
Abstract
The oxygen reduction reaction (ORR) catalyzed by efficient and economical catalysts is critical for sustainable energy devices. Although the newly-emerging atomically dispersed platinum catalysts are highly attractive for maximizing atomic utilization, their catalytic selectivity and durability are severely limited by the inflexible valence transformation between Pt and supports. Here, we present a structure by anchoring Pt atoms onto valence-adjustable CuOx/Cu hybrid nanoparticle supports (Pt1-CuOx/Cu), in which the high-valence Cu (+2) in CuOx combined with zero-valent Cu (0) serves as a wide-range valence electron reservoir (0‒2e) to dynamically adjust the Pt 5d valence states during the ORR. In situ spectroscopic characterizations demonstrate that the dynamic evolution of the Pt 5d valence electron configurations could optimize the adsorption strength of *OOH intermediate and further accelerate the dissociation of O = O bonds for the four-electron ORR. As a result, the Pt1-CuOx/Cu catalysts deliver superior ORR performance with a significantly enhanced four-electron selectivity of over 97% and long-term durability.
Similar content being viewed by others
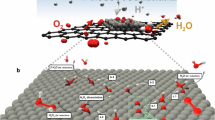


Introduction
Addressing the kinetic obstacles inherent in the oxygen reduction reaction (ORR) has been a significant barrier to advancing sustainable energy conversion technologies, such as fuel cells and metal-air batteries1,2. To date, platinum-based catalysts are still the most promising materials to catalyze the four-electron ORR pathway, but their high cost and insufficient activity have immensely limited their application3,4,5. Downsizing the loaded platinum to single atom level is a feasible strategy to maximize the utilization efficiency of platinum atoms6,7,8. Nevertheless, most platinum single sites anchored on nitrogen-carbon supports (Pt-N/C) possess insufficient 5d electrons to participate in charge interactions with O 2p, which depresses the dissociation of *OOH intermediate and favors the production of undesired H2O29,10. More seriously, the Pt 5d states are difficult to dynamically adjust in the reaction cycles11,12, which destroys the structural stability of the Pt-N/C moiety and degrades the durability of the catalysts rapidly13,14,15.
To date, many studies have focused on improving the d-band structures of M-Nx moieties involved in nitrogen-carbon support (M-N-C)16,17, but the activity is still limited by the easy charge transfer from metal to surrounding support atoms because of the strong and non-freely regulated metal-support interactions18. Recently, different types of single atom catalysts supported on metal oxides have been extensively studied19,20. Generally, the loaded catalytic metal atoms are passively in a high valence state and thus unfavorable for oxygen reduction. Even worse, the valence of most metal oxides is non-flexible during the reaction process, which directly makes the valence evolution of the active centers irreversible in the reaction cycles. As a result, severe dissolution of the supports and accordingly appreciable leaching of active sites during catalytic reactions occured21,22. To overcome these dilemmas, therefore, it is highly anticipated that the valence of metal oxide supports is flexibly adjustable and the electronic structure of loaded metals is easily regulated in the reactions23,24,25. However, few investigations seek to develop a coherent design strategy to precisely tune the valence states of the loaded metal atoms on the supports under working conditions, and a fundamental understanding of the critical mechanism that govern the performance of the catalytic sites is yet to be elucidated.
Intriguingly, small metal clusters or particles are oxidizable and can easily form amorphous oxide layers on their surface. The as-formed amorphous metal oxide/metal (MOx/M) hybrids possess a wide valence range (0 ↔ +n) and can thus serve as an ideal valence electron reservoir (VER) for tuning the valence states of the loaded catalytic atoms such as platinum26,27,28. Furthermore, the MOx/M hybrids is promise to undergo valence evolution under reaction conditions, which is crucial to realize a dynamic electron supply from supports to catalytic metal atoms. Herein, taking advantage of the easily oxidizable property of small Cu particles, we successfully designed a type of catalysts with Pt atomically anchored on the valence-flexible CuOx/Cu hybrid nanoparticles (Pt1-CuOx/Cu) via an in situ partial oxidation strategy. The wide adjustable range of valence states (Cu0 ↔ Cu+2) renders CuOx/Cu an ideal VER for sustainable electron exchange with Pt 5d states. By using in situ X-ray absorption fine structure (XAFS) and synchrotron fourier transform infrared (SR-FTIR) techniques, we followed the catalytic process and unraveled that the 5d electron occupancy of single Pt sites are effectively increased by 0.53 units at the initial ORR stage based on the electron flow from Cu 3d to Pt 5d as the elevated oxidation state of CuOx/Cu supports. Accordingly, the key intermediate *OOH rapidly dissociates to form *O species due to the optimized adsorption on the highly 5d electron-occupied Pt sites. Consequently, the Pt1-CuOx/C exhibits improved durability and electrocatalytic activity with a high half-wave potential of 0.92 V vs. reversible hydrogen electrode (RHE) and a significantly enhanced selectivity of four-electron pathway (>97%). Our strategy contributes to the design of efficient electrocatalysts and addressing the catalytic stability by manipulating the valence evolutions of active sites and supports.
Results and discussion
Synthesis and morphology characterizations
The synthesis of Pt dispersed on hybrid CuOx/Cu catalysts (Pt1-CuOx/Cu) is schematically illustrated in Fig. 1. Typically, the porous carbon supports originated from the alkaline hydrothermal treatment of 1,3,6-trinitropyrene. The monatomic Pt dispersed on amorphous CuOx/Cu hybrid structure was synthesized by temperature-controlled pyrolysis and subsequent in situ partial oxidation. Moreover, the control samples, including Pt single atoms or Cu nanoparticles dispersed on carbon supports (PtSA-CN, CuNP-CN), bare carbon supports (CN), etc., were also synthesized through similar treatments in addition to replacing different metal sources (see the “Methods” section for more details).

Schematic of the synthetic process. The Pt1-CuOx/Cu dispersed on carbon nanosheets was synthesized through sequential hydrothermal and pyrolytic processes.
The overall morphologies of Pt1-CuOx/Cu and control samples were characterized by transmission electron microscopy (TEM). In Supplementary Fig. 1, the Pt1-CuOx/Cu maintains the same nanosheet morphology as the carbon supports (CN). In the power X-ray diffraction (XRD) patterns (Supplementary Fig. 2a), neither PtSA-CN nor CN support exhibits metal diffraction peaks. TEM images of PtSA-CN reveal no obvious nanoparticles, and the aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) image of PtSA-CN directly confirms the single-atom phase of Pt in PtSA-CN (Supplementary Figs. 2b–d). In contrast, small nanoparticles apparently existed in the Pt1-CuOx/Cu catalyst, as identified by the high-resolution transmission electron microscopy (HRTEM) images in Fig. 2a, b. It can be seen from Fig. 2a and the size statistics results that the nanoparticles are evenly dispersed on the carbon supports with an average particle size measuring 3.57 nm. The HRTEM image in Fig. 2b and Supplementary Fig. 3 reveal that the nanoparticles possess lattice stripes inside, whose interplanar spacing was about ~0.20 nm, attributing to the Cu (111) plane. The amorphous feature at the periphery of the nanoparticles may be related to the formation of a CuOx shell layer by the partial oxidation strategies. The two distinct atomic distributions of the nanoparticles are color-marked in AC-HAADF-STEM images, which suggest the existence of CuOx/Cu shell/core structures (Fig. 2c).

a TEM image of Pt1-CuOx/Cu; inset is the corresponding size distribution; (b) HRTEM image of Pt1-CuOx/Cu; (c–e) AC-HAADF-STEM image (c) EDS elemental mapping (d) and line-scan analysis, scale bar: 2 nm (e), of Pt1-CuOx/Cu. f XRD results of Pt1-CuOx/Cu and the control samples.
The results of energy dispersive spectroscopy (EDS) elemental mapping in Fig. 2d and line-scan analysis in Fig. 2e further reveal that the Pt atoms were evenly dispersed on the surface of the CuOx/Cu hybrid nanoparticles, and Cu is the main component of the nanoparticles. The contents of Cu and Pt in Pt1-CuOx/Cu were determined by inductively coupled plasma mass spectrometry (ICP-MS) to be 3.4 wt% and 0.65 wt%, respectively. The XRD patterns of Pt1-CuOx/Cu show rather weak diffraction peaks of metallic Cu relative to the CuNP-CN samples, and no diffraction peaks of oxide species appear, which is attributed to the amorphous CuOx species in Pt1-CuOx/Cu. Moreover, the lack of metallic Pt diffraction peaks suggests that the atomic-level doping of Pt in CuOx/Cu hybrid nanoparticles (Fig. 2f). Hence, these above characterization results clearly demonstrate that the catalysts composed of the evenly dispersed Pt on hybrid CuOx/Cu nanoparticles were successfully obtained.
Electronic and local structure characterizations
The X-ray absorption near edge structure (XANES) spectra at O K-edge were obtained to evaluate the local structure of oxygen (Fig. 3a). The characteristic peaks at ~530 and ~534 eV can be assigned to the overlapping bands between the d bands of metal atoms and the 2p orbitals of oxygen29. The appearance of the characteristic peaks in the Pt1-CuOx/Cu sample relative to carbon support suggests the M-O interactions, which are directly related to the formation of amorphous oxide layers. To elucidate the local coordination structure of Pt1-CuOx/Cu, the Fourier transformed EXAFS (FT-EXAFS) spectra at Cu K-edge and Pt L3-edge were obtained in Fig. 3b, c. As shown in Fig. 3b, the Pt1-CuOx/Cu sample displays two dominant peaks at approximately 1.5 Å and 2.30 Å, which corresponds to the Cu-O and Cu-Cu coordination of the CuO and Cu foil, respectively. It is suggested that the CuOx species are formed after the oxidation process, which results in the coexistence of CuOx and Cu nanoparticles in the Pt1-CuOx/Cu sample. In the Pt L3-edge k2-weighted FT-EXAFS spectra in Fig. 3c, the characteristic peak at the first shell of Pt1-CuOx/Cu may be attributed to Pt-O scattering according to the fact that Pt atoms are dispersed on the CuOx/Cu surface. The characteristic peak at the second coordination shell, which is distinct from the Pt-Pt characteristic peak found in Pt foil, is likely attributable to the Pt-O-M scattering. Moreover, the Pt-O-M scattering peak is different from the double characteristic peaks of Pt-O-Pt in PtO2, ruling out the presence of agglomerated Pt particles and PtO2 species. Thus, the results of FT-EXAFS spectra demonstrate that the Pt species are atomically isolated by O atoms within amorphous CuOx layers, corresponding to the phase characterization in Fig. 2. The local structure around the Pt site was also evaluated by the wavelet transforms (WT) of FT-EXAFS (Supplementary Fig. 4). The Pt1-CuOx/Cu only shows the intensity maximum at 4.2 Å−1, suggesting the monoatomic dispersion of Pt on the surface of CuOx/Cu nanoparticles30.

a XANES spectra at O K-edge of Pt1-CuOx/Cu and CN; (b) Cu K-edge k3-weighted FT-EXAFS spectra of Pt1-CuOx/Cu, Cu foil and CuO; (c) Pt L3-edge k2-weighted FT-EXAFS spectra of Pt1-CuOx/Cu, Pt foil and PtO2; Cu 2p (d) and Pt 4 f (e) XPS spectra of Pt1-CuOx/Cu; (f) XANES spectra at Pt L3-edge of different samples.
To ascertain the valence states of Cu and Pt in Pt1-CuOx/Cu, X-ray photoelectron spectroscopy (XPS) was performed. Prior to XPS spectral analysis, the binding energies of all elements in the samples were calibrated according to charge correction of the C1s spectrum (Supplementary Fig. 5). As illustrated in the Cu 2p XPS spectra results (Fig. 3d), both the peaks of Cu 2p3/2 and Cu 2p1/2 can be fitted into two peaks, representing the existence of Cu0/Cu+ and Cu2+ states, which is different from the result of CuNP-CN sample (Supplementary Fig. 6a)31,32. The associated Cu LMM spectra of Pt1-CuOx/Cu was employed to further evaluate the proportion of different valence states. In Supplementary Fig. 6b, the Cu LMM spectra of Pt1-CuOx/Cu were deconvoluted into three peaks at 564.8, 566.4 and 570.6 eV, representing the Cu0, Cu2+ and Cu+ species, respectively. It can be seen that a wide valence range (0‒2+) exists in CuOx/Cu species. Figure 3e shows the Pt 4 f XPS spectra. The binding energy of Pt 4f7/2 in PtSA/CN control sample registers at 72.6 eV, closely resembling that of Pt2+ species33. The binding energy of Pt 4f7/2 in Pt1-CuOx/Cu slightly negatively shifted by ~0.3 eV relative to that of PtSA-CN, implying that the valence state of Pt is decreased by the surrounding Cu-based supports. As shown in the XANES spectra of Pt L3-edge, the intensity of the white-line (HA) for Pt1-CuOx/Cu presents a slightly weaker than that of PtSA-CN, indicating a lower oxidation state of Pt atoms in Pt1-CuOx/Cu (Fig. 3f), which is consistent with the Pt 4 f XPS analysis. As the fact that the HA of Pt L3-edge XANES spectra corresponds to the transition from the Pt 2p3/2 core-electron to empty 5d and reflects the occupation level of the Pt 5d states, the lower oxidation state of Pt in Pt1-CuOx/Cu suggests the more occupied states of Pt 5d. Besides, the intensity of the oscillation hump (HP) was significantly attenuated for Pt1-CuOx/Cu, confirming that the local structures around Pt atom in Pt1-CuOx/Cu are obviously different from those of the PtSA-CN catalyst. In addition, the valence state of Pt will increase significantly as the CuOx/Cu hetero-structure undergoes further oxidation, which is unfavorable for the adsorption and dissociation of oxygenated intermediates during the electrocatalytic process (Supplementary Fig. 7). These results clearly suggest a modified valence state of Pt 5d in the Pt1-CuOx/Cu hybrid nanoparticles.
Electrochemical oxygen reduction performance
To elucidate the electrochemical performance of Pt1-CuOx/Cu, electrocatalytic measurements were conducted on the rotating disk electrode instrument34. The linear sweep voltammetry (LSV) curves were measured for Pt1-CuOx/Cu and control samples (commercial Pt-C, PtSA-CN and CuOx/Cu). In the Fig. 4a, the Pt1-CuOx/Cu catalyst shows the best ORR performance with a half-wave potential (E1/2) of 0.92 V vs. RHE, and a diffusion limited current density (JL) of 6.1 mA cm–2 (Fig. 4a). The E1/2 of the Pt1-CuOx/Cu catalyst is 60 mV higher than that of Pt-C (inset of Fig. 4b). The kinetic current density (Jk) at 0.85 V vs. RHE of the Pt1-CuOx/Cu catalyst is three times that of Pt-C and far exceeds that of PtSA-CN and CuOx/Cu (Fig. 4b). The outstanding ORR kinetics of Pt1-CuOx/Cu is also demonstrated by the Tafel slope in Fig. 4c and Supplementary Fig. 8. The Pt1-CuOx/Cu exhibits the lowest Tafel slope (with and without iR correction) compared to that of control samples. The turnover frequency (TOF) achieved in Pt1-CuOx/Cu far exceeds the control samples in this work, implying that the intrinsic activity of Pt1-CuOx/Cu was obviously improved (Fig. 4d and Supplementary Fig. 9). Mass activity (MA) is a crucial indicator for assessing the effectiveness of electrocatalysts. The MA of Pt1-CuOx/Cu was calculated in Fig. 4d. Apparently, the Pt1-CuOx/Cu catalyst (6.1 A mgPt-1) significantly surpasses Pt-C and PtSA-CN catalysts. The E1/2 and MA of Pt1-CuOx/Cu are superior to most excellent catalysts (Supplementary Table 1). The electrochemically accessible surface area (ECSA) of Pt1-CuOx/Cu and the PtSA-CN control sample were measured by cyclic voltammetry curves at the scanning rates ranging from 10 to 50 mV s−1. The results plotted in Supplementary Fig. 10, the Pt1-CuOx/Cu catalyst possesses a high ECSA of 152 m2 g−1, which is twice that of PtSA-CN.

a LSV curves of Pt1-CuOx/Cu, Pt-C, PtSA-CN and CuOx/Cu samples, the resistance measured to be 20 ± 5 Ω, no iR correction was applied in the measurements. b The comparison of kinetic current density (Jk) and the inset is the locally amplified polarization plots. c Tafel slopes for Pt1-CuOx/Cu and the reference catalysts (The Tafel slopes were obtained at the scan rate of 10 mV s–1 without iR correction). d Turnover frequency and mass activity of Pt1-CuOx/Cu and the reference catalysts at 0.9 V vs. RHE. e The selectivity parameters of ORR pathway for the catalysts involved. f Stability tests of Pt1-CuOx/Cu and Pt-C. The error bars were estimated by the standard deviations of three individual calculations.
The LSV curves collected at regular rotation speed were employed for Pt1-CuOx/Cu catalyst to evaluate the selectivity of electrocatalytic ORR. As depicted in Supplementary Fig. 11, the JL of the Pt1-CuOx/Cu catalyst is increased with regular rotating speed from 400 to 2500 rpm gradually, indicating the efficient transfer of oxygen species at increased rotating speed. Based on the Levich equation, the fitting results of the LSV curves show good linear parallelism, and the electron transfer number (n) was about 3.98, implying the satisfactory 4e- selectivity of the ORR pathway. Moreover, the rotating ring-disk electrode (RRDE) was also employed to reveal the disk and ring currents. The analysis results for the RRDE measurements were plotted in Fig. 4e and Supplementary Fig. 12. The average n of Pt1-CuOx/Cu and PtSA-CN were 3.95 and 3.32 in the potential range of 0.2 to 0.7 V vs. RHE, respectively, corresponding to the calculated results from Levich equation. Meanwhile, the Pt1-CuOx/Cu possesses the lowest average peroxide species (H2O2) yield (below 3%), which suggests the four-electron selectivity (production of H2O or OH-) was enhanced to >97% for Pt1-CuOx/Cu, significantly larger than those of Pt-C and PtSA-CN (~70%) (Supplementary Fig. 12d). The above results evidently reveal that the atomically doped Pt on the CuOx/Cu surface has significantly enhanced catalytic performance due to the modified electronic structure. The catalytic stability of the ORR process was assessed by chronoamperometry for 100 h. The results in Fig. 4f show that the performance of commercial Pt-C is attenuated by 20%, while the Pt1-CuOx/Cu catalyst remains essentially unchanged after long-term operation. The cyclic voltammetry curves before and after 5000 cycles demonstrate the good durability of Pt1-CuOx/Cu (Supplementary Fig. 13). The ICP-MS monitoring results in Supplementary Fig. 9 reveal that only about 1.6% of Pt was leached from Pt1-CuOx/Cu after the electrochemical operation (Supplementary Fig. 14). And the XRD patterns and Pt 4 f XPS spectra of Pt1-CuOx/Cu show similar crystal phase and electronic structure information before and after the electrochemical measurement, indicating the satisfactory stability of the Pt active sites in Pt1-CuOx/Cu (Supplementary Fig. 15). Moreover, the performance of Pt1-CuOx/Cu catalyst was almost undisturbed by methanol, which is better than that of the Pt-C catalyst (Supplementary Fig. 16).
Exploration of dynamic valence evolution during the reaction
To deeply uncover the dynamic changes in the valence states and valence electron configurations of the catalysts, in situ XAFS characterizations were employed for Pt1-CuOx/Cu and PtSA-CN catalysts35,36. The normalized Pt L3-edge XANES spectra, which reflect the electronic structure evolution, are depicted in Fig. 5a, b. When the ORR potential was applied (1.00 V vs. RHE), the HA of Pt1-CuOx/Cu decreases obviously in relation to the open-circuit condition (immersed in electrolyte), indicating an enhanced electron occupation of Pt 5d at the initial reaction stage. In contrast, the HA of PtSA-CN was almost unchanged from open-circuit to the ORR potential (0.80 and 0.60 V vs. RHE), suggesting that the electronic state of the atomically dispersed Pt on general CN supports is relatively stable at the initial stage. The valence state of Pt in Pt1-CuOx/Cu increases gradually during the reaction process (1.00 → 0.75 V vs. RHE), and similarly, the valence state of Pt in PtSA-CN also shows a slight tendency to increase, which may be related to the interaction with the oxygen-related species. The formal d-band electron counts and valence states of Pt were calculated to quantify the valence electron evolution during the catalytic process based on the integral area of the white-line peak in differential XANES (ΔXANES) spectra (Supplementary Fig. 17). The fitting results show that the average valence state of Pt decreases from +2.61 to +1.92 as the conditions change from open-circuit to +1.00 V vs. RHE (Supplementary Fig. 18). Correspondingly, the formal d-band occupied density was estimated using a linear relationship obtained from the standards of Pt foil (5d96s1) and PtO2 (5d66s0). Figure 5c reflects that the occupied electron counts of Pt 5d bands for the Pt1-CuOx/Cu catalyst at open-circuit was 5d7.03 and then increased to 5d7.56 under 1.00 V vs. RHE, suggesting that the d-band electron quantity dynamically increased by 0.53 units.

In situ Pt L3-edge XANES spectra of Pt1-CuOx/Cu (a) and PtSA-CN (b). c The d-band electrons at different conditions of Pt1-CuOx/Cu and PtSA-CN. d In situ XANES spectra at Cu K-edge of Pt1-CuOx/Cu. e In situ Pt L3-edge EXAFS spectra of Pt1-CuOx/Cu. The error bars were estimated by the standard deviations of three individual calculations.
Furthermore, the dynamic evolution of Cu in Pt1-CuOx/Cu was analyzed by the in situ Cu K-edge XAFS to correlate with the changes of valence electrons at the Pt sites. In the Cu K-edge XANES spectra of Fig. 5d, as the ORR potential of 1.00 V was applied, the shift of the absorption edge towards higher-energy by approximately 0.8 eV compared to the open-circuit condition, which suggests an obviously increased Cu oxidation state at the initial reaction stages. To determine whether the increased oxidation state of Cu is influenced by the adsorption of oxygen-related species, the in situ XAFS measurements of the CuOx/Cu control sample were conducted (Supplementary Fig. 19). In contrast to those of Pt1-CuOx/Cu, the in situ Cu K-edge XANES spectra of CuOx/Cu show essentially no changes in the absorption edge at the initial reaction stage (before 0.80 V vs. RHE), and the slightly positive shift during the reaction process may be related to the adsorption of oxygen species on the surface of CuOx/Cu. The above results prove that the changes in the valence electron quantity for Pt and Cu in Pt1-CuOx/Cu are originated from the electron interaction between the Pt sites and CuOx/Cu supports at the initial reaction stage. The increased oxidation state of Cu corresponds to a decrease in the valence electron quantity, suggesting that the valence electron transformation is characterized by the CuOx/Cu support serving as a VER to rapidly increase the quantity of Pt 5d valence electrons at the initial reaction stage, which is beneficial for the orbital hybridization with O 2p and increasing the adsorption strength of oxygen species at the Pt sites during the ORR process.
The facilitated flow of valence electrons from the surrounding Cu atoms to single Pt sites must be driven by the dynamic interaction between the Pt and the valence-flexible CuOx/Cu supports. Thus, the corresponding local structure evolution of the CuOx/Cu hybrid nanoparticles and single-Pt sites were further analyzed by in situ FT-EXAFS. The typical FT-EXAFS spectra of Cu K-edge at different reaction stages are displayed in Supplementary Fig. 20. As the working conditions changed from open-circuit to the potential of 1.00 V vs. RHE, the intensity of the dominant peak (about 2.43 Å), which corresponds to the Cu-Cu coordination, is notably decreased, along with the increase of the Cu-O coordination peak at 1.50 Å. This phenomenon implies that the oxidation degree on the surface of core-Cu particles is deepened at the initial reaction stage, which well coincides with the in situ XANES results at Cu K-edge, demonstrating the possible electron exchange between the surrounding CuOx species and single Pt sites through the Cu-O-Pt coordination. The local structure evolution around Pt sites was further identified by Pt L3-edge FT-EXAFS. In the FT-EXAFS spectra of Fig. 5e and Supplementary Fig. 21, all curves at different conditions present a dominant peak attributed to the Pt-O coordination at about 1.60 Å. Notably, the intensity of this dominant peak shows a damping by 20% as the potential changed from open-circuit to 1.00 V vs. RHE, which suggests distinct variations in the local coordination structures of Pt sites at initial reaction stage. The peak displays an increase in intensity as the applied potential drops to 0.90 and 0.75 V vs. RHE, likely due to the interaction with oxygen-related species during the electrocatalytic oxygen reduction process. Quantitatively, the FT-EXAFS fitting results exhibit that the coordination number of Pt-O is four under open-circuit condition (Fig.5e, Supplementary Fig. 22, and Supplementary Table 2). Notably, the coordination number of Pt-O bonds is reduced to three as the potential of 1.00 V vs. RHE was applied, indicating that the lower-saturated coordination active site was formed under ORR conditions. The local structure with lower coordination (Pt-O3) highly mitigates the electron grabbing by surrounding O atoms and favors the valence electron transfer from CuOx/Cu support to Pt centers at the initial stage. In the whole process of dynamic regulation, the CuOx/Cu support with elevated valence state acts as an effective VER and completes an in situ electron transportation to Pt 5d states. As the potential decreases to 0.90 V vs. RHE, an additional Pt-O coordination emerges, which corresponds to the adsorption of oxygen-related intermediates (*O, *OOH) on Pt sites. Meanwhile, the coordination of Cu-O in Cu K-edge FT-EXAFS spectra changed weakly as the ORR potential decreased from 1.00 to 0.90 V vs. RHE, which implies that the evolution of oxygen-related species primarily occurred at Pt sites after the rapid supplementation of valence electrons in the initial stage (Supplementary Fig. 20). These results indicate that because of the hybrid CuOx/Cu as an effective VER, highly occupied Pt 5d states are formed during the ORR, which is highly favorable for the hybridization of O 2p with Pt 5d orbitals. Moreover, as shown in the Pt L3-edge XAFS spectra, the oxidation state and the local coordination structure of Pt sites return to the initial state when the condition reversed to the open-circuit condition, indicating that the dynamically valence transformation between Pt atom and CuOx/Cu support is reversible (Supplementary Fig. 23), which can prevent the dissolution of the supports and leaching of active sites in the process of ORR. Hence, the dynamic valence evolution during the catalytic reactions promotes the evolution of reactive oxygen species and maintains the structural integrity for boosting activity and durability of the catalysts.
Understanding of the catalytic enhancement mechanism
To reveal the nature of the catalytic performance enhanced by dynamic valence evolution, in situ SR-FTIR and electrochemical impedance spectroscopy (EIS) measurements were performed37. The results of Pt1-CuOx/Cu in Fig. 6a reflect that an obvious IR absorption band located at 890 cm−1 are observed during the ORR at various applied potentials. The absorption bands within the range of 800‒900 cm−1 are associated with the stretching vibration of oxygen species (*O)38,39. Therefore, the phenomenon of oxygen species (*O) accumulation can be attributed to the rapid dissociation of *OOH at the CuOx-isolated Pt sites to form *O intermediates, which is the key step of four-electron ORR40. Moreover, the absorption bands around 1200 cm–1 are usually associated with the stretching vibration of *OOH. The relatively weak vibration peak at 1180 cm–1 in Fig. 6a also demonstrates the rapid dissociation of *OOH intermediates by Pt1-CuOx/Cu. For comparison, in situ SRIR measurement was performed for PtSA-CN within the range of the corresponding catalytic ORR potentials. The results in Fig. 6b are obviously different from those for Pt1-CuOx/Cu. The absence of *O vibration peaks at 800‒900 cm−1 and the accumulation of *OOH at 923 cm-1 indicate that the dissociation of *OOH intermediates at the Pt-N4 active site is restricted. Meanwhile, the absorption bands at 1405 cm-1, which can be attributed to the surface-adsorbed *HOOH, are observed in Fig. 6b, implying the production of the H2O2 by-product. The variation of transmission intensity with ORR potential is plotted in Fig. 6c. It is clear that the *HOOH and *OOH intermediates accumulate continuously on the surface of PtSA-CN catalyst as the oxygen reduction progresses, indicating the occurrence of two-electron side reactions due to the difficulty of *OOH dissociation, which is in consistence with the results of selectivity testing41,42,43.

In situ SR-FTIR spectra in the range of 700–1500 cm–1 of Pt1-CuOx/Cu (a) and PtSA-CN (b). c The corresponding infrared signal fluctuations. In situ EIS measurements of Pt1-CuOx/Cu (d) and PtSA-CN (e). f The corresponding adsorption resistance (Rct) of oxygen species.
To demonstrate the facilitating effect of Pt1-CuOx/Cu on the dissociation of *OOH intermediates, in situ SR-FTIR was also measured for the Pt-C control sample by the same three-electrode system (Supplementary Fig. 24a). The results in Supplementary Fig. 24b are quite different from those of Pt1-CuOx/Cu catalyst. The presence of only absorption bands located at 1128 cm-1 indicates the formation of *OOH on the Pt nanoparticle sites within Pt-C, and the continuous accumulation of *OOH at low potentials was attributed to the difficulty of further cleavage of *OOH for the Pt-C catalyst. In comparison with PtSA-CN, Fig. 6c shows that *OOH intermediates possess imperceptible fluctuations on the surface of Pt1-CuOx/Cu, implying the rapid decomposition. Correspondingly, the *O species accumulated rapidly as the potential decreased from 1.00 V vs. RHE. Combined with the in situ XAFS results, the mechanism diagram of the catalytic process is shown in Supplementary Fig. 25, and it can be concluded that the valence electron transport from the VER (CuOx/Cu) to Pt active sites at the initial reaction stage (open circuit → 1.00 V vs. RHE) promotes the proton coupling and electron transfer process of intermediate evolution, thereby accelerating the kinetics of the four-electron ORR.
To further clarify the kinetics process of the species evolution over the electrode surface, in situ EIS was performed for the Pt1-CuOx/Cu and PtSA-CN catalysts under different ORR potentials44,45. In the Nyquist plots of Fig. 6d, e, the Pt1-CuOx/Cu possesses the distinct smaller semicircle size than that of PtSA-CN at a series of ORR potentials, which suggests a faster adsorption and evolution rate of reactants on the single Pt active sites. Furthermore, the Nyquist plots were simulated based on the equivalent circuit model in Supplementary Fig. 26. The fitted EIS parameter are displayed in Supplementary Tables S3, S4. Generally, the parameters of Rct represent the charge transfer resistance, and Cd are the capacitance. As shown in Fig. 6f, the Rct values of Pt1-CuOx/Cu are smaller than that of PtSA-CN in the whole reaction process and decrease rapidly at the kinetic control region, suggesting significantly accelerated adsorption dynamics of oxygen-related intermediates at the electrode/electrolyte interfaces. Moreover, the higher Cd values at various ORR potentials of Pt1-CuOx/Cu further demonstrates that the optimum adsorption of oxygen species on valence-reduced Pt sites dominates a highly selective four-electron ORR pathway. Therefore, the above in situ SR-FTIR and EIS results reveal that the dynamic valence transformation between Pt and CuOx/Cu at the reaction stage facilitates the formation and conversion of oxygen intermediates.
In summary, a strategy of using valence-adjustable metal oxide/metal (CuOx/Cu) hybrid nanoparticles as an VER was proposed to tune the Pt 5d valence states for efficient ORR. Benefiting from the flexible valence electron exchange between the dynamically valence-reconstructed CuOx/Cu supports and single Pt center, the obtained Pt1-CuOx/Cu catalyst delivers improved four-electron selectivity and long-term stability of ORR. Using combined in situ synchrotron radiation characterization technologies, we uncovered that the increase of oxidation state in CuOx/Cu nanoparticles can effectively elevate the Pt 5d electron quantity by 0.53 units at the initial reaction stage, which effectively tunes the adsorption strength of the *OOH intermediates thereby improving the selectivity of ORR. Our work provides a universal wide-range valence-regulatory strategy for the development of advanced electrocatalysts.
Methods
Materials
All chemicals were used without further purifications. The chloroplatinic acid hexahydrate (H2PtCl6·6H2O, ≥97%), aqueous ammonia (NH4OH, 25‒28%), hydrochloric acid (HNO3, 65‒68%), potassium hydroxide (KOH, ≥85%) and urea (99%) were acquired from Sinopharm Reagent Co., Ltd. Copper (II). Pyrene (C16H10, 97%), acetylacetonate (Cu(acac)2, 97%), and Nafion (5%) solution were acquired from Aladdin Biochemical Technology Co., Ltd.
CN support synthesis
Typically, trinitropyrene (300 mg, Nitrated pyrene) dissolved in deionized water (55 mL) under ultrasonic conditions. Then add 5 mL ammonia water and mix thoroughly, continue ultrasound for 4 h. After hydrothermal reaction at 200 °C for 10 h, it was concentrated to about 20 mL. The amino-functionalized CN support was obtained.
Synthesis of Pt1-CuOx/Cu
The Pt1-CuOx/Cu nanoparticles supported on CN support were prepared by wet-impregnation of aqueous solution containing H2PtCl6·6H2O and Cu(acac)2. Specifically, two metal salts of 100 μL H2PtCl6·6H2O (20 mg mL-1) and 15 mg Cu(acac)2 were added to the concentrated 20 mL CN support solution. Ultrasonically dispersed for 30 minutes and then freeze-dried. The power was ground with urea (mass ratio of 1:10), and heated in a tubular furnace (5 °C/min rate of heating, H2/N2 atmosphere, and held at 700 °C for 2 h). Finally, the product was heated in muffle furnace at 200 °C for 60 min to obtain the final products of Pt1-CuOx/Cu.
Synthesis of control samples
The synthesis processes for Pt single-atoms and Cu nanoparticles supported on C-N (PtSA-CN, CuNP-CN) catalyst were similar to that of the preparation of the Pt1-CuOx/Cu electrocatalyst. The notable difference is the use of the H2PtCl6·6H2O for PtSA-CN, and the Cu(acac)2 for CuNP-CN and CuOx/Cu control samples. The CuNP-CN was heated in muffle furnace for 60 minutes to obtain the CuOx/Cu-CN samples. And the Pt1-CuOx/Cu was heated in muffle furnace for 200 minutes to obtain the Pt-CuO samples.
Characterization
The AC-HAADF-STEM was conducted on a JEM-ARM200F instrument at 200 kV. TEM images were conducted on JEM−2100F at 200 kV. The XRD analysis was performed on miniflex600 of multifunctional rotating-anode X-ray diffractometer. The surface chemical state analysis by XPS were performed on thermo scientific ESCALAB 250Xi with Al kα 150 W. ICP-MS analysis was employed at iCAP RQ of inductively coupled plasma-mass spectrometry. The soft X-ray absorption spectra was collected at soft X-ray imaging line station (BL07W) in Hefei, China.
Electrochemical measurements
All electrochemical experiments in this study were conducted using a CH instruments (CHI 760E). In the standard three-electrode cell, the alkaline electrolyte used in electrochemical testing was O2-saturated 0.1 M KOH at room temperature. The fresh 0.1 M KOH was prepared prior to the electrochemical tests by dissolving 2.81 g KOH in 500 mL deionized water, and the pH was ca. 12.8 ± 0.1. The counter electrode and reference electrode were graphite rods and Hg/HgO, respectively. The reference Hg/HgO was calibrated in H2-saturated 0.1 M KOH electrolyte, and Pt wires was served as counter and working electrodes. The average potential at the zero current in the CV test curves is defined as the thermodynamic potential relative to Hg/HgO. The potentials relative to reversible hydrogen electrode (E(vs. RHE)) was calculated by E(vs. Hg/HgO) + 0.891 V. The resistance (R) was measured by the current interrupt method of the electrochemical workstation. The rotating (ring) disk electrode (RDE/RRDE, disk diameter of 5 mm and 4 mm, respectively) was used as working electrode. For better comparison, no iR correction was applied in the electrochemical measurements, except for the polarization curve test and Tafel analysis in Supplementary Fig. 8. The linear sweep voltammetry was conducted at regular rotation rates (400‒2500 rpm) with the scan rate of 10 mV s-1, and O2 was continuously pumped into the electrolyte for 30 minutes before the experiment, and a constant rate of O2 was kept pumping to ensure oxygen saturation. The catalyst ink was prepared as follows: Catalyst powder (5 mg) was dissolved in the solution (900 μL isopropyl alcohol, 100 μL DI water and 20 µl Nafion). Continuously sonicate for 20 min. Afterward, the sample ink was coated on the surface of the polished glassy carbon working electrode with the mass of sample deposited on the electrode is 1.53 g m–2. The JM 20 wt% Pt-C was used as the control sample in this work. The morphology, phase and polarization curves of Pt-C are displayed in Supplementary Fig. 27, and the mass loading on the electrode is 20 μgPt cm-2.
The electron transfer number (n) was calculated according to the equation below:
where J, Jk and JL are the current density, the kinetics and the diffusion-limited current densities, respectively. ω is the rotational speed of the RDE (rad·s−1), and the Faraday constant (F) is 96485 C·mol−1. Both C0 and D0 are the constant, which conducted as bulk concentration (1.2 × 10–6 mol·cm−3) and diffusion coefficient (1.9 × 10–5 cm2·s−1) of oxygen, respectively. V is the viscosity of the electrolyte, consider as 0.01 cm2·s−1.
The yield of hydrogen peroxide (H2O2, %) and the n based on RRDE measurements can be calculated according to the equation below:
where the disk and ring current are marked as Id and Ir, respectively. N is the Pt ring current collection efficiency.
The mass activity of the noble metal-based catalysts is usually measured by the amount of noble metal used. Here, the mass activity (MA, A gmetal-1) was evaluated by the equation:
the kinetic current density is marked as jk, A is the geometric area of electrode, m is the noble metal loading mass on the electrode.
The turnover frequency (TOF) of the Pt1-CuOx/Cu and control samples are calculated by the formula of TOF = (jk × A × Ne)/(4 × Mmetal × NA/M), where jk is the kinetic current density, mA cm-2; A is the surface of the electrode, cm-2; Ne is the electron number per coulomb, 6.24 × 1018 C-1; Mmetal is the mass of active metal leading on the electrode, mg; The Avogadro constant is 6.02 × 1023 mol-1; and M is the Pt molar mass, 195 g mol–1.
In situ SR-FTIR measurements
Typically, the in situ SR-FTIR measurements were conducted at BL01B of the National Synchrotron Radiation Laboratory (NSRL, China). The reflection mode of the SR-FTIR light was adopted. The three-electrode system was applied in the homemade FTIR electrochemical cell with three electrodes in one chamber (~3 mL) (Supplementary Fig. 24a). The infrared spectra at different potential conditions were measured after a certain potential applied for 5 min to obtain the steady signal. The catalyst ink was dissolved as follows: Sample (5 mg) was dissolved in a 1 mL solution (0.9 mL isopropyl alcohol, 0.1 mL DI and 20 µL Nafion), ultrasound for 20 min. The cleaned carbon cloth with area of 1 × 1.5 cm−2 was employed as the working electrode, a certain amount of the catalyst ink was coated onto carbon cloths evenly. After complete drying, the carbon cloth was pressed into the ZnSe window of the electrochemical cell. The O2-saturated 0.1 M KOH electrolyte was circulated into the electrochemical cell through a peristaltic pump.
In situ XAFS measurements
The in situ XAFS measurements were carried out at the 1W1B station in the Beijing Synchrotron Radiation Facility (BSRF, China). The in situ XAFS tests for electrochemical process were measured in a self-designed in situ cell with three electrodes in one chamber (~150 mL) (Supplementary Fig. 28). The in situ XAFS spectra at different electrochemical conditions were recorded using a Lytle detector (fluorescence mode), the working electrode is close to the X-ray window, 0.1 M KOH solution (O2-saturated) was used as electrolytes. To prepare the catalyst electrode for use as working electrode, 5 mg sample was dispersed in a 1 mL mixed solution (0.9 mL isopropyl alcohol, 0.1 mL DI and 20 µL Nafion), ultrasound for 20 min. A certain amount of the catalyst ink was uniformly coated onto the carbon cloths (1 × 1.5 cm–2) as the working electrode. The spectra labeled “open-circuit” were collected under open-circuit condition with the catalyst immersed in electrolyte. In situ spectra were collected by chronoamperometry measurements at a series of representative potentials (1.0–0.75 V vs. RHE for Pt1-CuOx/Cu and 1.0‒0.60 V vs. RHE for PtSA-CN).
Strategies of XAFS curve fittings
To analyze the local structure information of Pt atoms in the Pt1-CuOx/Cu samples, the Athena and Artemis program was used to fit the EXAFS data. The k2-weighted χ(k) data (2.5‒11.5 Å−1 of k-range) were Fourier-transformed to R-space under the hanning windows of dk = 1.0 Å–1. The fitted R-range was 1.0‒2.2 Å. Accordingly, the number of independent points (Nipt) can be evaluated by the formula: Nipt = 2Δk × ΔR/π = 2 × (11.5 − 2.5) × (2.3 − 1.0)/π = 7. For the fitting of Pt L3-edge spectra, the amplitude reduction factor S02 for Pt foil was first obtained by fitting the Pt foil data and the value was conducted as a fixed parameter in the fitting procedure of R-space data for Pt1-CuOx/Cu. Based on structural characterization information, the Pt–O scattering path was considered to fit the R-space data under open-circuit and 1.00 V vs. RHE conditions. The coordination numbers (CNs), bond length (R), Debye–Waller factors (σ2) and energy shift (ΔE0) were conducted as adjustable parameters (Npara) according to the fitting criterion, and the Npara was Npara = 4 <Nipt in this condition. As for the in situ spectrum under 0.90 V vs. RHE, the intensity of characteristic peak increased obviously compared to the 1.00 V vs. RHE. Taking into account the possible evolution of oxygen-related intermediates on the Pt sites, an additional Pt–O path was incorporated in the fitting procedure. And the energy shift (ΔE0) for different paths was set equal to reduce the Npara, and the Npara was Npara = 7 ≤ Nipt in this condition, which is reasonable in the fitting procedure.
Data availability
The data that support the findings of this work are available within the manuscript, Supplementary information files, and Source Data File. Source data are provided with this paper.
References
Debe, M. K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 486, 43–51 (2012).
Luo, M. et al. PdMo bimetallene for oxygen reduction catalysis. Nature 574, 81–85 (2019).
Kodama, K. et al. Challenges in applying highly active Pt-based nanostructured catalysts for oxygen reduction reactions to fuel cell vehicles. Nat. Nanotechnol. 16, 140–147 (2021).
Huang, J. et al. Experimental Sabatier plot for predictive design of active and stable Pt-alloy oxygen reduction reaction catalysts. Nat. Catal. 5, 513–523 (2022).
Chen G. et al. Modulation of electronic states in bimetallic-doped nitrogen-carbon based nanoparticles for enhanced oxygen reduction kinetics. Chin. J. Chem. Phys. 37, 513–521 (2024).
Liu, J. et al. High performance platinum single atom electrocatalyst for oxygen reduction reaction. Nat. Commun. 8, 15938 (2017).
Liang, X. et al. The progress and outlook of metal single-atom-site catalysis. J. Am. Chem. Soc. 144, 18155–18174 (2022).
Zhao, C. X. et al. Intrinsic electrocatalytic activity regulation of M-N-C single-atom catalysts for the oxygen reduction reaction. Angew. Chem. Int. Ed. 60, 4448–4463 (2021).
Kim, H. E. et al. Palladium single‐atom catalysts supported on C@C3N4 for electrochemical reactions. ChemElectroChem 6, 4757–4764 (2019).
Xiang, Z. et al. Catalyst with a low load of platinum and high activity for oxygen reduction derived from strong adsorption of Pt−N4 moieties on a carbon surface. Electrochem. Commun. 127, 107039 (2021).
Liu, S. & Huang, S. Theoretical insights into the activation of O2 by Pt single atom and Pt4 nanocluster on functionalized graphene support: Critical role of Pt positive polarized charges. Carbon 115, 11–17 (2017).
Guo, X. et al. Simultaneously achieving high activity and selectivity toward two-electron O2 electroreduction: The power of single-atom catalysts. ACS Catal. 9, 11042–11054 (2019).
Choi, C. H. et al. Tuning selectivity of electrochemical reactions by atomically dispersed platinum catalyst. Nat. Commun. 7, 10922 (2016).
Stamenkovic, V. R. et al. Improved oxygen reduction activity on Pt 3 Ni(111) via increased surface site availability. Science 315, 493–497 (2007).
Zhou, W. L. et al. Regulating the scaling relationship for high catalytic kinetics and selectivity of the oxygen reduction reaction. Nat. Commun. 13, 6414 (2022).
Li, W.-H. et al. Striding the threshold of an atom era of organic synthesis by single-atom catalysis. Chem 8, 119–140 (2022).
Zhai, H. et al. Electronic regulation of Pt single-atom catalysts via local coordination state adjustment for enhanced photocatalytic performance. ACS Catal. 13, 8063–8072 (2023).
Su, H. et al. In-situ spectroscopic observation of dynamic-coupling oxygen on atomically dispersed iridium electrocatalyst for acidic water oxidation. Nat. Commun. 12, 6118 (2021).
Yang, L. et al. Stabilizing Pt electrocatalysts via introducing reducible oxide support as reservoir of electrons and oxygen species. ACS Catal. 12, 13523–13532 (2022).
Lang, R. et al. Single-atom catalysts based on the metal-oxide interaction. Chem. Rev. 120, 11986–12043 (2020).
Therrien, A. J. et al. An atomic-scale view of single-site Pt catalysis for low-temperature CO oxidation. Nat. Catal. 1, 192–198 (2018).
Song, J. et al. Dispersion and support dictated properties and activities of Pt/metal oxide catalysts in heterogeneous CO oxidation. Nano Res. 14, 4841–4847 (2021).
Liu, J. et al. Reconstructing the coordination environment of platinum single-atom active sites for boosting oxygen reduction reaction. ACS Catal. 11, 466–475 (2020).
Liao, H. et al. Unveiling role of sulfate ion in nickel‐iron (oxy)hydroxide with enhanced oxygen‐evolving performance. Adv. Funct. Mater. 31, 2102772 (2021).
Liu, X. et al. Cationic oxidative leaching engineering modulated in situ self-reconstruction of nickel sulfide for superior water oxidation. Nano Lett. 23, 5027–5034 (2023).
Chen, S. et al. Reconstructed Ir‒O‒Mo species with strong Bronsted acidity for acidic water oxidation. Nat. Commun. 14, 4127 (2023).
Kang, H. et al. Generation of oxide surface patches promoting H-spillover in Ru/(TiOx)MnO catalysts enables CO2 reduction to CO. Nat. Catal. 6, 1062–1072 (2023).
Zhou, J. et al. The role of the metal core in the performance of WOx inverse catalysts. Chem. Catal. 3, 100756 (2023).
Grimaud, A. et al. Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat. Commun. 4, 2439 (2013).
Ye, C. et al. Electrical pulse induced one-step formation of atomically dispersed Pt on oxide clusters for ultra-low-temperature zinc-air battery. Angew. Chem. Int. Ed. 61, e202213366 (2022).
Jiang, Z. et al. Modulating the electronic metal-support interactions in single-atom Pt(1) -CuO catalyst for boosting acetone oxidation. Angew. Chem. Int. Ed. 61, e202200763 (2022).
Luo, J. et al. Efficient Cu/FeOx catalyst with developed structure for catalytic transfer hydrogenation of furfural. J. Catal. 413, 575–587 (2022).
Liang, L. et al. Cobalt single atom site isolated Pt nanoparticles for efficient ORR and HER in acid media. Nano Energy 88, 106221 (2021).
Wang, X. et al. Review of metal catalysts for oxygen reduction reaction: From nanoscale engineering to atomic design. Chem 5, 1486–1511 (2019).
Liao, H. et al. Oxyanion engineering suppressed iron segregation in nickel-iron catalysts toward stable water oxidation. Adv. Mater. 35, e2300347 (2023).
Liu, M. et al. In situ modulating coordination fields of single-atom cobalt catalyst for enhanced oxygen reduction reaction. Nat. Commun. 15, 1675 (2024).
Su, H. et al. Tensile straining of iridium sites in manganese oxides for proton-exchange membrane water electrolysers. Nat. Commun. 15, 95 (2024).
Cheng, W. R. et al. Tracking the oxygen dynamics of solid-liquid electrochemical interfaces by correlative in situ synchrotron spectroscopies. Acc. Chem. Res. 55, 1949–1959 (2022).
Zandi, O. & Hamann, T. W. Determination of photoelectrochemical water oxidation intermediates on haematite electrode surfaces using operando infrared spectroscopy. Nat. Chem. 8, 778–783 (2016).
Li, Y. et al. Operando infrared spectroscopic insights into the dynamic evolution of liquid-solid (photo)electrochemical interfaces. Nano Energy 77, 1–13 (2020).
Wanlin, Z. et al. In-situ synchrotron radiation infrared spectroscopic identification of reactive intermediates over multiphase electrocatalytic interfaces. Chin. J. Struct. Chem. 41, 2210004–2210015 (2022).
Tang, C. et al. Tailoring acidic oxygen reduction selectivity on single-atom catalysts via modification of first and second coordination spheres. J. Am. Chem. Soc. 143, 7819–7827 (2021).
Nayak, S. et al. Adsorbed intermediates in oxygen reduction on platinum nanoparticles observed by in situ IR spectroscopy. Angew. Chem. Int. Ed. 57, 12855–12858 (2018).
Xiao, Z. et al. Operando identification of the dynamic behavior of oxygen vacancy-rich Co(3)O(4) for oxygen evolution reaction. J. Am. Chem. Soc. 142, 12087–12095 (2020).
Zhang, H. et al. Symbiotic synergy enabling moderate oxo-hydroxy adsorption capacity for high-selectivity oxygen reduction. Nano Energy 101, 107587 (2022).
Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFA1502903 (Q.L.)), the National Natural Science Foundation of China (22241202 (Q.L.), 12135012 (S.W.) and 22361132539 (S.W.)), the Natural Science Foundation of Anhui Province (2208085J01 (Q.L.)), the Russian Science Foundation (project No. 24-43-00215 (M.S.)), the Postdoctoral Science Foundation of China (BX20230345 (W.Z.) and 2023M743366 (W.Z.)) and the Key Program of Research and Development of Hefei Science Center, CAS (2022HSC-KPRD003 (S.W.)). Part of this work was supported by the Instruments Center for Physical Science, University of Science and Technology of China.
Author information
Authors and Affiliations
Contributions
Q.L. conceived the project. W.Z. designed the experimental scheme and conducted the in situ experiments. B.L. prepared the samples and conducted the electrochemical experiments. Q.L., J.J., X.L., S.B., C.Y., Q.A., Y.Z., M.S., H.W. and S.W. provided constructive ideas to the experiment results. W.Z., B.L. and Q.L. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wei Li, Jun Pan and Gabor Samjeske for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, W., Li, B., Liu, X. et al. In situ tuning of platinum 5d valence states for four-electron oxygen reduction. Nat Commun 15, 6650 (2024). https://doi.org/10.1038/s41467-024-51157-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51157-4
