
Abstract
It is challenging to attain strong near-infrared (NIR) emissive gold nanoclusters. Here we show a rod-shaped cluster with the composition of [Au28(p-MBT)14(Hdppa)3](SO3CF3)2 (1 for short, Hdppa is N,N-bis(diphenylphosphino)amine, p-MBT is 4-methylbenzenethiolate) has been synthesized. Single crystal X-ray structural analysis reveals that it has a rod-like face-centered cubic (fcc) Au22 kernel built from two interpenetrating bicapped cuboctahedral Au15 units. 1 features NIR luminescence with an emission maximum at 920 nm, and the photoluminescence quantum yield (PLQY) is 12%, which is 30-fold of [Au21(m-MBT)12(Hdppa)2]SO3CF3 (2, m-MBT is 3-methylbenzenethiolate) with a similar composition and 60-fold of Au30S(S‑t‑Bu)18 with a similar structure. time-dependent DFT(TDDFT)calculations reveal that the luminescence of 1 is associated with the Au22 kernel. The small Stokes shift of 1 indicates that it has a very small excited state structural distortion, leading to high radiative decay rate (kr) probability. The emission of cluster 1 is a mixture of phosphorescence and thermally activated delayed fluorescence(TADF), and the enhancement of the NIR emission is mainly due to the promotion of kr rather than the inhibition of knr. This work demonstrates that the metal kernel and the surface structure are both very important for cluster-based NIR luminescence materials.
Similar content being viewed by others
Introduction
Because of the reduced photon scattering and minimal tissue autofluorescence, fluorophores with emission at NIR area exhibit excellent performance with regard to in vivo fluorescence imaging1,2,3. NIR organic dyes often display poor stabilities in organism and many other inorganic NIR emissive materials such as quantum dots show notable cytotoxicity4,5. Recently, gold nanoclusters (NCs) have emerged as a promising class of NIR emissive materials that can be used in biological applications after appropriate post-processing6,7,8,9, due to their good biocompatibility10,11,12,13,14, reliable photostability5 and tunable luminescence15,16,17,18,19,20,21,22,23,24,25,26,27. However, low PLQY of gold NCs impeded their practical use, only a few has NIR PLQY exceeding 5% in solution at room temperature21,22,23,24, due to energy gap law28. Therefore, it is highly desirable to attain gold NCs with strong NIR emission, and it is quite important to establish the structure-property relationships of luminescent gold NCs in terms of rational synthesis of high performance NIR emitters.
Gold NCs have well-defined structures, which is beneficial to the study of underlying luminescence mechanisms15,22,29,30,31,32,33. Generally, the mechanisms of cluster luminescence can be simply categorized in three types: 1) ligand-to-metal charge transfer6,18,34; 2) aggregation-induced-emission35,36,37; 3) metal-centered emission19,38. Regarding gold NCs with metal-centered emissions, it is difficult to map out the mechanism, because of their complex structures consisting many metal and ligand components. In 2015, Gao et al.24 and we reported an NIR-emissive alkynyl-protected gold nanocluster [Au24(PhC≡C)14(Ph3P)4](SbF6)2 with λemmax at 920 nm. The NIR emission was found to originate from the gold kernel according to TDDFT calculations. Aikens et al.39,40,41 also came to a similar conclusion that the kernel emission plays an important role in the luminescence of thiolate-protected gold nanoclusters. Jin et al.29 mapped out a kernel-origin mechanism through study on a series of mono-cuboctahedral kernel clusters, and elucidated that the suppression of the ultrafast kernel relaxation via surface tailoring is important for attaining high PLQY in solution.
Since the energy gap between highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) decreases as the number of kernel gold atoms increases42,43, it is envisioned that gold nanoclusters with a big metal kernel may have better chance to emit NIR light. However, many large gold nanoclusters have been reported and they are weakly emissive or even nonemissive44. Therefore, the determining factor is not simply about the size of the cluster core. To investigate the factors affecting the NIR emission of gold NCs, we have paid great effort in synthesizing NIR emissive gold NCs with a mixed ligand strategy45,46,47,48,49. Our attempts have led to the discovery of an [Au28(p-MBT)14(Hdppa)3] (SO3CF3)2 (1) with enhanced NIR emission. In this work, we demonstrate that the shape of the metal kernel and the surface structure are both very important in terms of attaining bright NIR luminescence in solution at room temperature. It is noteworthy that the enhancement of the NIR emission of 1 is mainly due to the promotion of kr rather than the inhibition of knr. It provides important information for the rational design and synthesis of strong NIR-emitting materials.
Results
Synthesis and characteration
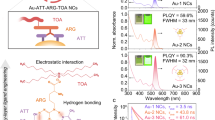
The synthesis of 1 is based on a “halide ion-free strategy” that we developed previously for preparing gold NCs co-protected by thiolate and phosphine ligands45. To increase the rigidity of surface ligand shell, we chose the combination of the biphosphine Hdppa and thiolates to generate highly emissive gold nanoclusters. In addition to 1, a closely related cluster [Au21(m-MBT)12(Hdppa)2]SO3CF3 (2) also has been synthesized. Generaly, the chloride ion in Me2SAuCl was removed in the presence of Hdppa by adding AgSO3CF3 to form the gold precursor HdppaAu2(SO3CF3)2. The reduction of a suspension containing HdppaAu2(SO3CF3)2 and RS-Au by NaBH4 led to the formation of the clusters. It is important to assure the precursors contain no halide, otherwise 1 and 2 could not be obtained. In addition, the isolation of 1 or 2 depends on the steric hindrance of the thiolate. The ligand m-MBT has a larger stereospecific resistance than p-MBT. Thiolates with larger steric hindrance favor the formation of smaller metal clusters50. Therefore, m-MBT gives Au21 while p-MBT affords Au28.
The compositions of the clusters were determined by high resolution electro-spray ionization time of flight mass spectrometry (ESI-TOF-MS) in positive mode (Fig. 1). The peaks at m/z 4197.40 in Fig. 1a and 6384.85 in Fig. 1b are corresponding to molecular ions [Au28(p-MBT)14(Hdppa)3]2+ and [Au21(m-MBT)12(Hdppa)2]+, respectively. The observed isotopic patterns are in good agreement with the simulation (inset in Fig. 1). The 31P NMR spectrum of 1 shows two singlets at δ = 70.22 and 73.53 ppm in a ratio of 2:1, indicating that the phosphines are in two kinds of chemical environment in 1 (Suppl. Fig. 1a). 1H NMR analysis of 1 was carried out (Suppl. Fig. 1b). We attribute the peaks at δ = 1.82–2.31, 5.58–5.59, and 6.21–8.09 ppm to the CH3 of p-MBT, NH of Hdppa and H on the aromatic ring of p-MBT and Hdppa in 1, respectively. As shown in Suppl. Fig. 3a, the 14 p-MBT on the surface of 1 include 6 bridged p-MBT (highlighted in turquoise) and 8 p-MBT in the trimeric staples (highlighted in yellow) with a ratio of 3: 4, indicating that peaks at δ = 2.15–2.19 ppm correspond to CH3 of 6 bridged p-MBT and δ = 1.82, 2.07 and 2.31 ppm correspond to CH3 of 8 p-MBT in the trimeric staples. The integration ratio of phenyl H in the p-MBT and Hdppa ligands and methyl H in p-MBT is calculated to be 2.77:1, which is consistent with the theoretical value of 2.76:1 in 1. 13C NMR spectrum of 1 (Suppl. Fig. 1c) shows peaks at δ = 21.08 and 128.10–137.90 ppm correspond to the methyl C in p-MBT and phenyl C of the p-MBT and Hdppa ligands in 1, respectively, and the integration ratio is 1:11.06, which is consistent with the theoretical value of 1:11.10 in 1. Similarly for 2, 31P NMR spectrum shows two singlets at δ = 69.24 and 71.70 ppm in a ratio of 1:1, suggesting that the numbers of phosphines in two different chemical environments are the same (Suppl. Fig. 2a). The 1H NMR spectrum (Suppl. Fig. 2b) and crystal structure (Suppl. Fig. 3b) analysis of 2 show that the peaks at δ = 2.02–2.38, 4.01, and 6.00–8.00 ppm correspond to the CH3 of m-MBT, NH of Hdppa and phenyl H of m-MBT and Hdppa in 2 respectively, δ = 2.36–2.38 ppm corresponds to CH3 of 4 bridged m-MBT (Suppl. Fig. 3b highlighted in turquoise) and δ = 2.02–2.12 ppm corresponds to 8 m-MBT in the trimeric staples (Suppl. Fig. 3b, highlighted in yellow). The integration ratio of phenyl H in the m-MBT and Hdppa ligands and methyl H in m-MBT is calculated to be 2.44:1, which is consistent with the theoretical value of 2.42:1 in 2. 13C NMR spectrum of 2 (Suppl. Fig. 2c) shows peaks at δ = 21.45 and 128.92–138.48 ppm correspond to the methyl C in m-MBT and phenyl C of the m-MBT and Hdppa ligands in 2 respectively, and the integration ratio is 1:10.23, which is consistent with the theoretical value of 1:10.00 in 2. It is notable, in the 1H (13C) NMR spectra of 2 at δ = 0.88, 1.27 and 30.12 ppm correspond to vacuum grease51, which was introduced during the purification of 2. Thermogravimetric analysis (TGA) reveals that 1 and 2 have similar thermostability, and they are stable below 190 °C (Suppl. Fig. 4).

a [Au28(p-MBT)14(Hdppa)3]2+ in 1. b [Au21(m-MBT)12(Hdppa)2]+ in 2. Inset: The measured (black trace) and simulated (red trace) isotopic patterns. c Chemical structures of p-MBT, m-MBT and Hdppa. Source data are provided as a Source Data file.
Molecular structure
The structures of 1 and 2 have been determined with single-crystal X-ray diffraction52 (Suppl. Tables 3, 4) and their molecular structures are shown in Fig. 2. As shown in Fig. 2a, 1 consists of a Au22 kernel surrounded by two trimeric staples (RS-Au-SR-Au-SR-Au-SR), three Hdppa and six p-MBT ligands. This Au22 kernel can be viewed as two interpenetrating bicapped cuboctahedral Au15 units (Fig. 2c) sharing eight Au atoms (Fig. 2d, highlighted in green). The remaining six Au atoms form two trimeric Au3(SR)4 staples in two ends of the Au22 kernel (Fig. 2e, highlighted in blue). The other six thiolates and three Hdppa are bound to the Au22 kernel as bridges (Fig. 2f highlighted in turquoise and pink). The Au22 kernel exhibits layer-by-layer fcc packing with A-B-C layers being planes (Fig. 2e). The Au-Au bond lengths in Au22 kernel are divided into two groups with shorter ones 2.660(4)–2.917(3) Å and longer ones 2.928(3)–3.359(3) Å. The Au-Au distances between the trimeric Au3(SR)4 staples and Au22 kernel are in the range of 2.939(4)–3.378(3) Å, indicating strong interactions between the staples and Au22 kernel. The Au-Au distances of those Au atoms bridged by thiolates and Hdppa ligands have average values of 3.225(3) and 2.941(3) Å, respectively.

a [Au28(p-MBT)14(Hdppa)3]2+ in 1. b [Au21(m-MBT)12(Hdppa)2]+ in 2. c Bicapped cuboctahedra Au15 at different viewpoint. d The model of interpenetrated bi-bicapped-cuboctahedron Au22. e The Au22 kernel capped with two trimeric staple-like motifs and ABC denotes the fcc packing form. f The Au22 kernel capped with three Hdppa and six bridging p-MBT. g The structure construction of [Au21(m-MBT)12(Hdppa)2]+ cation. Phenyl groups were omitted for clarity. Color labels: orange/green/violet/blue, Au atoms; yellow/turquoise, S atoms; pink, P atoms; dark blue, N atoms. Source data are provided as a Source Data file.
Cluster 2 contains a Au15 kernel consisting of a cuboctahedral Au13 (Fig. 2g highlighted in orange) and two capping Au atoms (Fig. 2g, highlighted in violet) on two opposite square faces. There are two trimeric staples (Fig. 2g, highlighted in blue), four simple bridging thiolates (Fig. 2g, highlighted in turquoise) and two bridging Hdppa surrounding the Au15 kernel. The Au-Au bond lengths in Au13 cuboctahedron can be divided into shorter ones 2.6447(13)–2.8746(12) Å and longer ones 2.9048(12)–3.3737(12) Å. However, the Au-Au distances between the two capped Au atoms and Au13 core are 2.6447(13)–3.1582(14) Å, indicating the two capping Au atoms are truly a part of the Au15 kernel. The Au-Au bond lengths between the trimeric Au3(SR)4 staple-like motifs and Au15 kernel are 2.8879(13)–3.3680(13) Å, which is similar to that of 1 (2.939(4)– 3.378(3) Å.
Electronic spectra and theoretical calculations
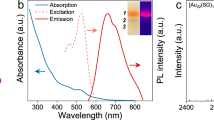
Clusters 1 and 2 are brown and green in CH2Cl2, respectively (Fig. 3, inset). As shown in Fig. 3, the UV–vis spectrum of 1 has four absorption peaks at 430, 543, 650, and 760 nm, and 2 has a prominent absorption peak at 612 nm and two weak shoulders at 320 and 415 nm. The optical energy gaps of 1 (Suppl. Fig. 5a, inset) and 2 (Suppl. Fig. 5c, inset) are determined to be 1.51 and 1.87 eV, respectively. The absorptions are intense, as indicated by their molar extinction coefficients, for 1 at 760 nm (ε = 21688.1 M−1 cm−1, Suppl. Fig. 5b) and 2 at 610 nm (ε = 39878.6 M−1 cm−1, Suppl. Fig. 5d). These two nanoclusters are quite stable, as monitored by UV–vis spectroscopy. The initial and final spectra are almost identical, indicating that no decomposition occurred after the solution of 1 or 2 was stored under ambient conditions for four weeks and that the title nanoclusters have good stability in solution (Suppl. Fig. 6).

Red and black lines represent 1 and 2 respectively. Inset: their photos of solutions. Source data are provided as a Source Data file.
TDDFT calculation on simplified 1, [Au28(SH)14(H2PNHPH2)3]2+, was carried out to understand the electronic structure of 1. As shown in Fig. 4a, the calculated absorption spectrum agrees well with the experimental, and the absorption peak at 760 nm was attributed to HOMO- LUMO transition accordingly. From the Kohn–Sham molecular orbitals (Fig. 4b) we can see that HOMO-LUMO transition has significant metal centered transition. The main component is Au sp←sp transition, and the secondary component is Au sp ← d transition. The molecular orbitals distribution for ground state in Fig. 4c directly reflect the HOMO-LUMO transition of the Au22 kernel.

a Experimental absorption spectrum of 1 and simulated spectrum of [Au28(SH)14(H2PNHPH2)3]2+, I/II/III correspond to the absorption peaks of 543, 650 and 760 nm. b Kohn–Sham molecular orbital energy levels diagram and the associated populations of atomic orbitals in each KS molecular orbital for [Au28(SH)14(H2PNHPH2)3]2+. c Frontier orbitals including HOMO and LUMO distribution for ground state (S0) and the excited states (S1 and T1) of [Au28(SH)14(H2PNHPH2)3]2+. Source data are provided as a Source Data file.
NIR Luminescence
The luminescence properties of 1 and 2 have been studied in CH2Cl2. The excitation and emission spectra of 1 and 2 were shown in Fig. 5a,b. The excitation spectra of 1 and 2 are similar to their absorption spectra. Clusters 1 and 2 show NIR emissions at about 920 and 860 nm, respectively. According to the research of luminescent quantum dots, the full-width at half-maximum (FWHM, \({{\boldsymbol{\triangle }}}\,{\bar{\upsilon }}_{1/2}\)) is related to electron–phonon coupling which has connection with structural vibration and distortion53,54. Considering that electron-phonon coupling exists in Au NCs55,56, we attribute the broadening PL bands of 1 and 2 to the structural distortion of excited state. The FWHM of 1 and 2 in UV–vis absorption spectra is 1688 and 2575 cm−1 respectively. The FWHM of 1 and 2 in emission spectra is 1947 and 2987 cm−1. The broadening from absorption bands to PL bands of 1 (259 cm−1) is smaller than that of 2 (412 cm−1), indicating that structural distortion of the excited state in 1 is smaller than that of 2. The PLQYs of 1 and 2 were 12% and 0.4% as measured with integrating sphere. The PL lifetime at 920 nm of 1 has two components, 0.40 μs (67%) and 1.43 s (33%), and the τavg (average lifetime) is 1.05 μs (Suppl. Fig. 7a). 2 shows a three components PL lifetime for 860 nm, 77 ns (58%), 0.301 μs (16%) and 1.55 μs (26%), and the τavg is 1.30 μs (Suppl. Fig. 7b). We do not rule out the possibility of the mixing of S1 and T1 states in terms of the broadening PL bands of 1 and 2.

a Cluster 1. b Cluster 2. Black and red lines represent excitation and emission spectra, respectively. Source data are provided as a Source Data file.
To confirm the origin of the luminescence of 1, we measured the emission spectra of 1 under different excitations (Suppl. Fig. 8), and found that the emission is excitation independent (Kasha’s rule). As shown in Suppl. Fig. 9a, the emission wavelength of 1 shifts from 920 to 890 nm with the increase of the solvent polarity. The NIR emission of the cluster 1 was enhanced as the temperature decreased. When the temperature was lowered from 300 to 190 K, the main emission wavelength shifts from 890 to 920 nm (Suppl. Fig. 9b). The NIR emission wavelength of the crystal of 1 is red-shifted from 920 to 1000 nm in comparison to its dichloromethane solution (Suppl. Fig. 9c). These facts indicate that the NIR emission of cluster 1 is highly sensitive to surrounding environments. We futher carried out TDDFT calculations on 1 without simpilification, and found that the phenyl groups of ligands do not participate in HOMO and LUMO states (Suppl. Fig. 10). We also found that both phosphorescence and TADF are present in cluster 1. As shown in Fig. 4c, the frontier orbitals of the HOMO and LUMO distributions for the excited states (S1 and T1) of [Au28(SH)14(H2PNHPH2)3]2+ are similar each other, and they are mainly distributed on the metal kernel. The energy gaps of S1 to S0 and T1 to S0 are 1.13 and 1.09 eV respectively, and the gap between S1 and T1 is 0.04 eV, which favors the reverse intersystem crossing57. The small gap between S1 and T1 and the redshift and enhancement of the NIR emission with decreasing temperature (Suppl. Fig. 9b) suggest the presence of TADF in cluster 1. In addition, when the solution was saturated with O2, the PL intensities of cluster 1 was somewhat suppressed compared to N2 (Suppl. Fig. 11a), implying the existence of a triplet state in cluster 1. As shown in Suppl. Fig. 7a, the PL lifetime at 920 nm of 1 includes 0.40 μs (67%, τ1) and 1.43 μs (33%, τ2), and the τ1 and τ2 can be assigned as TADF and phosphorescence (Suppl. Fig. 11b). As shown in Suppl. Fig. 16 and Table 1, the average Aucenter–Aushell, Aushell–Aushell and Aushell–Austaple bond lengths of the S0 state structure decrease by 0.085/0.089 Å, 0.091/0.089 Å and 0.228/0.239 Å upon formation of the optimized S1/T1 state geometry, respectively. The average Aushell–P, Aushell–Sbridge and Austaple–Scentral/terminal bond lengths of the S0 state structure decrease by 0.040/0.040 Å, 0.020/0.017 Å and 0.014/0.011 Å upon formation of the optimized S1/T1 state geometry, respectively. In contrast, the average Aushell–Sterminal bond lengths of the S0 state structure increased by 0.079/0.071 Å upon formation of the optimized S1/T1 state geometry. These data indicate that the excited state dynamics of cluster 1 are likely due to the shrinkage of the Au-Au bonds within the whole structure during excited state relaxation, suggesting that the NIR luminescence of cluster 1 is metal core related emission.
We measured the PLQY of 1 relative to IR-125 (Indocyanine green). The value was determined to be 10.0%, which is close to 12% as measured by the absolute method. It is quite surprising to see that 1 has a PLQY nearly 30-fold that of 2. They have similar ligand shells including Au3SR4 staples, but their kernel structures are quite different, that is, rod shaped Au22 for 1 and cuboctahedral Au13 for 2. Therefore, we believe that the Au22 rod plays a key role in having high PLQY. It is important to have identical ligands even the R group of the SR ligand, as proved by Jin et al.58 that the surface ligand significantly affects the NIR PLQY of Au52. To illustrate the effect of the rod-shaped structure on cluster luminescence, we chose a series of clusters with the same surface ligands but different metal-core structures for comparison. As shown in Suppl. Fig. 12, when the surface ligands and coordination structures are the same, the nanoclusters with rod-shaped metal cores have higher PLQY than the basic units, indicating the importance of the rod-shaped structures. We believe that this type of rod-shaped molecule is prosperous NIR emitters.
By comparing the Stokes shifts of 1 and 2, we found that 1 has much smaller value of 0.28 eV versus 0.59 eV of 2 (Table 1). This indicates that the elongated close packing kernel in 1 results in smaller excited state deformation 124,41. As the kernel is elongated from 2 to 1, the delocalization system is expanded, leading to the smaller energy gap and reduced excited state distortion, that is, the smaller excited state distortion leads to higher radiative decay rates (kr) (1.2 × 105 for 1, 3.1 × 103 for 2), the kr and non-radiative decay rates (knr) are calculated according to Eqs. (1) and (2). According to energy gap law, knr is related to energy gap term (E0) and excited state distortion, and the distortion is related to γ28,59,60. The simplified form of energy gap law is shown in Eq. (3), and expressions for the related parameters are shown in Eqs. (4)–(6).
Here, \(\Delta \bar{r}\) is average bond displacement, which represents the distortion of excited state. E0 is energy gap term which is related to the energy difference between the ground and excited state in their υ = 0 vibration level. ħ is the reduced Planck constant. ωM is angular frequency of medium vibration. ħωM usually ranges from 1000 to 1600 cm−1. SM is electron-vibrational coupling constant also called Huang-Rhys factors, and it is related to equilibrium displacement (ΔQe) by Eq. (5). M is reduced mass. ΔQe is equilibrium displacement between the ground state and excited state, it is related to average bond displacement \(\Delta \bar{r}\) by Eq. (6). b is the number of bonds with medium vibration. Parameter γ is defined by Eq. (4), which reflects the distortion of the excited state.
With the elongation from 2 to 1, the delocalization system is expanded. This elongation makes energy gap term of 1 smaller than that of 2 (E01 < E02). This elongation also makes the average bond displacement of 1 smaller than that of 2 (\(\Delta {\bar{r}}_{1}\) < \(\Delta {\bar{r}}_{2}\)). According to Eqs. (5) and (6), SM is proportional to the square of \(\Delta \bar{r}\). Since \(\Delta {\bar{r}}_{1}\) is samller than \(\Delta {\bar{r}}_{2}\), the SM of 1 smaller than that of 2 (SM1 < SM2). According to the Eq. (3), the decrease of E0 and SM shall result in the increase of knr. As shown in Table 1, the experimental results show that the knr of 1 is larger than that of 2 (8.3 × 105 for 1, 7.7 × 105 for 2). However, 1 has a higher PLQY than 2. According to energy gap law, we find that elongating Au NCs with expanded delocalization system can suppress the excited state distortion, thus keeping kr in a much higher level (1.2 × 105 for 1, 3.1 × 103 for 2). In other words, knr1 and knr2 are in the same order of magnitude, but kr1 is two orders of magnitude greater than kr2, which leads to much larger PLQY of 1 than 2.
Discussion
The Au22 kernel in 1 is similar to the Au22 core in Au30S(S-t-Bu)18 (3, t-BuSH is tert-butylthiol)61,62. These two Au22 kernels are almost identical and the ligand shells are also quite similar. The structure of 1 can be virtually generated from Au30S(S‑t‑Bu)18 via the replacement of two Au(S-t-Bu)2 staples and a S2− ion with three Hdppa ligands (Fig. 6b). Therefore, 1 and 3 are a pair of ideal analogs for investigating the role of surface structures on luminescence. We measured the absorption and emission spectra of 362.

a, b are the structure of [Au28(p-MBT)14(Hdppa)3]2+ and Au30S(S-t-Bu)18, respectively. The phenyl and alkyl group of thiolate and phosphenlate ligands are omitted to clarify. Color labels: orange/green, Au atoms; yellow, S atoms; pink, P atoms; dark blue, N atoms. c The optical absorption spectra of 1 (red trace) and 3 (blue trace) in CH2Cl2. d The emission spectrum of 3 in CH2Cl2. Source data are provided as a Source Data file.
As shown in Fig. 6c, the first absorption band (760 nm) of 1 is in much lower energy region in comparison to 630 nm of 3. The emission spectrum of 3 is shown in Fig. 6d, the emission peak at 860 nm is energetically higher than that of 1 (920 nm). PL lifetime at 860 nm of 3 has three components, 0.144 μs (18%), 0.842 μs (16%) and 4.28 μs (66%), and the τavg is 4.09 μs (Suppl. Fig. 13). The PLQY of 3 is only 0.2%, which is 60-fold lower than that of 1, despite their quite similar kernels. This fact indicates that the surface structure is also very important for PLQY in addition to the kernel structure. It was argued that the kernel relaxation of mono-cuboctahedral clusters can be suppressed via the replacement of Au(SR)2 staples with PCP ligands as demonstrated in [Au21(SC6H11)12(PCP)2]+ (C6H11SH = cyclohexanethiol, PCP = PPh2CH2PPh2)29. However, in the present work the replacement of Au(S-t-Bu)2 staples with Hdppa ligands plays a different role, it is not about suppressing kernel relaxation, it is about the enhancement of radiative decay.
The kr and knr of 1, 3 and other related clusters are summarized in Table 1. The knr values of 1 and 3 are in the same order of magnitude, which means that the change of surface structure from 3 to 1 does not suppress the kernel relaxation. However, the kr of 1 is three orders of magnitude higher than 3, which indicates that the high PLQY of 1 is mainly attributed to its fast radiative decay process. Replacing negative staples with neutral Hdppa ligands from 3 to 1 lead to the big difference in electronic structures. According to TDDFT calculations, the HOMO to LUMO transition in 3 is forbidden43, but the HOMO-LUMO transition of 1 is allowed. Due to the different electronic structures of 1 and 3, their PLQYs are differentiated by two orders of magnitude. As shown in Suppl. Table 2, compared to Au28(CHT)20 and Au28(TBBT)20, the knr of 1 is of the same order of magnitude, but the kr is one and two orders of magnitude larger, respectively. Compared to Au28(TBPT)22, the kr of 1 is of the same order of magnitude, but its knr is one order of magnitude smaller. Cluster 1 has much higher PLQY than the thiolated Au28 clusters. In addition, the emission wavelength of 1 is longer than thiolated Au28 clusters but similar to Au29. All these comparisons demonstrate that the surface structure can deeply influence the electronic structure of the cluster, that is, one can modulate the luminescence behavior of a nanocluster through controlling the surface structure.
Due to quantum size effects, gold NCs with similar number of gold atoms but different structures display significantly different electronic structures and optical properties. The enhanced NIR emission of 1 draws our attention to rod-shaped molecule. Rod-shaped gold NCs usually are NIR emissive21,63. For example, [Au24(C≡CPh)14(PPh3)4](SbF6)224 (4) has a Au22 core consisting of two cuboctahedral Au13 sharing a square face. [Au25(PPh3)10(SC2H4Ph)5Cl2]2+ (5)21 possesses a Au25 kernel which is constructed by two icosahedral Au13 sharing a vertex atom. Clusters 1, 4 and 5 (Fig. 7) are all rod-shaped and all have enhanced NIR emission of 12%, 12%, and 8%, respectively. However, as shown in Suppl. Fig. 14, the phosphine/thiolate-protected rod-shaped [Au24(PPh3)10(SC2H4Ph)5Cl2]+(6)47 shares a similar structure to 5 except for the absence of a sharing vertex shared, but the PLQY of 6 (1%) is eightfold lower than that of 5 (8%)21. Due to the absence of a sharing vertex in 6, the two Au12 units forming the Au24 core are bridged by thiolate ligands, and the distance between the two Au12 units is 2.93, resulting in the electrons being localized in each Au12 unit rather than delocalized in the rod-shaped Au24 kernel. On the contrary, since the two Au13 units in 5 form the final rod-shaped Au25 kernel through the shared vertex (Suppl. Fig. 14, highlighted in turquoise), the distance between the two Au13 units is zero, which favors electron delocalization throughout the rod-shaped Au25 kernel rather than in each Au13 unit. Comparing clusters 5 and 6, the extended electron delocalization system favors the reduction of excited state distortion to suppress knr (2.9 × 105 s−1 for 5, 3.3 × 106 for 6) and thus enhanced PLQY are attained. Although the phosphine/thiolate-protected rod-shaped Au37 has a similar structure to 5, its electrons are not delocalized throughout the rod-shaped structure (Suppl. Fig. 15)64, which leads to its low PLQY. This suggests that the rod-shaped structure is insufficient for a high PLQY; it also requires that the electrons in the cluster can be delocalized throughout the rod-shaped structure.

a The structure of [Au28(p-MBT)14(Hdppa)3]2+, [Au24(C≡CPh)14(PPh3)4]2+ and [Au25(PPh3)10(SC2H4Ph)5Cl2]2+ respectively. The phenyl and alkyl group of thiolate, phosphenlate and alkyne ligands are omitted to clarify. b The gold kernel of [Au28(p-MBT)14(Hdppa)3]2+, [Au24(C≡CPh)14(PPh3)4]2+ and [Au25(PPh3)10(SC2H4Ph)5Cl2]2+ respectively. Color labels: orange/green/light blue, Au atoms; yellow, S atoms; pink, P atoms; indigo, N atoms; gray, C atoms. Source data are provided as a Source Data file.
Here, we have synthesized a rod-shaped gold nanocluster 1 using the combination of thiolate and biphosphine ligands. This cluster shows enhanced NIR luminescence quantum yield compared to nanocluster with similar composition or structure. We demonstrate that the shape of the metal kernel and the surface structure are both very important in terms of attaining bright NIR luminescence, i.e., rod-shaped metal kernel with expanded electron delocalization systems and rigid surface structures contribute to increase the NIR PLQY of metal nanocluster-based materials. Due to the good biocompatibility of gold nanoclusters, 1 with enhanced NIR emission will be very promising in biological applications. We are taking the halide ion-free strategy to synthesize nanoclusters with various ligand combinations to attain highly NIR emissive materials.
Methods
Chemicals and materials
4-methylbenzenethiol (p-MBT, 98%), 3-methylbenzenethiol (m-MBT, 98.0%) and sodium trifluoromethanesulfonate (NaSO3CF3, 98%) were purchased from Energy Chemical Inc., N,N-bis(diphenylphosphino)amine (Hdppa, 98%), and silver trifluoromethanesulfonate (AgSO3CF3, 98.0%) were purchased from J&K; sodium borohydride (NaBH4, 98%) and other reagents employed were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). tert-Butylthiol (t-BuSH, 99.0%) were purchased from Meryer (Shanghai) Chemical Technology Co., LTD. All reagents were used as received without further purification. TLC plates were purchased from Yantai Jiangyou Silicone Development Co., LTD (silica gel, 0.4–0.5 mm). Me2SAuCl was prepared according to ref. 65
Synthesis of p-MBT-Au45
To 15.0 mL of acetone solution containing Me2SAuCl (294.6 mg, 1.0 mmol), 5.0 mL of acetone solution containing p-MBT (149.0 mg, 1.2 mmol) and NEt3 (166.5 µL, 1.2 mmol) were added under vigorous stirring. The mixture was stired for 0.5 h at room temperature in air in the dark. Then the solution was evaporated as dryness as possible to give a white solid, which was washed with water (3 × 10 mL), ethanol (3 × 10 mL) and ether (2 × 10 mL). Then the white solid was dried up in a vacuum oven to give 281.3 mg white power of p-MBT-Au (88% yield based on Au).
Synthesis of m-MBT-Au
The reaction conditions are the same as the synthesis of p-MBT-Au, except the thiol used here is m-MBT.
Synthesis of [Au28(p-MBT)14(Hdppa)3](SO3CF3)2 (1)66
3.0 mL CH2Cl2 solution containing Me2SAuCl (14.8 mg, 0.05 mmol) and Hdppa (9.7 mg, 0.025 mmol) was stirred for 3 min and then 0.1 mL of methanol solution containing AgSO3CF3 (12.8 mg) was added under vigorous stirring, and the solution immediately appeared as a white precipitate. The reaction continued for 20 min at room temperature in air in the dark. The resulting solution was centrifuged for 4 min at 10,280 × g, the supernatant was filtered, and the filtrate was evaporated to dryness and washed with ether (2 × 5 mL). The white solid was obtained by removing the washing solution ether, then mixed with p-MBT-Au (32.1 mg, 0.10 mmol) and CH2Cl2 (4.0 mL) to obtain a white turbid solution, to which 1.0 mL freshly prepared ethanol solution containing NaBH4 (1.42 mg) was added drop-wise under vigorous stirring, during this process, the color of the solution changed from white to pale brown and finally to brown, and the mixture was stirred for 24 h at room temperature in air in the dark. Stop stirring, a small amount of black precipitate appeared at the bottom of flask, and the mixture was evaporated to dryness to give a dark solid, which was first washed with pentane (2 × 5 mL) then dissolved in CH2Cl2 (1.6 mL), followed by centrifugation at 10,280 × g for 4 min. The supernatant solution was subject to diffusion of ether to afford 14.2 mg black crystals after 15 days (31% yield based on Au).
Synthesis of [Au21(m-MBT)12(Hdppa)2]SO3CF3 (2)
The reaction conditions are the same as the synthesis of [Au28(p-MBT)14(Hdppa)3](SO3CF3)2, except the thiolate-Au used here is m-MBT-Au. Unfortunately, the crystallization of Au21 is always accompanied by a small amount of Au28 and further purification is required to obtain pure Au21. Therefore, Thin-layer chromatography (TLC) was used for the isolation and purification of Au21. The mixture of Au NCs was first dissolved in a small amount of CH2Cl2, and then pipetted on the TLC plate, and the separation was conducted in a developing tank (developing solvent 30:1 (v/v) CH2Cl2: methanol). The green band corresponding to Au21 was cut off and dissolved in CH2Cl2/methanol (v/v = 20/1). To easily wash off Au21 from the silica gel a small amount of NaSO3CF3 was added. Centrifugation at 10280× g for 3 min, filtration of the supernatant to obtain a green solution. Repeat the washing, centrifugation, and filtration several times until the silica gel is white. The collected green solution was evaporated to dryness to give a dark solid 12.3 mg that is pure Au21 (26% yield based on Au).
Anal for 1. UV–vis (λ, nm): 250 nm; 330 nm; 435 nm; 550 nm; 650 nm; 760 nm Eg = 1.51 eV. NMR: 31P NMR (δ, ppm): δ = 70.22 ppm; 73.53 (d, J = 97.5 Hz) ppm (external reference 85% H3PO4). ESI-MS (m/z): 4197.4 ([Au28(p-MBT)14(Hdppa)3]2+). NIR-PL (λ): 920 nm (Exc: 436 nm) in CH2Cl2, PLQY, 0.12. Molar extinction coefficient (ε) at peak 760 nm, 21688.1 M−1cm−1.
Anal for 2. UV–Vis (λ, nm): 315 nm; 380 nm; 415 nm; 490 nm; 610 nm; Eg = 1.87 eV. NMR: 31P NMR (δ, ppm): δ = 69.24 (d, J = 95.6 Hz) ppm; 71.70 ppm (external reference 85% H3PO4). ESI-MS (m/z): 6384.85 ([Au21(m-MBT)12(Hdppa)2]+). NIR-PL (λ): 860 nm (Exc: 335 nm) in CH2Cl2, PLQY, 0.004. Molar extinction coefficient (ε) at peak 610 nm, 39878.6 M−1 cm−1.
Physical measurements
UV-vis-NIR absorption spectrum was recorded on cary5000. Mass spectrum was recorded on a Waters Synapt G2-Si mass spectrometer. 31P NMR data were recorded on a Bruker Avance II spectrometer (500 MHz). 1H and 13C NMR data were recorded on a JNM-ECA600 spectrometer (600 MHz). TGA was recorded on STDQ600. Near-infrared photoluminescence (NIR-PL) spectra and absolute PLQY tests were recorded on an Edinburgh Analytical Instruments FLS980 equipped with an integrating sphere (150 mm, PTFE inner surface), with a PMT R5509-73 detector for NIR emission and a PMT R928 for visible emission. PLQYs were determined with the FLS980 were evaluated according to Eq. (7):
where \({A}_{{{\rm{em}}}}\) is the integrated area of the sample’s emission (the emission spectral of CH2Cl2 was tested and was set as baseline); \({A}_{{{\rm{scatter}}}}^{{{\rm{ref}}}}\) and \({A}_{{{\rm{scatter}}}}^{{{\rm{sample}}}}\) are the integrated areas under the Rayleigh scattering peaks of the reference sample (CH2Cl2) and the sample under study; and \({k}_{{{\rm{R}}}928/{{\rm{R}}}5509}\) is the ratio of the sensitivities of the two detectors. The value of \({k}_{{{\rm{R}}}928/{{\rm{R}}}5509}\) was determined by testing the scattering peak within the range of 700-800 nm and calculating using Eq. (8):
Before the PLQY test of 1, the \({{{\boldsymbol{k}}}}_{{{\bf{R}}}{{\bf{928}}}/{{\bf{R}}}{{\bf{5509}}}}\) was determined as 0.11. After that the excitation wavelength was set at 382 nm according to excitation spectral of 1 and emission detection was arranged from 650 to 1200 nm (the integration of emission was arranged from 750 to 1050 nm). A 10% neutral density filter (NDF) was used during detection. The sensitivities of NIR and visible PMT detector was measured before testing as mentioned above. And to identify the reliability of visible PMT detector, we use rhodamine B as a standard. The emission decay curves were recorded using the time-correlated single-photon counting (TCSPC) method. The phosphorescence decay curves of 1, 2, and 3 were collected using a 447.2 nm Spectra LED laser as the light source. Temperature-dependent PL measurements of cluster 1 were carried out with an Edinburgh spectrofluorimeter (FLS980) and the solvent used was 2-methyltetrahydrofuran (2-Me-THF). The fluorimeter is coupled with an Optistat DN cryostat (Oxford Instruments), and the ITC temperature controller and a pressure gauge were used to conduct the temperature-dependent experiments from 80 to 300 K. The vacuum in the cryostat was maintained with a leybold turbo molecular pump. Spectra were taken at different temperatures after a wait period of 10 min. The error in temperature setting is ± 0.5 K.
X-ray crystallography
Intensity data of compounds 1 and 2 were collected on an Agilent SuperNova Dual system (Cu Kα). Absorption corrections were applied by using the program CrysAlis (multi-scan). The structures of 1 and 2 were solved by direct methods, all non-hydrogen atoms except solvent molecules and counteranions were refined anisotropically by least-squares on F2 using the SHELXTL program. The diffuse electron densities resulting from the residual solvent molecules were removed from the data set using the Olex2 solvent mask.
Computational methods
Density functional theory (DFT) calculations were performed with the quantum chemistry program Gaussian 1667. The 6-31 G(d) basis set was used for N, H, P, and LANL2DZ for Au68,69. Geometry optimizations were performed with the B3LYP functional, and time-dependent DFT calculations of the UV-vis absorption spectrum were performed with the PBE functional. One hundred singlet states (nstates = 100, singlet) are chosen in the calculations of the UV-vis absorption spectra. The molecular orbitals were visualized via the Multiwfn software and the VMD package70,71.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding author upon request. The X-ray crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 2181997 to 2181998. Source data are provided with this paper.
References
Hong, G., Antaris, A. L. & Dai, H. Near-infrared fluorophores for biomedical imaging. Nat. Biomed. Eng. 1, 0010 (2017).
Huang, J. & Pu, K. Near-infrared fluorescent molecular probes for imaging and diagnosis of nephro-urological diseases. Chem. Sci. 12, 3379–3392 (2020).
Huang, J. & Pu, K. Activatable molecular probes for second near-infrared fluorescence, chemiluminescence, and photoacoustic imaging. Angew. Chem. Int. Ed. 59, 11717–11731 (2020).
Altinoğlu, E. I. & Adair, J. H. Near infrared imaging with nanoparticles. WIREs Nanomed. Nanobiotechnol. 2, 461–477 (2010).
Xu, J. & Shang, L. Emerging applications of near-infrared fluorescent metal nanoclusters for biological imaging. Chin. Chem. Lett. 29, 1436–1444 (2018).
Gan, Z. et al. Fluorescent gold nanoclusters with interlocked staples and a fully thiolate-bound kernel. Angew. Chem. Int. Ed. 55, 11567–11571 (2016).
Chen, J. et al. Atomically precise photothermal nanomachines. Nat. Mater. 23, 271–280 (2024).
Ma, H. et al. Bioactive NIR-II gold clusters for three-dimensional imaging and acute inflammation inhibition. Sci. Adv. 9, eadh7828 (2023).
Ma, H., Wang, J. & Zhang, X.-D. Near-infrared II emissive metal clusters: from atom physics to biomedicine. Coord. Chem. Rev. 448, 214184 (2021).
Goswami, N., Zheng, K. & Xie, J. Bio-NCs – the marriage of ultrasmall metal nanoclusters with biomolecules. Nanoscale 6, 13328–13347, (2014).
Zhang, H. et al. Bacteria photosensitized by intracellular gold nanoclusters for solar fuel production. Nat. Nanotechnol. 13, 900–905 (2018).
Li, D. et al. Gold Nanoclusters for NIR-II Fluorescence Imaging of Bones. Small 16, e2003851 (2020).
Yang, Y. et al. Illuminating platinum transportation while maximizing therapeutic efficacy by gold nanoclusters via simultaneous near-infrared-I/II imaging and glutathione scavenging. ACS Nano 14, 13536–13547 (2020).
Zheng, K., Setyawati, M. I., Leong, D. T. & Xie, J. Antimicrobial gold nanoclusters. ACS Nano 11, 6904–6910 (2017).
Kang, X. & Zhu, M. Tailoring the photoluminescence of atomically precise nanoclusters. Chem. Soc. Rev. 48, 2422–2457 (2019).
Lei, Z., Pei, X.-L., Jiang, Z.-G. & Wang, Q.-M. Cluster linker approach: preparation of a luminescent porous framework with NbO topology by linking silver ions with gold(I) clusters. Angew. Chem., Int. Ed. 53, 12771–12775 (2014).
Lei, Z., Zhang, J.-Y., Guan, Z.-J. & Wang, Q.-M. Intensely luminescent gold(I) phosphinopyridyl clusters: visualization of unsupported aurophilic interactions in solution. Chem. Commun. 53, 10902–10905 (2017).
Yu, Y. et al. Identification of a highly luminescent Au22(SG)18 nanocluster. J. Am. Chem. Soc. 136, 1246–1249 (2014).
Han, X.-S. et al. Structure determination of alkynyl-protected gold nanocluster Au22(tBuC≡C)18 and its thermochromic luminescence. Angew. Chem., Int. Ed. 59, 2309–2312 (2020).
Ito, S., Takano, S. & Tsukuda, T. Alkynyl-protected Au22(C≡CR)18 clusters featuring new interfacial motifs and R-dependent photoluminescence. J. Phys. Chem. Lett. 10, 6892–6896 (2019).
Li, Q. et al. Photoluminescence in rod-shaped icosahedral gold nanoclusters. Small 17, 2007992 (2021).
Li, Q., Zeman, C. J., Schatz, G. C. & Gu, X. W. Source of bright near-infrared luminescence in gold nanoclusters. ACS Nano 15, 16095–16105 (2021).
Takano, S. et al. Photoluminescence of doped superatoms M@Au12 (M = Ru, Rh, Ir) homoleptically capped by (Ph2)PCH2P(Ph2): efficient room-temperature phosphorescence from Ru@Au12. J. Am. Chem. Soc. 143, 10560–10564 (2021).
Wan, X.-K. et al. A near-infrared-emissive alkynyl-protected Au24 nanocluster. Angew. Chem. Int. Ed. 54, 9683–9686 (2015).
Zhu, C. et al. Fluorescence or phosphorescence? The metallic composition of the nanocluster kernel does matter. Angew. Chem. Int. Ed. 61, e202205947 (2022).
Wang, X. et al. Ligand-protected metal nanoclusters as low-loss, highly polarized emitters for optical waveguides. Science 381, 784–790 (2023).
Shen, H. et al. Photoluminescence quenching of hydrophobic Ag29 nanoclusters caused by molecular decoupling during aqueous phase transfer and emissionrecovery through supramolecular recoupling. Angew. Chem. Int. Ed. 63, e202317995 (2024).
Englman, R. & Jortner, J. The energy gap law for radiationless transitions in large molecules. Mol. Phys. 18, 145–164 (1970).
Li, Q. et al. A mono-cuboctahedral series of gold nanoclusters: photoluminescence origin, large enhancement, wide tunability, and structure-property. Correlation. J. Am. Chem. Soc. 141, 5314–5325 (2019).
Zhou, M., Lei, Z., Guo, Q., Wang, Q.-M. & Xia, A. Solvent dependent excited state behaviors of luminescent gold(I)–silver(I) cluster with hypercoordinated. Carbon J. Phys. Chem. C. 119, 14980–14988 (2015).
Shi, W.-Q. et al. Near-unity NIR phosphorescent quantum yield from a room-temperature solvated metal nanocluster. Science 383, 326–330 (2024).
Luo, L., Liu, Z., Du, X. & Jin, R. Near-infrared dual emission from the Au42(SR)32 nanocluster and tailoring of intersystem crossing. J. Am. Chem. Soc. 144, 19243–19247 (2022).
Liu, Z. et al. Elucidating the near-infrared photoluminescence mechanism of homometal and doped M25(SR)18 nanoclusters. J. Am. Chem. Soc. 145, 19969–19981 (2023).
Wu, Z. & Jin, R. On the ligand’s role in the fluorescence of gold nanoclusters. Nano Lett. 10, 2568–2573, (2010).
Goswami, N. et al. Luminescent metal nanoclusters with aggregation-induced emission. J. Phys. Chem. Lett. 7, 962–975 (2016).
Kang, X. et al. Bimetallic Au2Cu6 nanoclusters: strong luminescence induced by the aggregation of copper(I) complexes with Gold(0) species. Angew. Chem., Int. Ed. 55, 3611–3614 (2016).
Luo, Z. et al. From aggregation-induced emission of Au(I)-thiolate complexes to ultrabright Au(0)@Au(I)-thiolate core-shell nanoclusters. J. Am. Chem. Soc. 134, 16662–16670 (2012).
Dong, J. et al. Synthesizing photoluminescent Au28(SCH2Ph-tBu)22 nanoclusters with structural features by using a combined method. Angew. Chem., Int. Ed. 60, 17932–17936 (2021).
Weerawardene, K. L. & Aikens, C. M. Theoretical insights into the origin of photoluminescence of Au25(SR)18- nanoparticles. J. Am. Chem. Soc. 138, 11202–11210, (2016).
Aikens, C. M. Electronic and geometric structure, optical properties, and excited state behavior in atomically precise thiolate-stabilized noble metal nanoclusters. Acc. Chem. Res. 51, 3065–3073 (2018).
Weerawardene, K. et al. Luminescence and electron dynamics in atomically precise nanoclusters with eight superatomic electrons. J. Am. Chem. Soc. 141, 18715–18726 (2019).
Li, J.-J., Guan, Z.-J., Yuan, S.-F., Hu, F. & Wang, Q.-M. Enriching structural diversity of alkynyl-protected gold nanoclusters with chlorides. Angew. Chem. Int. Ed. 60, 6699–6703 (2021).
Luo, L. et al. Three-atom-wide gold quantum rods with periodic elongation and strongly polarized excitons. Pro. Natl Acad. Sci. 121, e2318537121 (2024).
Wang, G., Huang, T., Murray, R. W., Menard, L. & Nuzzo, R. G. Near-IR luminescence of monolayer-protected metal clusters. J. Am. Chem. Soc. 127, 812–813 (2005).
Wan, X.-K., Wang, J.-Q. & Wang, Q.-M. Ligand-protected Au55 with a novel structure and remarkable CO2 electroreduction performance. Angew. Chem. Int. Ed. 60, 20748–20753 (2021).
Shichibu, Y. et al. Biicosahedral gold clusters [Au25(PPh3)10(SCnH2n+1)5Cl2]2+ (n = 2−18): a stepping stone to cluster-assembled materials. J. Phys. Chem. C 111, 7845–7847 (2007).
Das, A. et al. Total structure and optical properties of a phosphine/thiolate-protected Au24 nanocluster. J. Am. Chem. Soc. 134, 20286–20289 (2012).
Wan, X.-K., Wang, J.-Q., Nan, Z.-A. & Wang, Q.-M. Ligand effects in catalysis by atomically precise gold nanoclusters. Sci. Adv. 3, e1701823 (2017).
Wang, J.-Q., Guan, Z.-J., Liu, W.-D., Yang, Y. & Wang, Q.-M. Chiroptical activity enhancement via structural control: the chiral synthesis and reversible interconversion of two intrinsically chiral gold nanoclusters. J. Am. Chem. Soc. 141, 2384–2390 (2019).
Chen, Y., Zeng, C., Kauffman, D. R. & Jin, R. Tuning the magic size of atomically precise gold nanoclusters via isomeric methylbenzenethiols. Nano Lett. 15, 3603–3609 (2015).
Fulmer, G. R. et al. NMR chemical shifts of trace impurities: common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 29, 2176–2179 (2010).
Crystal Data for 1: C172H161Au28F6N3O6P6S16, a = 59.872(3), b = 35.5197(10), c = 19.6889(4) Å, V = 41871(2) Å3, orthorhombic, space group Aba2, Z = 8, T = 100(2) K, 58676 reflections measured, 25703 unique (Rint = 0.0813), final R1 = 0.0815, wR2 = 0.2148 for 22569 observed reflections [I > 2 sigma(I)]. CCDC 2181997; Crystal Data for 2: C136H136Au21Cl6F3N2O5P4S13, a = 17.7872(4), b = 19.5178(6), c = 24.3621(5) Å, α = 94.019(2)°, β = 101.145(2)°, γ = 98.866(2)°, V = 8155.4(4) Å3, triclinic, space group P-1, Z = 2, T = 100(2) K, 51697 reflections measured, 24899 unique (Rint = 0.0707), final R1=0.0782, wR2=0.2130 for 19832 observed reflections [I > 2 sigma(I)]. CCDC 2181998.
Yuan, F. et al. Bright high-colour-purity deep-blue carbon dot light-emitting diodes via efficient edge amination. Nat. Photon. 14, 171–176 (2019).
Balan, A. D. et al. Effect of thermal fluctuations on the radiative rate in core/shell quantum dots. Nano Lett. 17, 1629–1636 (2017).
Zhou, M. et al. Three-orders-of-magnitude variation of carrier lifetimes with crystal phase of gold nanoclusters. Science 364, 279–282 (2019).
Wang, J.-Q. et al. Total structure determination of the largest alkynyl-protected fcc gold nanocluster Au110 and the study on its ultrafast excited-state dynamics. J. Am. Chem. Soc. 142, 18086–18092 (2020).
Luo, L., Liu, Z., Du, X. & Jin, R. Photoluminescence of the Au38(SR)26 nanocluster comprises three radiative processes. Commun. Chem. 6, 22 (2023).
Wang, Y. et al. Tailoring carbon tails of ligands on Au52(SR)32 nanoclusters enhances the near-infrared photoluminescence quantum yield from 3.8 to 18.3%. J. Am. Chem. Soc. 145, 26328–26338 (2023).
Strouse, G. F. et al. Influence of electronic delocalization in metal-to-ligand charge transfer excited states. Inorg. Chem. 34, 473–487 (2002).
Treadway, J. A. et al. Effect of delocalization and rigidity in the acceptor ligand on MLCT excited-state decay. Inorg. Chem. 35, 2242–2246 (1996).
Yang, H., Wang, Y., Edwards, A. J., Yan, J. & Zheng, N. High-yield synthesis and crystal structure of a green Au30 cluster co-capped by thiolate and sulfide. Chem. Commun. 50, 14325–14327 (2014).
Crasto, D., Malola, S., Brosofsky, G., Dass, A. & Häkkinen, H. Single crystal XRD structure and theoretical analysis of the chiral Au30S(S-t-Bu)18 cluster. J. Am. Chem. Soc. 136, 5000–5005 (2014).
Zeng, C. et al. Gold quantum boxes: on the periodicities and the quantum confinement in the Au28, Au36, Au44, and Au52 magic series. J. Am. Chem. Soc. 138, 3950–3953 (2016).
Nobusada, K. & Iwasa, T. Oligomeric gold clusters with vertex-sharing Bi- and triicosahedral. Struct. J. Phys. Chem. C. 111, 14279–14282 (2007).
Hooper, T. N. et al. Synthesis, structure and reactivity of stable homoleptic gold(i) alkene cations. Chem. Eur. J. 15, 12196–12200 (2009).
Wan, X.-K., Tang, Q., Yuan, S.-F., Jiang, D.-E. & Wang, Q.-M. Au19 nanocluster featuring a V-shaped alkynyl-gold motif. J. Am. Chem. Soc. 137, 652–655 (2015).
M. J.Frisch. et al. Gaussian 16, Revision B.01, (Gaussian Inc. 2016).
Hay, P. J. & Wadt, W. R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 82, 299–310 (1985).
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 (1988).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (92361301, 22201188 and 223B2111). X.-K.W. acknowledge financial support from the Fundamental Research Funds for the Central Universities. The authors thank Dr. Tianyu Huang, Dr. Qi Li, Prof. Juan Qiao, and Prof. Lian Duan at Tsinghua University for the help with luminescence measurements; Mingxing Chen at Peaking University for the help with PLQY measurements; Yuanheng Wang and Tong Jiang at Tsinghua University for helpful discussion.
Author information
Authors and Affiliations
Contributions
Q.-M.W. supervised and guided the project. X.-K.W. conceived and carried out the synthesis, crystallization of the clusters, characterization and analyzed the data, X.-S.H. conducted the NIR characterization, analyzed the data and assisted in the synthesis and characterization. Z.-J.G. conducted the density functional theory calculations. W.-Q.S. and J.-J.L. assisted in the synthesis and NIR characterization. X.-K.W., X.-S. H. and Q.-M.W. wrote the manuscript. All authors discussed the results and contributed to the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wan, XK., Han, XS., Guan, ZJ. et al. Interplay of kernel shape and surface structure for NIR luminescence in atomically precise gold nanorods. Nat Commun 15, 7214 (2024). https://doi.org/10.1038/s41467-024-51642-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51642-w
