidemic on 5 July 2023. For clear visualization, a time-resolved phylogenetic tree was built by randomly selecting 500 sequences from Guangdong and 500 contemporary sequences from outside China (Fig. 1a). The sequences from Guangdong (terminal branches highlighted with dots) to show the different transmission patterns in two waves. During the initial epidemic wave in Guangdong, from December 2022 to March 2023, the majority of sequences belonged to two BA.5 subvariants including BA.5.2.48* and BF.7.14* (Fig. 1a, the asterisk represents the BA.5.2.48 or BF.7.14 including their sublineages). Only a few sequences from outside China (branches without dots) were detected within these two lineages. In the second wave, the XBB.1 and its sublineages became dominant, with sequences from regions outside China intermingling with those from Guangdong within various phylogenetic clusters (Fig. 1a). The structure of the phylogeny could provide clues for virus and transmission16. In the initial wave, the distance tree showed that most internal nodes of the BA.5.2.48* sequences were positioned near the root node of the clade (Fig. S1). This phylogenetic pattern suggested that the early BA.5.2.48 strains, characterized by limited genetic diversity, were responsible for the rapid, exponential growth of the epidemic, with the majority of genetic mutations emerging within a brief timeframe. In contrast, the Guangdong XBB.1 variant sequences were constructed to a deeper tree in which internal nodes of Guangdong sequences were more diversified and far from the ancestor node of the XBB.1 clade (Fig, S1). This finding suggested the persistence of XBB.1 lineages and the possibility of multiple introductions into the Guangdong region.

a A time-resolved phylogenetic tree was estimated with TreeTime by including (i) 500 sequences randomly selected from Guangdong, China; (ii) 500 contemporary sequences randomly selected from regions outside China; and (iii) representative reference sequences that provide a framework for the phylogenetic tree’s structure. The times when Non-Pharmaceutical Interventions (NPIs) were lifted are indicated with dashed vertical lines. Sequences from Guangdong were highlighted, and specific lineages, including BA.5.2.48* (including BA.5.2.48, DY.1, DY.2, DY.3 and DY.4 variants according to the Pangolin nomenclature), BF.7.14* (including BF.7.14.1-BF.7.14.7 variants), and XBB.1 (including XBB.1.5, XBB.1.16, XBB.1.9, EG.5.1, XBB.1.5.70 and HK.3), were marked with different color. b–d The fluctuating frequencies of SARS-CoV-2 lineages identified in Guangdong province (b), mainland China (c), and regions outside of mainland China (d) were shown, respectively. e A heatmap depicting amino acid mutations in the spike protein. All sequences were obtained from GISAID, with sample collection dates between 1 October 2022 and 31 July 2023. By comparing to the ancestral sequence of BA.2.48*, BF.7.14*, and XBB.1 clades, mutations with frequencies over 3% were represented with a gradient color scale, transitioning from yellow to deep blue.
To illustrate the difference in variant distribution, we sourced all contemporary sequences from other regions within mainland China as well as from regions outside of China from public databases. As shown in Fig. 1b–d, while BA.5.2.48* lineages were dominant in Guangdong during the first wave, globally, BQ.1 and XBB.1 had become the dominant strains, replacing the BA.5 variants. The limited detection of BA.5.2.48* and BF.7.14* lineages outside mainland China suggests these strains had either limited competency compared to variants elsewhere or there were scarce opportunities for them to spread during the initial wave. This result also illustrated a picture of rapid spread, primarily driven by a founder effect, where a few initial strains sparked the first epidemic wave. The subsequent wave, from April to July 2023, was dominated by the XBB.1 lineages (Fig. 1b). The genetic landscape of strains in Guangdong became more diverse in this wave. We also quantify the potential transmission events through the phylogenetic trees by including randomly selected sequences from mainland China and regions out of China (Fig. S2). As anticipated, the increasing transmission evens between mainland China and international regions was observed after lifting NPIs, consistent with the fluctuations in the number of inbound and outbound travelers (Fig. S2).
We further analyzed the emergence and vanishing of mutations throughout these two major COVID-19 waves in mainland China by examining the frequency of amino acid mutations in the BA.5.2.48* (12,296 sequences) and BF.7.14* (5469 sequences) identified in mainland China and XBB.1 cluster sequences isolated in mainland China (17,695 sequences) and outside of China (339,038 sequences). The heatmap of amino acid mutations in the spike protein revealed limited non-synonymous mutations within the spike coding sequences of BA.5.2.48* and BF.7.14* lineages, despite the extensive spread of infections (Fig. 1e). Notably, while a few adaptive mutations emerged, their restricted spread suggests that the rapid transmission across a highly susceptible population surpassed the evolutionary pressure for these variants to persist. In contrast, the following XBB.1 wave witnessed relatively more non-synonymous changes, particularly E180V, F456L, and K478R found in spike protein. When compared to contemporary sequences from other regions, these spike mutations occurred at a relatively higher frequency in XBB.1 lineage sequences in mainland China (Fig. 1e and Fig. S3). For instance, the F456L and K478R mutations were found in 22% (3891 in 17,695) and 25 % (4455 in 17,695) of XBB.1 variant sequences in mainland China between 10 October 2022 and 31 July 2023, whereas they accounted for 5% (16,355 in 339,038) and 9% (30,016 in 339,038) of contemporary XBB.1 variant sequences outside of China. Additionally, mutations such as E180V and K478R co-occurred in sequences from mainland China, forming a genetic cluster with the majority of sequences from this area (Fig. S3). The persistence of these mutations was observed through the XBB.1 epidemic among multiple regions of mainland China (Fig. S3). The differences in mutation frequency during the XBB.1 epidemic wave could be attributed to the founder effect, a transmission/growth advantage in the context of heterogeneous population immunity, or both.
Sewage surveillance and serosurveys before and after the lifting of NPIs
The phylogenetic analysis indicated viral variations across the two epidemic waves and distinct patterns in virus transmission. Following the lifting of NPIs, there has been a decreased willingness among individuals to undergo molecular testing for SARS-CoV-217, even in the presence of clinical symptoms. This reluctance poses significant challenges in assessing the viral prevalence. To address this limitation, we conducted cross-sectional serosurveys of the general population in Guangzhou, prior to and after each epidemic wave (Fig. 2a). The dynamic of the epidemiology was determined according to the surveillance data from sewage and national sentinel hospitals. Sewage samples obtained from the Wastewater Treatment Plant in Guangzhou showed that viral loads peaked (9117.75 copies/mL) on 11 January 2023 and subsequently declined to a low level (50.86 copies/mL) in samples collected on 6 March 2023. An uptick in viral loads was noted starting in April 2023, with a peak (2888 copies/mL) observed on 9 May 2023 (Supplementary Table 5). The decrease was more gradual from June to July 2023. A highly consistent trend was also observed in the SARS-CoV-2 positive rate among Influenza-Like Illness (ILI) cases reported by national sentinel hospitals (Fig. 2a). These findings underscore the role of the BA.5 and XBB.1 variants in driving the nationwide epidemic in mainland China.

a The scheme of serosurvey was conducted before and after each epidemic wave, with the corresponding timing marked by dashed vertical lines. The SARS-CoV-2 viral loads detected in Guangzhou’s sewage (marked by purple line) and nationally reported SARS-CoV-2 positive percentage in influenza-like illnesses (ILIs) (sourced from https://www.chinacdc.cn/jkzt/crb/zl/szkb_11803/jszl_13141/202401/t20240110_271902.html, in Chinese) (marked by orange line) were plotted to show the epidemiological dynamics. b The cumulative percentage of 2nd and 3rd serosurvey population that had been vaccinated. A total of 1446 individuals with immunological backgrounds were included in the count. The vaccinated population is color-coded as follows: the first dose in orange, the second dose in green, the third dose in red, and the fourth dose in purple. c The overview of vaccination status among the surveyed subpopulations (n = 1408). Inactivated-virus vaccination (individuals received one to four doses of vaccination with the primary does being an inactivated-virus vaccine); unvaccinated (individuals who had not received any vaccination); others (those who had primarily received recombinant protein vaccine or adenovirus vaccine). A stacked column format on the right side of the panel showed the percentage of individuals with various vaccination statuses within each age group as of December 2023.
The first serosurvey of our study commenced from 12–31 October 2022, during a period when SARS-CoV-2 was rarely detected in the population with the stringent ‘zero-COVID’ policy. The second serosurvey was executed from 14 to 22 February 2023, a time when the viral load in sewage water was close to the detection threshold and the positive percentage of SARS-CoV-2 in ILI cases was extremely low (Fig. 2a). The third serosurvey was conducted from 1–17 July 2023, nearly two months following the second epidemic peak (Fig. 2a). The targeted collection aimed to include 1900 sera, from which 1696 samples (89.26%) were ultimately incorporated into the study, as detailed in Table 1. A total of 204 sera were excluded from the analysis for reasons including insufficient serum volume and the absence of demographic information.
In alignment with China’s COVID-19 vaccination strategy (see details in Methods), the study population was categorized into four age groups. Across all three serosurveys, women were found to be in the majority compared to men, accounting for over 50% of the participants. The median ages for the surveyed population were 39, 34, and 32 for the respective surveys. Individuals aged 18–59, made up the largest proportion of the study population, with 71.00%, 67.11%, and 56.97% participation in each of the three serosurveys, respectively (Table 1).
The initial serosurvey, comprising 100 participants, aimed to assess the immune status of the population before the emergence of the Omicron variants. Subsequent 2nd and 3rd serosurveys, with a total of 1,596 participants, were conducted to explore potential changes in immune response following the BA.5 and XBB.1 subvariant epidemic waves, respectively. The vaccination status for 2nd and 3rd serosurvey populations was illustrated in Fig. 2b, c. Among individuals with known vaccination status (n = 1446), 91.84% followed the inactivated-virus vaccination strategy (Fig. 2c). By October 2022, over 80% of the population had received their second vaccine dose, and 56.02% had received their third dose (Fig. 2b). Over 70% of individuals aged 3–17 had completed the two-dose primary immunization series, while nearly 70% of those over the age of 18 had completed the three-dose inactivated-virus vaccination course (Fig. 2c).
The association between nAb titers and variant infections
Molecular epidemiology data showed two Omicron BA.5 sublineages (including BA.5.2.48* and BF.7.14*), and XBB.1, were the main drivers of the first and second epidemic waves, respectively. Assessing neutralization antibody (nAb) levels of the population before and following each wave would provide insights into the dynamic of population immunity and the incidence of infections.
Pseudovirus-based serum neutralization tests, which utilize spike proteins from various SARS-CoV-2 variants, have been extensively employed to track the corresponding nAbs against these strains18. To validate the reliability of this method in our study, its accuracy was initially compared with the gold-standard microneutralization test (MNT). This evaluation involved nAb titers against the BA.5 and XBB.1.9 strains in 100 serums collected from RT-PCR and viral sequencing confirmed individuals from the respective waves. A robust linear regression analysis yielded a highly significant correlation between pseudovirus neutralization test (PNT) and MNT log2 values (Fig. 3a, b), with a coefficient of determination (R²) exceeding 0.95 for both strains, underscoring a high degree of methodological agreement.

a Presented the neutralization titers of 50 reference sera collected post the first wave, confirming BA.5* infections. b Illustrated the neutralization titers from 50 reference sera post the second wave, confirming XBB.1.9* infections. All neutralization titer values were determined through neutralization experiments with authentic viruses and pseudoviruses for BA.5 and XBB.1.9 variants. The figure includes the regression equation and the coefficient of determination (R²) for each set of data. Abbreviations: MNT, microneutralization test; PNT, pseudovirus neutralization test. c, d The ROC curves depict the area under the curve (AUC) and the cut-off points for discriminating between BA.5 (c) and XBB.1 (d) infections based on pseudovirus serum neutralization titers. The optimal cut-off value, indicated in the upper left corner of each ROC curve plot, is determined using the Youden index and reflects the balance between sensitivity and specificity. The AUC values along with their 95% confidence intervals (95% CI) are displayed in the figure.
We proceeded to evaluate the population’s nAb levels against BA.5 and XBB.1.9 to estimate the potential infection rate during each wave, based on the following hypotheses: (1) Despite the high coverage of inactivated-virus vaccination in the population, a limited nAb titer may be generated in response to Omicron variants19. Consequently, a possible threshold of BA.5 nAb level could be established to differentiate between individuals infected and not infected with BA.5 following the first epidemic wave. (2) The noted antigenic divergence between the XBB.1 variants and the BA.5 variants20,21,22,23 suggested the potential to evaluate infections caused by XBB.1 following the second epidemic wave.
To test the hypothesis, we analyzed 100 serum samples collected before BA.5* epidemic wave. Our findings are in line with those of other studies: the majority of these serum samples had very low or undetectable levels of nAbs against the BA.5 variant (Fig. 4a), despite the presence of nAbs against the wildtype strain. To establish the nAb threshold for distinguishing infections caused by the BA.5 and XBB.1 variants following the two epidemic waves, we used data from individuals with confirmed infections of these variants post-epidemic waves as the positive reference group (for details, see Table S4). Serum samples collected prior to each wave served as the negative reference. We compared the pVNT50 (half-maximal plaque reduction neutralizing titer) levels against BA.5 or XBB.1.9 between the positive and negative reference serums.

The nAb levels to different Omicron variants were measured and the corresponding infection rates were estimated by age groups prior to (a) and post the first (b–d) and second (e–g) epidemic waves. a The population nAb levels against BA.5 variant pre-epidemic wave (n = 100 biologically independent samples). b The estimated infection rate of BA.5 variants after the first wave (n = 836 biologically independent samples); c, d The corresponding nAb levels to BA.5 (c) and XBB.1.9 (d) from the BA.5 variants infected individuals (n = 765 biologically independent samples). e The estimated infection rate of Omicron XBB.1 variants during the second wave (n = 760 biologically independent samples). f, g The corresponding nAb levels to XBB.1.9 from XBB.1 variants infected (n = 339 biologically independent samples) (f) and uninfected (n = 421 biologically independent samples) individuals (g). The number of individuals in each age group is shown below the x axis. b, e Solid horizontal and vertical lines are used to show the corresponding 95% credible intervals (CrI) of the estimated infection rate based on Bayesian analysis on serosurvey samples. c, d, f, g Pseudovirus assays for each serum sample are technically replicated twice. Multiple two-tailed Mann–Whitney tests were used to compare the pVNT50 differences between the two groups. All exact P values are noted. Error bars represent geometric mean and its 95% confidence interval. Values above the symbols denote the geometric mean pVNT50 values. Source data are provided as a Source Data file.
Using the ROC curve analysis, we determined the optimal threshold for SARS-CoV-2 nAbs to differentiate between BA.5 and XBB.1 infections. As illustrated in Fig. 3c, d, the BA.5 infection prediction accuracy was exceptional, with an area under the ROC curve (AUC) of 0.994, signifying a highly accurate discrimination capability. A pVNT50 threshold of ≥28.63 achieved 95.0% sensitivity and 100% specificity, confirming the method’s effectiveness in detecting BA.5 post first wave infections. This finding indicated that the relative nAb titers to BA.5 can accurately identify individuals infected with the BA.5 variant following the first epidemic wave. Similarly, the antigenic differences between BA.5 and XBB.1 enabled the detection of XBB.1 infections through nAb levels against XBB.1.9. The predictive testing of reference samples for XBB.1 infection showed a strong area under the curve (AUC) of 0.960, indicating substantial discrimination power. A pVNT50 cut-off of ≥177.19 corresponded to 94.0% sensitivity and 85.9% specificity, reinforcing the method’s accuracy in identifying XBB.1 infections. These results underscored the practicality of pVNT50-based serosurveys as a reliable method for infection rate estimation, following the expansive BA.5 and XBB.1 epidemic waves in Guangdong, China.
The population neutralization immune response and infection rate following the Omicron waves
The above results showed the corresponding nAb levels to BA.5 and XBB.1.9 could infer the potential infection rates of the population following the epidemic waves. As shown in Fig. 4a, the first serosurvey conducted in October 2022, prior to the first epidemic wave, showed that the population exhibited very low levels of nAbs against BA.5 (GMT = 4.3, 95% CI: 3.6–5.3). This susceptibility, coupled with the lifting of NPI measures on 7 December 2022, resulted in a rapid and intense surge in infections24. The high prevalence of Omicron variants could be inferred from the elevated positive rate of SARS-CoV-2 among ILI cases reported by national sentinel hospitals (Fig. 2a). However, the precise infection rates across the entire population, and particularly within different age groups, remain largely unknown. Utilizing the ROC curves depicted in Fig. 3, we estimated the potential infection rates of BA.5* and XBB.1* following the first and second epidemic waves. On February 2023, one month after the peak of the first epidemic wave, the serosurvey data indicated that the entire population had estimated infection rates as high as 96.23% (95% CrI: 94.13–98.11%), highlighting the extensive spread of the virus after the releasing of NPIs.
For stratified age groups, the estimated infection rate was slightly lower in the older population (88.46%, 95% CrI: 81.31–94.57%) (Fig. 4b). The youngest age group, children aged 3–11 years, displayed the highest absolute nAb level against BA.5 after the first epidemic, with a pVNT50 of 604.8 (95% CI: 435.7–839.6). This was closely followed by the 12–17 years group, with the nAb level remaining high at 535.7 pVNT50 (95% CI: 427.9–670.7). Both these young age groups exhibited significantly higher nAb levels compared to the 18–59 years old group (P = 0.001), highlighting a potentially stronger immune response in children and adolescents (Fig. 4c). Intriguingly, the individuals over 60 years had the lowest estimated infection rates (88.46%, 95% CrI: 81.31–94.57%) among four age groups, possibly due to the lower risk of exposure compared to other age groups25.
We additionally assessed the nAb response to the XBB.1.9 variant in the population presumed to have been infected with BA.5 variants (n = 765) following the first epidemic wave (Fig. 4d). Despite the expected reduction in nAb levels due to antigenic differences, a notably higher nAb response to XBB.1.9 was detected in the 3–11 years age group among those with prior BA.5 infections (GMT = 65.9, 95% CI: 51.0–85.1), which was statistically higher than all other age categories (both P < 0.05). This suggests that BA.5 infections in young children may elicit a broader/stronger nAb response. In contrast, in the 18–59 age group, the level of nAb against XBB.1.9 induced by BA.5 infection was lowest, and was ~1.88 times and 1.84 times lower than that in adolescents (aged 12–17) and the elderly (aged ≥ 60), respectively (both P < 0.0001).
Following the establishment of the broad population immunity after the BA.5 wave, a combination of waning immunity and viral evasion led to a second epidemic wave driven by XBB.1 variants, ~4 months later. Serosurvey data collected after the second wave indicated significant differences in nAb levels across age demographics. Notably, despite widespread XBB.1 variants infections, children aged 3–11 years exhibited a substantially lower infection rate of 10.51% (95% CrI: 0.83–24.89%), compared to other age groups, which had rates of 40.26% (95% CrI: 29.01–51.39) in those aged 12-17 years, 41.40% (95% CrI: 35.68–47.24%) in individuals aged 18–59 years, and 37.72% (95% CrI: 28.32–47.06%) in those over 60 years (Fig. 4e). This pronounced disparity in infection rates between the two waves for young children may indicate either a reduced susceptibility to XBB.1 or an inherently higher nAb level against it in this age group. The enhanced cross-reactive nAb response observed in younger individuals post-BA.5 infection (Fig. 4d) could potentially account for these variations.
To determine the protective threshold of nAb levels against XBB.1 variants infection, we assessed the relative nAb levels to XBB.1.9 in a subgroup of the population presumed to be infected (n = 339), defined by a pVNT50 above 177.19. Notably, older individuals, aged over 60 years, exhibited significantly higher levels of nAbs (GMT = 1285.0, 95% CI: 1053.0–1569.0) compared to other age groups after reinfections with the XBB.1 variant (both P < 0.05) (Fig. 4f). This pattern contrasts with observations from the previous BA.5 wave, where the nAb levels induced by BA.5 infections in individuals over 60 years old were not significantly different from those in other age groups. Given that the severity of illness may be correlated with the nAb response level, further epidemiological investigation is warranted to explore potential differences in illness severity following the reinfections among various age groups.
While in the uninfected subgroup (n = 421), the nAb levels against XBB.1.9 was not significantly varied by ages, except for the younger age group (12–17 years, GMT = 30.9, 95% CI: 24.3–39.4) and the older age group (over 60 years, GMT = 27.9, 95% CI: 21.7–35.8), which exhibited slightly higher nAb levels compared to the adult population (18–59 years, GMT = 19.6, 95% CI: 16.9–22.8) (Fig. 4g). These findings will help establish a baseline for nAb levels in future serosurvey, crucial for assessing risks from emerging variants.
Discussion
The lifting of China’s ‘dynamic zero-COVID’ policy represents a significant shift in the country’s management of the COVID-19 pandemic. A previous modeling study suggested that the Omicron variant could spread very quickly, with an epidemic doubling time of between 1.6 and 2.0 days following the full exit from zero-COVID in mainland China. The study estimated an infection rate among the population of between 95% and 99%26. In this study, we provide real-world data obtained through an integrated approach of molecular and serological surveillance in Guangdong, China, highlighting key aspects of viral evolution and changes in population immunity during this pivotal transition.
Firstly, the study investigated the dynamic interplay between population immunity and viral evolution. During the first wave, the dominance of BA.5.2.48 and its sublineages highlights the importance of the founder effect, where specific lineages introduced early on can disproportionately shape transmission dynamics. The BA.5.2.48 variants with a limited genetic diversity (Fig. S1), rapidly drove infection rates above 90%, thereby rapidly establishing a robust population immunity barrier (Fig. 4b, c). This exerted strong selective pressure, which hindered the establishment of other potentially more transmissible lineages, leading to a disparity in variant prevalence between mainland China and other regions. The concurrent establishment of broad population immunity results in a negligible accumulation of potential adaptive mutations in the BA.5.2.48* as well as BF.7.14* spike protein by April 2023 (Fig. 1e). This suggests that rapid transmission within a highly susceptible population outpaced evolutionary pressure for the accumulation of adaptive mutations. In contrast, the second wave driven by XBB.1 encountered a more heterogeneous immune landscape, resulting in varied infection rates across different age groups (Fig. 4e). This allowed for some selective pressure and the emergence of potentially adaptive mutations.
Although the SARS-CoV-2 variants distribution was similar to the global pattern during the second wave, some local genetic clusters were observed to lead higher frequency of some potential adaptive mutations detected, including F456L and K478R (Fig. S3). F456L was rarely detected before 14 July 2023 based on the public dataset (up to 28 Jan 2024). However, a phylogenetic cluster of XBB.1 with the F456L mutation was identified from sequences collected across multiple regions of mainland China between April and July 2023 (Fig. S3). The dominance of F456L has been observed in the following omicron epidemic led by XBB.1.9, EG.5.1, and HK.3 appearing to enhance neutralization escape and ACE2 binding affinity of these variants27,28,29,30. The K478R mutation also observed in diverse Omicron sublineages (BA.2, BA.5, XBB.1.5, and XBB.2.3), presents an intriguing case. Studies suggest it enhances viral binding to host cells, potentially increasing infectivity, while its location within a key antibody target region may also facilitate immune escape31. These findings highlight the dynamic interplay between population immunity and viral evolution in shaping Omicron’s trajectory. The initial high-transmission wave, coupled with the robust and broad population immunity it induced, significantly constrained BA.5.2.48 evolution. In contrast, the immunological heterogeneity during the XBB.1 wave may provide opportunities for selective pressure and facilitate the emergence, spread, and persistence of adaptative strains. As shown in the surveillances from both sewage water and sentinel hospitals, the moderate prevalence of the XBB.1 variant persisted two months after the peak of the second epidemic wave (Fig. 2a).
Cross-sectional serosurvey studies conducted after the BA.5 and XBB.1 epidemic waves provide valuable insights into the variability of immune responses across different age groups, particularly in the context of reinfection. While earlier reports, including those from the United State32 and Canada33, consistently documented higher infection rates in younger age groups and lower rates in older adults, likely due to vaccination disparities and pre-existing immunity resulting from infections, our findings present a more nuanced picture in mainland China.
During the first epidemic wave, there is little variance in infection rates among different age groups with estimated infection rates ranging from 88.46% to 97.54%. This finding challenges the age-based seroprevalence in early SARS-CoV-2 epidemic34 suggesting that the Omicron variant effectively may circumvent pre-existing immunity elicited by vaccination across all age groups. The subsequent XBB.1 epidemic wave revealed a distinct age-related pattern, with young children exhibiting a significantly lower infection rate compared to other age groups (Fig. 4e). As shown in Fig. 4d, quantitative testing of the nAb level suggests that this shift might be attributed to the enhanced immune response in children. Following the BA.5 variants infections, only a significant difference was observed between 3 and 11 and 18 and 59 age groups for BA.5 nAb (Fig. 4c). By contrast, a significantly higher level of nAb level to XBB.1.9 was observed in the young age group than in other groups. These results suggest the young age population may develop a broader immune response after BA.5 infection, offering them greater protection against subsequent infection with XBB.1 variants. Reinfection of XBB.1 variants was found to elicit a high level of XBB.1.9-specific nAb across all age groups, with the GMT of pVNT50 titer ranging from 612 to 1285 (Fig. 4f). These results are consistent with the recent findings in mouse models and human cohorts, which indicate that double Omicron exposure can induce a large proportion of matured Omicron-specific antibodies. These antibodies possess distinct receptor-binding domain epitopes to WT-induced antibodies and immune imprinting caused by vaccination of the ancestral strain was largely mitigated35. The disparity in infection rates and neutralization response elicited after the infection among different age groups will have significant implications for understanding immune dynamics and informing targeted public health interventions in the face of evolving viral variants.
The transition of COVID-19 out of pandemic to a localized epidemic phase marks a new chapter in our relationship with the virus. However, the epidemic pattern of COVID-19 is not homogenous. Our findings, alongside data from other regions, highlight the significant regional heterogeneity in terms of viral landscape, rate of severe cases, and the distribution of etiological factors. While mainland China has been dealing with BA.5*, XBB.1* dominated elsewhere. Concurrently with this paper’s submission, the newly emerged KP.2, the subvariant of JN.1, has become one of the major circulating variants in the U.S. but remains rarely detected in mainland China. This disparity, likely shaped by vaccination schemes and the history of prior infections, underscores the necessity for continuous serosurveys, region-specific data analysis, and international data exchange. These efforts are crucial for accurate risk assessment, targeted interventions, and effective management of the diverse localized landscape of COVID-19.
Limitations
Some limitations of our study should be acknowledged. Firstly, our research utilized a multiple cross-sectional approach to assess antibody levels among different age groups before and after each Omicron wave. While this design provides valuable insights into the alterations in population immunity at different time points, it does not offer the longitudinal perspective of a cohort study, which may limit the depth of our interpretation regarding the dynamics of immune response over time. Secondly, we measured the nAb levels in 1596 individuals against Omicron variants BA.5 and XBB.1. These measurements are instrumental in estimating infection rates and the immune status post-epidemic waves. However, our study does not include data on cellular immune responses. The absence of this information may result in an incomplete characterization of the immune response to Omicron variants across different age demographics. Thirdly, the serosurvey was conducted in the capital city of Guangdong province. While the national vaccination strategy and the nationwide trajectory of COVID-19 during 2022–2023 suggest a uniform spread (as illustrated in Fig. 2a), the generalizability of our findings to other regions in China requires validation through additional research in diverse geographical settings.
Methods
Ethics statement
This study was approved by the institutional ethics committee of the Guangdong Provincial Center for Disease Control and Prevention (Approval protocol number: W96-027E-202214). Written consent was obtained from patients or their guardian(s) when samples were collected. Patients were informed about the surveillance before providing written consent, and data directly related to disease control were collected and anonymized for analysis.
Study design and serum sampling
Three cross-sectional serosurveys were conducted in Guangzhou between October 2022 and July 2023. The timeline of the SARS-CoV-2 serosurveys in correlation with epidemiological waves is illustrated in Fig. 2a.
Given the strict NPIs implementation, it is reasonable to assume that the majority of people in China were naive to SARS-CoV-2 infections before the lifting of NPIs. It is well-established that the inactivated-virus vaccination based on the wildtype SARS-CoV-2 provided limited nAb response specific to Omicron variants35. To ascertain the baseline level of population immunity before the Omicron wave, 100 serum samples were collected in October 2022, prior to the release of NPIs. All samples were obtained from individuals confirmed not to be infected with SARS-CoV-2 by the Guangdong Province 24-hour nucleic acid testing reporting system36.
For the subsequent two serological surveys, serum specimens were obtained using a multi-stage sampling approach, reflecting the community population. The sample size was calculated using the formula:
Where, N is the population of Guangzhou City, n1 is the number of samples calculated through simple random sampling (i.e., the middle part of formula (1)). Deff (design effect) is 3, the significance level α is 0.05, and the positive rate P in the first serosurvey was set to 0.8, and 0.6 in the second. The admissible error δ was 0.1 P. The minimum required sample size was calculated as 768. However, considering the practical challenges of survey and sampling, the number of survey samples in each round was set at 900. The sampling process categorized regions into two groups based on the population density of Guangzhou’s 11 administrative districts. Haizhu, Yuexiu, Tianhe, and Liwan, with a population density of over 22,000 people/km², were considered central areas, while Baiyun, Panyu, Conghua, and seven other districts were non-central areas with a density of 5000–21,000 people/km². Three areas were randomly selected in a 1:2 ratio of central to non-central areas. Finally, 300 serum samples were chosen from 4 to 5 streets or towns in each area, providing a representative sample size for the serological survey across Guangzhou city.
For the second and third serosurveys, individuals with fever or those who had received immunotherapy within the past three months were excluded. Serums were separated after the blood collection and transported to the laboratory of the Guangdong Provincial Center for Disease Control and Prevention via a complete cold chain. Participants were asked to complete an online survey recording their sociodemographic and vaccination history, as well as information on SARS-CoV-2 infections if molecular testing had been conducted.
The participants were categorized into four age groups corresponding to distinct vaccination strategies. For individuals aged 3-17, the basic immunization required the completion of two doses of an inactivated virus vaccine. Those 18 and older were to receive a booster immunization, which could include an additional dose—either inactivated-virus, recombinant protein, or adenovirus vector vaccine—following their initial two doses of inactivated-virus vaccine, or alternatively, completing a two-dose series of the adenovirus vector vaccine.
Pseudovirus neutralization tests
HEK 293 T cells stably expressing human ACE2 were kindly provided by Dr. Hu from Guangzhou Medical University37. The Pseudotype-based neutralization assay was conducted according to the published method18. The neutralization activity was assessed by measuring the reduction in luciferase gene expression. Briefly, a 20 μl aliquot of serum sample was then continuously diluted in the medium, in a series of 1:4 to 1:4096 dilutions. The SARS-CoV-2 pseudovirus, adjusted to a titer of 3 × 104 TCID50/mL based on the prior titration results, was added to the diluted serum in a volume of 60 μL. This serum-pseudotype mixture was incubated at 37 °C and 5% CO2 for one hour. Subsequently, 100 μL of cell suspensions containing human 293T-hACE2 cells (5 × 105 cells/mL) were added to the mixtures, and the experiments were carried out in duplicate. Following a 24-hour incubation at 37 °C, the neutralizing potency of the plasma samples was evaluated using a luciferase assay. The 50% pseudovirus neutralization titer (pVNT50) was defined as the plasma dilution that reduced the relative light unit (RLU) by 50% compared to the virus control wells, with the background RLU from the cell control wells subtracted.
Microneutralization tests
Microneutralization assays were conducted in the Biosafety Level III Laboratory (BSL-3) of the Guangdong Center for Disease Control and Prevention. In brief, Omicron BA.5 (GDPCC 2.00303) and XBB.1.9 (GDPCC 2.01543) were used as representative viral strains. Serum samples were serially diluted four times with the minimal required medium, ranging from a dilution of 1:4 to 1:1024. Each diluted serum sample was mixed with 100 SARS-CoV-2 TCID50 and incubated at 37 °C for 1 hour. The serum-virus mixture was then added to VeroE6 cells. After incubation for 7 days, the cytopathic effect was observed. The highest dilution of the nAb that resulted in a 50% inhibition rate of the cytopathic effect was recorded as the titer.
Sewage surveillance
During the study period, from November 2022 to July 2023 (Supplementary Table 5), we collected raw sewage samples monthly from the primary sedimentation tanks at the Liede Wastewater Treatment Plant (WWTP) in Guangzhou City, China, in accordance with previously described methods38. This WWTP serves a population of ~2,150,000. The raw water samples, 10 mL in volume, were processed using the MagMAX™ Wastewater Ultra Nucleic Acid Isolation Kit (A52606, Thermo) to 100 μL concentrated nucleic acids. 5 μL extracted RNA was quantified with a SARS-CoV-2 RT-PCR commercial kit (2201001460, Mingde Biotech). For viral load quantification, a reference pseudovirus was utilized, as detailed in a previous study36. The pseudovirus was ten-fold serially diluted, ranging from 1.67 × 103 to 1.67 × 107 copies/mL, and viral RNA was extracted and PCR quantified to construct a standard curve. The relative viral loads in the extracted RNA of sewage samples (VL_RNA, copies/ml) were converted to viral RNA copies per mL based on the Ct value of the ORF1ab gene. The concentration efficiency (Eff, 45% in our study) had been previously determined according to the pre-processing method of this study39. Ultimately, the relative SARS-CoV-2 viral RNA loads in the sewage samples (VL_sewage, copies/mL) were calculated using the following equation: \({{{\bf{VL}}}}\_{{{\bf{sewage}}}}{{=}}\frac{{{{{\bf{VL}}}}}\_ _{{{{\bf{RNA}}}}}{{\times }}{{{\bf{100}}}}{{\mu }}{{{\bf{L}}}}}{{{{\bf{Eff}}}}{{\times }}{{{\bf{10}}}}{{{\bf{mL}}}}{{\times }}{{{\bf{1000}}}}}\).
Statistical analyses
For the comparison between MNT and PNT, all the titers experienced initial base-2 logarithmic transformation. We utilized the logarithm base-2 values of PNT as the independent variable and the corresponding logarithm base-2 values of MNT as the dependent variable. Employing a linear regression model, we calculated the R-squared value to quantify the strength of the relationship between PNT and MNT. In the serosurvey result, all titers are reported as geometric mean and its 95% confidence interval. Multiple Mann-Whitney tests were used to compare the pVNT50 differences between the two groups. All analyses were conducted using R (version 4.2.2) and GraphPad Prism (version 10.1.2). A two-sided p value of <0.05 was considered to indicate statistical significance.
Infection rate estimation
We used ROC curve analysis to predict infection rate, establishing cutoff points with the Youden index for the measured variables. This was complemented by the calculated AUC, which served as a quantitative measure of the markers’ discrimination power between the two groups.
To integrate uncertainty arising from ROC cut-off values sensitivity and specificity, we used a Bayesian model to produce a posterior distribution of infection rate based on serological data. It accounted for the uncertainty associated with finite sample size, as referenced in a previous study40. Age-stratified infection rate estimates were computed for each phase independently using a Bayesian model. This approach, described in detail in the previous study40, considers uncertainty due to finite laboratory validation data and produces estimates using typical choices of uninformative or weakly informative prior distributions. The estimates are presented as posterior means and 95% CrI based on 10,000 samples from a Bayesian posterior distribution. All calculations were conducted using Python, specifically utilizing the code found at (github.com/LarremoreLab/covid_serological_sampling).
Whole-genome sequencing
Whole-genomic sequencing of SARS-CoV-2 was performed as previously described36, 41. Briefly, cDNA was generated from extracted viral RNA by using SuperScript IV (Thermo Fisher, 18091). Multiplex PCR was performed by using different primer schemes for the following sequencing. The library was constructed with Illumina Nextera XT DNA Library Preparation Kit (Illumina, FC-131-1096) and was sequenced with PE150 (Illumina, FC-420-1003) or SE100 (Illumina, 20044338) on Illumina Miniseq. Molecular surveillance of SARS-CoV-2 was performed by Guangdong provincial CDC and 21 prefecture-level CDCs as previously described34. Briefly, once virus-positive samples were confirmed, the oropharyngeal swab samples preserved in 3 mL of viral transport medium were sent to Guangdong CDC in 24 h. Patients were informed about the surveillance before providing written consent. In total, 6183 samples were collected between 1 October 2022 and 31 July 2023 and subjected to next-generation sequencing. From this dataset, we ultimately reported 5813 high-quality genome sequences (>95% genome coverage), spanning the period from 22 October 2022, to 5 July 2023.
Dataset construction and sampling procedure
We downloaded all contemporary sequences available on GISAID (from 1st October 2022 to 31 July 2023). We followed the criteria used in the previous study to construct the dataset. Briefly, we filtered sequences <27,000 bases long or flagged by the Nextclade tool (36) for suspiciously clustered single-nucleotide polymorphisms (SNPs) [quality control (QC) SNP clusters status metric not “good”; ≥6 mutations in 100 bases], too many private mutations (QC private mutations status metric not good; ≥10 mutations from the nearest tree node), or overall bad quality (Nextclade QC overall status “bad”). Three datasets were generated corresponding to the SARS-CoV-2 genome sequences collected from different regions between 1 October 2022 and 31 July 2023. Dataset A: 5813 SARS-CoV-2 genome sequences generated in this study representing the circulating strains in Guangdong; Dataset B: 32,142 SARS-CoV-2 genome sequences collected from other regions of mainland China; Dataset C: 1,078,904 SARS-CoV-2 genome sequences collected outside of mainland China.
Phylogenetic analysis
To visualize the phylogenetic distribution of Guangdong sequences during the study period, we randomly selected 500 sequences from Dataset A and 500 sequences from Dataset C and combined them with reference sequences of major lineages (according to the reference sequences in the nextclade dataset, https://clades.nextstrain.org/). The phylogenetic tree reconstruction was performed by using NEXTSTRAIN’s (https://www.nextstrain.org/) augur (https://www.docs.nextstrain.org/projects/augur/en/stable/) pipeline. Multiple sequence alignment was performed by using Mafft by using MN908947 as the reference. We followed the Nextstrain pipeline’s recommendation to mask the first 100 and last 50 sites of the alignment because the start and end of SARS-CoV-2 sequences are prone to sequencing errors. Maximum-likelihood trees were estimated by using FasttreeMP42 and refined with TreeTime43 to infer the molecular-clock phylogeny. Finally, the collection of all annotated nodes and metadata was exported to the interactive phylodynamic visualizing tool Auspice (https://auspice.us/) in JSON format44. The potential transmissions between mainland China, Guangdong, and regions out of China were inferred by using TreeTime.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The findings of this study are based on metadata associated with a total of 32,142 sequences from other regions of mainland China and 1,078,904 sequences outside of mainland China. All data were sourced from the Global Initiative on Sharing All Influenza Data (GISAID) and were downloaded on 27 Jan 2024 via gisaid.org (GISAID: EPI_SET_240726sr and EPI_SET_240726ta). For a detailed list of contributors associated with each sequence utilized in this study, please refer to the respective DOIs: https://doi.org/10.55876/gis8.240726sr and https://doi.org/10.55876/gis8.240726ta. All Guangdong sequences generated in this study have been submitted to the GISAID (https://platform.epicov.org/) and Genbank (https://www.ncbi.nlm.nih.gov/genbank/). The submission numbers for these sequences, along with scripts for phylogenetic analysis and figures, are accessible at the following GitHub repository: tyumen001/molecular-epi-ncov and provided in Supplementary Data 1. Data to reproduce Fig. 1 can be found in Source Data File (sheet 1–4). All Serosurvey data is available in Source Data File (sheet 5&7–15). Additionally, the sewage data have been documented in sheet 6 of the Source Data File and are further elaborated in Supplementary Table 5. Source data are provided in this paper.
Code availability
Code is available at Github: https://github.com/tyumen001/molecular-epi-ncov.
References
Statement on the fifteenth meeting of the IHR. Emergency Committee on the COVID-19 pandemic [Available from: https://www.who.int/news/item/05-05-2023-statement-on-the-fifteenth-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-coronavirus-disease-(covid-19)-pandemic] (2005).
Vitiello, A., Ferrara, F., Auti, A. M., Di Domenico, M. & Boccellino, M. Advances in the Omicron variant development. J. Intern. Med. 292, 81–90 (2022).
Cumulative Number of COVID-19 Cases by Region [Available from: https://outbreak.info/regions].
Yu, J. et al. Neutralization of the SARS-CoV-2 omicron BA.1 and BA.2 variants. N. Engl. J. Med. 386, 1579–1580 (2022).
Xu, R., Wang, W. & Zhang, W. As the SARS-CoV-2 virus evolves, should omicron subvariant BA.2 be subjected to quarantine, or should we learn to live with it? Front. Public Health 10, 1039123 (2022).
Pan, Y. et al. Characterisation of SARS-CoV-2 variants in Beijing during 2022: an epidemiological and phylogenetic analysis. Lancet 401, 664–672 (2023).
Uriu, K. et al. Enhanced transmissibility, infectivity, and immune resistance of the SARS-CoV-2 omicron XBB.1.5 variant. Lancet Infect. Dis. 23, 280–281 (2023).
Kaku, Y. et al. Virological characteristics of the SARS-CoV-2 JN.1 variant. Lancet Infect. Dis. 24, e82 (2024).
Kaku Y. et al. Virological characteristics of the SARS-CoV-2 KP.2 variant. Lancet Infect. Dis. 24, e416 (2024).
Legros, V. et al. A longitudinal study of SARS-CoV-2-infected patients reveals a high correlation between neutralizing antibodies and COVID-19 severity. Cell Mol. Immunol. 18, 318–327 (2021).
Garcia-Beltran, W. F. et al. COVID-19-neutralizing antibodies predict disease severity and survival. Cell 184, 476–488.e11 (2021).
Nathanielsz, J., Toh, Z. Q., Do, L. A. H., Mulholland, K. & Licciardi, P. V. SARS-CoV-2 infection in children and implications for vaccination. Pediatr. Res. 93, 1177–1187 (2023).
Anda, E. E. et al. Seroprevalence of antibodies against SARS-CoV-2 in the adult population during the pre-vaccination period, Norway, winter 2020/21. Eur. Surveill. 27, 2100376 (2022).
Collier, D. A. et al. Age-related immune response heterogeneity to SARS-CoV-2 vaccine BNT162b2. Nature 596, 417–422 (2021).
Released by the Taskforce for Joint P. Control mechanism for C-utSCoPRCoD. Prevention and control measures to further optimize COVID-19 response. Health Care Sci. 2, 7–9 (2023).
Attwood, S. W., Hill, S. C., Aanensen, D. M., Connor, T. R. & Pybus, O. G. Phylogenetic and phylodynamic approaches to understanding and combating the early SARS-CoV-2 pandemic. Nat. Rev. Genet. 23, 547–562 (2022).
Li, X. et al. Knowledge and willingness toward SARS-CoV-2 rapid antigen testing among older adults in China: a nationwide cross-sectional study. Aging Clin. Exp. Res. 35, 3127–3136 (2023).
Nie, J. et al. Quantification of SARS-CoV-2 neutralizing antibody by a pseudotyped virus-based assay. Nat. Protoc. 15, 3699–3715 (2020).
Cao, Y. et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 608, 593–602 (2022).
Yang, J. et al. Low levels of neutralizing antibodies against XBB Omicron subvariants after BA.5 infection. Signal Transduct. Target. Ther. 8, 252 (2023).
Zhu, A. et al. Antigenic characterization of SARS-CoV-2 Omicron subvariants XBB.1.5, BQ.1, BQ.1.1, BF.7 and BA.2.75.2. Signal Transduct. Target. Ther. 8, 125 (2023).
Delphine P. et al. Distinct evolution of SARS-CoV-2 Omicron XBB and BA.2.86/JN.1 lineages combining increased fitness and antibody evasion. Nat. Commun. 13, 2254 (2024).
Mykytyn, A. Z. et al. Antigenic mapping of emerging SARS-CoV-2 omicron variants BM.1.1.1, BQ.1.1, and XBB.1. Lancet Microbe 4, e294–e295 (2023).
Prevention CCfDCa. National situation of COVID-19 infection 2024. Available from: https://www.chinacdc.cn/jkzt/crb/zl/szkb_11803/jszl_13141/202401/t20240110_271902.html.
Liang, Y. et al. Estimating changes in contact patterns in China over the first year of the COVID-19 pandemic: implications for SARS-CoV-2 spread - four cities, China, 2020. China CDC Wkly. 5, 113–119 (2023).
Goldberg, E. E., Lin, Q., Romero-Severson, E. O. & Ke, R. Swift and extensive omicron outbreak in China after sudden exit from ‘zero-COVID’ policy. Nat. Commun. 14, 3888 (2023).
Faraone, J. N. et al. Immune evasion and membrane fusion of SARS-CoV-2 XBB subvariants EG.5.1 and XBB.2.3. Emerg. Microbes Infect. 12, 2270069 (2023).
Focosi, D., Spezia, P. G., Gueli, F. & Maggi, F. The era of the FLips: how spike mutations L455F and F456L (and A475V) are shaping SARS-CoV-2 evolution. Viruses [Internet]. 16, 3 (2024).
Qu P. et al. Immune evasion, infectivity, and fusogenicity of SARS-CoV-2 BA.2.86 and FLip variants. Cell 187, 585–595.e6 (2024).
Jian, F. et al. Convergent evolution of SARS-CoV-2 XBB lineages on receptor-binding domain 455-456 synergistically enhances antibody evasion and ACE2 binding. PLoS Pathog. 19, e1011868 (2023).
Yamasoba, D. et al. Virological characteristics of the SARS-CoV-2 omicron XBB.1.16 variant. Lancet Infect. Dis. 23, 655–656 (2023).
Freedman, N. D. et al. COVID-19 SeroHub, an online repository of SARS-CoV-2 seroprevalence studies in the United States. Sci. Data. 9, 727 (2022).
Skowronski, D. M. et al. Serial cross-sectional estimation of vaccine-and infection-induced SARS-CoV-2 seroprevalence in British Columbia, Canada. CMAJ 194, E1599–E1609 (2022).
Le Vu, S. et al. Prevalence of SARS-CoV-2 antibodies in France: results from nationwide serological surveillance. Nat. Commun. 12, 3025 (2021).
Yisimayi, A. et al. Repeated Omicron exposures override ancestral SARS-CoV-2 immune imprinting. Nature 625, 148–156 (2024).
Li, B. et al. Viral infection and transmission in a large, well-traced outbreak caused by the SARS-CoV-2 Delta variant. Nat. Commun. 13, 460 (2022).
Hu, L. et al. The E484K substitution in a SARS-CoV-2 spike protein subunit vaccine resulted in limited cross-reactive neutralizing antibody responses in mice. Viruses 14, 854 (2022).
Lu, J. et al. Capturing noroviruses circulating in the population: sewage surveillance in Guangdong, China (2013–2018). Water Res. 196, 116990 (2021).
Yang, H. et al. Research on high-throughput enrichment method of sewage viruses based on magnetic bead adsorption. Chin. J. Virol. 39, 1623–1629 (2023).
Larremore, D. B. et al. Estimating SARS-CoV-2 seroprevalence and epidemiological parameters with uncertainty from serological surveys. eLife 10, e64206 (2021).
Lu, J. et al. Genomic epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell 181, 997–1003.e9 (2020).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010).
Sagulenko, P., Puller, V. & Neher, R. A. TreeTime: maximum-likelihood phylodynamic analysis. Virus Evol. 4, vex042 (2018).
Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018).
Acknowledgements
This work was supported by the Natural Science Foundation of Guangdong Province (2023A1515011927, J.L.), the Guangzhou Science and Technology Project (202102080590, Z.L.), the Guangdong Science and Technology Program (2021B1212030007, J.H.) and Shenzhen San-Ming Project (SZSM201809085, S.X.). We thank Oliver Pybus for insightful discussions.
Author information
Authors and Affiliations
Contributions
J.L., L.S., and B.Li designed the study. L.Q., M.Y., C.X., H.Y., L.Y., H.L., H.Z., S.X., and Pengwei Hu undertook fieldwork and experiments. Z.L., P.H., and L.Z. provided epidemiological and genetic information. J.L., M.Q., Z.L., and J.C. designed and performed genetic analyses. L.Q., M.Y., and C.X. designed and performed epidemiological analyses. J.L., L.Q., M.Y., and M.Q. drafted the manuscript. Y.Y., L.C., J.H., and J.S. edited the manuscript. All authors contributed to the manuscript. All authors read and approved the contents of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Lorenzo Subissi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Z., Hu, P., Qu, L. et al. Molecular epidemiology and population immunity of SARS-CoV-2 in Guangdong (2022–2023) following a pivotal shift in the pandemic. Nat Commun 15, 7033 (2024). https://doi.org/10.1038/s41467-024-51141-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51141-y
This article is cited by
-
Genome surveillance of SARS-CoV-2 Omicron XBB subvariants in Wuhan in June 2023
Archives of Virology (2026)
-
Estimating the two consecutive epidemic waves of SARS-CoV-2 Omicron in Shenzhen, China from November 2022 to July 2023: a modeling study based on multi-source surveillance and mobility data
Advances in Continuous and Discrete Models (2025)
