
Abstract
After an RNA polymerase reaches a terminator, instead of dissociating from the template, it may diffuse along the DNA and recommence RNA synthesis from the previous or a different promoter. Magnetic tweezers were used to monitor such secondary transcription and determine the effects of low forces assisting or opposing translocation, protein roadblocks, and transcription factors. Remarkably, up to 50% of Escherichia coli (E. coli) RNA polymerases diffused along the DNA after termination. Force biased the direction of diffusion (sliding) and the velocity increased rapidly with force up to 0.7 pN and much more slowly thereafter. Sigma factor 70 (σ70) likely remained associated with the DNA promoting sliding and enabling re-initiation from promoters in either orientation. However, deletions of the α-C-terminal domains severely limited the ability of RNAP to turn around between successive rounds of transcription. The addition of elongation factor NusG, which competes with σ70 for binding to RNAP, limited additional rounds of transcription. Surprisingly, sliding RNA polymerases blocked by a DNA-bound lac repressor could slowly re-initiate transcription and were not affected by NusG, suggesting a σ-independent pathway. Low forces effectively biased promoter selection suggesting a prominent role for topological entanglements that affect RNA polymerase translocation.
Similar content being viewed by others

Introduction
A transcription cycle comprises recognition of the promoter sequence by RNA polymerase (RNAP), initiation, elongation, and termination. In canonical termination, RNAP dissociates from the DNA template at a terminator and diffuses away, predominantly in 3-D1,2. However, previous reports indicate that upon reaching a terminator RNAP may remain on the DNA template3,4 and diffuse one-dimensionally to a nearby promoter to execute another cycle of transcription in the same or opposite direction5,6,7. The ability of RNAP to repetitively transcribe the same DNA sequence by sliding back to the promoter might contribute significantly to gene regulation. Indeed, cycles of transcription by the same RNAP enzyme would efficiently accumulate transcript, eliminate the need to recruit multiple RNAPs, and reduce the probability of collisions among different RNAPs.
While this alternative to canonical termination has been reported4,5,6,7, the biological significance and mechanism of repetitive transcription remains poorly understood. Force could influence this process by directing RNAP diffusion, which might in turn be blocked by DNA-bound proteins. The role of sigma factor (σ) in repetitive transcription is also unclear. In order to initiate transcription, RNAP requires a σ factor for open complex formation8. While the σ factor is thought to detach from RNAP after initiation, there is evidence that σ may remain associated with transcription elongation complexes and promote open bubble formation during secondary transcription events9. Moreover, it is unclear what domains of RNAP, sliding along DNA after completing a “primary" round of elongation in one direction, are required to initiate transcription at a “secondary" promoter oriented in the opposite direction as previously observed5.
In this study, magnetic tweezers (MTs) are used to monitor translocation by RNAP along single DNA templates following arrival at terminators, in different buffer conditions, with various forces assisting or opposing translocation. From 20% to 50% of RNAPs exhibit non-canonical termination, and multiplexed MT assays produce statistically significant data for analyses of the effect of force, roadblock proteins, and different terminators on repetitive transcription. External force directs and modifies the velocity of diffusion, biasing the search for a secondary promoter. The σ factor is likely required for relatively rapid initiation of repetitive transcription from promoters but not for slower re-initiation from a non-promoter site. While sliding RNAPs re-initiate from promoters oriented in either direction with respect to a previous round of elongation, deletion of the C-terminal domains of the α subunits (α-CTD), known to interact with DNA, nearly abolishes the switch of direction.
Results
Force directs RNA polymerase diffusion and repetitive transcription
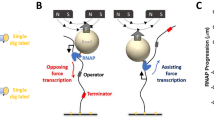
To investigate the mechanism underlying repetitive transcription by RNAP, we used MTs to apply mechanical force assisting or opposing the translocation of single transcription complexes along DNA containing one or two promoters, a high affinity lac repressor binding site (Lac O1), and a terminator and simultaneously monitor multiple DNA templates (Supplementary Data 1-4). The orientation of the promoter sequence(s) determined whether external force opposed or assisted transcription, and the magnitude of force was dependent on the separation between the magnet and the paramagnetic particle linked to RNAP, as illustrated in Fig. 1a. The experiments were conducted at forces ranging from -5 (opposing) to +5 (assisting) pN. After reaching a terminator, opposing force caused RNAPs to slide back to the promoter where some enzymes re-initiated transcription (Fig. 1b, cycles 1 & 2). Single RNAPs repetitively transcribed the same sequence as many as seven times. Finally, recycling RNAPs usually slid to the promoter and paused briefly before either dissociating directly from the promoter or sliding off the end of the DNA template (Fig. 1b, cycle 3). These two pathways of RNAP release could not be distinguished in the assay, because the untethered bead rapidly left focus in either case.

a A diagram of the experimental setup for transcription against opposing force. b A representative recording of multiple rounds of transcription under opposing force includes a temporary roadblock-associated pause during transcription (shaded region 1), pauses at the terminator (shaded regions 2), RNAP temporarily roadblocked during backward sliding (shaded region 3), and pauses at the promoter prior to re-initiation (shaded regions 4). The inset shows data points corresponding to RNAP sliding back from the terminator in cycle 1. c On templates with a dual terminator sequence, the percentage of RNAP that slid backward was twice that on templates with single terminators. The total number of events are listed above each bar. d Opposing force (negative values) significantly raised the probability that the post-terminator complex slid toward the promoter from which the previous cycle of transcription initiated. The total number of events are listed above each bar. e RNAP sliding rates increased rapidly as opposing (–) or assisting (+) force increased from 0 to 0.7 pN but plateaued thereafter. The red center line denotes the 50th percentile, while the blue box contains the 25th to 75th percentiles of the dataset. The black whiskers mark the 5th and 95th percentiles, and values beyond these upper and lower bounds are considered outliers, marked with red crosses. The numbers of sliding events (N) are 128, 1086, 131, 104, 10, 4, 16, 5 for the -5, -2, -0.7, -0.2, 0.2, 0.7, 2, 5 pN force conditions, respectively. Source data are provided as a Source Data file.
Repetitive transcription is an intriguing alternative to canonical termination in which RNAP dissociates from the DNA template. Under opposing force, slightly more than 20% of RNAPs slid backward from single λT1 or T500 terminators. However, a pair of consecutive terminators (Fig. 1c) doubled the probability of sliding to ~50%. This suggests that termination efficiency influenced transitions from transcribing to sliding conformations. Although sliding most often began at a terminator, occasionally it started from a position beyond the terminator (Fig. 1b Cycle 3). In these cases, conformational changes associated with termination that promote sliding might have occurred slowly during run-on elongation. This is consistent with a reported two-stage post-elongation complex (post-termination complex)10.
Repetitive transcription was sensitive to the direction but not the magnitude of force. Under opposing forces up to 25% of post-termination complexes rapidly slid backwards to the previously utilized promoter (Fig. 1d) and ~ 10% of these re-initiated transcription (Supplementary Table 1, Single promoter records). Even under forces as low as 0.2 pN, post-termination complexes almost always slid in the direction of applied force. To further test the effect of force on sliding, a reversed DNA construct was employed (Fig. 1d right). After transcribing under assisting force, post-termination complexes rarely slid backward against the applied force, regardless of the magnitude. This susceptibility to force indicates that post-termination complexes traveled along the DNA template instead of diffusing through space or transferring between juxtaposed segments. Furthermore, while the rates of transcription were consistent with literature data11,12,13, the distributions of rates measured in successive cycles associated with one template were almost identical and distinct from those associated with another identical template (Supplementary Fig. 1e) as reported previously13. Altogether these facts strongly indicate that the same RNAP enzyme produced the repetitive cycles of transcription in any given recording.
Unidirectional sliding of RNAP would be expected if force were to bias one-dimensional diffusion. While bidirectional diffusion reported previously persisted for minutes5,7, fast, unidirectional sliding toward a promoter required <3 s even under extremely low force, 0.2 pN. The magnitude of applied force did not change the probability of sliding, but higher force increased the sliding velocity (Fig. 1e). It was estimated to be at least 200 bp/s at the weakest forces applied and increased linearly with the magnitude of low forces (<2 pN) before plateauing at higher forces. The conformational entropy of the free end of the DNA template feeding into a sliding RNAP might be one factor limiting the sliding velocity. For DNA threading into a 5 nm long, 10 nm diameter pore, the associated entropic force of uncoiling for threading has been estimated to be ~ 0.7–1.9 pN14, which coincides well with the inflection point in the sliding velocity versus force data. Forces opposing or assisting transcription clearly bias the direction of post-termination complex sliding and reduce delays between successive encounters with nearby promoters.
Secondary transcription from a roadblock site
When LacI protein was introduced, RNAP paused frequently at the O1 binding site during transcription, confirming that LacI acts as a “roadblock” (Fig. 2a Cycle 1). LacI also blocked post-termination sliding producing a pause followed by continued sliding to the promoter (Fig. 2a gray-shaded region of cycle 2, Supplementary Fig. 2a) or re-initation from the roadblock (Fig. 2a gray-shaded region of Cycle 1, Fig. 2b). The distribution of dwell times associated with roadblock-induced pauses did not depend on the magnitude of the force (Supplementary Fig. 2b). This result further supports the idea that force-driven post-termination complexes slide along the DNA template, pausing until roadblocks dissociate from the DNA.

a In this representative recording, the shaded regions indicate pauses at the roadblock during post-termination sliding followed by re-initiation ① or continued sliding past the roadblock to the promoter (recapture) ②. b Heat maps indicate probabilities associated with locations at which post-termination complexes started and stopped sliding under opposing force in buffer without (upper) or with (lower) LacI. Sliding primarily began at the terminator ( ~ -0.4 μm) and ended at promoter ( ~ 0 μm) (green box), unless LacI was present to block sliding and induce terminator-to-LacI binding site (red box) and LacI binding site-to-promoter (blue box) sliding events. c Adding LacI to the buffer increased the probability of roadblocking and re-initiation at the roadblock. d Although NusG diminished the probability of sliding to promoters, it did not change the ratio of re-initiation, ~ 0.1 (yellow/red). e A minor population of RNAPs, less than 10%, exhibited multiple cycles of transcription. f Sub-saturating levels of NTPs (100 μM and 20 μM) reduced average end-to-end transcription rates, but not pause-free rates, of both initial and re-initiating transcriptions, compared to a saturating NTP level (1000 μM). The red center line denotes the 50th percentile, while the blue box contains the 25th to 75th percentiles of datasets. The black whiskers mark the 5th and 95th percentiles, and values beyond these upper and lower bounds are considered outliers, marked with red crosses. N: number of events; P values from two-sided two sample t-test are shown in figures (red: p < 0.1; black: p > 0.1).
After pauses at the roadblock, most post-termination complexes slid past to the promoter where ~4% recommenced transcription independently of whether LacI was present (Supplementary Fig. 3a). Unexpectedly, when LacI was present, ~2.4% of sliding post-termination complexes recommenced transcription at the roadblock (Fig. 2a gray-shaded region 1) while significantly less, ~ 0.6%, did so in the absence of LacI (Fig. 2c). Remarkably, the dwell times of post-termination complexes that re-initiated transcription at the roadblock were much longer than those that paused and then continued sliding (Supplementary Fig. 2b). Evidently, after sufficient time at roadblocks, post-termination complexes may form open complexes and recommence transcription. Thus, protein roadblocks on DNA templates can block elongation complexes to delay transcription or produce 3′-truncations15 as well as sliding, post-termination complexes that may produce 5′-truncated transcripts.
Sigma factor promoted sliding and secondary promoter recognition
Promoter recognition by RNAP and transition to an open complex involves σ factors. In a buffer without free σ70, secondary initiation from the promoter would require σ factor to remain bound to RNAP throughout primary transcription and subsequent sliding of the post-termination complex to the promoter. To test this hypothesis, NusG, a transcription elongation factor that competes with σ70 for binding to RNAP16, was introduced. NusG diminished the frequency of repetitive transcription events from the promoter site (Fig. 2d) as expected for competition that would release σ70 from elongation and post-termination complexes. More precisely, NusG decreased the probability of post-termination complexes sliding along DNA template, but a similar fraction of sliding post-termination complexes re-initiated after reaching the promoter (Fig. 2d). This is consistent with reports that σ70 often remains associated with post-termination complexes on DNA7,9, and another report describing experiments in which optical tweezers were used to apply forces to core RNAP and found only brief, force-sensitive dwell times at t500, his, and λT2 terminators17 but no sliding and secondary transcription events. In the current experiments, the probability of re-initiation dropped precipitously after one round (Fig. 2e). With a measured dissociation constant of 3.8 ± 0.8 × 10−3 9, few RNAPs were likely to retain σ70 through multiple cycles of transcription lasting several hundred seconds (Fig. 1b).
NusG negligibly changed re-initiation at the roadblock (Supplementary Fig. 3a), although post-termination complexes lingered much longer at roadblocks than at promoters before re-initiating transcription (Supplementary Fig. 2b). Thus, with or without associated sigma factor, sliding post-termination complexes slowly formed transcription bubbles for elongation at a non-promoter sequence.
To verify that the secondary cycles of RNAP translocation were indeed associated with transcription, we compared the translocation rates of RNAPs as a function of NTP concentration (Fig. 2f). With saturating NTP the initial round of transcription was relatively slower than the secondary cycle due to pauses at LacI roadblocks. However, the pause-free translocation rates were similar for both cycles and for translocation from either promoters or roadblocks. With no LacI, sub-saturating NTP negligibly affected the pause-free rates for translocation but significantly lowered the overall translocation rate of both initial and secondary cycles, producing more frequent and lengthier pauses associated with incorporation of NTPs and synthesis of transcripts18. Recapitulating, for translocation events from promoters, similar pause free translocation rates and sensitivity to NTP concentration are evidence of transcription in both primary and secondary cycles. At saturating NTP, translocation rates from promoters and roadblocks were indistinguishable, which transitively shows that transcription re-initiated from both. Re-initiation probability also dropped with lower NTP concentrations. Since sub-saturating NTP level is known to mitigate the efficiency of initiation19, we postulate that re-initiation consists of the mechanistically identical initiation stage followed by synthesis of transcripts as in the canonical cycle.
Force biased re-initiation between converging or diverging promoters
The applied force directed sliding by post-termination complexes and determined at what promoter re-initiation occurred. Consequently, post-termination complexes slid back to re-initiate multiple times at promoters oriented against the force but were directed away from a promoter oriented with the force (Fig. 3a–d). In a template with convergent promoters, the promoter aligned with the force was utilized only if an RNAP located this promoter at the initial round (Fig. 3a, b). In a template with divergent promoters, the promoter aligned with the force was utilized either during the initial round or during the last round of recycling (Fig. 3c, d). This result suggests that very slight forces affecting post-termination complex sliding might bias transcription from nearby but oppositely oriented promoters in vivo.

a In a recording for a template with converging promoters, after one transcription event from P1 under assisting force, RNAP slid to P2 and completed three cycles of transcription opposing force before finally dissociating from the promoter. b A time-based catalog of the transcription events involving promoter P2 observed along the templates with converging promoters shows the beginning and end of each transcription event with different rounds depicted in different colors. Transcription from P1, the promoter oriented in the direction of force, never repeated. c In a recording for a template with diverging promoters, transcription from P1, opposing the direction of force, repeated four times before RNAP slid past P1 to re-initiate once from P2 assisted by force. d A time-based catalog of the transcription events involving promoter P2 for templates with diverging promoters shows repetitive events from promoter P1, oriented against the force but only single primary or final repetitive events from P2 oriented with the force. e In figures prepared with Chimera version 1.2, α-CTDs are shown to interact with promoter DNA in a closed rrnB promoter complex22 (left). An enlarged view of the α-CTD shows contacts with the UP element (right). f Deletion of α-CTD diminished the probability that RNAP turned around to re-initiate in the direction opposite to the primary transcription event (anti-sense re-initiation) on templates with converging or diverging promoters.
As reported previously6, sliding post-termination complexes readily recognized promoters with either orientation. Remarkably, the experiments described here with convergent or divergent pairs of promoters revealed equivalent dwell times before recommencing transcription in either direction (Supplementary Fig. 2d). To re-initiate secondary transcription from a promoter oriented in the opposite direction, a post-termination complex, which likely has lost all or part of the transcription bubble20, rapidly slides in the direction of applied force until it recognizes a promoter and switches polarity, seizing as the template what was previously the non-template DNA strand to form an open bubble. During this switch, nonspecific DNA contacts made by core RNAP and σ factor must be transiently lost. How does the enzyme remain associated with the DNA while making a U-turn? The C-terminal domains of the α subunits (α-CTD) could mediate the polarity switch. The α-CTDs are connected to the α N-terminal domains (NTDs) via long flexible linkers; the NTDs interact with the β (α1) and β’ (α2) subunits, serving as a scaffold for core enzyme assembly21. The α-CTDs make direct contacts to AT-rich sequences (UP elements) upstream of the core promoter (Fig. 3e)22, and are required for the exceptional strength of rRNA promoters, but are dispensable for initiation at many promoters23. A “consensus” UP element is composed of T- and A-tracks centered at the -50 and -40 promoter positions23. T-tracks are also key signature motifs of intrinsic terminators, which may also contain a matching A-track upstream of a hairpin24. Thus, the α-CTDs may maintain contacts with their preferred DNA elements in the course of polarity switch, anchoring RNAP on the DNA despite the loss of other protein-DNA contacts. To explore this possibility, we measured the probability of transcription from divergent or convergent secondary promoters using an RNAP variant lacking the α-CTD. This mutant RNAP rarely turned around to re-initiate transcription at promoters oriented in the direction opposite to the preceding transcription event (Fig. 3f, Supplementary Fig. 3c, d). However, the missing α-CTD did not affect RNAP’s ability of re-initiating transcription from a promoter aligned with preceding transcription event (Supplementary Table 1 convergent and divergent promoter records summary).
Discussion
In this study, we demonstrate an alternative to canonical transcription termination. The ability of RNAP to slide along DNA and re-initiate transcription without fully dissociating would efficiently accumulate transcripts and reduce the need to recruit RNAPs, which is especially advantageous for rapid and repeated transcription from a single promoter. The one-dimensional search by sliding is also a much faster way to locate nearby promoters if there are no intervening roadblocks. This behavior, tuned by transcription factors like sigma, might significantly modulate RNAP production in certain cellular environments. For example, molecular crowding, and phase separation, or intracellular tethering25, could produce forces that influence RNAP behavior and skew gene expression patterns. It is intriguing that cells could exploit mechanical forces to regulate transcription dynamically and instantly respond to environmental changes or stress.
These experiments indicate four distinct steps to repeat transcription consisting of RNAP (1) remaining associated with the DNA template after elongation; (2) RNAP sliding or diffusing along the DNA; (3) RNAP stopping at obstacles or promoters, and (4) RNAP re-initiating transcription at these sites. In step (1), the RNAP-DNA complex likely undergoes some conformational change that allows the RNAP to slide freely along DNA. This may resemble a final stage of transcription termination in which the DNA strands of the transcription bubble have re-hybridized20. In step (2), RNAP either diffuses along the DNA as previously reported5,6, or slides rapidly in one direction driven by as little as sub-piconewton levels of force. In step (3), RNAP (i) recognizes promoters in either orientation or only aligned in the direction of the previous elongation if the α-CTDs are missing or (ii) becomes blocked by DNA-bound proteins. In step (4), RNAP can form open complexes and initiate transcription at promoters with a characteristic delay of about a minute, or even at a non-promoter site if roadblocked for five-fold longer. The presence of DNA-bound proteins, σ factors and tension would significantly bias the products of the secondary transcription, as illustrated in Fig. 4.

After termination, RNAP (blue) may release mRNA (green) and dissociate from the DNA, or slide along the template. If blocked by a protein roadblock (yellow), RNAP may re-open a transcription bubble at the roadblock site and exhibit roadblock-induced recycling, a process independent of the presence of σ factor (cyan). Upon recognition of a distant promoter with the help of σ factor, wild-type RNAP may re-initiate transcription at promoters oriented in either direction.
While re-initiation at the original promoter could beneficially boost in the RNA yield, spurious initiation at a (protein) roadblock might trigger the synthesis of incomplete or cryptic transcripts from non-promoter sites leading to dysregulation of gene expression in any living cell and contributing to aging in humans26. Note, however, that spurious transcription might also be a principal mechanism for stability, particularly in stressful conditions. Pervasive RNA synthesis serves as a key means to survey the genome for DNA lesions prior to the next round of replication, with RNAP stalling at, and directly delivering DNA repair factors to, the site of damage27. A burst of noisy transcription within a “problem” region would thus be expected to promote repair therein. In the experiments described in this report, transcription re-initiated at a protein roadblock in vitro. However, other obstacles, e.g., damaged deoxyribonucleotides or misincorporated ribonucleotides, may also obstruct RNAP translocation and support transcription restart, thereby enabling the recruitment of respective repair machineries, such as the Uvr complex28 or RNaseHII29.
Whereas in previous experiments, the rate and direction of the transcription elongation was remarkably resistant to externally applied force30, the 2D sliding of post-termination complexes reported here is not. Subtle force opposing or assisting RNAP translocation and DNA-bound proteins (roadblocks) along the template could significantly alter the efficiency and output of repetitive transcription. Even a tiny amount of force greatly reduces the time required to slide to a re-initiation site. Moreover, force directs sliding in only one direction along a DNA sequence. Opposing force increases the frequency of repetitive transcription of a single promoter by an individual RNAP and necessarily diminishes gene expression from other promoters further downstream. Assisting force prohibits repetitive transcription and drives a sliding post-termination complex toward downstream promoters. Thus, the direction of force acting on RNAP can coordinate the expression of one or more genes; it biased transcription between convergent promoters without transcriptional interference between RNAPs initiating from opposing promoters (RNAP collision model)31,32. While the collision model would not predict similar levels of interference between divergent genes, force-directed RNAP diffusion produces preferential expression of one transcript independently of the arrangement of promoters or additional regulatory factors33. Even factors like RapA, which was recently shown to accelerate dissociation of post-termination complexes10 and would therefore diminish post-elongation sliding, would not weaken preferential, force-driven expression. A transcribing RNAP constitutes a formidable mechanochemical motor, whereas terminated RNAP (PTC) could be easily and speedily pushed along DNA in any direction by other molecular motors such as other transcribing RNAPs, replisome and associated DNA helicases and translocases (e.g. Rep, DinG, FtsK)34,35, and other DNA helicases (UvrD, Mfd, RecBCD, etc)36,37. This would dramatically accelerate the search for nearby promoters during recycling of PTCs and spurious re-initiation at intervening roadblocks.
In absence of free σ factor in solution, less than 10% of post-termination complexes exhibited repetitive transcription at a promoter site, which is likely the portion of post-termination complexes retaining σ factor9. Adding NusG reduced that ratio to ~ 2% suggesting that retention of σ factor enhanced sliding and repetitive transcription. In the absence of force, high affinity roadblocks would limit the sequences accessible to diffusing RNAPs and increase dwell times. However, a sliding RNAP driven by force against a high affinity roadblock might pause long enough to start transcription with or without σ factor. This mechanism may differ from canonical transcription initiation in which the σ factor assists promoter recognition and DNA strand separation. How a core RNAP enzyme might form an open bubble remains to be investigated.
These experiments indicate that post-termination sliding occurs without a transcription bubble. First, sliding rates far exceeded measured in vitro transcription rates even under the lowest external mechanical forces, and a transcription bubble traveling at these speeds would intermittently encounter large energy barriers when base pairs to be denatured ahead are far more stable than those that re-hybridize behind. Second, sliding of a transcription bubble cannot explain our findings that (a) sigma factor, which helps form the transcription bubble, is crucial for promoter re-initiation, and (b) re-initiation from a non-promoter site requires 5-fold longer dwell time (very likely for opening a transcription bubble). Third, RNAPs re-initiated with the same delay at forward and reverse promoters, likely associated with the times for RNAPs to form transcription bubbles and re-initiate in either direction.
It is curious that post-termination sliding has not been previously reported in force spectroscopy assays of transcription12,13,17,38,39 in which post-termination complexes mostly dissociated from the template. One might hypothesize that in these experiments, RNAP slid too fast to seize the promoter and ran off the end of the template. However, the current experiments show that post-termination complexes often pause for several seconds at the terminator before sliding at as much as 700 bp/s driven by forces as high as 5 pN. At this sliding rate they also readily seize promoters and re-initiate transcription. This finding contradicts the previous assumption that 3 pN of tension could make sliding too fast and transient to be detected5. Alternatively, higher forces employed in much of the previous work might disrupt weak interactions between a sliding RNAP and the DNA backbone, although optically resolved sliding was not observed in transcription interference assays without force either40. Most previous measurements utilized optical trapping with a feedback mechanism to maintain constant force. This may be suitable to monitor slower processive steps of molecular motors (transcribing RNAPs), but high bandwidth feedback may be critical for faster, continuous, non-processive events (sliding RNAPs)41.
It remains unclear if other RNA polymerases exhibit post-termination sliding. There is no experimental evidence of such sliding by eukaryotic polymerases. However, eukaryotic cells evolved a specialized polymerase (Pol I) with a higher loading rate to amplify rRNA production42, which might be an efficient substitute.
Force significantly directed sliding of post-termination complexes to accelerate repetitive transcription and modulate the relative utilization of adjacent promoters. Stringent control of the constituents afforded by the single molecule assembly revealed that σ70 often remained associated with the RNAP core enzyme to enhance re-initiation after sliding. Furthermore, DNA-binding proteins acting as roadblocks to sliding established non-promoter locations at which RNAP re-initiated transcription with five-fold greater delays than those observed for promoters. Sliding post-termination complexes indiscriminately utilized promoters oriented in either direction but could not easily switch template strands once the α-CTDs were deleted. These experiments highlight how very slight forces affecting post-termination diffusion of RNAP significantly impact transcription regulation.
Method
Protein expression and purification
Plasmids encoding wild-type E. coli core RNAP [pIA1202; α-β-β’[AVI][His]-ω) or Δ-αCTD RNAP [pIA1558; α1-235-β-β′[AVI][His]-ω) under control of the phage T7 promoter were transformed into E. coli BL21(λDE3) and grown in LB at 37 °C to OD600 ~ 0.5. Protein expression was induced with 0.5 mM IPTG for 5 hours at 30 °C. To increase the efficiency of RNAP biotinylation, we separately expressed E. coli biotin ligase BirA from the T7 promoter (Addgene#109424) in E. coli BL21(λDE3) using the same induction conditions. Cells were pelleted by centrifugation (6000 x g, 4 °C, 10 min). Cell pellets containing overexpressed RNAP and BirA were mixed and resuspended in Lysis Buffer (10 mM Tris-OAc pH 7.8, 0.1 M NaCl, 10 mM ATP, 10 mM MgOAc, 100 μM d-biotin, 5 mM β-ME) supplied with Complete EDTA-free Protease Inhibitors (Roche) per manufacturer’s instructions.
Cells were lysed by sonication. Cell debris was pelleted by centrifugation (20,000 x g, 40 min, 4 °C). The cleared cell extract was incubated with Ni Sepharose 6 Fast Flow resin (Cytiva, Marlborough, MA) for 40 min at 4 °C with agitation. The resin was washed with Ni-A Buffer (25 mM Tris pH 6.9, 5% glycerol, 500 mM NaCl, 5 mM β-ME, 0.1 mM phenylmethylsulfonyl fluoride (PMSF) supplemented with 10 mM, 20 mM, and 30 mM imidazole. Protein was eluted in Ni-B Buffer (25 mM Tris pH 6.9, 5% glycerol, 5 mM β-ME, 0.1 mM PMSF, 100 mM NaCl, 300 mM imidazole).
The sample was diluted 1.5 times with Hep-A Buffer (25 mM Tris pH 6.9, 5% glycerol, 5 mM β-ME) and then loaded onto Heparin HP column (Cytiva). A linear gradient between Hep-A and Hep-B Buffer (25 mM Tris pH 6.9, 5% glycerol, 5 mM β-ME, 1 M NaCl) was applied. The biotinylated RNAP core was eluted at ~40 mS/cm.
The eluate from Heparin HP column was diluted 2.5 times with Hep-A Buffer and loaded onto Resource Q column (Cytiva). A linear gradient was applied from 5% to 100% Hep-B Buffer. The biotinylated RNAP core was eluted at ~25 mS/cm with a final concentration 2.5 μM.
Fractions from the elution peaks were analyzed by SDS-PAGE. Those containing purified protein were combined and dialyzed against Storage Buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 45% glycerol, 5 mM β-ME, 0.2 mM EDTA).
E. coli RNAP holoenzymes were formed by mixing core RNAP (2.5 μM) and a two-fold molar excess of σ70 (5 μM)43 in buffer (20 mM Tris-HCl pH 8.0, 0.1 mM EDTA, 100 mM K-acetate, 100 μg/ml BSA, 10 mM DTT, 5% glycerol, and 0.025% Tween-20) and incubating at 37 °C for 10 min.
Lac repressor protein (LacI) was prepared in the laboratory of Kathleen Matthews44.
NusG factor was prepared in the laboratory of Irina Artsimovitch45.
Transcription templates
The DNA templates for single promoter opposing and assisting force transcription assays were PCR amplicons from a plasmid template containing the T7A1 promoter followed by 23 bp without G in the template strand, a 1.2 kb downstream elongation region including a lac operator site (O1), and finally a terminator sequence (λT1, t500, or trpA1-λT1; Supplementary Information & Supplementary Data 1-4). The plasmid template was amplified with single-digoxigenin labeled forward and unlabeled reverse primer pairs, and Q5 Hot Start High-Fidelity 2X PCR Master Mix (New England Biolabs, Ipswich, MA). The transcribed region had the following spacings: Promoter-713 bp-Lac operator-612 bp-Terminator, as illustrated in Fig. 1b. For the opposing force experiments, primers D-A/JBOIDO1/5096 5′-dig-ATCGTTGGGAACCGGAG and S/JBOIDO1/2086 5′-AGCTTGTCTGTAAGCGGATG were used to generate a 3k bp amplicon with 1021 bp between the chamber surface anchor point and the transcription start site. For the assisting force experiments, primers D-S/YY-400/103 5′-dig-GCTTGGTTATGCCGGTACTG and A/pUC18-nuB104/2043 5′-ACGACCTACACCGAACTGAG were used to generate a 4k bp amplicon with 2014 bp between the anchor point and transcription start site. The longer separation in the assisting force amplicon reduces adhesion of promoter-bound magnetic beads to the chamber surface at the beginning of transcription.
In preparation of the templates with the convergent promoters, we used a digoxigenin-labeled primer D-S/YY-400/103 and a primer with the palindromic restriction site ApaI A/pUC18-nuB104/1983-ApaI 5′-gccacggggcccAAGGGAGAAAGGCGGACAG to generate amplicons with digoxigenin label on one end and ApaI restriction site on the other end, using the plasmid in Supplementary Data 3. We repeated the PCR with unlabeled primer S/YY-400/103 and the same ApaI containing primer to generate amplicons with the same restriction site but no digoxigenin label. Both amplicons were then digested by ApaI enzymes (New England BioLabs) in rCutSmart buffer (New England Labs) per manufacturer’s instruction. To produce a satisfactory amount of constructs with a digoxigenin-labeled end and an unlabeled end, the digoxigenin-labeled and unlabeled amplicons were ligated with T7 ligase (New England BioLabs) in a specific ratio of 1:2. Similarly, in preparation of the templates with the divergent promoters, we paired a digoxigenin-labeled primer D-A/JBOIDO1/5096 and its unlabeled version with a primer with palindromic SalI restriction site S/JBOIDO1/2086-SalI 5′-ggagacgtcgacAGCTTGTCTGTAAGCGGATG to generate two sets of amplicons, which were then digested by SalI (New England Labs) restriction enzymes in NEBuffer r3.1 (New England Labs) and ligated in 1:2 ratio to generate the divergent promoter templates. The information about plasmids and primers used in these experiments is listed in Supplementary Table 2 and Supplementary Data.
Preparation of halted TEC and tethers
Microchambers were assembled with laser-cut parafilm gaskets between two glass coverslips46,47. The volume of a microchamber was about 10 μL. Polyclonal anti-digoxigenin (Roche Diagnostics, catalog number 11333089001, lot number 66890900) was introduced to coat the inner surface of the chamber at a concentration of 8 μg/mL in PBS for 90 min at room temperature. The surface was then passivated with Blocking buffer (PBS with 1% caesin, GeneTex, Irvine, CA) for 20 min at room temperature.
Transcription tethers were assembled by mixing 15 nM of biotinylated RNAP holoenzyme and 1.5 nM linear DNA template in Transcription Buffer (20 mM Tris glutamate pH 8, 50 mM potassium glutamate, 10 mM magnesium glutamate, 1 mM DTT, 0.2 mg/ml casein) and incubated 20 min at 37 °C. Afterward, 50 μM ATP, UTP, GTP (NewEngland Biolabs, Ipswich, MA), and 100 μM GpA dinucleotides (TriLink, San Diego, CA) were added to the solution and incubated for additional 10 min at 37 °C to allow the ternary complex to initiate transcription and stall at the first G in the template. The solution of ternary complex was diluted to a final concentration of 250 pM RNAP:DNA complex, flushed into the passivated microchambers, and incubated for 10 min. Then, 20 μL of streptavidin-coated superparamagnetic beads (MyOneT1 Dynabeads, Invitrogen/Life Technologies, Carlsbad, CA), washed in PBS per manufacturer instruction and then diluted 1:100 in Transcription Buffer, were flushed into microchambers to attach beads to biotinylated RNAP stalled on the DNA. After 5 min incubation, excess superparamagnetic beads, untethered DNA molecules and proteins in solution were flushed out with 50 μL Transcription Buffer.
Magnetic tweezers assays
Transcription complexes were prepared with E. coli RNAP holoenzyme biotinylated on the C-terminus of the β’ subunit and DNA molecules labeled at one end with digoxigenin for anchoring to the microchamber surface. These molecules included a T7A1 promoter, a binding site for lac repressor, and a terminator in a 1.2 kbp sequence. Each RNAP enzyme in a magnetic tweezing assembly was coupled to a streptavidin-coated magnetic bead and transcription elongation complexes were stalled after 23 bp by supplying only ATP, GTP, and UTP before tethering to the surface of an anti-digoxigenin-coated microchamber. Then the flow chamber was placed on the MT microscope stage, and a field of view with several freely moving tethered beads was selected. The real-time tracking was acquired at least 30 Hz and up to 120 Hz frame rate, depending on the number of tracked tethers in the camera view. After a brief period of tracking (< 3 min) the selected beads to establish a baseline tether length, 1 mM NTPs including CTP were introduced to produce transcription that increased or decreased tether lengths for the assisting, or opposing, force configurations respectively (Fig. 1a). NusG and LacI proteins, when used, were added together with NTPs at 1 μM and 20 nM, respectively. Recording lasted typically ~ 1 h. Since the magnetite content of individual beads varies slightly and DNA tethers become extended to differing degrees, the transcription (tether length) records were scaled to assemble a data set for statistics (See Supplementary Fig. 1 and Data Processing in SI).
Statistics & reproducibility
No statistical method was used to predetermine sample sizes. At the beginning of an experiment, a randomly selected view with approximately twenty or more particles exhibiting movement, which appeared to be tethered by DNA and not stuck on the surface, were selected for tracking. Randomization was inherent, because DNA tethers with repetitive transcription cycles could not be predicted a priori. Every transcription record was included in the initial data set48. Then, an automated algorithm, as described in Supplementary Information, was utilized to detect and categorize recycling behaviors and summarize the statistics. Due to this automation, the investigators were not blinded to conditions during experiments and analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data underlying the findings of this study are available in a “figshare” repository at https://doi.org/10.6084/m9.figshare.2547889648. Source data are provided with this paper.
Code availability
The codes used for data analysis, figure generation are available in a “figshare” repository together with the transcription records data at https://doi.org/10.6084/m9.figshare.2547889648. The codes of MT and softwares used for data acquisition in this study will be available upon request to the corresponding author.
References
Berg, O., Winter, R. & Von Hippel, P. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry 20, 6929–6948 (1981).
Wang, F. et al. The promoter-search mechanism of Escherichia coli RNA polymerase is dominated by three-dimensional diffusion. Nat. Struct. Mol. Biol. 20, 174–181 (2013).
Arndt, K. & Chamberlin, M. Transcription termination in Escherichia coli: measurement of the rate of enzyme release from rho-independent terminators. J. Mol. Biol. 202, 271–285 (1988).
Bellecourt, M., Ray-Soni, A., Harwig, A., Mooney, R. & Landick, R. RNA polymerase clamp movement aids dissociation from DNA but is not required for RNA release at intrinsic terminators. J. Mol. Biol. 431, 696–713 (2019).
Harden, T. et al. Alternative transcription cycle for bacterial RNA polymerase. Nat. Commun. 11, 448 (2020).
Kang, W., Hwang, S., Kang, J., Kang, C. & Hohng, S. Hopping and flipping of RNA polymerase on DNA during recycling for reinitiation after intrinsic termination in bacterial transcription. Int. J. Mol. Sci. 22, 2398 (2021).
Kang, W. et al. Transcription reinitiation by recycling RNA polymerase that diffuses on DNA after releasing terminated RNA. Nat. Commun. 11, 450 (2020).
Paget, M. & Helmann, J. The σ70family of sigma factors. Genome Biol. 4, 1–6 (2003).
Harden, T. et al. Bacterial RNA polymerase can retain σ70 throughout transcription. Proc. Natl Acad. Sci. USA 113, 602–607 (2016).
Inlow, K., Tenenbaum, D., Friedman, L., Kondev, J. & Gelles, J. Recycling of bacterial RNA polymerase by the Swi2/Snf2 ATPase RapA. Proc. Natl Acad. Sci. USA 120, e2303849120 (2023).
Herbert, K., Zhou, J., Mooney, R., Porta, A., Landick, R. & Block, S. E. coli NusG inhibits backtracking and accelerates pause-free transcription by promoting forward translocation of RNA polymerase. J. Mol. Biol. 399, 17–30 (2010).
Forde, N., Izhaky, D., Woodcock, G., Wuite, G. & Bustamante, C. Using mechanical force to probe the mechanism of pausing and arrest during continuous elongation by Escherichia coli RNA polymerase. Proc. Natl Acad. Sci. USA 99, 11682–7 (2002).
Adelman, K. et al. Single molecule analysis of RNA polymerase elongation reveals uniform kinetic behavior. Proc. Natl Acad. Sci. USA 99, 13538–13543 (2002).
Chen, L. & Conlisk, A. Forces affecting double-stranded DNA translocation through synthetic nanopores. Biomed. Microdevices 13, 403–414 (2011).
Qian, J. et al. Reciprocating RNA Polymerase batters through roadblocks. Nat. Commun. 15, 3193 (2024).
NandyMazumdar, M. et al. RNA polymerase gate loop guides the nontemplate DNA strand in transcription complexes. Proc. Natl Acad. Sci. USA 113, 14994–14999 (2016).
Larson, M., Greenleaf, W., Landick, R. & Block, S. Applied force reveals mechanistic and energetic details of transcription termination. Cell 132, 971–982 (2008).
Janissen, R., Eslami-Mossallam, B., Artsimovitch, I., Depken, M. & Dekker, N. High-throughput single-molecule experiments reveal heterogeneity, state switching, and three interconnected pause states in transcription. Cell Rep. 39, 110749 (2022).
Sojka, L. et al. Rapid changes in gene expression: DNA determinants of promoter regulation by the concentration of the transcription initiating NTP in Bacillus subtilis. Nucleic Acids Res. 39, 4598–4611 (2011).
You, L. et al. Structural basis for intrinsic transcription termination. Nature 613, 783–789 (2023)
Murayama, S., Ishikawa, S., Chumsakul, O., Ogasawara, N. & Oshima, T. The Role of α-CTD in the genome-wide transcriptional regulation of the Bacillus subtilis cells. PLoS ONE 10, e0131588 (2015).
Shin, Y. et al. Structural basis of ribosomal RNA transcription regulation. Nat. Commun. 12, 528 (2021).
Gourse, R., Ross, W. & Gaal, T. UPs and downs in bacterial transcription initiation: the role of the alpha subunit of RNA polymerase in promoter recognition. Mol. Microbiol. 37, 687–695 (2000).
Roberts, J. Mechanisms of bacterial transcription termination. J. Mol. Biol. 431, 4030–4039 (2019).
Shaban, H. A., Barth, R. & Bystricky, K. Navigating the crowd: visualizing coordination between genome dynamics, structure, and transcription. Genome Biol. 21, 278 (2020).
McCauley, B. et al. Altered chromatin states drive cryptic transcription in aging mammalian stem cells. Nat. Aging 1, 684–697 (2021).
Nudler, E. Transcription-coupled global genomic repair in E. coli. Trends Biochem. Sci. 48, 873–882 (2023).
Bharati, B. et al. Crucial role and mechanism of transcription-coupled DNA repair in bacteria. Nature 604, 152–159 (2022).
Hao, Z. et al. RNA polymerase drives ribonucleotide excision DNA repair in E. coli. Cell 186, 2425-2437.e21 (2023).
Yin, H. et al. Transcription against an applied force. Science 270, 1653–1657 (1995).
Hao, N., Palmer, A., Ahlgren-Berg, A., Shearwin, K. & Dodd, I. The role of repressor kinetics in relief of transcriptional interference between convergent promoters. Nucleic Acids Res. 44, 6625–6638 (2016).
Brophy, J. & Voigt, C. Antisense transcription as a tool to tune gene expression. Mol. Syst. Biol. 12, 854 (2016).
Wu, A. et al. Repression of divergent noncoding transcription by a sequence-specific transcription factor. Mol. Cell 72, 942-954.e7 (2018).
Boubakri, H., Septenville, A., Viguera, E. & Michel, B. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 29, 145–157 (2010).
Lee, J., Finkelstein, I., Arciszewska, L., Sherratt, D. & Greene, E. Single-molecule imaging of FtsK translocation reveals mechanistic features of protein-protein collisions on DNA. Mol. Cell 54, 832–843 (2014).
Terakawa, T., Redding, S., Silverstein, T. & Greene, E. Sequential eviction of crowded nucleoprotein complexes by the exonuclease RecBCD molecular motor. Proc. Natl Acad. Sci. USA 114, E6322–E6331 (2017).
Epshtein, V. et al. UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature 505, 372–377 (2014).
Davenport, R., Wuite, G., Landick, R. & Bustamante, C. Single-molecule study of transcriptional pausing and arrest by E. coli RNA polymerase. Science 287, 2497–500 (2000).
Shaevitz, J., Abbondanzieri, E., Landick, R. & Block, S. Backtracking by single RNA polymerase molecules observed at near-base-pair resolution. Nature 426, 684–687 (2003).
Wang, L., Watters, J., Ju, X., Lu, G. & Liu, S. Head-on and co-directional RNA polymerase collisions orchestrate bidirectional transcription termination. Mol. Cell 83, 1153-1164.e4 (2023).
Rice, S., Purcell, T. & Spudich, J. [6] Building and using optical traps to study properties of molecular motors. Methods Enzymol. 361, 112–133 (2003).
Albert, B. et al. RNA polymerase I-specific subunits promote polymerase clustering to enhance the rRNA gene transcription cycle. J. Cell Biol. 192, 277–293 (2011).
Mekler, V. & Severinov, K. Use of RNA polymerase molecular beacon assay to measure RNA polymerase interactions with model promoter fragments. In bacterial Transcriptional Control: Methods and Protocols. (eds Artsimovitch, I. & Santangelo, T.) vol. 1276, pp. 199–210 (Humana Press, New York, NY, 2015).
Xu, J., Liu, S., Chen, M., Ma, J. & Matthews, K. Altering Residues N125 and D149 Impacts Sugar Effector Binding and Allosteric Parameters in Escherichia coli Lactose Repressor. Biochemistry 50, 9002–9013 (2011).
Sevostyanova, A. & Artsimovitch, I. Functional analysis of Thermus thermophilus transcription factor NusG. Nucleic Acids Res. 38, 7432–7445 (2010).
Ding, Y. et al. DNA supercoiling: A regulatory signal for the λ repressor. Proc. Natl Acad. Sci. USA 111, 15402–15407 (2014).
Yan, Y., Leng, F., Finzi, L. & Dunlap, D. Protein-mediated looping of DNA under tension requires supercoiling. Nucleic Acids Res. 46, 2370–2379 (2018).
Qian, J. Data for “Force and the alpha-C-terminal domains bias RNA polymerase recycling” https://doi.org/10.6084/m9.figshare.25478896 (2024).
Acknowledgements
LacI was a generous gift from Kathleen Matthews, Rice University. Plasmids for these experiments were created by Derrica McCalla. We thank Wenxuan Xu and Yan Yan for preliminary measurements. This work was supported by the National Institutes of Health (NIH) grants R01 GM084070 and R35GM149296 to L.F. and R01 GM067153 to I.A.
Author information
Authors and Affiliations
Contributions
J.Q. developed the experiments, collected, and analyzed data, and wrote the manuscript. B.W. prepared proteins. I.A. supervised the preparation of proteins and participated in writing of the manuscript. D.D. designed the plasmids, participated in the writing of the manuscript, and led the project with L.F. L.F. conceived and co-led the project, contributed to writing and revising the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Seth Darst and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qian, J., Wang, B., Artsimovitch, I. et al. Force and the α-C-terminal domains bias RNA polymerase recycling. Nat Commun 15, 7520 (2024). https://doi.org/10.1038/s41467-024-51603-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51603-3
