
Abstract
Electrosynthesis of adipic acid (a precursor for nylon-66) from KA oil (a mixture of cyclohexanone and cyclohexanol) represents a sustainable strategy to replace conventional method that requires harsh conditions. However, its industrial possibility is greatly restricted by the low current density and competitive oxygen evolution reaction. Herein, we modify nickel layered double hydroxide with vanadium to promote current density and maintain high faradaic efficiency (>80%) within a wide potential window (1.5 ~ 1.9 V vs. reversible hydrogen electrode). Experimental and theoretical studies reveal two key roles of V modification, including accelerating catalyst reconstruction and strengthening cyclohexanone adsorption. As a proof-of-the-concept, we construct a membrane electrode assembly, producing adipic acid with high faradaic efficiency (82%) and productivity (1536 μmol cm−2 h−1) at industrially relevant current density (300 mA cm−2), while achieving >50 hours stability. This work demonstrates an efficient catalyst for adipic acid electrosynthesis with high productivity that shows industrial potential.
Similar content being viewed by others
Introduction
Adipic acid (AA) is one of the most important aliphatic dicarboxylic acids, which is mainly used to produce nylon-66 and other polyamides or polymers1. In industry, AA is synthesized via oxidation of cyclohexanol and cyclohexanone mixture (that is, KA oil) using 50 ~ 60 vol.% nitric acid as the oxidant, which is suffering from environmental issues related to the concentrated nitric acid and the emission of nitrous oxides (N2O and NOx) as greenhouse gases2,3. Although H2O2 can be used as an alternative greener oxidant, its high cost and harsh synthesis conditions hinder practical applications, calling for a more cost-effective and sustainable method4,5,6.
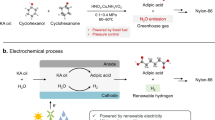
In the last decade, the electrocatalytic approach for chemical and fuel synthesis has gained increasing academic attention, for its advantages of utilizing renewable energy and operation under mild conditions (such as room temperature and ambient pressure)7,8,9,10. In this regard, the development of electrocatalytic KA oil-to-AA conversion is important for the above advantages and also for avoiding nitric acid usage and nitrous oxide emission encountered in its conventional production (Fig. 1a). A pioneering work was contributed by Petrosyan et al., who reported electrocatalytic cyclohexanone oxidation reaction (COR; cyclohexanone or cyclohexanol were often investigated to represent KA oil) over a nickel oxyhydroxide (NiOOH), but low current density (6 mA cm−2) and moderate AA yield (52%) were obtained11,12. Since then, significant progress has been made in developing Ni-based catalysts for enhancing activity in COR. For instance, a copper-doped nickel hydroxide (Cu-Ni(OH)2) catalyst was synthesized to facilitate Cα–Cβ cleavage in cyclohexanol13 We recently reported a Ni(OH)2 catalyst modified with sodium dodecyl sulfonate (SDS) to create a hydrophobic microenvironment that enriches cyclohexanone14.

a Challenges in the KA oil electrooxidation to produce AA. b Comparison of previously reported Ni-based catalysts and our catalyst for electrocatalytic COR both in three-electrode system and flow cell system11,13,14,16,26. The reaction parameters and performances in details are shown in Supplementary Tables 1 and 2. c Catalytic performances of our NiV-LDH-NS catalyst for COR in H-cell reactor and MEA that work in a wide potential window.
Despite the process with improved COR activity in the forgoing reports, the reported Ni-based catalysts showed high faradaic efficiency (FE) of AA ( > 80%) only at relatively low potentials, often below 1.6 V versus reversible hydrogen electrode (RHE, abbreviated as VRHE). As a result, the reported partial current density (that is, total current density multiplied by FE) of AA was always lower than 60 mA cm−2 (Fig. 1b and Supplementary Table 1). The low current density is far below the industrial requirement (>200 mA cm−2)15, thereby greatly hindering electrocatalysis technology for high-productivity AA synthesis (Fig. 1a; top). One can apply a more positive potential (for a three-electrode system) or larger cell voltage (for a two-electrode system) to increase the current density, which are straightforward approaches for many electrocatalytic transformations, especially oxygen evolution reactions (OER). However, for COR under high anodic potentials, OER may become a dominant competitor that decreases the FE of AA thus diminishing energy efficiency (Fig. 1a; bottom). For instance, in retrospect of the preceding progress (Fig. 1b and Supplementary Table 1), we unfortunately found that the FE of AA decreased from 93% to 76% over SDS-modified Ni(OH)2 as the applied potential increased from 1.5 VRHE to 1.7 VRHE14, and decreased from 93% to 69% over CuxNi1-x(OH)2/CF when the potential was from 1.52 VRHE to 1.62 VRHE16. As a result, the reported partial current density of AA was not proportionally increased at higher potential, largely restricting AA productivity increase, not to mention the high energy consumption owing to the low FE of AA. Besides Ni-based catalysts, cobalt-based one has also demonstrated its catalytic activity in COR17,18,19. However, they also suffer from diminished FE at higher potential, with more potential limitations for industrial applications compared with Ni-based catalysts such as more fluctuating prices and lower abundance. Therefore, it is desirable to develop Ni-based catalysts with high current density and FE in COR, aiming for high productivity of AA with practical possibility.
Herein, we reported vanadium (V)-modified nickel layered double hydroxide nanosheet (NiV-LDH-NS) as an efficient electrocatalyst of COR for AA production, which works in a wide potential window with largely suppressed OER, reaching high FEs and current densities in both H-cell and membrane electrode assembly (MEA; Fig. 1b). We first showed that over a typical Ni(OH)2 nanosheet catalyst (Ni(OH)2-NS), FE of AA expectedly decreased at higher potential—from 80% (at 1.5 VRHE) to 42% (1.9 VRHE). By evident contrast, after modifying Ni(OH)2 with V, the NiV-LDH-NS showed higher current density at a given potential and more importantly, maintained high FEs in a wide potential window. For example, it showed a current density of 170 mA cm−2 with FE of 83% at 1.9 VRHE, representing a more advantageous catalyst for COR in the three-electrode system (Fig. 1c and Supplementary Table 1). Experimental and theoretical evidence revealed that V modification promotes the reconstruction kinetics from Ni(OH)2 to high-valence Ni oxyhydroxide (Ni3+xOOH1-x), with the latter serving as the active phase for COR. Furthermore, V modification enhances cyclohexanone adsorption over the catalyst surface, which plays a pivotal role in suppressing OER at high anodic potentials. To show the possibility of NiV-LDH-NS in a more practical scenario, we constructed a flow MEA reactor, showing FE of AA (82%) at an industrially relevant current density (300 mA cm−2), greatly outpacing our previous results in a flow membrane-free reactor (Fig. 1b and Supplementary Table 2). The corresponding AA productivity (1536 μmol cm−2 h−1) is even higher than that via a thermal-catalytic process (<30 mmol gcatalyst−1 h−1)4. Moreover, by employing the MEA, a good stability of the catalyst was demonstrated by maintaining >80% FE of AA at 200 mA cm−2 for 60 h and >70% FE of AA at 300 mA cm−2 for 58 h. Eventually, preliminary techno-economic analysis (TEA) showcases the profitability of the electrocatalytic strategy for AA production.
Results
A typical Ni(OH)2 catalyst shows decreased FE at more positive potentials
According to previous literature, Ni(OH)2 is a typical catalyst that exhibits promising activity for COR, hence Ni(OH)2-NS was first synthesized via a co-precipitation method13,14. The sample exhibited a β-Ni(OH)2 structure as evidenced by X-ray diffraction (XRD; Fig. 2a), ultrathin nanosheets (thickness: 2–3 nm, lateral size: 20–50 nm) as shown by high-resolution transmission electron microscopy (HRTEM; Supplementary Fig. 1) and atomic force microscopy (AFM) measurement (Supplementary Fig. 2). Owing to the ultrathin nature, aggregation of nanosheets was also observed.

a XRD patterns of the Ni(OH)2-NS and the NiV-LDH-NS. FE, productivity, and current densities of AA over b Ni(OH)2-NS and c NiV-LDH-NS at different potentials. The error bars represent the standard deviation of three independent measurements by using the same kind of catalyst. d HRTEM images of NV-LDH-NS. Scale bar, 20 nm. e HAADF-STEM image of NiV-LDH-NS and corresponding element maps showing the distribution of Ni (green), V (yellow), and O (blue). Scale bar, 100 nm. f Ni 2p3/2, g O 1 s, and h V 2p3/2 XPS data for Ni(OH)2-NS (top) and NiV-LDH-NS (bottom). i FE and j productivity of AA over two catalysts in 7 cycles. The error bars represent the standard deviation of three independent measurements by using the same kind of catalyst, which is within 10%. Source data for a–c and f–j are provided in Source Data file.
We then evaluated Ni(OH)2-NS for COR. By employing a constant-potential electrolysis, we obtained an 80% FE of AA at a low potential (1.5 VRHE) with no occurrence of OER (Fig. 2b), suggesting COR is energetically more favorable than OER at low anodic potential. Glutaric acid (GA) was observed as a main by-product with an FE of 3%. The presence of trace amounts of succinic acid (SA), malonic acid (MA), and oxalic acid (OA) was additionally quantified by HPLC (see Supplementary Fig. 3 for product distribution). No formic acid was detected in the products, suggesting that carbonate may potentially serve as the C1 by-product. To verify this assumption, the electrolyte after complete electrolysis of 0.4 M cyclohexanone was acidified, and the gas product was passed through a Ca(OH)2 solution. As a result, the solution became turbid, confirming the formation of carbonate after electrolysis. However, due to the low overall electric quantity during electrolysis (Fig. 2b, c), the concentration of carbonate was relatively low, making it difficult to quantify. In addition, other C2-C5 products may also be generated but not be quantified. Although the difficulty for total product quantification, a total FEs of 90% suggests that the majority of electrochemical processes are determined, providing us the basis for mechanism understanding. Owing to the low current density (20 mA cm−2), AA productivity was measured to be 97 μmol cm−2 h−1 (Fig. 2b), equivalent to 19 mmol h−1 g−1 based on the mass loading of catalyst (5 mg cm−2), which was lower than a thermal-catalytic performance (~30 mmol h−1 g−1)1. As the applied potential was increased from 1.5 VRHE to 1.9 VRHE, despite the increasement in overall current density (from 20 to 114 mA cm−2), a severe sacrifice of FE of AA took place simultaneously—decreasing from 80% to 42%. The decreased FE at more positive potential was largely attributed to the competition of OER. Particularly, the competition of OER at 1.7 VRHE gave rise to a significant decrease in FE of AA and thus slightly lowered the productivity of AA when the total current density increased. As a consequence, although the partial current density of AA increased from 16 to 48 mA cm−2 with AA productivity increasement (from 97 to 298 μmol cm−2 h−1), huge additional energy was consumed (2.5 Wh gAA−1 higher from 1.5 to 1.9 VRHE), resulting in an increase of carbon emission by 2.7 g CO2 gAA−1 (see the Supplementary Note 1 for calculation details). The preceding observation of OER as the reaction competitor with COR under high anodic potentials is consistent with previous reports, posing a ubiquitous challenge for enhancing AA productivity14,17.
Introducing V in Ni(OH)2 enhances current density and FE at a wide potential window
To develop a more efficient catalyst for COR based on Ni(OH)2-NS, we first analyzed the active phase. We observed the peaks at 473 cm−1 and 553 cm−1 in in situ Raman results (Supplementary Fig. 4), which corresponded to the bending and stretching of Ni3+−O bonds in NiOOH, respectively. It was documented that NiOOH is generated as a result of Ni(OH)2 reconstruction and following Ni(OH)O accumulation under anodic potentials, representing the intrinsic active phase in electrocatalytic oxidations20,21. Therefore, we anticipate that accelerating the phase reconstruction process of Ni(OH)2-to-NiOOH may enhance the catalytic activity of COR.
We sought to modify Ni(OH)2 with different metals since heteroatom-modifying was reported to facilitate phase reconstruction in transition metal oxides/hydroxides22,23,24. The samples were synthesized via co-precipitation of Ni and the second metal’s precursors. Among different metal-modified samples, V-modified one with V:Ni atomic ratio of 1:8 (entitled as NiV-LDH-NS) gave a superior current density in COR (Supplementary Fig. 5) and more importantly, high FEs of AA within a wide potential window. Specifically, at low potential (1.5 VRHE), the current density of NiV-LDH-NS was 1.9-fold higher than that of Ni(OH)2-NS (39 vs. 20 mA cm−2), and FE of AA was comparable over these two catalysts (83% vs. 80%). As a result of higher current density and similar high FE of AA, NiV-LDH-NS exhibited 2.1-fold higher productivity compared with Ni(OH)2-NS (204 vs. 97 μmol cm−2 h−1), demonstrating the promoting effect of V modification on current density at low potentials (Fig. 2c).
When the applied potential was increased (for example, 1.9 VRHE), the current density over NiV-LDH-NS exhibited 1.5-fold higher compared with that over Ni(OH)2-NS (170 vs. 114 mA cm−2), with a similar increasing extent as that at lower potential (1.9-fold higher). Notably, NiV-LDH-NS maintained a high FE of AA (83%) with OER being greatly suppressed (O2 FE of 4%; Fig. 2c), outpacing Ni(OH)2-NS and previously reported catalysts showing much lower FEs of AA at high anodic potentials (Supplementary Table 1). Benefiting from the high FE of AA at wide potential window (1.5 ~ 1.9 VRHE), an AA production rate of 867 μmol cm−2 h−1 (corresponding to 174.3 mmol g−1 h−1) was achieved at 1.9 VRHE, showing advantageous performance among electrocatalytic systems and even thermal-catalytic systems when the activity was normalized with the total mass loading of NiV-LDH-NS sample (Supplementary Fig. 6).
Catalyst characterizations
To understand the high current density and high FE within a wide potential range after modifying Ni(OH)2 with V, we characterized the structure of NiV-LDH-NS. XRD results show that V modification induces phase transformation from β-Ni(OH)2 to α-Ni(OH)2, without observation of V-related crystalline species (Fig. 2a). HRTEM results show that NiV-LDH-NS inherits the ultrathin nanosheet morphology of Ni(OH)2-NS with a similar lateral size (Fig. 2d). AFM measurement reveals a strong tendency for nanosheets aggregation, resulting in a measurable thickness of approximately 7 nm (Supplementary Fig. 7), larger than that of Ni(OH)2-NS (thickness: 2–3 nm). V and Ni elements are well dispersed in the nanosheet, as identified by energy-dispersive X-ray spectroscopy (EDS) mapping analysis (Fig. 2e). To elucidate the electronic structure of V and its effect on Ni, X-ray photoelectron spectroscopy (XPS) was employed (Fig. 2f–h). Ni(OH)2-NS displays characteristic spin-orbit peaks of Ni2+ (fitting peak at 855.6 eV and satellite at 861.1 eV, Fig. 2f)25. The O 1 s XPS spectrum of Ni(OH)2-NS could be divided into three peaks, such that the peaks at 529.9, 530.9 and 532.8 eV assigned to the lattice oxygen (OL), hydroxyl groups (Ni-OH), and adsorbed oxygen on the surface defects (OAds), respectively (Fig. 2g)26,27,28,29. After V modification, the V 2p3/2 peak appears which can be deconvoluted into three peaks located at 517.1 eV (V5+), 516.6 eV (V4+), and 515.8 eV (V3+), indicating that the V species exist predominantly in high oxidation states in the structure (Fig. 2h)25,30,31. Moreover, the Ni 2p peaks at 855.4 eV in NiV-LDH-NS exhibit negative shifts (approximately 0.2 eV) compared with that in Ni(OH)2-NS, implying an electron transfer from V to Ni. The observation of a relatively lower valence state of Ni after V modification was corroborated with the result of Ni K-edge X-ray absorption near edge structure (XANES) spectra (more details are given in the later” V modification promotes catalyst reconstruction” section). NiV-LDH-NS after COR for 1 h was denoted as NiV-LDH-POST, and it was also comprehensively characterized by using TEM, EDS mapping, XRD, Raman spectroscopy, and XPS measurements (Supplementary Fig. 8 and 9). The catalyst maintains as aggregates with ultrathin nanosheet morphology (Supplementary Fig. 8a–c). The crystallinity of the sample decreased, accompanied by a reduction in V content due to the reconstruction of the catalyst with V leaching (Supplementary Fig. 8d–f). XPS spectra exhibited a decrease in the intensity of V peaks (Supplementary Fig. 9), resulting from the leaching of V. Moreover, the analysis of O 1s spectra (Supplementary Fig. 9d) and electron paramagnetic resonance (EPR) measurement (Supplementary Fig. 10) demonstrated the increased amount of oxygen vacancy after 1 h electrolysis over NiV-LDH-NS, which may lead to a negative shift of the binding energy of Ni 2p (More details are shown in Supplementary Fig. 9 and 10)26,27,32,33. As a result, NiV-LDH-NS exhibited slight structural changes after 1 h of COR.
To verify the important role of V in promoting COR, we synthesized NiV-LDH catalysts with different V:Ni atomic ratios other than 1:8 (1:32, 1:16, and 1:4, which are denoted as NiV-32, NiV-16, and NiV-4, respectively) using the same co-precipitation method. The EDS mapping results indicate that the V:Ni atomic ratio in the catalysts is close to that in the precursors (Supplementary Fig. 11a–e). With the increase of V modification, the intensity of V 2p spectra was enhanced and constant negative shifts were observed in the binding energy of Ni 2p region (Supplementary Fig. 12). Meanwhile, the proportion of OL was progressively rising. The catalytic results show that even with minimal V modification (V:Ni atomic ratio of 1:32), OER was effectively suppressed, displaying O2 FE reduction from 27% to 11% at 1.8 VRHE after V modification (Supplementary Fig. 11f). When V:Ni ratio increased from 1:32 to 1:8, the catalytic activity was enhanced. However, the current density decreased with further increasing V modification amount (V:Ni ratio of 1:4), which we suppose is attributed to a reduction in the density of Ni active sites (specifically, NiOOH active phase; Supplementary Fig. 11f). As a result of the promoting effect of V modification and the maintenance of Ni active sites, the catalyst with a V:Ni ratio of 1:8 exhibited the highest FE and productivity of AA among the V:Ni ratio screening tests. To investigate whether the V:Ni ratio was maintained after electrolysis, the composition of the used catalysts was characterized. The results show that V:Ni ratio decreased to approximately 1:22 after the reaction for the catalysts with initial V:Ni ratios of 1:16 ~ 1:4, presumably owing to catalyst reconstruction with V leaching (Supplementary Fig. 13). Note that when the initial V:Ni ratio is equal to or higher than 1:16, comparable FEs of AA were observed (Supplementary Fig. 11f), which can be explained by the catalysts reconstruction, by which similar V:Ni ratio was evolved in the catalysts showing comparable catalytic performance.
To further verify the importance of V modification in Ni(OH)2 for enhancing COR performance, we designed two additional synthetic methods to introduce V into Ni(OH)2-NS material. One is via a mixing method, and the sample is denoted as NiV-MIX; another is via a sequential spraying method, and the sample is denoted as NiV-SP. The synthetic details are shown in Methods. The successful modification of V onto the Ni(OH)2-NS surface of these two samples was evidenced by SEM-EDS mapping (Supplementary Fig. 14). The electrolysis results show that the FEs of AA on NiV-MIX and NiV-SP electrodes were 78% and 79% at 1.8 VRHE, respectively, both exhibiting higher FEs compared with that of Ni(OH)2-NS (51%). Moreover, OER over NiV-MIX and NiV-SP electrodes were suppressed (FEs of O2: 7% and 2%, respectively) compared with that over Ni(OH)2-NS (FE of O2: 27%). These results verify the positive effect of V modification in Ni(OH)2 on suppressing OER (Supplementary Fig. 14). However, the stability of the catalysts was compromised as evidenced by a decrease in FE of AA to 45% on NiV-MIX and 35% on NiV-SP after seven-cycle COR, implying the necessity of employing an appropriate methodology to stabilize V species, such as modification of V within the lattice of Ni(OH)2 in NiV-LDH-NS, that is, the key catalyst in this work.
We also evaluated the stability of the Ni(OH)2-NS and NiV-LDH-NS by performing multiple cycles for COR. The reaction was conducted for 1 h per cycle, and the electrolyte was refreshed after each cycle. The FE and productivity of AA over Ni(OH)2-NS were reduced by 50% and 60% after the 7th cycle, respectively, while an increased OER was observed (Fig. 2i, j). After each cycle, we analyzed the cyclic voltammetry (CV) curves of the catalysts and observed a gradual decline in the oxidation peaks of Ni2+, indicating a deterioration in the redox capacity of Ni (Supplementary Fig. 15a-c). Together with the increasing Ni cation concentrations in the electrolyte during electrolysis (Supplementary Fig. 15d), we attribute the performance degradation (lower FE and productivity of AA) to Ni leaching from the catalyst that results in more exposure of Ni foam substrate showing OER activity. In contrast, NiV-LDH-NS mitigates the decline in FE of AA and productivity to 10% (Fig. 2i, j), suggesting that the modification of V effectively inhibits Ni leaching (Supplementary Fig.15d). To understand the increased stability by V modification, we performed theoretical calculations. According to the previous literature34,35, an enthalpy change for the demetallation process of a metal atom from the active facet of the catalyst can be used as a rational descriptor to evaluate the stability of the catalyst. Therefore, the enthalpy changes for the demetallation processes of Ni atom on the surface of (100) facet of reconstructed Ni(OH)2-NS and NiV-LDH-NS (which are NiOOH and NiVOOH, respectively) were evaluated (details of model construction are described in Supplementary Note 2 and Supplementary Fig. 16). The Ni demetallation processes for NiOOH and NiVOOH are illustrated (Supplementary Fig. 17). The energy cost for Ni demetallation over NiVOOH (0.0325 eV) is higher than that over NiOOH (0.0005 eV), suggesting that V modification enhances the stability of NiOOH.
Reaction kinetics studies
To confirm the suppression of OER over NiV-LDH-NS especially at high anodic potentials, we performed differential electrochemical mass spectrometry (DEMS) to study the potential-dependent O2 formation over different samples. The results show that, without cyclohexanone, O2 appeared at an onset potential of 1.53 VRHE over NiV-LDH-NS, which was slightly lower than that over Ni(OH)2-NS (1.62 VRHE) (Supplementary Fig. 18). This result implies that OER suppression during COR over NiV-LDH-NS may not be due to its intrinsically inferior OER activity, in agreement with the slightly higher current density in linear sweep voltammetry (LSV) curves over NiV-LDH-NS compared with that over Ni(OH)2-NS in the absence of cyclohexanone (Supplementary Fig. 19). Upon the introduction of cyclohexanone, the delay in O2 evolution—probably owing to the thermodynamic advantage of COR—explains the high FE of AA at low potential window. More importantly, the onset potential of OER over NiV-LDH-NS (1.73 VRHE) delayed more significantly compared with that over Ni(OH)2-NS (1.65 VRHE), in accordance with the high FE of AA and low FE of O2 over NiV-LDH-NS at more positive potentials (Fig. 2c).
To further understand the promoting effect of V modification, we analyzed the reaction kinetics of OER and COR over Ni(OH)2-NS and NiV-LDH-NS by measuring their Tafel slopes. Notably, the current density within the Tafel region arises from the oxidation of Ni2+ to Ni3+ during the low-to-high potential LSV testing. To mitigate the interference of Ni2+ oxidation to the Tafel slope measurement of COR, we first implemented catalyst oxidation at 1.8 VRHE for 10 min, followed by conducting LSV tests in a reverse scanning mode, that is from high to low potential (Supplementary Fig. 20). The original LSV curves were corrected with 100% iR compensation to derive Tafel slope. In the absence of cyclohexanone, the Tafel slope over NiV-LDH-NS (41.6 mV dec−1) is lower than that over Ni(OH)2-NS (65.5 mV dec−1), indicative of a facilitated OER kinetics through V modification (Supplementary Fig. 20c). After introducing cyclohexanone, the Tafel slope over NiV-LDH-NS (37.3 mV dec−1) is lower than that over Ni(OH)2-NS (127.4 mV dec−1), indicating V modification induced a kinetically more pronounced effect on COR compared to OER (Supplementary Fig. 20d). These findings suggest that, although V modification facilitates OER to a certain extent, it significantly accelerates COR kinetics thus leading to high FE of AA.
V modification facilitates catalyst reconstruction
To understand the foregoing promoted FE and productivity of AA by V modification, we diverted our attention to mechanistic studies. Several previous reports have demonstrated that heteroatom modification can reduce catalyst crystallinity with higher electrochemically active surface area (ECSA), leading to an augmentation of active sites and consequently enhancing the catalytic activity36,37. To investigate this possibility, we conducted ECSA measurements both before and after electrochemical activation, revealing that the ECSAs of Ni(OH)2-NS and NiV-LDH-NS were comparable (Supplementary Fig. 21), excluding the impact of active sites density on catalytic enhancement after V modification.
According to well-established knowledge, during electrooxidation of alcohols or other nucleophilic substrates catalyzed by Ni(OH)2, Ni(OH)2 first loses electrons and protons following reconstruction to NiOOH at certain anodic potentials via an electrochemical step38,39,40,41. Then, the formed NiOOH serves as the authentic active species for COR, by abstracting hydrogen and electron from the nucleophilic substrates to generate oxidative products via a chemical step20,41. However, it was recently reported that although the reconstruction to NiOOH may serve as the rate-determining step (RDS) for alcohol electrooxidation over Ni(OH)2, oxidation of alcohols by Ni3+ can be a spontaneous process with a non-redox electron transfer via unoccupied Ni3+ orbitals as proposed in a recent literature41,42. Enlightened by the mechanistic study of the same literature, dimethylglyoxime disodium salt octahydrate (C4H6N2Na2O2·8H2O) was adopted as a probe molecule to in situ capture any Ni2+ formation that is generated from Ni3+ reduction during COR (Supplementary Fig. 22 and Supplementary Note 3). The results show Ni2+ formation, confirming that the chemical reduction of NiOOH and the electrooxidation of Ni(OH)2 occur simultaneously during COR. Thereby, the catalytic activity might be greatly affected by reconstruction kinetics from Ni(OH)2 to NiOOH. Based on this rationale, we next investigated if V modification accelerates the reconstruction of Ni(OH)2 thus improving COR.
We first demonstrated that NiOOH serves as the active phase for COR over both Ni(OH)2-NS and NiV-LDH-NS using in situ Raman technique, by observing NiOOH formation at positive potentials and its subsequent consumption after introducing cyclohexanone, following the aforementioned “electrochemical-chemical” process (Fig. 3a). Moreover, the reactivity of the reconstructed NiV-LDH-NS surpasses that of Ni(OH)2-NS, as evidenced by the accelerated disappearance of the Raman signal for Ni3+–O. We then showed that NiV-LDH-NS exhibits a lower positive potential for NiOOH generation compared with Ni(OH)2-NS under the conditions with or without cyclohexanone (Fig. 3b, c and Supplementary Fig. 4c, d). Notably, the superior OER performance of NiV-LDH-NS led to more intense bubbles adhering to the front lens of the objective for Raman measurement and thus the disappearance of Raman peaks at 1.55 VRHE (Supplementary Fig. 4d). According to the DEMS results (Supplementary Fig. 18), the current densities at low potentials (<1.58 VRHE for Ni(OH)2-NS and <1.53 VRHE for NiV-LDH-NS) primarily arise from the reconstruction of Ni2+ species rather than OER in the absence of cyclohexanone. Therefore, the more intense oxidation peak of Ni2+ in LSV curve over NiV-LDH-NS suggests that V modification endows NiV-LDH-NS with enhanced reconstruction ability (see Supplementary Fig. 19 for detailed analysis).

a In situ Raman spectra of Ni(OH)2-NS (left) and NiV-LDH-NS (right) at OCP condition after pre-oxidized 60 seconds at 1.5 VRHE in 0.5 M KOH and 0.4 M cyclohexanone. In situ Raman spectra of b Ni(OH)2-NS and c NiV-LDH-NS in 0.5 M KOH + 0.4 M cyclohexanone under different potentials. In situ XANES spectra at Ni K-edge for Ni(OH)2-NS and NiV-LDH-NS in d 0.5 M KOH and e 0.5 M KOH with 0.4 M cyclohexanone. Insets are the magnified region of the spectra between 8342 and 8446 eV. f Valence states of Ni in Ni(OH)2-NS and NiV-LDH-NS at different potentials. g In situ Ni EXAFS spectra for NiV-LDH-NS before and after introducing cyclohexanone at different potentials. h Theoretical models of Ni(OH)2-NS and NiV-LDH-NS. Top: over Ni(OH)2-NS, slow reconstruction from Ni(OH)2-NS to NiOOH serves as RDS and the cyclohexanone reduces high-valence Ni species to maintain a low valence state of Ni via a chemical step to produce AA. Bottom: over NiV-LDH-NS, the reconstruction step is facilitated by V modification, leading to a shift of RDS from the reconstruction step to the chemical step. i Gibbs free energy changes for reconstruction processes of Ni(OH)2-NS and NiV-LDH-NS. Source data for a–j, and i are provided in Source Data file.
To probe the evolution of the atomic and electronic structures in the reconstruction process of catalyst, in situ X-ray absorption spectroscopy (XAS) experiments were employed, providing a powerful tool to probe the dynamic process of Ni species during successive three steps: OER, cyclohexanone injection at open circuit potential (OCP), and COR. The XANES spectra at the Ni K-edge with potential rise before and after cyclohexanone introduction are depicted (Fig. 3d, e). The absorption edge energy of NiV-LDH-NS exhibits a significantly more positive shift than that of Ni(OH)2-NS at the same potential (Fig. 3d, e, insets). The average valence of Ni at each condition was estimated by regressing the shifts in absorption edge energy of Ni K-edge through linear combination fitting of the XANES spectra (Fig. 3f), and the reference spectra were acquired from reported literature (Supplementary Fig. 23)43.
In the first step (before cyclohexanone was injected, corresponding to OER process; Fig. 3f, left), the valence state of Ni in NiV-LDH-NS ( + 1.83) was slightly lower than that in Ni(OH)2-NS ( + 1.97) at potential when catalysts were not reconstructed (<1.3 VRHE), which can be attributed to electron transfer from V to Ni, in accordance with aforementioned XPS results (Fig. 2f). When the potential exceeded the reconstruction point (1.5 VRHE), the valence state of Ni in NiV-LDH-NS ( + 3.28) exhibited a more pronounced increasement compared to that observed in Ni(OH)2-NS ( + 2.49). At an even higher potential (1.8 VRHE), a higher valence of Ni species (+3.64) was generated over NiV-LDH-NS than that over Ni(OH)2-NS ( + 3.47). According to recent reports, this process corresponds to high-valence Ni4+ species generation within Ni3+xOOH1-x structure (Ni3+x represents the mixture species of Ni3+ and Ni4+), which previously exhibited boosted catalytic activity in alcohols dehydrogenation38,39,44. Therefore, the superior performance of NiV-LDH-NS in COR may be due to the facilitated reconstruction ability that forms catalytically active high-valence Ni species.
In the second step (after cyclohexanone was injected at OCP; Fig. 3f, middle), the Ni valance states over both catalysts decreased obviously, corresponding to the process of Ni3+xOOH1-x reduction by cyclohexanone, consistent with in situ Raman spectroscopy results (Fig. 3a). Moreover, the Ni valence states were nearly revered to their initial states (the first step at low potential), indicating the reversibility of Ni redox in Ni3+xOOH1-x.
In the third step (COR process), at COR potential (1.5 and 1.8 VRHE; Fig. 3f, right), the valence state of Ni in Ni(OH)2-NS exhibited only slightly increased (+2.16 and +2.40), which were much lower than that at the same potential in the first step (+2.49 and +3.47). These results imply that after cyclohexanone was injected, COR was kinetically limited by the slow Ni2+-to-Ni3+x oxidation (that is, Ni reconstruction) rather than the chemical step between NiOOH and cyclohexanone over Ni(OH)2-NS, leaving Ni positing at low-valence states. Thus, we deduce that Ni reconstruction may serve as the RDS during COR over Ni(OH)2-NS. By contrast, NiV-LDH-NS maintained a relatively high valence Ni species (>3) during COR, with much lower valence reduction (less than 0.2) compared with that in the first step at the same potential (1.65 and 1.8 VRHE), demonstrating that Ni2+-to-Ni3+x oxidation was kinetically boosted by V modification, driving Ni reconstruction a faster process than chemical step reduced by cyclohexanone. Extended X-ray absorption fine structure (EXAFS) results also revealed a complete transformation of Ni−O (from 1.6 to 1.4 Å) and Ni−Ni(V) (from 2.8 to 2.4 Å) bonds in the presence of cyclohexanone. This corresponds to the reconstruction of Ni(OH)2 phase into NiOOH phase and the chemical reduction of NiOOH phase by cyclohexanone (Fig. 3g). However, the reconstruction kinetics of Ni(OH)2-NS is significantly impeded by cyclohexanone (more details in Supplementary Note 4 and Supplementary Fig. 24).
Collectively, over Ni(OH)2-NS (Fig. 3h, top), the slow reconstruction step from Ni(OH)2 phase to NiOOH phase may serve as the RDS of the whole COR process rather than the chemical step in which AA is generated from cyclohexanone while NiOOH is chemically reduced. Over NiV-LDH-NS (Fig. 3h, bottom), V modification enhances the kinetics of Ni2+-to-Ni3+x oxidation, thus accelerating the formation of NiVOOH compared to its consumption by chemical reduction, leading to a shift of RDS to the chemical step. To understand the facilitated Ni reconstruction by V modification, we conducted more theoretical calculations. As depicted in Fig. 3h, the reconstruction processes for Ni(OH)2-NS and NiV-LDH-NS were modeled. The lattice hydroxyls over Ni(OH)2-NS and NiV-LDH-NS were deprotonated by the abstraction of OH‒ in electrolyte, generating an electron-deficient lattice oxygen, with the corresponding chemical reactions given as follows:
The Gibbs free energy changes for reconstruction were calculated (Fig. 3i), and NiV-LDH-NS (0.81 eV) displays a much lower one than Ni(OH)2-NS (1.66 eV), suggesting that V modification decreases the required voltage for Ni reconstruction. We rationalize that the facilitated reconstruction may lower the energy barrier of the overall COR (see the reaction mechanism study later in details), which in turn accelerates the reaction with higher current density.
V modification strengthens cyclohexanone adsorption
The above analysis shows that V modification induces rapid phase reconstruction of Ni(OH)2 that promotes reaction rate thus current density of COR. However, Ni3+x sites can also promote OER activity, as evidenced by the boosted current density over NiV-LDH-NS versus Ni(OH)2-NS from LSV curves in the absence of cyclohexanone (Supplementary Fig. 19), making COR and OER competitive reactions. Therefore, the significantly higher FE of AA over NiV-LDH-NS cannot be fully explained by the promoted phase reconstruction by V modification.
It is widely accepted that for electrooxidation of nucleophilic substrates in an alkaline medium, the reaction often proceeds via a Langmuir-Hinshelwood (L-H) model. Specifically, the substrate and OH− anions are competitively co-adsorbed on the catalyst surface, and the adsorbed OH− are oxidized to reactive hydroxyl species (OH*) as the electrophile for nucleophile oxidation, with the mechanism being previously demonstrated by experimental data and/or theoretical calculations45,46,47. Therefore, the concentration of reactants and their ratio (the organic substrate and OH−) can regulate the reactant coverage over the catalyst surface, thus the FE and productivity of the desired product can be influenced14,48,49,50. In our case, we posit that a high surface coverage of cyclohexanone was achieved in NiV-LDH-NS that favors COR process, and conversely, a low coverage of cyclohexanone in Ni(OH)2-NS favors OER process.
To verify the above hypothesis, we initially conducted two sets of reactant concentration (Ccyclohexanone and COH−)-dependent experiments. The first one was constant-potential electrolysis (1.8 VRHE) with varied Ccyclohexanone (0.05 ~ 0.45 M) and fixed COH− (0.5 M) over Ni(OH)2-NS and NiV-LDH-NS catalysts. The FE and productivity of AA were then calculated. For NiV-LDH-NS catalyst, the AA productivity versus the Ccyclohexanone reveals a typical “volcano-type” curve in an L-H mode (Fig. 4a), demonstrating that the high coverage of cyclohexanone causes competition with OH− adsorption. While for Ni(OH)2-NS, AA productivity monotonically rises as Ccyclohexanone increases from 0.05 to 0.45 M, indicating the surface coverage of cyclohexanone is still relatively low although the bulk concentration is high (0.45 M). Moreover, by increasing the COH− to 1.5 M, the Ccyclohexanone-dependent “volcano-type” curve on Ni(OH)2-NS was observed, and the inflection point of productivity delayed compared to that on NiV-LDH-NS, providing further evidence for the weak adsorption of cyclohexanone over Ni(OH)2-NS (Supplementary Fig. 25a and Note 5). Furthermore, the FE of AA over NiV-LDH-NS was sensitive to Ccyclohexanone and climbed rapidly to >80% when Ccyclohexanone rose from 0.05 M to 0.3 M, indicating the readily enrichment of cyclohexanone on NiV-LDH-NS (Fig. 4b). In contrast, elevating Ccyclohexanone did not suppress OER significantly over Ni(OH)2-NS, which is likely due to the inadequate adsorption of cyclohexanone. Vice versa, further investigation on the dependence of COH− on catalytic performance also confirmed that cyclohexanone adsorption is enhanced over NiV-LDH-NS, which endures higher COH− without decreasing FE of AA during COR (Supplementary Fig. 25b, c and Note 5).

AA a productivity and b FE over Ni(OH)2-NS and NiV-LDH-NS at different Ccyclohexanone in 0.5 M KOH. c Adsorption energies of cyclohexanone over NiOOH and NiVOOH. d FE of AA over Ni(OH)2-NS and NiV-LDH-NS using intermittent potential and constant potential strategies at 1.80 VRHE in 0.5 M KOH with 0.4 M cyclohexanone. The error bars represent the standard deviation of three independent measurements by using the same sample, which is within 10%. e Top: over Ni(OH)2-NS, weak cyclohexanone adsorption occurs with low surface Ccyclohexanone, inducing serious OER competition. Bottom: over NiV-LDH-NS, enhanced cyclohexanone adsorption occurs with high surface Ccyclohexanone, leading to suppressed OER. Source data for a–d are provided in Source Data file.
To validate the enhanced adsorption of cyclohexanone over NiV-LDH-NS, we employed electrochemical combined quartz crystal microbalance (E-QCM) to monitor the mass change of the adsorbed species in real-time. The results indicate that the initial adsorption capacity of cyclohexanone on NiV-LDH-NS is 1.6-fold higher compared to Ni(OH)2-NS at OCP status, and this adsorption capacity discrepancy further increases as the potential increases to 1.5 VRHE (Supplementary Fig. 26). Spin-polarized DFT calculation was performed to investigate the adsorption behavior of cyclohexanone over NiOOH and NiVOOH (Fig. 4c). Cyclohexanone was adsorbed at Ni site over NiOOH with adsorption energy (Eads) of ‒0.57 eV, while that can be either adsorbed at Ni site or V site over NiVOOH, with V site affording much lower Eads (‒0.69 eV), in consistent with the observed stronger cyclohexanone adsorption over NiVOOH.
To further verify that enhancing cyclohexanone adsorption boosts AA formation and suppresses OER, we used an intermittent-potential strategy to enrich cyclohexanone over the catalyst surface (for both Ni(OH)2-NS and NiV-LDH-NS) inspired by previous reports51,52. Specifically, we applied a potential at 1.8 VRHE for COR, and switched it to an OCP status, and then switched it back to 1.8 VRHE. In this case, cyclohexanone can be accumulated over catalyst surface during the OCP status between electrolysis (see Method section for detailed procedure). The results show that for both Ni(OH)2-NS and NiV-LDH-NS, the catalytic performances were improved by using the intermittent-potential electrolysis compared with the constant-potential one (Fig. 4d). Notably, Ni(OH)2-NS displays a more pronounced improvement of COR (AA FE: from 51% to 82%) and suppression of OER (O2 FE: from 27% to 4%) than NiV-LDH-NS, which is because cyclohexanone accumulation can be ameliorated more greatly over a catalyst with weaker adsorption ability (that is, Ni(OH)2-NS) by the intermittent-potential electrolysis.
Collectively, the inhibited OER over NiV-LDH-NS can be explained by enhanced adsorption of cyclohexanone (Fig. 4e). Over Ni(OH)2-NS (Fig. 4e, top), the weak adsorption of cyclohexanone results in relatively low Ccyclohexanone on the catalyst surface, while the coverage of OH* is comparatively higher. Consequently, an excess of OH* species gives rise to serious OER competition and diminished FE of AA. By contrast, over NiV-LDH-NS (Fig. 4e, bottom), V modification enhances the adsorption capacity of cyclohexanone and thus increases surface Ccyclohexanone, effectively utilizing adsorbed OH* species for COR with promoted AA production and inhibited OER.
Reaction pathway analysis
In addition to investigating the impact of V modification on Ni species reconstruction and cyclohexanone adsorption, we also examined whether V alters the pathway of COR toward AA formation. In the existing literature, several different paths of COR were proposed in previous literature, and we analyzed their possibilities in our reaction system (see more details in Supplementary Fig. 27 and Supplementary Note. 6)13,14,26. Firstly, it was reported that the first step of COR pathway may involve the initial oxidation of cyclohexanone to generate a key intermediate, 2-hydroxycyclohexanone (2)13,14. To verify this process, EPR was investigated by employing 5,5-dimethyl-1-pyrroline N-oxide (DMPO) for capturing active intermediates adsorbed on the catalyst surface. The EPR results identified the existence of C-centered radicals (R·) and hydroxyl radicals (OH·) over both catalysts during COR, indicating the formation of enol radical intermediate (1) via the dehydrogenation of Cα − H of cyclohexanone and the further oxygenation of 1 by OH* to afford 2 (Fig. 5a and Supplementary Fig. 28). Despite the same intermediate was identified on both catalysts, the proportion of R· signal area over NiV-LDH-NS was relatively high compared to Ni(OH)2-NS, potentially attributed to the enhanced adsorption capability of cyclohexanone (Supplementary Table 3 and Note 7). We further employed 2 and 1,2-cyclohexanedione (3) as the starting reactant for electrolysis to validate if V alters the following oxidation step. The electrolysis results of the potential intermediates (2 and 3) over Ni(OH)2-NS and NiV-LDH-NS exhibited comparable product selectivity, indicating that COR over Ni(OH)2-NS or NiV-LDH-NS proceed via a similar pathway (Fig. 5b). Moreover, AA was the main product only when 2 was used as the reactant, suggesting that AA is generated by a direct oxidation process through Cα − Cβ bond cleavage of 2 rather than subsequent oxidation to form 3 over both catalysts, because 3 was mainly converted to GA when it was used as the starting reactant (Supplementary Figs. 29, 30).

a EPR signals over NiV-LDH-NS in 0.5 M KOH + 0.4 M cyclohexanone. b Results of electrocatalysis of 2-hydroxycyclohexanone (2) and 1,2-cyclohexanedione (3). The electrolysis was conducted in 0.5 M KOH and 0.1 M 2 or 3 at 1.8 VRHE for an hour. The error bars represent the standard deviation of two independent measurements by using the same kind of catalyst. c Proposed reaction pathway for COR over both catalysts. d Schematic illustrations of COR pathways over Ni(OH)2-NS (left) and d NiV-LDH-NS (right). The red arrows indicate the promoted step during COR process by V modification. Source data for a and b are provided in Source Data file.
Collectively, we demonstrated that Ni(OH)2-NS and NiV-LDH-NS catalyzed COR via a similar pathway: cyclohexanone was adsorbed over catalyst surface and dehydrogenated with an electron loss to form 1, which was then oxygenated by OH* to yield 2, followed by subsequent multi-step transformations leading to the production of AA (Fig. 5c). However, OER competition was exclusively observed on Ni(OH)2-NS when cyclohexanone was used as the reactant, while minimal oxygen was collected with both 2 and 3 as reactants. Hence, the observed disparity in catalytic performance may owe to changes in both the energy barrier of RDS and the cyclohexanone adsorption capacity by V modification, rather than arising from changes in the reaction pathway. Therefore, we analyzed the RDS of the reaction pathway over two catalysts. The aforementioned in situ XAS results have demonstrated that V modification induces RDS shifting from reconstruction step to chemical step during COR, maintaining complete NiOOH phase and high-valence Ni species over NiV-LDH-NS (Fig. 3f, Supplementary Fig. 24 and Note 4). We further analyzed the reaction processes represented by each part of current density in different potential regions during CV measurements (see details in Supplementary Fig. 31 and Note 8), and carried out kinetic isotope H/D exchange experiments, together demonstrating that the RDS of COR over NiV-LDH-NS involves the cleavage of Cα − H bonds in a chemical step, rather than the reconstruction step (see more discussions in Supplementary Fig. 32 and Note 8).
Based on the above analysis, the total effects of V modification are depicted in Fig. 5d. Ni(OH)2-NS and NiV-LDH-NS catalysts experienced surface reconstruction under elevated anode potentials and catalyzed COR via a similar pathway. Over Ni(OH)2-NS (Fig. 5d, left), the reconstruction step is the RDS during COR process; Whereas, over NiV-LDH-NS (Fig. 5d, right), V modification significantly accelerates the reconstruction process and shifts the RDS to the dehydrogenation of Cα − H on cyclohexanone to form 1. Moreover, cyclohexanone adsorption occurs at V sites with enhanced adsorption over NiV-LDH-NS, which contributes to the inhibition of OER.
Continuous AA production in MEA
In light of the excellent electrocatalytic performance of NiV-LDH-NS with high FE within a wide potential window, we constructed an MEA for the pursuit of continuous production of AA. The MEA was assembled by using NiV-LDH-NS as the anode, commercial PtRu/C as the cathode53, and an anion exchange membrane (type: FAA-3-50) (Fig. 6a and Supplementary Fig. 33)54. The concentration of anolyte was optimized to be 1 M KOH owing to the reduced cell voltage and comparable FE of AA compared to 0.5 M KOH in the above studies (Supplementary Fig. 25c). The LSV curves were recorded as shown in Supplementary Fig. 34, indicating NiV-LDH-NS has significantly greater COR performance than Ni(OH)2-NS. To demonstrate the superiority of NiV-LDH-NS, constant-current electrolysis was performed at stepped current densities from 50 to 500 mA cm−2, and corresponding cell voltages were recorded. The results show that NiV-LDH-NS exhibited a cell voltage of 1.76 V at the current density of 300 mA cm−2, approximately a 16% reduction in cell voltage compared to Ni(OH)2-NS (2.09 V), showing energetically more efficient for AA production (Fig. 6b).

a Schematic illustration of an MEA. b Cell voltage without iR compensation over Ni(OH)2-NS and NiV-LDH-NS in 1 M KOH with 0.4 M cyclohexanone at different current densities. c AA productivity and FE over Ni(OH)2-NS and NiV-LDH-NS at different current densities. The error bars represent the standard deviation of two independent measurements by using the same kind of catalyst. d Comparison of catalytic performance between our work and other reported flow cell systems14,17,19. The reaction parameters and performances in details are shown in Supplementary Table 2. e Cell voltage and FE of AA over NiV-LDH-NS at 200 and 300 mA cm−2, respectively, during long-term tests. Source data for b–e are provided as a Source Data file.
Meanwhile, as shown in Fig. 6c, NiV-LDH-NS largely maintained good FEs (from 83% to 61%) at higher current densities (from 200 to 500 mA cm−2), delivering higher productivity of AA (from 1031 to 1900 μmol cm−2 h−1). Meanwhile, only 0.8% of adipate anion was observed in the cathode chamber after electrolysis, suggesting that cyclohexanone crossover was not significant in our case (Supplementary Fig. 35). In contrast, the FE of AA over the Ni(OH)2-NS diminished from 61% to 34% at the same extent of current density increasement, thereby impeding AA productivity enhancement (from 762 to 1050 μmol cm−2 h−1). In particular, there was even a slight decrease in productivity of AA, which is because of the intense competition from OER and thus the substantial reduction of FE of AA when the current density increased (from 200 to 250 mA cm−2, Supplementary Fig. 5). To our best knowledge, the catalytic results using the MEA with NiV-LDH-NS catalyst significantly surpass previously reported performances in flow reactors with Ni-based catalysts (Supplementary Table 2). Moreover, as shown in Fig. 6d, NiV-LDH-NS exhibits significant advantages in terms of current density, cell voltage, and FE of AA compared to the best-performing Cobalt-based catalyst (that is, Co3O4 supported on graphdiyne (Co3O4/GDY))17. Furthermore, we estimated the energy consumption during AA production, revealing a remarkably low value of 2.4 Wh gAA−1 at the current density of 300 mA cm−2 and cell voltage of 1.76 V (detailed calculation in Supplementary Note 1). Compared to the optimal result of 4.1 Wh gAA−1 by Co3O4/GDY in previous report, the energy consumption of AA production was reduced by 42% in our work, while a 4-fold increasement in production rate was simultaneously attained (1536 vs. 319 μmol cm−2 h−1)17.
The stability of the NiV-LDH-NS catalyst for long-term AA production in MEA was assessed under a current density of 200 and 300 mA cm−2, respectively (Fig. 6e). Because OH− is consumed faster at higher current densities, the electrolyte at 300 mA cm−2 was refreshed more frequently than at 200 mA cm−2 (for more details, see “Electrochemical measurements” subsection). At current density of 200 mA cm−2, the average FE during the initial 6-hour period of COR is 93%, which subsequently slightly decreased to 81% after 60 hours, along with a slight increase of 7% in cell voltage (from 1.62 V to 1.73 V), indicating a good stability. When the current density was increased to 300 mA cm−2, the FE of AA largely maintained (decreased from 85% to 72%), but the cell voltage obviously increased (from 1.71 to 2.09 V, corresponding to 22%) during a 46-hour test (Fig. 6e). We postulated that the primary reason for the decreased performance was the corrosion of the anion exchange membrane (AEM) by cyclohexanone that increased the resistance of the electrolyzer and the cell voltage (Supplementary Fig. 36), accompanied by a slight leakage of electrolyte from the anode to the cathode, leading to a reduction in anolyte volume, and thus the electrolysis had to be terminated. In addition, the decline of FE of AA can be also attributed to catalyst leaching and thus the exposure of Ni foam that favors OER. To demonstrate the contribution of corroded AEM for the decreased stability at 300 mA cm−2, we replaced a new AEM after 46-hour electrolysis. To our expectation, the catalytic performance evidently recovered, with the cell voltage being significantly decreased to its pristine value (from 2.09 to 1.71 V) with following slight increase in the next 12-hour electrolysis (5% from 1.71 to 1.79 V; Fig. 6e).
Overall, we achieved 60-hour stability for continuous production of AA at 200 mA cm−2, showing large maintenance of FE of AA and cell voltage. We also attempted higher current density of 300 mA cm−2, and realized overall 58-hour stability by replacing a new AEM at 46 h. The above studies demonstrate the stability of the catalyst, and clearly point out the necessity of developing more robust AEM in the future to improve the long-term stability of MEA for continuous AA production at desirable industrially relevant current density.
Techno-economic analysis
Based on the performance of our MEA, we proposed a complete AA production process, involving substrate feeding, electrolysis, neutralization, and separation units (Supplementary Fig. 37). A preliminary TEA was conducted to assess the economic viability of this system by using a model of electrocatalytic carboxylate production in alkaline electrolytes55. In this context, the costs include capital, operating, and materials (Fig. 7a and Supplementary Fig. 38), while the revenue comes from AA and H2 production. The TEA results show that under our operation condition (current density of 300 mA cm−2, cell voltage of 1.76 V, FE of 82%), the total cost and revenue are $2429 and $2564, respectively, which means that a net profit of $135 is achievable when 1 ton of AA is produced (See details in Supplementary Note 9).

a Total cost of the AA electrochemical process under the base case scenario at conditions of the FE of 82%, current density of 300 mA cm−2, and cell voltage of 1.76 V. Sensitivity analysis of three kinds of costs to b FE and c Current density. In the sensitivity analysis, only the parameters under investigation were varied while keeping the remaining parameters constant based on the TEA model. d The impact of different FEs and current densities on the profit of AA electrosynthesis and the profits by using Ni(OH)2-NS and NiV-LDH-NS, assuming the cell voltage remains constant at 1.76 V. Source data for a–d are provided in Source Data file.
Based on this premise, we further investigated the impact of FE and current density on the profitability of AA electrosynthesis. We found that the profitability is highly sensitive to FE of AA, since a decrease in FE results in a significant increase in operational costs, thereby substantially augmenting the total cost (Fig. 7b). Regarding current density, a higher one (> 200 mA cm−2) mainly contributes to the reduction of capital cost and plant construction cost by minimizing the electrolytic cell area, thus contributing to increased profit (Fig. 7c). Compared to current density, the impact of FE on profits is more significant. By depicting the impact of FE and current density on profit, we clearly discern the significance of reaching high FE ( > 60%) at industrially relevant current density (>200 mA cm-2) for profitability. Benefiting from the high FE of AA, the reaction system using NiV-LDH-NS as the catalyst keeps profitable within the range of 100–500 mA cm−2 (pentagram dots; Fig. 7d). However, for Ni(OH)2-NS, the decrease of FE at high current densities (>200 mA cm−2) leads to unprofitable result (round dots; Fig. 7d), highlighting the importance of catalyst with high FE at high current density.
Besides the importance of catalyst for reducing the capital and operational costs, our TEA assessment shows that the profitability can be further augmented in the following two ways. The first one is to co-sell potassium sulfate (K2SO4) to the market, a by-product in the neutralization unit but with a potential revenue of 828 $ tonAA−1 (Supplementary Note 9). The second one is to optimize the processing, including recycling the materials or developing a more cost-efficient AA separation technology (to substitute neutralization and separation units). The currently adopted acid-base neutralization process may cause high material cost (that accounts for the largest proportion of 85.3%), in which cyclohexanone and KOH contribute to 94% (2069 $ tonAA−1; Fig. 7a), although the overall process is still profitable as discussed above. We expect that by recycling KOH and the unreacted cyclohexanone via more advanced methods, such as electrodialysis to completely recycle KOH14, the material cost might be further reduced (estimated to be 1,073 $ tonAA−1 by electrodialysis; Supplementary Note 9).
Discussion
In conclusion, we successfully achieved the high FE productivity of AA at high current density by introducing V into Ni(OH)2 nanosheets. The AA FE over NiV-LDH-NS reaches 83–88% at a wide potential range from 1.5−1.9 VRHE and a high current density of 170 mA cm−2, while OER is effectively suppressed to 3%. The modification of V promotes the reconstruction from Ni2+ to Ni3+x and enhances cyclohexanone adsorption. Experimental and theoretical data demonstrated that the promoted reconstruction enhances the current density of cyclohexanone oxidation and shifts the RDS of COR from reconstruction to dehydrogenation involving Cα − H cleavage, and the enhanced adsorption of cyclohexanone inhibits OER. By constructing an MEA, continuous AA production at an industrial current density of 300 mA cm−2 was achieved, presenting a record-high AA FE of 82% with a productivity of 1536 μmol cm−2 h−1. The >50-hour tests demonstrated a good stability of NiV-LDH-NS by maintaining high FEs of AA ( > 80% at 200 mA cm−2 for 60 h; >70% at 300 mA cm−2 for 58 h) in the MEA. We should point out that it is necessary to develop more robust AEM for long-term stability at desirable industrially relevant current density. Furthermore, TEA highlights the profitability of the reaction strategy for AA production and the significance of high-performance catalyst and advanced separation technology for further reducing the costs.
Methods
Chemicals and materials
Except noted, all chemicals were purchased and used without further purification. Deionized water was used throughout the experiments. Nickel (II) nitrate hexahydrate (Ni(NO3)2·6H2O, 99%), vanadium (III) chloride (VCl3, 98%), adipic acid (C6H8O4, 99.5%), glutaric acid (C5H6O4, 99.5%), cyclohexanone−2,2,6,6-d4 (C6H6D4O, 98%,98 atom. %D) were purchased from Aladdin Reagent Co. Ltd (Shanghai, China). Potassium hydroxide (KOH, 95%) was obtained from Macklin Reagent Co. Ltd (Shanghai, China). Hydrochloric acid (HCl, 36.5-38 wt%) was obtained from Bei Jing TongGuang Fine Chemicals Company. Cyclohexanone (C6H10O, 99.5%) was purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). Potassium deuteride (KOD 99.8 atom. %D, 30 wt. %in D2O) was purchased from Energy Chemical (Shanghai, China). 2-Oxoadipic acid (96.61%) was purchased from TargetMol, USA. Platino-ruthenium/carbon (Pt 40%, Ru 20%, C 40%), nickel foam (thickness 3.2 mm), nickel felt (thickness 0.25 mm), proton exchange membrane (Nafion 117: thickness 183 μm), Fumasep anion exchange membrane (FAA-3-50: thickness 50 μm) were purchased from Shengernuo Co. Ltd. (Suzhou, China).
Synthesis of Ni(OH)2-NS
A 20 mL aqueous solution containing 75 mM Ni(NO3)2·6H2O was added dropwise to 20 mL deionized water under magnetic stirring at 80 °C. Simultaneously, 0.5 M KOH was added dropwise into the solution to maintain a pH of 10. The reaction was completed within 10 min. After cooling to room temperature, the product was collected by centrifugation, washed with deionized water twice and ethanol once continuously. Then the obtained wet Ni(OH)2-NS was dispersed in 10 mL ethanol to form colloids.
Synthesis of NiV-LDH-NS
The synthesis of NiV-LDH-NS was similar to that described above for Ni(OH)2-NS, except that the aqueous solution containing 75 mM Ni(NO3)2·6H2O and 9.38 mM VCl3.
Synthesis of NiV-MIX
A 2 mL prepared Ni(OH)2-NS colloid was mixed with 0.5 mL aqueous solution containing 2 mg VCl3 and then was ultrasonicated for 1 h. The ink was sprayed on a commercial nickel foam with the size of 2 cm × 2 cm using an airbrush to obtain NiV-MIX. Then, it was cut to 1 cm × 1 cm as the working electrode after washing with deionized water.
Synthesis of NiV-SP
A 2 mL prepared Ni(OH)2-NS colloid was sprayed on a commercial nickel foam with the size of 2 cm × 2 cm using an airbrush to obtain Ni(OH)2-NS/nickel foam. Subsequently, 0.5 mL aqueous solution containing 2 mg VCl3 was sprayed on the Ni(OH)2-NS/nickel foam to obtain NiV-SP. Then, it was cut to 1 cm × 1 cm as the working electrode after washing with deionized water.
Characterizations
XRD patterns were measured using a D8 ADVANCE (Bruker, USA) equipped with Cu Kα radiation source (λ = 1.5406 Å) at 40 kV and 40 mA, with a scan angle of 5°–80° and a scan rate of 2 ° min-1. HRTEM and EDS mapping were performed on a JEM−2100F, JEOL (Japan) at an accelerating voltage of 200 kV. XPS was measured on Thermo escalab 250XI with a monochromatic Al Kα X-ray source. All XPS spectra were calibrated using C 1 s line at 284.8 eV. EPR measurement was conducted in an electron paramagnetic resonance spectrometer (FA−200 (JEOL)). The SEM analysis was conducted using a HITACHI SU-8010 microscope. In situ Raman spectroscopy was conducted on a confocal Raman microscope (LabRAM HR-800, Horiba Jobin Yvon, France) at a 532 nm laser wavelength. Prior to the measurements, the Raman shift was calibrated using the Raman band of Si at 520.7 cm−1. The catalysts supported on nickel foam was immersed in 0.5 M KOH with or without 0.4 M cyclohexanone in the custom-built three-electrode polytetrafluoroethylene (PTFE) cell with Hg/HgO (in 1.0 M KOH) as the reference electrode and Pt wire as the counter electrode. Each spectrum was collected by accumulating 2 scans with acquisition time of 20 seconds. ICP-MS was conducted on Agilent ICPOSES730 with axial detector mode. In online DEMS measurements with QAS100/PrismaPro DEMS system (PFEIFFER VACCUM and LingLu Instruments (Shanghai) Co., Ltd.), the Hg/HgO (1.0 M KOH) electrode and Pt wire were used as reference electrode and counter electrode, respectively. The XAS of Ni K-edge was measured in the fluorescence mode at the BL1W1B beamline of Beijing Synchrotron Radiation Facility (BSRF). The in situ XAS experiments were conducted in 0.5 M KOH solution with or without introducing cyclohexanone on the custom-built three-electrode PTFE cell with catalysts supported on carbon paper as the working electrode, Hg/HgO (in 1.0 M KOH) as the reference electrode and Pt wire as the counter electrode. Each spectrum was collected with an acquisition time of 100 s at certain applied potentials from open circuit potential (OCP).
Electrochemical measurements
The electrochemical measurements of the samples in an H-type reactor were performed with a CHI 760e electrochemical workstation (Shanghai Chenhua Instrument Limited)) except noted. The three-electrode system consists of Hg/HgO (in 1.0 M KOH) as the reference electrode, Pt foil as the counter electrode, and the as-prepared catalysts supported on nickel foam as the working electrode. Each chamber of the H-cell was filled with 40 mL electrolyte. Specifically, commercial nickel foam was pretreated with 1 M HCl, deionized water, and acetone each for 15 min. The ink for working electrode consisted of 2 mL prepared catalyst colloid that was sprayed on the nickel foam with the size of 2 cm × 2 cm using an airbrush and total mass loading of 5 mg cm−2. The mass loading was determined by the mass change of nickel foam before and after the ink sprayed. Then it was cut to 1 cm × 1 cm as the working electrode. All potentials measured against Hg/HgO (EHg/HgO) were converted to the reversible hydrogen electrode (ERHE) scale in this work using ERHE = EHg/HgO + 0.097 V + 0.059 × pH, where pH values of the electrolytes were determined with a pH meter. Proton exchange membrane (Nafion 117) was used as the membrane. All the tests for electrochemical reduction of benzaldehyde were conducted under magnetic stirring of 800 rpm. The LSV and CV curves with a scan rate of 10 mV s−1 ranging from 1.0 to 1.8 VRHE in 0.5 M KOH with or without 0.4 M cyclohexanone. Unless otherwise specified, all curves are reported without iR compensation. The solution resistance was determined by electrochemical impedance spectroscopy technique at open circuit potential in a frequency range from 105 to 1 Hz with a perturbation of 5 mV and the LSV curves for Tafel analysis were corrected with 100% iR compensation at scan rate of 10 mV s−1 (Supplementary Fig 39)56. The Tafel slopes were derived from LSV curves in Supplementary Fig. 20. The electrolysis was monitored at the potentials of 1.5, 1.6, 1.7, 1.8 and 1.9 VRHE without iR compensation by applying constant potential for 1 h. All tests were performed at room temperature.
The electrochemical measurements of the samples in an MEA were performed with a CS1350 electrochemical analyzer (CS Instruments, Inc., Wuhan). The two-electrode system consists of commercial PtRu/C supported on a nickel felt as the counter electrode and the as-prepared catalysts supported on a nickel felt as working electrode. The ink for counter electrode consisted of 10 mg PtRu/C commercial catalyst and 40 μL of 5 wt% Nafion solution in 2 mL ethanol by ultrasonic for 30 min. The ink was sprayed on a commercial nickel felt with the size of 2 cm × 2 cm using an airbrush. Then it was cut to 1 cm × 1 cm as the counter electrode. The ink for working electrode consisted of 2 mL prepared catalyst colloid and 80 μL of 5 wt% Nafion solution by ultrasonic for 30 min. The ink was evenly sprayed on both sides of commercial nickel felt with the size of 2 cm × 2 cm using an airbrush. The mass loading was ca. 5 mg cm−2 determined by the mass change of nickel felt before and after ink sprayed. Then it was cut to 1 cm × 1 cm as the working electrode. Anion exchange membrane (FAA-3-50) was used as the membrane. The LSV and CV curves with a scan rate of 10 mV s−1 ranging from 1.0 to 2.5 V in 1 M KOH with or without 0.4 M cyclohexanone. The electrolysis was monitored by applying constant current density at a flow rate of 50 mL min−1 until 300 C passes at the current densities of 100, 150, 200, 250, 300, 400 and 500 mA cm−2 in 1.0 M KOH and 0.4 M cyclohexanone. The NiV-LDH-NS/NF anode for long-term stability test was produced by using an ultrasonic sprayer (USD-300P-120S, Nasonic (Suzhou) Co., Ltd). The flow rate during long-term stability test was 80 mL min−1. Considering that the electrocatalytic conversion of cyclohexanone to adipate is an OH−-consuming process, we refreshed the electrolyte after a specified period. At the current density of 200 mA cm−2, the electrolyte was refreshed every 24 h. At a current density of 300 mA cm−2, the electrolyte was refreshed every 12 h due to the faster consumption of OH−. The AEM and electrolyte were replaced at 46 h due to a drastic cell voltage increase and electrolyte leakage from anode to cathode caused by AEM corrosion. All tests were performed at room temperature.
The intermittent potential electrolysis of the sample in an H-type cell was performed with a CS1350 electrochemical analyzer (CS Instruments, Inc., Wuhan). The three-electrode system is same as mentioned before. The anode potential was controlled at 1.8 VRHE with a duration of 2 s and open circuits were applied for 5 s, which was repeated for 1800 cycles until the cumulative electrolysis time equaled that of continuous electrolysis, 3600 s. All tests were performed at room temperature.
Product analysis
To analyze liquid products, 400 μL of electrolyte solution was collected during a chronoamperometry test, and was neutralized by 400 μL of 0.5 M H2SO4 solution. The samples were then analyzed by an HPLC (Agilent 1200 Infinity Series) equipped with a variable wavelength detector. The oxidation products, AA, GA, SA, MA, and OA were detected using an organic acid column (Coregel 87H3) and aqueous H2SO4 (5 mM) as the mobile phase (0.6 mL min−1). The standard curves of products for external calibration are shown in Supplementary Fig. 40. O2 was collected by a drainage gas collection method at room temperature. The current density, productivity, and FE were calculated with the following equations:
Where area was the geometric area of electrodes (1.0 cm2) and reaction time was 1 h (3600 s).
Where area was the geometric area of electrodes (1.0 cm2) and reaction time is 1 h.
where n is the number of electron transfers for each product formation, n = 4 for water oxidation to O2, n = 6 for cyclohexanone oxidation to AA, GA, SA, MA, OA or CO32−, F is Faraday constant of 96485 C mol−1.
The relationship between current density and productivity was:
Computational method
All calculations were carried out within the context of periodic density functional theory (DFT), as implemented in CASTEP57, with the Perdew-Burke-Ernzerhof (PBE) / generalized gradient approximation (GGA) functional58. The core electrons and Coulomb potential were described by the ultrasoft pseudopotential59. The k-points and cutoff energy were set as 3 × 3 × 1 and 400 eV. The potential energy surface in the procedure of optimization was searched via BFGS algorithm60, and three convergence criterion were set as follows: (1) energy tolerance of 1.0 × 10−5 eV per atom; (2) force tolerance of 0.03 eV/Å; (3) displacement tolerance of 1.0 × 10−3 Å. The atomic coordinates of the optimized computational models in this work are provided in CIF format (Supplementary data 1).
The adsorption energies (Eads) of cyclohexanone were calculated as Eq. (7):
where Etotal, Efacet, and Ecyclohexanone represent the energy of the total system after adsorption, the isolated (100) facet of \({{{\rm{NiOOH}}}}\) or \({{{\rm{NiVOOH}}}}\), and the isolated cyclohexanone, respectively.
The Gibbs free energy changes (ΔG) of the reconstruction processes were calculated by Eq. (8) and (9):
where \({G}_{{{{\rm{NiOOH}}}}},\,{G}_{{{{\rm{NiVOOH}}}}}\), \({G}_{{{{\rm{Ni}}}}{({{{\rm{OH}}}})}_{2}}\), and \({G}_{{{{\rm{NiV}}}}{({{{\rm{OH}}}})}_{2}}\) represent the Gibbs free energies (G) of NiOOH, NiVOOH, Ni(OH)2, and NiV(OH)2, respectively.
The G were obtained by analyzing the phonon density of states (Eq. 10):
where E is the total energy, ZPE is the zero-point energy, and \(kT\int F(\omega )\, {{\mathrm{ln}}}\, [1-\exp (-\frac{\hslash \omega }{kT})d\omega ]\) is the correction of Gibbs free energy.
The enthalpy changes (ΔH) for Ni demetallation processes were calculated by Eq. (11) and (12):
where HNiOOH_vacancy, HNiVOOH_vacancy, HNiOOH, HNiVOOH, and HNi represent the enthalpy (H) of NiOOH with Ni vacancy, NiVOOH with Ni vacancy, NiOOH (100), NiVOOH (100), and Ni, respectively. Besides, the bulk Ni with cubic-close-packing was used to compute the H of Ni (Supplementary). Phonon calculations were used to evaluate the temperature dependence of the H.
Data availability
All data generated or analyzed during this study are included in this Article (and its Supplementary Information). Source data are provided with this paper. The data that support the findings of this study are available in the paper and its Supplementary Information or from the corresponding author upon request. Source data are provided with this paper.
References
Sato, K., Aoki, M. & Noyori, R. A “Green” route to adipic acid: direct oxidation of cyclohexenes with 30 percent hydrogen peroxide. Science 281, 1646–1647 (1998).
Van de Vyver, S. & Román-Leshkov, Y. Emerging catalytic processes for the production of adipic acid. Catal. Sci. Technol. 3, 1465–1479 (2013).
Yan, W. et al. Recent progress in adipic acid synthesis over heterogeneous catalysts. Front. Chem. 8, 185 (2020).
Schaub, T. Producing adipic acid without the nitrous oxide. Science 366, 1447–1447 (2019).
Bhanja, P. et al. New hybrid iron phosphonate material as an efficient catalyst for the synthesis of adipic acid in air and water. ACS Sustain. Chem. Eng. 4, 7147–7157 (2016).
Bhanja, P., Chatterjee, S., Patra, A. K. & Bhaumik, A. A new microporous oxyfluorinated titanium(IV) phosphate as an efficient heterogeneous catalyst for the selective oxidation of cyclohexanone. J Colloid Interface Sci. 511, 92–100 (2018).
Wu, J. et al. Ligand hybridization for electro-reforming waste glycerol into isolable oxalate and hydrogen. Angew. Chem. Int. Ed. 62, e202216083 (2023).
Tang, C., Zheng, Y., Jaroniec, M. & Qiao, S.-Z. Electrocatalytic refinery for sustainable production of fuels and chemicals. Angew. Chem. Int. Ed. 60, 19572–19590 (2021).
Lu, Y. et al. Aqueous electrocatalytic small-molecule valorization trilogy. Chem 10, 1371–1390 (2024).
Jin, B., Gao J., Zhang Y. & Shao M. Deprotonated of layered double hydroxides during electrocatalytic water oxidation for multi-cations intercalation. Smart Mol., e20230026 (2024).
Lyalin, B. V. & Petrosyan, V. A. Electrosynthesis of adipic acid by undivided cell electrolysis. Russ. Chem. Bull. 53, 688–692 (2004).
Lyalin, B. & Petrosyan, V. Electrosynthesis of glutaric acid and regularities of electrocatalytic oxidation of cycloalkanones at a NiOOH anode in aqueous NaOH. Russ. Chem. Bull. 58, 2426–2431 (2009).
Wang, R. et al. Electrifying adipic acid production: copper-promoted oxidation and C-C cleavage of cyclohexanol. Angew. Chem. Int. Ed. 61, e202214977 (2022).
Li, Z. et al. Electrocatalytic synthesis of adipic acid coupled with H2 production enhanced by a ligand modification strategy. Nat. Commun. 13, 5009 (2022).
Luo, Y., Zhang, Z., Chhowalla, M. & Liu, B. Recent advances in design of electrocatalysts for high‐current‐density water splitting. Adv. Mater. 34, 2108133 (2022).
Liu, L. et al. Self‐supported bimetallic array superstructures for high‐performance coupling electrosynthesis of formate and adipate. Exploration, 20230043 (2023).
Liu, F. et al. Graphdiyne as an electron modifier for boosting electrochemical production of adipic acid. Adv. Funct. Mater. 34, 2310274 (2023).
Zhou, H. et al. Selectively upgrading lignin derivatives to carboxylates through electrochemical oxidative C(OH)-C bond cleavage by a mn-doped cobalt oxyhydroxide catalyst. Angew. Chem. Int. Ed. 60, 8976–8982 (2021).
Liu, F., Gao, X., Guo, Z., Tse, E. C. M. & Chen, Y. Sustainable adipic acid production via paired electrolysis of lignin-derived phenolic compounds with water as hydrogen and oxygen sources. J. Am. Chem. Soc. 146, 15275–15285 (2024).
Chen, W. et al. Activity origins and design principles of nickel-based catalysts for nucleophile electrooxidation. Chem 6, 2974–2993 (2020).
Zhou, B. et al. Platinum modulates redox properties and 5-hydroxymethylfurfural adsorption kinetics of Ni(OH)(2) for biomass upgrading. Angew. Chem. Int. Ed. 60, 22908–22914 (2021).
Wang, M. H. et al. Operando high-valence Cr-modified NiFe hydroxides for water oxidation. Small 18, e2200303 (2022).
Du, Z. et al. Rapid surface reconstruction of pentlandite by high‐spin state iron for efficient oxygen evolution reaction. Angew. Chem. Int. Ed. 63, e202317022 (2024).
Zeng, Y. et al. Surface reconstruction of water splitting electrocatalysts. Adv. Energy Mater. 12, (2022).
Sun, H. et al. Highly efficient overall urea electrolysis via single-atomically active centers on layered double hydroxide. Sci Bull (Beijing) 67, 1763–1775 (2022).
Jia, Y. et al. Directional electrosynthesis of adipic acid and cyclohexanone by controlling the active sites on NiOOH. J. Am. Chem. Soc. 146, 1282–1293 (2023).
Wang, S. et al. New BiVO4 dual photoanodes with enriched oxygen vacancies for efficient solar-driven water splitting. Adv. Mater. 30, 1800486 (2018).
Asnavandi, M., Yin, Y., Li, Y., Sun, C. & Zhao, C. Promoting oxygen evolution reactions through introduction of oxygen vacancies to benchmark NiFe–OOH catalysts. ACS Energy Lett 3, 1515–1520 (2018).
Idriss, H. On the wrong assignment of the XPS O1s signal at 531–532 eV attributed to oxygen vacancies in photo- and electro-catalysts for water splitting and other materials applications. Surf. Sci. 712, (2021).
Karmakar, A. et al. Stabilization of ruthenium nanoparticles over NiV-LDH surface for enhanced electrochemical water splitting: an oxygen vacancy approach. J. Mater. Chem. A 10, 3618–3632 (2022).
Sun, H. et al. Atomic metal-support interaction enables reconstruction-free dual-site electrocatalyst. J. Am. Chem. Soc. 144, 1174–1186 (2022).
Vernekar, D. et al. Direct oxidation of cyclohexane to adipic acid by a WFeCoO(OH) catalyst: role of brønsted acidity and oxygen vacancies. ACS Catal 11, 10754–10766 (2021).
Qin, H. et al. Synergistic engineering of doping and vacancy in Ni(OH) 2 to boost urea electrooxidation. Adv. Funct. Mater. 33, 2209698 (2022).
Klyukin, K., Zagalskaya, A. & Alexandrov, V. Role of dissolution intermediates in promoting oxygen evolution reaction at RuO2(110) surface. J. Phys. Chem. C 123, 22151–22157 (2019).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2022).
Luo, H. et al. Role of Ni in PtNi bimetallic electrocatalysts for hydrogen and value-added chemicals coproduction via glycerol electrooxidation. ACS Catal 12, 14492–14506 (2022).
Voiry, D. et al. Best practices for reporting electrocatalytic performance of nanomaterials. ACS Nano 12, 9635–9638 (2018).
Bender, M. T., Lam, Y. C., Hammes-Schiffer, S. & Choi, K. S. Unraveling two pathways for electrochemical alcohol and aldehyde oxidation on NiOOH. J. Am. Chem. Soc. 142, 21538–21547 (2020).
Bender, M. T., Warburton, R. E., Hammes-Schiffer, S. & Choi, K.-S. Understanding hydrogen atom and hydride transfer processes during electrochemical alcohol and aldehyde oxidation. ACS Catal 11, 15110–15124 (2021).
Bender, M. T. & Choi, K. S. Electrochemical oxidation of HMF via hydrogen atom transfer and hydride transfer on NiOOH and the impact of NiOOH composition. ChemSusChem 15, e202200675 (2022).
Yan, Y., Zhong, J., Wang, R., Yan, S. & Zou, Z. Trivalent nickel-catalyzing electroconversion of alcohols to carboxylic acids. J. Am. Chem. Soc. 164, 4814–4821 (2024).
Yan, Y. et al. Nonredox trivalent nickel catalyzing nucleophilic electrooxidation of organics. Nat. Commun. 14, (2023).
Zhao, S. et al. Structural transformation of highly active metal–organic framework electrocatalysts during the oxygen evolution reaction. Nat. Energy 5, 881–890 (2020).
Goetz, M. K., Bender, M. T. & Choi, K.-S. Predictive control of selective secondary alcohol oxidation of glycerol on NiOOH. Nat. Commun. 13, 5848 (2022).
Zhu, Y. Q. et al. Identification of active sites formed on cobalt oxyhydroxide in glucose electrooxidation. Angew. Chem. Int. Ed. 62, e202219048 (2023).
Wang, Y. et al. Efficient electrocatalytic oxidation of glycerol via promoted OH* generation over single-atom-bismuth-doped spinel Co3O4. ACS Catal 12, 12432–12443 (2022).
Lu, Y. et al. Integrated catalytic sites for highly efficient electrochemical oxidation of the aldehyde and hydroxyl groups in 5-hydroxymethylfurfural. ACS Catal 12, 4242–4251 (2022).
Gao, X. et al. Boosting urea electrooxidation on oxyanion-engineered nickel sites via inhibited water oxidation. Nat. Commun. 14, 5842 (2023).
Yang, Y. et al. Defect-promoted ni-based layer double hydroxides with enhanced deprotonation capability for efficient biomass electrooxidation. Adv. Mater., e2305573 (2023).
Chen, D. et al. Highly efficient biomass upgrading by a ni-cu electrocatalyst featuring passivation of water oxidation activity. Angew. Chem. Int. Ed., e202309478 (2023).
Li, Z. et al. Alcohols electrooxidation coupled with H(2) production at high current densities promoted by a cooperative catalyst. Nat. Commun. 13, 147 (2022).
Huang, Y. et al. Pulsed electroreduction of low-concentration nitrate to ammonia. Nat. Commun. 14, 7368 (2023).
Chen, Y. et al. Facile synthesis of Mo2C nanocrystals embedded in nanoporous carbon network for efficient hydrogen evolution. Chin. J. Chem. 35, 911–917 (2017).
Wang, Y., Yang, Z., Wu, L., Ge, L. & Xu, T. Ion exchange membrane “ABC” – a key material for upgrading process industries. Chin. J. Chem. 39, 825–837 (2021).
Nabil, S. K. et al. Acid–base chemistry and the economic implication of electrocatalytic carboxylate production in alkaline electrolytes. Nat. Catal. 7, 330–337 (2024).
van der Heijden, O., Park, S., Eggebeen, J. J. J. & Koper, M. T. M. Non‐kinetic effects convolute activity and tafel analysis for the alkaline oxygen evolution reaction on NiFeOOH electrocatalysts. Angew. Chem. Int. Ed. 62, e202216477 (2023).
Clark, S. J. et al. First principles methods using CASTEP. Z. Kristallogr. Cryst. Mater. 220, 567–570 (2005).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892–7895 (1990).
Anglada, J. M. & Bofill, J. M. How good is a Broyden–Fletcher–Goldfarb–Shanno-like update Hessian formula to locate transition structures? specific reformulation of Broyden–Fletcher–Goldfarb–Shanno for optimizing saddle points. J. Comput. Chem. 19, 349–362 (1998).
Acknowledgements
This work was supported by the National Key R&D Program of China (2023YFA1507400), the National Natural Science Foundation of China (Grant No. 22325805, 21935001), the Beijing Natural Science Foundation (JQ22003), the Tsinghua University Initiative Scientific Research Program, the Haihe Laboratory of Sustainable Chemical Transformations. The authors thank the BL1W1B in the Beijing Synchrotron Radiation Facility (BSRF).
Author information
Authors and Affiliations
Contributions
X.L., Y.Z. and H.D. conceived the project and co-wrote the manuscript. X.L. carried out most of the experiments. Y.Z. carried out the theoretical calculation. J.L. provided much support for schooling the experiemnts and manuscript. Y.W. contributed to the in situ Raman spectra. Q.S. provided useful suggestions for figure editing. A.L. helped with EPR experiments. K.J. provided useful suggestions on experiment designs. X.W. contributed to the EDS mapping. J.Z. and X.Z helped with the analysis of XPS data and the construction of MEA.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sang Hyun Ahn, Jooyoung Lee and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, X., Zhu, YQ., Li, J. et al. Electrosynthesis of adipic acid with high faradaic efficiency within a wide potential window. Nat Commun 15, 7685 (2024). https://doi.org/10.1038/s41467-024-51951-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51951-0
This article is cited by
-
Imidazolium radical-mediated electron transfer enhances electrochemical C–N coupling for glycine synthesis
Nature Synthesis (2025)
-
Selective photoelectrochemical synthesis of adipic acid using single-atom Ir decorated α-Fe2O3 photoanode
Nature Communications (2025)
-
Synergistic enhancement of electrochemical alcohol oxidation by combining NiV-layered double hydroxide with an aminoxyl radical
Nature Communications (2025)
