
Abstract
Richter transformation (RT) is an aggressive lymphoma occurring in patients with chronic lymphocytic leukaemia. Here we investigated the anti-CD3/anti-CD19 T-cell-engager blinatumomab after R-CHOP (i.e. rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) in patients with untreated RT of diffuse large B-cell lymphoma histology (NCT03931642). In this multicentre phase 2 study, patients without complete response (CR) after two cycles of R-CHOP were eligible to receive an 8-week blinatumomab induction via continuous vein infusion with stepwise dosing until 112 μg/day. The primary endpoint was the CR rate after blinatumomab induction and secondary endpoint included safety, response duration, progression-free and overall survival. Thirty-nine patients started the first cycle of R-CHOP, 25 of whom received blinatumomab. After blinatumomab induction, five (20%) patients achieved CR, four (16%) achieved partial response, and six (24%) were stable. Considering the entire strategy, the overall response rate in the full-analysis-set was 46% (n = 18), with CR in 14 (36%) patients. The most common treatment-emergent adverse events of all grades in the blinatumomab-safety-set included fever (36%), anaemia (24%), and lymphopaenia (24%). Cytokine release syndrome (grade 1/2) was observed in 16% and neurotoxicity in 20% of patients. Blinatumomab demonstrated encouraging anti-tumour activity (the trial met its primary endpoint) and acceptable toxicity in patients with RT.
Similar content being viewed by others
Introduction
Richter transformation (RT) is defined as the onset of aggressive lymphoma, mostly diffuse large B-cell lymphoma (DLBCL), in patients with chronic lymphocytic leukaemia (CLL). Eighty percent of RT cases are clonally related to the prior CLL and result from a transformation process involving complex genomic events with dramatic lesions, resulting in high mutational load1. The outcome of patients with RT is usually very poor. Chemoimmunotherapy (CIT), analogous to the treatment of de novo DLBCL, results in low response rates with a median overall survival (OS) of 6–12 months2,3. The combination of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) remains a frequently used regimen for RT, modelled on its use in de novo DLBCL, and yields a complete response rate (CRR) of 7%, an overall response rate (ORR) of 67%, and median progression-free survival (PFS) of 10 months4. Bruton tyrosine kinase and BCL2 inhibitors have transformed the management of patients with CLL but fail to prevent the onset of RT. As single agents, these drugs demonstrate only a transient effect in some patients with RT5,6,7. The combination of venetoclax with CIT has provided promising efficacy results, but toxicities were limiting8. Haematopoietic stem cell transplantation (HSCT) can improve remission duration in selected patients able to receive this consolidation procedure; less than 15% of patients can undergo transplantation because of primary refractory disease9.
The modulation of antitumour immunity may be an appealing strategy for RT. Immune checkpoint inhibition has been shown to induce reasonable responses in RT but not in CLL10,11. Bispecific constructs that recruit T cells to tumour B cells are emerging as a promising approach for treating B-cell lymphoproliferative disorders. Blinatumomab is a CD19 × CD3 bispecific monoclonal antibody approved for the treatment of acute lymphoblastic leukaemia and has demonstrated clear efficacy in relapsed or refractory DLBCL. A phase II study involving stepwise dosing (9, 28, and 112 μg/d) of blinatumomab by continuous infusion has shown encouraging results, with an ORR of 43% (including 19% complete response (CR)) after a single 8-week cycle12.
We hypothesised that blinatumomab would improve the response of patients with RT who without CR after two cycles of R-CHOP. This debulking therapy aimed to induce a tumour response to minimise the risk of cytokine release syndrome (CRS) and neurologic events and to identify good responders to CIT without needing an experimental approach. Here, we report the results of the BLINART (BLINAtumomab after R-CHOP debulking therapy for patients with Richter Transformation) phase 2 study showing the efficacy and the acceptable safety profile of blinatumomab after R-CHOP in patients with previously untreated RT.
Results
Patient population
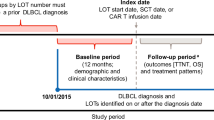
A total of 45 patients were screened, and 41 were subsequently enrolled in the study at 18 sites between 5 July 2019 and 19 July 2021 (Fig. 1, Supplementary Table 1). After 35 patients were initially included, the protocol was amended to include six more patients to achieve the estimated sample size. Of the 41 enrolled patients, two were excluded from the study before receiving any therapy because of misdiagnosis in one case and because of prohibited concomitant medication (high-dose cortico-therapy) in the other case. The full analysis set included 39 patients who started their first cycle of R-CHOP.

R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
As of the data cut-off date of 20 December 2022, a blinatumomab induction course had been initiated in 25 patients (blinatumomab efficacy/safety set). Fourteen patients did not receive blinatumomab for the following reasons: nine patients achieved CR after R-CHOP and did not pursue blinatumomab therapy according to the trial design, one patient progressed during the first cycle of R-CHOP, three patients died (two because of febrile neutropenia and one following intracranial haemorrhage) after R-CHOP, and one patient presented with severe pneumonia after R-CHOP. Seventeen (68%) patients received the planned blinatumomab induction regimen and doses. Blinatumomab was temporarily discontinued in three (12%) patients and permanently discontinued in seven (28%), which was due to progression in six patients and neurotoxicity in one. Four (16%) patients required dose modifications. Six patients underwent blinatumomab consolidation therapy, none of whom required dose modification or treatment interruption.
Baseline demographic and clinical characteristics of the study participants are summarized in Table 1. Regarding the full analysis set, the median age was 67 years (range, 38–83), 26 patients were male, and 13 were female. CLL genetic features at baseline were as follows: 19/35 (54%) had 17p deletion, 21/34 (62%) had TP53 mutations, and 20/32 (63%) had highly-complex karyotype (≥five chromosomal aberrations)13. The median number of prior therapeutic lines for CLL was two (range, 0–11). Twenty-two (56%) patients had previously undergone CIT, and 24 (62%) had been exposed to ibrutinib, 11 (28%) of whom had also been exposed to venetoclax.
Regarding RT features, 33 (85%) patients presented with advanced disease (stages 3–4 as per Ann Arbor staging), and the international prognostic index (IPI) was 0–1 in two (5%), 2–3 in 28 (72%), and 4–5 in nine (23%). All patients were previously untreated for RT according to the selection criteria. For all 35 patients with available pathology specimens, the central pathology review findings were concordant with an RT diagnosis of diffuse large B-cell histology (DLBCL) or high-grade B-cell lymphoma-not otherwise specified (NOS). Four cases with DLBCL-NOS (non-GC) were unavailable for central review. In the 8 (20.5%) patients with available data for clonal relatedness based on immunoglobulin heavy chain rearrangement analysis, 6 RT were related to the prior CLL.
Efficacy
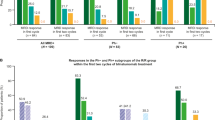
Among the 39 patients who started R-CHOP, 34 were evaluable at evaluation #1 after two cycles (Fig. 1). Nine patients achieved CR and did not receive blinatumomab. The remaining 25 patients, including 10 with partial response (PR), three with SD, and 12 with progressive disease, underwent blinatumomab induction therapy (Fig. 2). Of these 25 patients (blinatumomab efficacy/safety analysis set), five achieved CR, four achieved PR, and six were stable. The five patients who achieved CR after blinatumomab induction showed a partial response after the 2 cycles of R-CHOP. Four of these five patients carried aggressive CLL genetic features with three having TP53 alterations and a hypercomplex karyotype and one having an 11q deletion. Three had mutated IGHV. The CRR at the end of induction (primary endpoint) was 20% (95% confidence interval (CI), 10%–40.7%), and the ORR was 36% (95% CI, 12%–42%).

R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone. Source data are provided as a Source Data file.
Comparing the responses after R-CHOP (evaluation #1) with those obtained after blinatumomab induction (evaluation #2), blinatumomab induction increased the response in seven (28%) patients (Fig. 2). Considering the entire trial cohort (including all patients who started R-CHOP) (n = 39, evaluations #1 and #2), the ORR was 46% (95% CI, 30–63%) and the CRR was 36% (95% CI, 21–53%). Blinatumomab consolidation treatment was administered to six patients, including two with CR after blinatumomab induction, and three with PR. Blinatumomab consolidation therapy led to CR in one patient and PR in two patients.
After a median follow-up period of 23.2 months, the nine responding patients among the 25 treated with blinatumomab (blinatumomab efficacy/safety set) showed a median duration of response to blinatumomab of 14.9 months (95% CI, 1.9–non-estimable) (Fig. 3a). None of the patients received allogeneic HSCT as consolidation after blinatumomab. Of note, patient 31 experienced short-duration CR but eventually underwent allogeneic HSCT despite metabolic progression at the post-consolidation evaluation (evaluation #3), with no additional therapy prior to transplantation, and was still alive with CR 20 months at the last follow-up. In the blinatumomab efficacy/safety set, the median PFS was 2 months (95% CI, 1.5–4.1) with a 1-year rate of 20% (95% CI, 7.3–37.2) (Fig. 3b), and the median OS was 7.5 months (95% CI, 4–16.6) with a 1-year rate of 40% (95% CI, 21.3–58.1) (Fig. 3c). To date, four blinatumomab-treated patients have shown late CR during follow-up without other anti-lymphoma therapies. Two patients had TP53 disruption but mutated IGHV and one had unrelated RT. In the full analyses set, the median PFS was 3.8 months (95% CI, 3.3–13.5) with a 1-year rate of 35.9% (95% CI, 21.4–50.6), and the median OS was 9.1 months (95% CI, 5.8–18) with a 1-year rate of 48.4% (95% CI, 32–62.9) (Supplementary Fig. 2). Of the nine patients who achieved CR after R-CHOP, only 2 were dead at the last follow-up.

Related Kaplan-Meier curves are denoted by red lines, with plots representing censored data. Dashed grey lines indicate 95% confidence intervals. Source data are provided as a Source Data file.
Twenty-six patients were dead at the time of analysis, including 22 patients whose deaths were related to RT progression, three to infections (one febrile neutropenia with sepsis on day 9 of R-CHOP cycle 2, one with pneumonia on day 16 of R-CHOP cycle 1, one SARS-COV-2-related respiratory failure 14 months after second-line RT-directed therapy), and one to cerebral hematoma in the context of thrombocytopenia on day 20 of R-CHOP cycle 1.
Safety
In total, 134 treatment-emergent adverse events (TEAEs) were reported in 23 (92%) (Table 2) of the 25 patients who received at least one dose of blinatumomab (blinatumomab efficacy/safety analysis set). The most common TEAEs of all grades (>1 case), regardless of their relationship with blinatumomab, included fever (36%), anaemia (24%), decreased lymphocyte count (24%), and hyperglycaemia (24%). CRS was observed in four (16%) patients (grade 1 in three and grade 2 in one), and neurological events consistent with neurotoxicity occurred in five (20%) patients (grade 2 ataxia in one, grade 1 tremor in one and grade 2 tremor in one, grade 4 confusion in one, and grade 3 encephalopathy in one). All neurological events resolved, except in one patient in whom grade 1 confusion continued for two additional weeks and until the time of death due to disease progression. None of the patients received tocilizumab therapy. Grade > 2 haematological toxicities included anaemia (8%), neutropenia (4%), lymphopenia (16%), and thrombocytopenia (4%). Adverse events (AEs) led to transient blinatumomab interruption in three (12%) patients (confusion in one, gastrointestinal perforation in one, and cholestasis in one) and permanent interruption in only one (4%) patient (neurotoxicity). Moreover, AEs resulted in dose modification in four (16%) patients for the following reasons: confusion in one patient, intestinal perforation in one, and cholestasis in two.
Nine serious AEs (SAEs) were reported in eight (32%) patients during blinatumomab induction. These included encephalopathy, fever, sepsis (Staphylococcus aureus), catheter-related infections, thromboembolic events, hyperglycaemia, hip pain, tremors with dysgraphia, and ileal perforation. The investigators considered the SAEs as treatment-related in five (56%) patients. No fatal AEs were observed during blinatumomab therapy.
During blinatumomab consolidation, a total of 16 TEAEs were reported in five (83%) patients, and toxicities are shown in Supplementary Table 2.
Discussion
Highly effective targeted therapies are now available for treating patients with CLL; however, RT remains a major obstacle to long-term CLL control. This phase 2 trial evaluated the efficacy and safety of the first-in-class anti-CD3/anti-CD19 bispecific molecule blinatumomab in patients with RT. Our results showed that an 8-week course of blinatumomab induced CR in 20% of the patients without CR after R-CHOP debulking. The CR rate obtained in the whole population treated with R-CHOP was in the range of two recently reported RT trials (18.8–50%), whereas our population included a higher proportion of patients with adverse features (TP53 alterations, prior CLL-directed therapeutic lines)8,14. A durable clinical response lasting six months or more was observed in five (20%) patients with a unique induction course in four and an additional consolidation course in one. The role of allo-HSCT in prolonging the response in these patients remain to be determined. In a recent retrospective study, chimeric antigen receptor T-cell therapy yielded a limited median PFS of 4.7 months15.
Major challenges face clinical trials dedicated to patients with RT, such that fewer than 10 trials have been reported when the present study started. First, RT is a rare condition, and the annual rate of transformation from CLL to RT is estimated to range from 0.5% to 1%. Second, as urgent interventions are usually needed to combat RT owing to its aggressive features, enrolment in trials evaluating targeted therapies or modern immunotherapies is difficult. To overcome this challenge, we used R-CHOP as a bridging therapy to rapidly initiate a tumour response and subsequently minimise the risk of CRS and neurological events. This strategy also aimed to identify good responders to CIT without the need for an experimental approach. We have observed a higher proportion of patients achieving CR after debulking than expected, which may be due to the use of positron emissions tomography to assess the response or to the suggestion that certain RT patients rapidly respond to R-CHOP. How this translates into prolonged survival remains to be established, as a rapid response is not necessarily correlated with a long response in CLL16. Finally, the pathological features of RT can be confused with those of other conditions such as CLL progression and prolymphocytic evolution17. Therefore, we performed a meticulous central review of our cohort, and all patients with available specimens were confirmed to have RT.
Bispecific antibodies are currently emerging as effective strategies for treating patients with de novo DLBCL. The anti-CD3/anti-CD20 antibodies glofitamab and epcoritamab have both been very recently approved for the treatment of relapsed or refractory DLBCL after two or more lines of systemic therapy18,19. In line with our data, evidence supporting the efficacy of bispecific antibodies in RT has been increasing. In a recent report of nine cases treated with blinatumomab, four showed a reduction in nodal disease (including one CR)20. Preliminary and subgroup analyses also recently suggested the efficacy of epcoritamab (CR in 5/10 patients)21 and glofitamab (CR in 5/11 patients)22. There remains a need to improve the duration of response, which could result from a better detection of residual disease in RT through cell-free DNA approaches1. Regarding safety, the incidence of CRS during this trial was lower than that reported elsewhere for de novo DLBCL or RT with either epcoritamab or glofitamab, indicating a potential benefit for CIT bridging therapy in RT or a better safety profile of blinatumomab regarding CRS12. The incidence and severity of neurological events were comparable to those observed with glofitamab and higher than those with epcoritamab. However, cross-trial comparisons should be interpreted with caution. No grade 5 TEAEs were observed during blinatumomab induction, and the cause of death was disease progression in the vast majority of patients.
Recent data have provided evidence of the role of the immune microenvironment in RT. T-cell exhaustion is driven by immune checkpoint deregulation, including an increased expression of checkpoint inhibitory molecules such as programmed cell death protein 1 (PD-1). Neoplastic B-cell PD-1 expression was found to be weak in both CLL and de novo DLBCL and strong in RT23. Interestingly, this observation was linked to clinical responses to the PD-1 blocking antibodies such as pembrolizumab or tislelizumab in RT, whereas no clear anti-tumour activity was observed in CLL10,14. Furthermore, high tumour cell mutational burden, such as that observed in RT1, is emerging as a predictor of an improved therapeutic response to these agents. One potential underlying mechanism could be that the increased neoantigen load at transformation leads to a subsequent immune response. Further studies are warranted to explore the immune environment of RT and factors affecting the response to immunotherapies.
A limitation of our study is the lack of assessment of the clonal relationship between CLL and RT. Although this feature affects response and patient outcome, its timely assessment remains difficult owing to the limited size of the RT specimen and the need to start urgent therapy in the setting of clinical trials. To limit the consequences of this missing information, we propose excluding patients who achieve a CR from the investigation of blinatumomab effects, assuming that clonally unrelated cases would respond best to R-CHOP. Another limitation of our study is that the statistical power was insufficient to identify the determinants of response and outcome because of the small number of patients.
In summary, blinatumomab demonstrated encouraging antitumour activity and acceptable toxicity in patients with RT. Our study opens new avenues for further strategies employing bispecific antibodies against RT.
Methods
Study design and patients
The BLINART study was an investigator-sponsored phase 2, single-arm, open-label, prospective trial conducted across 28 French centres in the setting of the French Innovative Leukemia Organization. Eligible patients were ≥ 18 years old with previously untreated RT, defined as a confirmed diagnosis of CLL or small lymphocytic lymphoma with biopsy-proven DLBCL based on standard criteria24,25, had an Eastern Cooperative Oncology Group performance status of <3, and had to meet the following haematologic criteria at screening unless they had a biopsy-confirmed significant bone marrow involvement of either CLL or RT cells: an absolute neutrophil count of ≥ 1.0 G/L, a platelet count of ≥ 50 G/L, and independence of transfusion within seven days of screening. The exclusion criteria included having a of history or the presence of clinically relevant disorders affecting the central nervous system (CNS) or known active DLBCL in the CNS (confirmed by cerebrospinal fluid analysis). The full eligibility criteria are provided in the Supplementary Information (Study protocol).
For a patient to be included, a histologic diagnosis of RT had to be made by a local pathologist. A centralised pathology review was performed by a panel of expert hematopathologists from the Lymphoma Study Association (LYSA). Karyotypes were classified as simple, complex, or highly complex according to a previous report26.
Patients were enrolled in the study between 5 July 2019 and 19 July 2021.
The protocol was approved by the Comité de Protection des Personnes Nord-Ouest III (Caen, France) and the institutional review board or ethics committee of each participating institution in accordance with the Good Clinical Practice guidelines and the ethical principles originating from the Declaration of Helsinki. Informed consent was obtained from all patients. The study is registered with clinicaltrials.gov, number NCT03931642, and the European Union Drug Regulating Authorities Clinical Trials Database, EudraCT number 2018-003483-32. All authors had access to the study data.
Treatment and response assessment
The treatment and response assessment schedules are summarised in Supplementary Fig. 1. The response was assessed by investigators and defined according to the Lugano 2014 classification for the fluorodeoxyglucose-avid lymphomas using the Deauville Score27. First, the patients underwent debulking therapy with two cycles of standard R-CHOP administered every three weeks, which comprised intravenous (IV) rituximab at 375 mg/m2, cyclophosphamide at 750 mg/m2, doxorubicin at 50 mg/m2, and vincristine at 1·4 mg/m2 (capped at 2·0 mg) on day 1, and oral prednisone at 60 mg/m2/d on days 1–5. The first response evaluation (evaluation #1) was performed between days 15 and 21 of the second R-CHOP cycle. Patients who achieved CR were managed at the discretion of the treating physician. All patients without CR received blinatumomab induction according to a schema previously evaluated in patients with de novo DLBCL12. This consisted of a single 8-week course of blinatumomab by continuous vein infusion at a stepwise dose of 9 μg/d in the first week, 28 μg/d in the second week, and 112 μg/d thereafter. The second response evaluation (evaluation #2) was performed 8–10 weeks after the initiation of blinatumomab induction therapy. Patients who achieved an objective response (CR or PR) after induction were eligible to receive one further optional cycle of blinatumomab consolidation. This consisted of a single 4-week course of blinatumomab 9 μg/d by continuous vein infusion in the first week, 28 μg/d in the second week, and 112 μg/d thereafter. Any further salvage or consolidation strategies, including autologous or allogeneic HSCT, were to be performed at the discretion of the treating physicians.
Before each rituximab infusion, premedication, including an antihistaminic and paracetamol, was administered to minimise infusion-related reactions. Methylprednisolone was administered before the first infusion. During each R-CHOP cycle, granulocyte colony-stimulating factor was given from days 6 to 12 or until the neutrophil count reached ≥ 1 G/L. Before the first blinatumomab dose in each cycle and with every dose increase, the patients received 20 mg of prophylactic dexamethasone orally 6–12 h and 1 h prior to infusion to minimise the risk of CRS and neurologic events. If signs of CRS were noted, dexamethasone was administered at 8 mg orally three times daily for up to 72 h. Antipyretics were recommended to prevent fever within the first two days of blinatumomab administration. Recombinant erythropoietin was administered at the treating physician’s discretion. Prophylaxis for Pneumocystis jirovecii infection with sulfamethoxazole/trimethoprim and antiviral prophylaxis with valaciclovir were recommended.
Statistical analysis
The trial was designed to detect CRR improvement after blinatumomab induction from a baseline of 7% (as observed in a prospective study evaluating R-CHOP4). The blinatumomab efficacy and safety analysis sets included all patients in whom blinatumomab induction therapy was initiated. The sample size was calculated based on Simon’s two-stage design with the following assumptions: p0 = 0.10, p1 = 0.30, type I error of 5%, and type II error of 20%. According to Simon’s estimation, six or more CRs out of the 29 evaluable patients would be required to declare overall success. An evaluable patient was defined as one who started the first blinatumomab infusion. A maximum dropout rate of 15% after debulking therapy was considered based on the expected CRR reported with R-CHOP. Subsequently, a total of 35 patients were initially planned to be included at baseline; 10 were to be enrolled in the first stage, and the remaining 25 were to be enrolled in the second stage. If one or none of the ten patients showed CR at the first stage, the trial was to be stopped.
The dropout rate after the debulking therapy was higher than expected. Accordingly, six additional patients were included, and the sample size was re-estimated. Based on the sample size calculation by Simon’s two-stage design with the aforementioned assumptions for p0 and p1, five or more CRs out of the 25 evaluable patients guarantee a satisfactory statistical power, greater than 85%, for declaring overall success, with a one-sided type I error of 5%.
The primary analysis will be performed in intention-to-treat. The full analysis set included all patients who started their first cycle of R-CHOP. The blinatumomab efficacy/safety analysis set corresponded to all patients who started the blinatumomab induction course.
The primary endpoint was CRR according to the Lugano 2014 classification27 after an 8-week induction course of blinatumomab. CRR was the proportion of patients achieving CR after the blinatumomab induction course among those who started blinatumomab; patients who did not complete the induction course for any reason were considered non-responders at evaluation #2. The secondary endpoints were the ORR after blinatumomab induction and consolidation treatments according to the Lugano 2014 classification27, CRR after blinatumomab consolidation treatment according to the Lugano 2014 classification27, PFS, OS, duration of response, and blinatumomab toxicity. AEs were evaluated in accordance with the National Cancer Institute Common Terminology Criteria for Adverse Events, version 5.
All statistical analyses were performed using the Stata software (version 13, StataCorp, College Station, TX, USA), GraphPad Prism (version 9, GraphPad Software, Boston, MA, USA), and the R software (version 4.1.3, R Core Team 2023), according to the International Conference on Harmonisation-Good Clinical Practice guidelines.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data related to Figs. 2, 3, and Supplementary Fig. 2 are provided with this paper. The study protocol is available in the Supplementary Information file. Aggregated data underlying the findings described in this article and supporting clinical documents can be obtained within 12 weeks after requesting and for unlimited time. Requests will be made to the corresponding author (R.G.; rguieze@chu-clermontferrand.fr) and reviewed by the FILO CLL committee for scientific rationale and feasibility. The datasets generated and/or analysed during the current study are not publicly available due to proprietary considerations. All data provided are anonymized to respect the privacy of patients who have participated in the trial, in line with applicable laws and regulations. The remaining data are available within the Article, Supplementary Information, or Source Data file. Source data are provided in this paper.
References
Parry, E. M. et al. Evolutionary history of transformation from chronic lymphocytic leukemia to Richter syndrome. Nat. Med. 29, 158–169 (2023).
Thompson, P. A. & Siddiqi, T. Treatment of Richter’s syndrome. Hematology 2022, 329–336 (2022).
Smyth, E., Eyre, T. A. & Cheah, C. Y. Emerging therapies for the management of Richter transformation. J. Clin. Oncol. 41, 395–409 (2023).
Langerbeins, P. et al. Poor efficacy and tolerability of R-CHOP in relapsed/refractory chronic lymphocytic leukemia and Richter transformation. Am. J. Hematol. 89, E239–E243 (2014).
Hillmen, P. et al. Acalabrutinib monotherapy in patients with Richter transformation from the Phase 1/2 ACE-CL-001 clinical study. Blood 128, 60–60 (2016).
Davids, M. S. et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin Lymphoma. J. Clin. Oncol. 35, 826–833 (2017).
Bouclet, F. et al. Real-world outcomes following venetoclax therapy in patients with chronic lymphocytic leukemia or Richter syndrome: a FILO study of the French compassionate use cohort. Ann. Hematol. 100, 987–993 (2021).
Davids, M. S. et al. Venetoclax plus dose-adjusted R-EPOCH for Richter syndrome. Blood 139, 686–689 (2022).
Tsimberidou, A.-M. et al. Clinical outcomes and prognostic factors in patients with Richter’s syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J. Clin. Oncol. 24, 2343–2351 (2006).
Ding, W. et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood 129, 3419–3427 (2017).
Younes, A. et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: a phase 1/2a study. Lancet Haematol. 6, e67–e78 (2019).
Viardot, A. et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 127, 1410–1416 (2016).
Baliakas, P. et al. Cytogenetic complexity in chronic lymphocytic leukemia: definitions, associations, and clinical impact. Blood 133, 1205–1216 (2019).
Al-Sawaf, O. et al. Tislelizumab plus zanubrutinib for Richter transformation: the phase 2 RT1 trial. Nat. Med. 30, 240–248 (2024).
Kittai, A. S. et al. Anti-CD19 chimeric antigen receptor T-cell therapy for richter transformation: an international, multicenter, retrospective study. J. Clin. Oncol. 42, 2071–2079 (2024).
Munir, T. et al. Chronic lymphocytic leukemia therapy guided by measurable residual disease. N. Engl. J. Med. 390, 326–337 (2024).
Soilleux, E. J. et al. Diagnostic dilemmas of high-grade transformation (Richter’s syndrome) of chronic lymphocytic leukaemia: results of the phase II National Cancer Research Institute CHOP-OR clinical trial specialist haemato-pathology central review. Histopathology 69, 1066–1076 (2016).
Dickinson, M. J. et al. Glofitamab for relapsed or refractory diffuse large B-Cell Lymphoma. N. Engl. J. Med. 387, 2220–2231 (2022).
Thieblemont, C. et al. Epcoritamab, a Novel, Subcutaneous CD3xCD20 Bispecific T-cell-engaging antibody, in relapsed or refractory large B-cell lymphoma: dose expansion in a phase I/II trial. J. Clin. Oncol. 41, 2238–2247 (2023).
Thompson, P. A. et al. A phase two study of high dose blinatumomab in Richter’s syndrome. Leukemia 36, 2228–2232 (2022).
Kater, A. P. et al. Subcutaneous Epcoritamab in patients with Richter’s syndrome: early results from Phase 1b/2 Trial (EPCORE CLL-1). Blood 140, 850–851 (2022).
Carlo-Stella, C. et al. Glofitamab monotherapy induces durable complete remissions and has a manageable safety profile in patients with Richter’s transformation. Hematol. Oncol. 41, 63–65 (2023).
He, R. et al. PD-1 Expression in Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (CLL/SLL) and Large B-cell Richter Transformation (DLBCL-RT): A Characteristic Feature of DLBCL-RT and potential surrogate marker for clonal relatedness. Am. J. Surg. Pathol. 42, 843–854 (2018).
Hallek, M. et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 111, 5446–5456 (2008).
Swerdlow, S. H. et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127, 2375–2390 (2016).
Jondreville, L., Krzisch, D., Chapiro, E. & Nguyen-Khac, F. The complex karyotype and chronic lymphocytic leukemia: prognostic value and diagnostic recommendations. Am. J. Hematol. 95, 1361–1367 (2020).
Cheson, B. D. et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J. Clin. Oncol. 32, 3059–3068 (2014).
Acknowledgements
We thank the patients who participated in this trial, their families, investigators, and coordinators at each clinical site. This study was an investigator-sponsored study conducted by the French Innovative Leukemia Organization. The study was financially supported by Amgen, which was not involved in the study design, data collection and analysis or manuscript writing. We thank Dr. Serge Alfandari and Prof. Nicolas Boissel, members of the Data and Safety Monitoring Board. We thank Lamya Haddaoui for supervising the sample collection for this trial, Alexandra Fayault for coordinating the administrative aspects of this study, and Force Hemato for funding the genetic assays.
Author information
Authors and Affiliations
Contributions
R. G., P. F., B. P., and V. R. conceived and designed the study and wrote the manuscript. R.G., L.Y., D.R-W., L-M.F., E.F., L.M., T.A., A.C., S.D.G., A-S.M., A.S., B.D., P.Q., B.H., K.L., J.G., A.Q., O.T., and P.F. recruited the patients. J.B. coordinated the biological analyses. G.L. collected and interpretated the IGHV sequencing data. D.S. collected and interpreted the data. B.M. reviewed and interpreted the imaging data. L.V. reviewed and interpreted the cytogenetic analyses. B.P. and M.D.A. performed the statistical analyses. C.L. supervised and conducted the central pathological review. All authors had full access to the data and reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
R.G. reports research grants from Amgen, AstraZeneca, Roche, Janssen, Abbvie, and BeiGene; consulting fees; honoraria; and travel funds from AstraZeneca, Roche, Janssen, Abbvie, and BeiGene. J.G. reports consulting fees, honoraria, and travel funds from AstraZeneca, Janssen, Sanofi. E.F. reports consulting fees, honoraria, and travel funds from AstraZeneca, Janssen, Abbvie, and BeiGene. A.C. reports consulting fees, honoraria and travel funds from AstraZeneca, Roche, Janssen, and Abbvie. J.B. reports honoraria from AstraZeneca and Janssen, and travel funds from AstraZeneca and Abbvie. K.L. has received research grants from AbbVie, AstraZeneca, Novartis, BeiGene, Amgen, as well as personal fees from AbbVie, Novartis, BMS, BeiGene, Takeda, Janssen, AstraZeneca, and Amgen. P.F. reports, consulting fees, honoraria, and travel funds from AstraZeneca, Roche, Gilead, Janssen, BeiGene, and Lilly. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Udo Holtick, Philip Thompson, Bo Zhang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guièze, R., Ysebaert, L., Roos-Weil, D. et al. Blinatumomab after R-CHOP bridging therapy for patients with Richter transformation: a phase 2 multicentre trial. Nat Commun 15, 6822 (2024). https://doi.org/10.1038/s41467-024-51264-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51264-2
This article is cited by
-
Molecularly targeted therapy and immunotherapy in leukemias
Journal of Hematology & Oncology (2026)
-
The Evolving Therapeutic Landscape of Richter Transformation
Current Hematologic Malignancy Reports (2025)
-
Richter transformation in diffuse large B-cell lymphoma in patients with chronic lymphocytic leukemia receiving ibrutinib: risk factors and outcomes
Leukemia (2025)
