
Abstract
How intracellular bacteria subvert the major histocompatibility complex (MHC) class I pathway is poorly understood. Here, we show that the obligate intracellular bacterium Orientia tsutsugamushi uses its effector protein, Ank5, to inhibit nuclear translocation of the MHC class I gene transactivator, NLRC5, and orchestrate its proteasomal degradation. Ank5 uses a tyrosine in its fourth ankyrin repeat to bind the NLRC5 N-terminus while its F-box directs host SCF complex ubiquitination of NLRC5 in the leucine-rich repeat region that dictates susceptibility to Orientia- and Ank5-mediated degradation. The ability of O. tsutsugamushi strains to degrade NLRC5 correlates with ank5 genomic carriage. Ectopically expressed Ank5 that can bind but not degrade NLRC5 protects the transactivator during Orientia infection. Thus, Ank5 is an immunoevasin that uses its bipartite architecture to rid host cells of NLRC5 and reduce surface MHC class I molecules. This study offers insight into how intracellular pathogens can impair MHC class I expression.
Similar content being viewed by others


Introduction
Major histocompatibility complex (MHC) class I antigen presentation is essential for adaptive immunity against intracellular microbes1. Yet many intracellular pathogens have evolved strategies to undermine this pathway. Such approaches have been well studied for viruses, but considerably less is known about how intracellular bacteria subvert MHC class I-mediated immunity2,3,4,5,6,7. Orientia tsutsugamushi is an obligate intracytosolic bacterium and symbiont of trombiculid mites8. It causes scrub typhus, a disease known for millennia to be endemic to the Asia-Pacific where an estimated one million cases occur annually8,9,10. Locally acquired cases in Africa, the Middle East, and South America and the detection of Orientia species in mites in the United States signify scrub typhus as a globally emerging health threat11,12,13,14,15,16,17,18. O. tsutsugamushi infects leukocytes at the mite feeding site and disseminates to invade endothelial cells of multiple organs19,20. Acute scrub typhus symptoms are non-specific and include fever, rash, headache, and lymphadenopathy21,22. If untreated, O. tsutsugamushi infection of endothelial cells can progress to systemic vascular collapse, organ failure, and death23,24. O. tsutsugamushi strains are extensively diverse in terms of virulence in humans and animal models25,26,27,28,29,30,31,32. The molecular bases for variations in the virulence capacities of different O. tsutsugamushi strains are poorly defined.
MHC class I molecules and CD8+ T cells are important for restraining O. tsutsugamushi growth and protecting against lethal infection in mice26,33,34. Moreover, CD8+ T cell numbers are elevated during the convalescent but not acute phase in scrub typhus patients35. Hence, O. tsutsugamushi must counter the MHC class I pathway to maximize its survival in animal and human hosts. NLRC5 (NOD-, LRR- and CARD-containing 5/class I transactivator) is a master regulator of MHC class I gene expression36,37. It is upregulated by interferon-γ (IFNγ) and is the sole transactivator of MHC class I gene expression in endothelial cells and other non-professional antigen-presenting cells (APCs)37,38,39,40,41. The O. tsutsugamushi Ikeda strain lowers NLRC5 levels in non-professional APCs, even in the presence of IFNγ, to inhibit MHC class I gene expression and reduce the amounts of MHC class I molecules on the cell surface42. Ikeda, isolated from a patient in Japan, causes severe disease in humans and is fatal in laboratory mice43,44,45,46,47,48. It is the only microbe known to modulate MHC class I cellular levels by targeting NLRC5, although the mechanism is uncharacterized.
Ikeda and all other sequenced O. tsutsugamushi strains encode among the largest microbial arsenals of ankyrin repeat (AR)-containing effector proteins (Anks)43,49,50,51,52,53. ARs are protein-protein interaction motifs that are conserved across the Tree of Life52. Their acquisition by intracellular microbes is linked to increased virulence54,55. Most O. tsutsugamushi Anks carry a C-terminal eukaryotic-like F-box domain, which interacts with host Skp1 (S-phase kinase-associated protein 1) and Cul1 (cullin 1) to nucleate the SCF (Skp1-Cul1-F-box) E3 ubiquitin ligase complex that mediates polyubiquitination of substrates to destine them for 26S proteasomal degradation56,57. Twelve Ikeda Anks (Ank1, Ank2, Ank4, Ank5, Ank6, Ank8, Ank9, Ank10, Ank12, Ank13, Ank17, Ank20) have been verified to nucleate the endogenous SCF complex56. The Orientia AR-F-box bipartite architecture is exhibited by numerous other intracellular bacterial effectors and viral proteins54,58,59,60,61,62,63. Strikingly, however, nearly all of the host cell target proteins that get ubiquitinated and degraded due to interacting with microbial F-box-containing Anks remain unidentified. Thus, understanding of this evolutionarily conserved intracellular microbial pathogenesis strategy is limited.
Here, we report that O. tsutsugamushi Ikeda Ank5 employs a two-pronged mechanism to target NLRC5 and reduce MHC class I surface expression. Ank5 uses its fourth AR to bind NLRC5 at the N-terminus and sequester it in the cytoplasm, preventing MHC class I gene transactivation. The effector also utilizes its F-box to direct SCF-mediated ubiquitination of the NLRC5 leucine-rich repeat (LRR) domain and promote NLRC5 proteasomal degradation. Ectopic overexpression of a recombinant version of Ank5 capable of binding but not degrading NLRC5 protects the transactivator from degradation during Orientia infection, further establishing the respective contributions of the AR and F-box domains to Ank5 function. O. tsutsugamushi UT76, another strain that carries ank5, is capable of lowering NLRC5, whereas the Karp strain, which lacks ank5, cannot. Our findings elucidate a detailed mechanism of action for an immunoevasin that subverts the MHC class I pathway by both blocking the nuclear translocation of NLRC5 and orchestrating its degradation.
Results
O. tsutsugamushi promotes NLRC5 degradation by the 26S proteasome
O. tsutsugamushi Ikeda (hereafter referred to as O. tsutsugamushi unless discussed in the context of other strains) decreases NLRC5 protein levels in non-professional APCs even though it induces nlrc5 transcription42, which suggests that the pathogen targets the transactivator at a posttranscriptional step. As further support for this hypothesis, O. tsutsugamushi reduces levels of IFNγ-induced endogenous NLRC5 as well as ectopically expressed Flag- and myc-tagged NLRC5 (Fig. 1a–d). HeLa cells were used for this and other experiments herein because they are non-professional APCs that constitutively express MHC class I genes in an exclusively NLRC5-dependent manner37,64. HeLa cells are also established models for studying O. tsutsugamushi-host interactions, and their amenability to transfection allows for assessing if ectopically expressed oriential virulence factors phenocopy aspects of infection42,65,66,67,68,69,70,71,72,73,74.

a–d HeLa cells were mock transfected and treated with IFNγ or transfected to express Flag-NLRC5 or myc-NLRC5 and then mock infected [U] or infected with O. tsutsugamushi [I]. a Whole cell lysates were analyzed by immunoblot. b–d NLRC5 (b), Flag (c), or myc (d) densitometric signal was divided by corresponding GAPDH signal and normalized to uninfected cells (black bars). (n = 3 (b) or 5 (c, d) independent experiments). e–h HeLa cells were mock infected [U] or infected with O. tsutsugamushi [I] and treated with cycloheximide (CHX) (e, f) or MG132 (g and h) for 16 h prior to collection. (e and g) Whole cell lysates were analyzed by immunoblot. f, h NLRC5 densitometric signal was divided by corresponding GAPDH densitometric signal, normalized to uninfected cells, and multiplied by 100 (black bars) (n = 3 independent experiments). i HeLa cells were transfected to co-express Flag-NLRC5 and HA-Ub, mock infected [U] or infected with O. tsutsugamushi [I] and treated with MG132 for 24 h prior to collection. Input lysates and Flag IP complexes were analyzed by immunoblot (n = 3 independent experiments). j–m HeLa cells were transfected to express Flag-NLRC5, mock infected [U] or infected with O. tsutsugamushi [I] and treated with MG132 for 24 h prior to collection. Flag-NLRC5 was immunoprecipitated and the eluted complex was analyzed by mass spectrometry. j, l Fragmentation spectra of the post-translationally modified peptides K200 [184ATASLDTPEGAIMGDVKVEDGADVSISDLFNTR216] (j) and K1194 [1179SPFLLANTLSLCPRVK1194] (l). Spectra-matched b- (blue) and y-ion (red) fragments are displayed and summarized by the trypsinized peptide sequence inset above. k, m Peptide intensity presented as fold change (n = 3 independent experiments). Vertical lines between bands indicate where the blot was cropped or imaged separately. Data are means ± SD. Unpaired two-tailed t-test was used for data analysis (b–d, f, h). Source data are provided as Source Data file.
To determine if O. tsutsugamushi prevents translation of nascent NLRC5 protein or targets NLRC5 for proteasomal degradation, HeLa cells were infected or not followed by the addition of cycloheximide, a eukaryotic protein synthesis inhibitor, or MG132, a 26S proteasome inhibitor. At 24 h post-infection (p.i.), NLRC5 levels were decreased by approximately 50% in vehicle-treated cells (Fig. 1e–h). O. tsutsugamushi-mediated NLRC5 reduction persisted in cycloheximide-treated cells to the point that NLRC5 was undetectable (Fig. 1e, f). In contrast, NLRC5 levels were equivalent in MG132-treated infected and uninfected cells. Thus, O. tsutsugamushi promotes NLRC5 degradation by the 26S proteasome (Fig. 1g, h).
NLRC5 is ubiquitinated in O. tsutsugamushi infected cells
Proteins destined for proteasomal degradation are marked by the covalent addition of polyubiquitin. To assess whether O. tsutsugamushi promotes NLRC5 ubiquitination, HeLa cells transfected to co-express hemagglutinin (HA)-ubiquitin (Ub) and Flag-NLRC5 were infected for 24 h, after which MG132 was added for an additional 24 h to enrich for ubiquitinated proteins. Lysates were collected followed by immunoprecipitation of Flag-NLRC5. Successful MG132 treatment was confirmed by the upregulation of Nrf175 (Fig. 1i). NLRC5 levels were markedly increased in MG132-treated infected cells, likely due to the combinatorial effect of upregulated nlrc5 expression in response to O. tsutsugamushi infection42, Flag-NLRC5 overexpression, and 26S proteasome inhibition. Indeed, levels of HA-ubiquitinated proteins overall and HA-ubiquitinated NLRC5 were more abundant in MG132-treated infected cells versus MG132-treated uninfected cells (Fig. 1i).
To identify the NLRC5 residue(s) that becomes ubiquitinated during O. tsutsugamushi infection, HeLa cells expressing Flag-NLRC5 were infected and treated with MG132. Input lysates (Supplementary Fig. 1) were subjected to immunoprecipitation to recover Flag-NLRC5 and interacting proteins. Eluted complexes were processed for liquid chromatography with tandem mass spectrometry (LC-MS/MS) and peptide fragments were examined for diglycine motifs, the post-tryptic mark of ubiquitination. Sequences were mass matched ± four parts per million and aligned for analysis. The proteome precipitated by Flag-NLRC5 from O. tsutsugamushi infected cells was three times more complex than that from uninfected cells (Supplementary Fig. 2a). STRING (Search Tool for the Retrieval of Interacting Genes/Proteins)76 analysis of the infection specific NLRC5 interacting proteome revealed an enrichment for 26S proteasome subunits, proteins involved in unfolded protein binding, and other gene ontology categories (Supplementary Table 1). Filtering the infection-specific interactome to display only known interactions revealed that none of these proteins have been reported to form interacting networks with NLRC5 (Supplementary Fig. 2b). Of note, recovery of 26S proteasome subunits and proteins involved in unfolded protein binding is consistent with NLRC5 becoming marked for proteasomal degradation. Analysis of NLRC5 tandem mass spectra revealed the peptides 184ATASLDTPEGAIMGDVKVEDGADVSISDLFNTR216 and 1179SPFLLANTLSLCPRVK1194 to have b- and y-ions specifically positioning a diglycine motif on residues K200 and K1194, which were recovered at 17-fold and 10-fold greater abundances, respectively, from infected versus uninfected samples (Fig. 1j–m). Peptide 889LMAEAASQLHIARK902 with evidence of ubiquitination at K902 was recovered at comparable levels from infected and uninfected cells (Supplementary Fig. 3). Altogether, these data demonstrate that O. tsutsugamushi promotes ubiquitination of NLRC5 on K200 and K1194 as well as NLRC5 degradation by the 26S proteasome.
Orientia upregulates ank5 prior to extensive intracellular growth
NLRC5 degradation in O. tsutsugamushi infected cells is bacterial protein synthesis-dependent42, suggesting that an oriential protein mediates this phenomenon. O. tsutsugamushi Ikeda Ank5 is a 345-amino acid effector protein of unknown function that has an N-terminal domain consisting of four tandemly arranged ARs and a C-terminal F-box that occurs within a PRANC (pox proteins repeats of ankyrin-C terminal) domain (Supplementary Fig. 4a), the latter of which is a feature shared only with other O. tsutsugamushi Anks and Anks of vertebrate poxviruses, Wolbachia species, and wasps that Wolbachia species infect43,51,54,56,70,77,78. The Ikeda chromosome encodes three identical Ank5 copies (OTT_RS01000, OTT_RS01950, and OTT_RS02325)43,51,53. We assessed ank5 transcription over the course of O. tsutsugamushi growth in HeLa cells and EA.hy926 human vascular endothelial cells. Bacterial replication in HeLa cells lagged for the first 24 h after which it pronouncedly increased, whereas in EA.hy926 cells replication initiated during the first 12 h and became logarithmic between 12 and 24 h (Supplementary Fig. 4b). Although ank5 transcripts were detectable throughout infection of both cell types, ank5 upregulation occurred at 8 and 12 h p.i. in HeLa cells and at 2, 4, and 8 h p.i. in EA.hy926 cells (Supplementary Fig. 4c, d), indicating that O. tsutsugamushi strongly expresses ank5 prior to its exponential growth phase.
A yeast two-hybrid screen was performed using Ank5 lacking residues 301-344 (Ank5ΔF-box) as bait so that known F-box dependent interacting partners Skp1 and Cul156 would not be overrepresented among the prey proteins. One of the putative Ank5ΔF-box interactors was NLRC5 (Supplementary Table 2), a conspicuous finding given the effect of Ikeda on NLRC5 and because NLRC5 was not identified as a potential interactor of other Ikeda Anks by prior yeast two-hybrid screenings65,67. Hence, a domain within Ank5 residues 1-300 potentially mediates binding to NLRC5.
Orientia reducing NLRC5 levels correlates with genomic ank5 carriage
Because O. tsutsugamushi is genetically intractable79 we compared strains that do or do not carry ank5 for the ability to lower host cell NLRC5 and MHC class I levels. Of the more than 30 antigenically distinct O. tsutsugamushi strains, nine have been fully sequenced and the ank genomic content has been determined for eight43,49,50,53,80. Two of these strains, Ikeda and UT76 (NZ_LS398552.1), the latter of which originated from a patient in Thailand81, carry ank553. A search performed using the National Center for Biotechnology Information (NCBI) Nucleotide Basic Local Alignment Search Tool (BLASTN) (https://blast.ncbi.nlm.nih.gov/Blast.cgi) with ank5 (OTT_RS01000) unique nucleotides 400-546 as query identified an additional homolog in Wuj/2014 (NZ_CP044031.1), an isolate that had been recovered from a patient in China. The UT76 and Wuj/2014 ank5 homologs are single-copy and exhibit 96–97% nucleotide and 90–92% amino acid identity with Ikeda Ank5 (Supplementary Table 3).
Classical MHC class I molecules consist of a heavy chain (HLA-A, -B, or -C) and β2-microglobulin (β2 M)82. UT76 infected HeLa cells exhibited marked decreases in total NLRC5, HLA-ABC, and β2 M along with cell surface MHC class I (Fig. 2a–h), mirroring results reported for Ikeda infected cells42. O. tsutsugamushi Karp, which does not encode Ank553, induced an approximate three-fold increase in NLRC5 levels (Fig. 2i, j). This agrees with the transactivator being upregulated in response to intracellular pathogen infections39,41,83,84,85. Moreover, Karp did not alter HLA-ABC and β2 M amounts (Fig. 2k, l). Yet, Karp-infected cells still exhibited significantly reduced MHC class I surface levels (Fig. 2m–p). Thus, O. tsutsugamushi Ikeda and UT76, which carry ank5, reduce NLRC5 levels during infection while Karp, which lacks ank5, does not. Moreover, Karp inhibition of MHC class I surface expression is Ank5-independent.

HeLa cells were mock infected [U] or infected for 72–96 h with O. tsutsugamushi strain UT76 [I] (a–h) or Karp [I] (i–p) and treated with IFNγ. a, i Whole cell lysates were analyzed by immunoblot. b–d, j–i The densitometric signal of NLRC5 (b, j), HLA-ABC (c, k), or β2 M (d, l) was divided by corresponding GAPDH densitometric signal and normalized to uninfected cells (black bars) (n = 3 (j), 4 (b, k, l), or 5 (c, d) independent experiments). e, m Infected HeLa cells were fixed, immunolabelled for TSA56 (red), stained with DAPI, and visualized using LSCM and differential interference contrast microscopy (DIC). f, n The percentage of infected cells was quantified. g, h, o, p Surface levels of MHC class I from uninfected and infected cells were analyzed by flow cytometry. g, o Representative histograms of MHC class I levels normalized to mode. Unlabeled (black) samples served as the control. h, p The mean fluorescence intensity of each sample was divided by that of its unlabeled control per replicate (n = 3–4 independent experiments). Data are means ± SD. Unpaired two-tailed t-test was used for data analysis (b–d, h, j–l, p). Source data are provided as Source Data file.
Ank5 binds and decreases levels of NLRC5
We sought to determine if the predicted Ank5-NLRC5 interaction is valid. Given that Ank5-specific antibodies are unavailable, HeLa cells were transfected to express Flag-tagged Ikeda Ank5 and treated with IFNγ. As a specificity control, cells were also transfected to express Flag-tagged Ank9, an O. tsutsugamushi effector that destabilizes the Golgi and is not predicted to bind NLRC565. Western blot analysis of input lysates and Flag antibody-immunoprecipitated complexes confirmed that NLRC5 was upregulated in IFNγ treated cells and precipitated by Flag-Ank5 but not Flag-Ank9 (Fig. 3a). Conspicuously, NLRC5 levels were 64–70% lower in IFNγ-treated cells expressing Flag-Ank5 compared to mock-transfected or Flag-Ank9 expressing cells (Fig. 3a, b). Flag-Ank5 also significantly reduced NLRC5 levels in transfected EA.hy926 cells to phenocopy the effect observed in O. tsutsugamushi infected EA.hy926 cells (Supplementary Fig. 5a–d). NLRC5 was nearly depleted in infected EA.hy926 cells at 24 h p.i. (Supplementary Fig. 5c, d), which is 24–48 h earlier than what has been observed during infection of HeLa cells42 and correlates with the differential O. tsutsugamushi growth and ank5 expression kinetics in these cell types (Supplementary Fig. 4b–d).

a, b HeLa cells were mock transfected or transfected to express Flag-tagged Ank5 or Ank9 and treated with vehicle [-] or IFNγ [+]. a Inputs and Flag IP complexes were analyzed by immunoblot. b Densitometric quantification of NLRC5:GAPDH signal from input blots (n = 4 independent experiments). Indicators of significance between IFNγ-treated are blue. c–f HeLa cells were transfected to express indicated proteins, treated with IFNγ, and sorted on GFP positivity. c Whole cell lysates were analyzed by immunoblot. d–f Densitometric signal of NLRC5 (d), HLA-ABC (e), or β2 M (f) was divided by corresponding GAPDH signal and normalized to GFP-expressing IFNγ treated cells (black bars) (n = 3 independent experiments). g, h HeLa cells were mock transfected or transfected to co-express Flag-Ank5 or Flag-Ank9 with myc-NLRC5. g Input lysates and Flag IP complexes were analyzed by immunoblot. h Densitometric quantification of the NLRC5:GAPDH ratio from input blots normalized to mock samples (black bar) (n = 3 independent experiments). i–n HeLa (i, j, m, n) or EA.hy926 (k, l) cells were mock transfected or transfected to express indicated proteins and treated with IFNγ. (i, k) Whole cell lysates were analyzed by immunoblot. j, l Densitometric quantification of the NLRC5:GAPDH signal from input blots normalized to mock transfected cells treated with IFNγ (black bar, HeLa; red bar, EA.hy926) (n = 3 independent experiments). m, n MHC class I surface levels on GFP-positive cells were analyzed by flow cytometry. m Representative histograms of MHC class I levels normalized to mode. Unlabeled (black) samples were the control. [–] denotes vehicle control and [+] denotes IFNγ addition. n The mean fluorescence intensity of each sample was divided by that of its unlabeled control per replicate (n = 7 independent experiments). Data are means ± SD. Unpaired two-tailed t-test (b, d–f, h, j, l) and One-way ANOVA with Tukey’s post hoc test (n) were used for data analysis. Source data are provided as Source Data file.
To assess if Ank5-mediated reduction of NLRC5 inhibits expression of MHC class I components, HeLa cells were transfected to express GFP-Ank5, GFP-Ank9, or GFP and treated with IFNγ. Using GFP-Ank5 as a representative example, AlphaFold predicted that Ank tertiary structure is not altered by the N-terminal addition of GFP (Supplementary Fig. 5e). Fluorescence-activated cell sorting (FACS) was used to enrich for GFP-positive cells. GFP-Ank5 but not GFP or GFP-Ank9 decreased levels of NLRC5, HLA-ABC, and β2 M (Fig. 3c–f). The slight elevation of HLA-ABC levels in GFP-Ank9-expressing cells could be due to the inhibitory effect by Ank9 on the secretory pathway65. When myc-NLRC5 was co-expressed with Flag-tagged Ank5, Ank9, Ank4 (inhibits endoplasmic reticulum-associated degradation67), Ank6 (blocks NF-κB accumulation in the nucleus56,66), or Ank13 (nucleomodulin that dysregulates expression of more than 2000 host genes when ectopically expressed68), its levels were significantly lower exclusively in cells expressing Flag-Ank5 (Supplementary Fig. 5f, g). Flag-Ank5 also specifically co-precipitated and reduced levels of myc-NLRC5 (Fig. 3g, h). However, GFP-tagged Ank4, Ank5, Ank6, Ank9, and Ank13 each lowered MHC class I surface levels to varying degrees with GFP-Ank5, GFP-Ank4, and GFP-Ank6 yielding the most pronounced reductions (Supplementary Fig. 5h–k). When Flag- or GFP-tagged UT76 Ank5 was ectopically expressed in IFNγ stimulated HeLa or EA.hy926 cells, it functioned similarly to Flag- or GFP-tagged Ikeda Ank5 by reducing NLRC5 and cell surface MHC class I levels (Fig. 3i–n). Thus, ectopically expressed Ikeda and UT76 Ank5 proteins can bind and reduce cellular levels of NLRC5, which, in addition to the different actions of other ectopically expressed O. tsutsugamushi Ank effectors, lowers amounts of MHC class I proteins to recapitulate phenomena associated with O. tsutsugamushi infection.
Ank5 directs NLRC5 ubiquitination and 26S proteasomal degradation
NLRC5 is degraded by the 26S proteasome in O. tsutsugamushi infected cells. To test if Ank5-mediated reduction of NLRC5 is also proteasome-dependent, HeLa cells expressing myc-NLRC5 with or without Flag-Ank5 were treated with MG132. In mock transfected cells, proteasome inhibition resulted in higher levels of myc-NLRC5 (Fig. 4a). Because cells were transfected to express Flag-Ank5 prior to the addition of MG132 or vehicle, NLRC5 levels were considerably lower regardless of treatment. Nevertheless, Flag-Ank5 coprecipitated three-fold more NLRC5 in the presence of MG132 (Fig. 4b), indicating an interaction exists between Ank5 and NLRC5 that promotes 26S proteasomal degradation of the transactivator.

a, b HeLa cells were transfected to express myc-NLRC5 alone or together with Flag-Ank5 and treated with vehicle [−] or MG132 [+]. a Both input lysates and Flag IP complexes were analyzed by immunoblot. b Densitometric signal of immunoprecipitated NLRC5 divided by that of the corresponding Flag-Ank5 signal (n = 3 independent experiments). c, d HeLa cells were transfected with nontargeting (siNT) or Skp1-targeting (siSkp1) siRNA. The cells were then transfected to co-express myc-NLRC5 and either Flag-Ank5 or Flag-Ank5∆F-box. c Whole cell lysates were analyzed by immunoblot. (d) Densitometric quantification of the NLRC5:GAPDH signal ratio (n = 5 independent experiments). e, f HeLa cells were transfected to co-express myc-NLRC5 with indicated Flag-Ank5 proteins. e Whole cell lysates were analyzed by immunoblot. Vertical lines between bands indicate where the blot was cropped or imaged separately. f Densitometric quantification of NLRC5:GAPDH signal. (n = 6 independent experiments). g, h HeLa cells were transfected to co-express Flag-NLRC5 and either GFP-Ank5 or GFP-Ank5-F-boxAAAAA, treated with MG132, and cell sorted on GFP-positivity. Flag-NLRC5 was immunoprecipitated and the eluted complexes were analyzed by mass spectrometry. g Fragmentation spectra of the post-translationally modified K1194 [1179SPFLLANTLSLCPRVK1194] peptide. Spectra-matched b- (blue) and y-ion (red) fragments are displayed and summarized by the trypsinized peptide sequence inset above. h Peptide intensity presented as fold change (n = 2 independent experiments). Data are means ± SD. Unpaired two-tailed t-test (b, d, f) was used for data analysis. Source data are provided as Source Data file.
The Ank5 F-box domain binds Skp1 to assemble the SCF E3 ubiquitin ligase complex56. To confirm if the F-box and Skp1 contribute to the effector’s ability to reduce NLRC5 cellular levels, HeLa cells that had been treated with Skp1 targeting small interfering RNA (siRNA) or non-targeting siRNA were transfected to co-express myc-NLRC5 and either Flag-Ank5 or Flag-Ank5∆F-box. Flag-Ank5 failed to degrade myc-NLRC5 in Skp1 knockdown cells (Fig. 4c, d). Also, myc-NLRC5 levels remained elevated in cells expressing Flag-Ank5∆F-box whether Skp1 had been knocked down or not. As another means of validating the essentiality of the F-box for Ank5 to direct NLRC5 degradation, HeLa cells were transfected to express myc-NLRC5 together with Flag-Ank5∆F-box or Flag-tagged Ank5 having F-box residues L301, P302, E304, I309, and D317 that are required for binding Skp1 and nucleating the SCF complex replaced with alanine (Flag-Ank5-F-boxAAAAA)56. Flag-Ank5∆F-box and Flag-Ank5-F-boxAAAAA were similarly incapacitated for the ability to degrade NLRC5 (Fig. 4e, f).
To delineate the NLRC5 lysine(s) that Ank5 directs to become ubiquitinated, HeLa cells were transfected to co-express Flag-NLRC5 and GFP-tagged Ank5 or Ank5-F-boxAAAAA. The cells were treated with MG132 and GFP-positive populations enriched for via FACS. Lysates were verified for expression of proteins of interest and MG132 treatment (Supplementary Fig. 6a). LC-MS/MS analysis of precipitated Flag-NLRC5 revealed that the same NLRC5 peptide (1179SPFLLANTLSLCPRVK1194) that is ubiquitinated at K1194 in O. tsutsugamushi infected cells (Fig. 1l) is ubiquitinated at K1194 in cells expressing GFP-Ank5 (Fig. 4g), though the overall amount was lower than in infected cells, explaining the fewer product ions observed. That said, this peptide was recovered at seven-fold greater abundance from cells expressing GFP-Ank5 compared to cells expressing GFP-Ank5-F-boxAAAAA (Fig. 4h). Peptide 32MKFFLPNTDLDSR44 with evidence of ubiquitination at K33 was recovered at similar levels from both transfected cell populations (Supplementary Fig. 6b, c). Altogether, these data confirm that ectopically expressed Ank5 phenocopies O. tsutsugamushi infection by directing SCF ubiquitination of NLRC5 at K1194 and its proteasomal degradation in an F-box-dependent manner.
Ank5 AR four is critical for binding NLRC5
Guided by the principle that AR domains mediate protein-protein interactions86,87,88, we sought to identify the specific Ank5 AR(s) that bind NLRC5. Because individual ARs are modular and as long as a minimum of two repeats is maintained for stability, deletion of a single repeat within an AR domain does not significantly alter tertiary structure of the domain or the entire protein89,90,91. Accordingly, plasmids were generated that encoded Flag-Ank5-F-boxAAAAA proteins lacking each of the four ARs to allow for assessment of the ΔAR domain-NLRC5 interaction without the complication of NLRC5 being degraded. AlphaFold predicted that all the Ank5 ARs are tandemly arranged helix-turn-helix motifs that form a concave interaction interface consistent with known AR domain structure86,92 (Supplementary Fig. 7). The F-box of each was predicted to adopt a fold prototypical of other F-box containing proteins56,93,94,95. Importantly, when the ΔAR mutant models were overlaid on full-length Ank5-F-boxAAAAA, no structural alterations were predicted for any ∆AR protein’s AR domain (Supplementary Fig. 7). Flag-Ank5-F-boxAAAAA and the ∆AR1 and ∆AR2 versions thereof co-immunoprecipitated myc-NLRC5 with comparable efficiencies (Fig. 5a, b). The ability of Ank5-F-boxAAAAA to co-immunoprecipitate NLRC5 was impaired when AR3 was deleted and abolished when AR4 was removed. UT76 Ank5 AR4 and Ikeda Ank5 AR4 are identical (Supplementary Table 3). Whereas Flag-tagged UT76 Ank5∆F-box precipitated NLRC5, Flag-tagged UT76 Ank5∆AR4 could not (Supplementary Fig. 8a). Yet, neither were able to reduce myc-NLRC5 levels (Supplementary Fig. 8b), indicating that, like Ikeda Ank5, UT76 Ank5 requires its AR4 to bind NLRC5 and its F-box to direct NLRC5 degradation.

a–d HeLa cells were transfected to co-express myc-NLRC5 and indicated Flag-tagged Ank5 proteins. a, c Input lysates and Flag IP complexes were analyzed by immunoblot. b, d Densitometric signal of immunoprecipitated NLRC5 was divided by the corresponding Flag-Ank5 protein signal and multiplied by 100 to determine percent interaction (n = 3 independent experiments). e, f Schematic of wt NLRC5 (e) and isoform 3 (f). Amino acid positions at the beginning of each domain are indicated. g–j HeLa cells were transfected to co-express myc-tagged wt NLRC5 or isoform 3 with Flag-tagged Ank5 or Ank5-F-boxAAAAA (g and h) or transfected to express myc-tagged wt NLRC5 or isoform 3 and then mock infected [U] or infected with O. tsutsugamushi [I] (i, j). g, i Input lysates, Flag IP complexes, and whole cell lysates were analyzed by immunoblot. Red asterisks denote the bands of interest. h, j Densitometric quantification of the myc:GAPDH signal was normalized to Flag-Ank5-F-boxAAAAA (h) or uninfected (j) cells (black bars) (n = 3 independent experiments). k, l HeLa cells were transfected to express indicated proteins, sorted on GFP positivity, mock infected [U] or infected with O. tsutsugamushi [I], and treated with IFNγ. k Whole cell lysates were analyzed by immunoblot. l Densitometric quantification of the NLRC5:GAPDH signal (n = 3 independent experiments). Indicators of statistical significance between pairs of uninfected (black bars) and infected (white bars) samples are colored black. Indicators of statistical significance between transfected and uninfected samples compared to uninfected cells expressing GFP-Ank5 are colored blue. Vertical lines between bands indicate where the blot was cropped or imaged separately. Data are means ± SD. Unpaired two-tailed t-test (b, d, h, j, l) was used for data analysis. Source data are provided as Source Data file.
The β-hairpin and first α-helix of each 33-residue AR create the recognition surface. Within this region, conserved residues preserve tertiary structure while non-conserved or semi-conserved residues at positions 2, 3, and 5 in the β-turn mediate protein-protein interactions and dictate target specificity86,93,96. In agreement with pulldown studies (Fig. 5a, b), NovaDock predicted that Ank5 AR3 and AR4 bind NLRC5. Of the eight candidate interacting residues, two in AR3 (L68 and Y69) and two in AR4 (Y103 and S105) correspond to AR3 positions 2 and 3 and AR4 positions 3 and 5 (Supplementary Fig. 9). We investigated the contribution of these four amino acids to NLRC5 binding. The remaining four could not be evaluated because they occupy positions within the inner α-helix that would alter AR tertiary structure if mutated97,98,99,100. Flag-Ank5-F-boxAAAAA proteins with the residues of interest substituted with alanine were assessed for the ability to bind myc-NLRC5 compared to the positive control, Flag-Ank5-F-boxAAAAA, and negative control, Flag-Ank5∆AR4-F-boxAAAAA. Changing L68 or Y69 to alanine had no effect on the interaction while swapping S105 for alanine yielded a detectable but statistically insignificant reduction in NLRC5 recovery (Fig. 5c, d). The ability of the Y103A mutant to co-immunoprecipitate NLRC5 was nearly abolished. Thus, Ank5 requires AR4 to bind NLRC5, and this interaction critically involves Y103.
NLRC5 regions essential for Ank5-mediated binding and degradation
NLRC5 is a multi-domain protein101,102,103. Its N-terminal aCARD (atypical caspase activation and recruitment domain) is followed by NACHT (present in NAIP, CIITA, HET-E, and TP1), small helical (AxP), winged helix (WH), and superhelical (SH) domains103. Its C-terminal half consists of 26 tandemly arranged LRRs. There are six NLRC5 isoforms with varying LRR region lengths that result from differential splicing41. Relative to full-length NLRC5 (Fig. 5e), isoform 3 consists only of residues 1 to 720 and lacks the entire LRR domain (Fig. 5f). Because LRR regions can mediate protein-protein interactions104, we assessed the relevance of the NLRC5 LRR region in the Ank5 interaction by comparing the abilities of Flag-Ank5 and Flag-Ank5-F-boxAAAAA to co-immunoprecipitate myc-tagged versions of wild-type (wt) NLRC5 and isoform 3. A myc tag antibody was used to detect myc-NLRC5 proteins because the NLRC5 antibody used thus far recognizes a C-terminal epitope that isoform 3 lacks. The myc antibody detected a nonspecific host cell protein in all samples (Fig. 5g, i). Flag-Ank5 and Flag-Ank5-F-boxAAAAA precipitated both isoforms revealing that the LRR region is dispensable for Ank5 to bind NLRC5 proteins. Interestingly, Flag-Ank5 was unable to degrade myc-NLRC5 isoform 3 (Fig. 5g, h), insinuating that the LRR region is necessary for Ank5-mediated NLRC5 degradation. When cells expressing myc-tagged wt NLRC5 or isoform 3 were infected with O. tsutsugamushi, wt NLRC5 levels were severely reduced whereas isoform 3 levels were significantly elevated (Fig. 5i, j). Therefore, O. tsutsugamushi Ank5 binds to NLRC5 within residues 1 to 720 but requires the C-terminal LRR domain to orchestrate its degradation.
Ank5-F-boxAAAAA antagonizes Orientia-mediated NLRC5 degradation
To confirm whether bacterial-derived Ank5 promotes NLRC5 degradation in infected cells, we needed an approach that would circumvent O. tsutsugamushi genetic intractability. Since ectopically expressed Ank5 engages NLRC5 via AR4 and promotes its degradation using its F-box, we rationalized that overexpressed Ank5-F-boxAAAAA binding to NLRC5 would competitively antagonize the actions of Orientia-derived Ank5. HeLa cells were transfected to express GFP-tagged Ank5, Ank5-F-boxAAAAA, Ank5∆AR4, Ank5∆AR4-F-boxAAAAA, or GFP. Each population was sorted on GFP positivity and infected with O. tsutsugamushi or mock infected followed by the addition of IFNγ. At 24 h p.i., endogenous NLRC5 levels were significantly lower in infected versus uninfected cells expressing GFP and comparably reduced in uninfected and infected cells expressing GFP-Ank5 (Fig. 5k, l). NLRC5 amounts were also significantly lower in cells expressing GFP-Ank5∆AR4 and GFP-Ank5∆AR4-F-boxAAAAA during O. tsutsugamushi infection, indicating the inability of either protein to outcompete bacterial-derived Ank5. However, NLRC5 levels were unchanged between uninfected and infected cells expressing GFP-Ank5-F-boxAAAAA, validating the ability of the ectopically expressed protein to protect NLRC5 from degradation mediated by oriential Ank5. These results also verify that Ank5 expressed by O. tsutsugamushi during infection binds NLRC5 via its AR4 to direct its degradation in an F-box-dependent manner.
Ank5 inhibits MHC class I expression in an AR4-dependent manner
The ability of Ank5 to degrade NLRC5 is AR4- and F-box-dependent. Yet, it was unclear if Ank5 requires both domains to impair MHC class I expression. HeLa cells expressing GFP, GFP-Ank5, GFP-Ank5-F-boxAAAAA, or GFP-Ank5∆AR4 were treated with IFNγ to upregulate NLRC5 expression, sorted on GFP positivity to enrich for each transfected population, and measured for NLRC5 and MHC class I mRNA and protein expression. Compared to unstimulated controls, IFNγ treated cells exhibited significantly elevated transcript and protein levels of both NLRC5 and MHC class I as well as MHC class I cell surface amounts (Fig. 6). NLRC5 mRNA levels were unchanged in IFNγ stimulated cells expressing GFP-Ank5, GFP-Ank5-F-boxAAAAA, and GFP-Ank5∆AR4 compared to cells expressing GFP (Fig. 6a). Expression of GFP-tagged Ank5 or Ank5-F-boxAAAAA, but not Ank5∆AR4, resulted in lower transcript levels of HLA-A, the first gene in the HLA locus105,106, and β2 M despite GFP-Ank5 being the only one of the three proteins that promoted NLRC5 degradation (Fig. 6b–c). Similarly, HLA-ABC and β2 M total protein along with MHC class I cell surface levels were significantly reduced by GFP-Ank5 and GFP-Ank5-F-boxAAAAA but not GFP-Ank5∆AR4 (Fig. 6d–i). Overall, these data indicate that Ank5 binding NLRC5 by virtue of AR4 is sufficient to impair MHC class I gene expression in an F-box-independent manner, which translates to losses in overall and cell surface levels of MHC class I.

a–i HeLa cells were transfected to express indicated proteins, treated with IFNγ or vehicle control, and sorted on GFP-positivity. a–c Total RNA was collected and RT-qPCR was performed using gene-specific primers. Relative expression of NLRC5-to-GAPDH (a), HLA-A-to-GAPDH (b), and β2M-to-GAPDH (c) was determined using the 2-∆∆CT method in which conditional values were normalized to those values of GFP-expressing cells treated with IFNγ (black bars). (n = 5 independent experiments). d–g Whole cell lysates were collected from sorted cells and analyzed by immunoblot (d). Vertical lines between bands indicate where the blot was cropped or imaged separately. e–g The densitometric signal of NLRC5 (e), HLA-ABC (f), or β2 M (g) was divided by the corresponding GAPDH densitometric signal and normalized to GFP-expressing cells treated with IFNγ (black bars) (n = 3 independent experiments). h, i Surface levels of MHC class I from sorted cells were analyzed by flow cytometry. h Representative histograms of MHC class I levels normalized to mode. Unlabeled (black) samples served as the control. [–] denotes vehicle control and [+] denotes IFNγ addition. i The mean fluorescence intensity of each sample was divided by that of its unlabeled control per replicate (n = 4 independent experiments). Data are means ± SD. One-way ANOVA with Dunnett’s post hoc test (a–c), Unpaired two-tailed t-test (e–g), and One-way ANOVA with Tukey’s post hoc test (i) were used for data analysis. Source data are provided as Source Data file.
Ank5 impairs NLRC5 nuclear transit in an F-box-independent manner
A small portion of the NLRC5 cytoplasmic pool transits through the nucleus to transactivate MHC class I gene expression37,64,107. Given the observation that Ank5 does not necessarily need to direct NLRC5 degradation to downregulate MHC class I expression, we hypothesized that it interferes with NLRC5 nuclear translocation. Western blot analysis was performed on cytoplasmic and nuclear fractions of IFNγ-treated HeLa cells expressing GFP, GFP-Ank5, or GFP-Ank5-F-boxAAAAA. Hsp90 and lamin A/C served as cytoplasmic and nuclear fraction loading controls, respectively. Compared to untreated GFP-expressing HeLa cells, IFNγ-stimulated cells expressing GFP exhibited robust endogenous NLRC5 cytoplasmic levels with a small portion present in the nuclear fraction (Fig. 7a–c). NLRC5 was barely detectable in IFNγ-stimulated cells expressing GFP-Ank5. NLRC5 nuclear levels were significantly reduced in IFNγ-treated cells expressing GFP-Ank5-F-boxAAAAA versus GFP (Fig. 7a, c). Thus, in addition to promoting NLRC5 proteasomal degradation, Ank5 inhibits NLRC5 translocation into the nucleus and does so in an F-box-independent manner.

a–c HeLa cells expressing indicated proteins were treated with IFNγ or vehicle, sorted on GFP-positivity, and resolved into cytoplasmic [C] and nuclear [N] fractions. a Samples were analyzed by immunoblot. b, c The densitometric signal of cytoplasmic (b) or nuclear (c) NLRC5 was divided by that of Hsp90 or lamin A/C, respectively (n = 3 independent experiments). d, e HeLa cells were transfected to co-express indicated myc-NLRC5 proteins and Flag-tagged Ank5. d Inputs and Flag IP complexes were analyzed by immunoblot. e Densitometric quantification of myc:GAPDH signal from inputs (n = 3 independent experiments). f, g HeLa cells were transfected to express indicated GFP-NLRC5 proteins, mock infected [U] or infected with O. tsutsugamushi [I], and treated with IFNγ at 2 h p.i. (f) At 24 h p.i., whole cell lysates were immunoblotted. g Densitometric quantification of GFP:GAPDH signal from inputs was normalized to matched uninfected controls (n = 7 independent experiments). h HeLa cells were transfected to co-express Flag-BAP or Flag-Ank5 with indicated GFP-NLRC5 proteins and treated with IFNγ or vehicle (n = 4 independent experiments). MHC class I surface levels of GFP-positive cells were analyzed by flow cytometry. Indicators of statistical significance between pairs of untreated (black bars) and IFNγ-treated (white bars) samples are colored black, between untreated samples compared to BAP [–] are colored blue, and between IFNγ-treated samples compared to BAP [+] are colored red. i U and I HeLa cells were transfected to express indicated GFP-NLRC5 proteins and treated with IFNγ or vehicle for 24 h (n = 3 or 4 independent experiments). At 48 h p.i., samples were collected and assessed for MHC-I surface levels by flow cytometry. Fold change mean fluorescence intensity (MFI) was calculated by subtracting the MFI of each vehicle control-treated sample from that of its IFNγ-treated counterpart and then dividing that by its vehicle control. Data are means ± SD. One-way ANOVA with Dunnett’s post hoc test (b, c, h) and Unpaired two-tailed t-test (e, g, h, i) were used for data analysis. Source data are provided as Source Data file.
Generation of degradation-resistant NLRC5 proteins
We wanted to evaluate if Ank5 can still lower MHC class I surface levels in cells overexpressing NLRC5 that is resistant to proteasomal degradation. As an initial attempt, HeLa cells expressing myc-tagged versions of NLRC5 bearing K200R, K1194R, or K200R and K1194R substitutions were infected with O. tsutsugamushi. These lysines were chosen for replacement with arginine because they are preferentially ubiquitinated in cells infected with O. tsutsugamushi (K200 and K1194; Fig. 1j–m) and/or cells ectopically expressing Flag-Ank5 (K1194; Fig. 4g, h). K902R was included as a negative control because K902 is comparably ubiquitinated in infected and uninfected cells (Supplementary Fig. 3). Over five experiments, none of these proteins were consistently present at significantly greater levels in infected cells compared to degradation-sensitive myc-NLRC5 (Supplementary Fig. 10a, b). This is not surprising because lysines in many other host proteins exhibit high redundancy as acceptor sites for ubiquitination108,109,110,111,112,113,114,115,116. In other words, changing the NLRC5 lysines that are preferentially ubiquitinated during Orientia infection would not prevent other lysines from being ubiquitinated, which would still lead to proteasomal degradation. Accordingly, we took a more rigorous approach by generating constructs encoding myc-tagged NLRC5 bearing K-to-R substitutions at all lysines between residues 720 and 1866 (NLRC5720-1866 K*R), which is the C-terminal region that dictates susceptibility to proteasomal degradation during Orientia infection and in cells ectopically expressing Ank5 (Fig. 5g–j), or all lysines of the entire protein (NLRC5K*R). When Flag-Ank5 was co-expressed with myc-NLRC5720-1866 K*R or myc-NLRC5K*R, it interacted with both but could not promote the degradation of either (Fig. 7d, e). GFP-NLRC5720-1866 K*R and GFP-NLRC5K*R also resisted degradation in O. tsutsugamushi infected cells (Fig. 7f, g).
Next, we assessed if GFP-NLRC5 and the two GFP-tagged K-to-R substituted NLRC5 proteins can traffic into the nucleus and, if so, whether this is inhibited by ectopically expressed Ank5 or O. tsutsugamushi. To properly interpret this assay, one must bear in mind that because it focuses exclusively on GFP-positive HeLa cells it biases for cells in which GFP-tagged NLRC5 proteins have not been degraded. GFP-NLRC5 signal in the nucleus was reduced several-fold in cells expressing Flag-Ank5 compared to Flag-BAP and nearly abolished in infected versus uninfected cells regardless of IFNγ stimulation (Supplementary Fig. 10c–e). Thus, even in instances where GFP-NLRC5 had not been degraded in cells expressing Flag-Ank5 or infected with Orientia, its nuclear translocation was blocked. GFP-NLRC5720-1866 K*R and GFP-NLRC5K*R failed to accumulate in the nucleus for all conditions examined (Supplementary Fig. 10c–e) indicating that at least some NLRC5 lysines, particularly those between 720 and 1866, are important for its nuclear translocation. Overall, both K-to-R substituted NLRC5 proteins retain the ability to interact with Ank5, resist Ank5- and Orientia-induced degradation, and are incapable of nuclear translocation.
Ank5 uses a dual mechanism to lower MHC class I surface levels
GFP-NLRC5720-1866 K*R and GFP-NLRC5K*R were leveraged to functionally interrogate Ank5 further. Because both degradation-resistant proteins bind Ank5 they would retain the effector and competitively limit its access to endogenous NLRC5. If the degradation-resistant NLRC5:Ank5 ratio was substantial enough, at least some of the endogenous NLRC5 pool would be free to translocate to the nucleus, transactivate expression, and elevate MHC class I surface levels. HeLa cells were transfected to express Flag-Ank5 or Flag-BAP plus GFP-tagged NLRC5, NLRC5720-1866 K*R, or NLRC5K*R. The cells were stimulated with IFNγ or not followed by flow cytometry analyses of GFP-positive cells for MHC class I surface expression. Unstimulated cells co-expressing Flag-BAP and GFP-NLRC5 exhibited higher levels of surface MHC class I relative to cells co-expressing Flag-BAP and either GFP-NLRC5720-1866 K*R or GFP-NLRC5K*R (Fig. 7h). This is likely because GFP-NLRC5 is capable of nuclear translocation and hence would combine with endogenous NLRC5 to boost MHC class I expression whereas nuclear translocation-deficient GFP-NLRC5720-1866 K*R and GFP-NLRC5K*R would not. IFNγ stimulation increased surface MHC class I expression in cells expressing Flag-BAP and each GFP-NLRC5 protein. Importantly, the significant elevation in surface MHC class I observed for cells expressing Flag-BAP and either of the K-to-R-substituted nuclear translocation-deficient NLRC5 proteins confirmed that endogenous NLRC5 was capable of transactivating MHC class I expression. Flag-Ank5 pronouncedly lowered MHC class I surface levels regardless of which GFP-NLRC5 protein it was co-expressed with and did so in the presence of IFNγ. Thus, the abundance of overexpressed Flag-Ank5 was sufficient such that enough of it was present to interact with GFP-NLRC5, GFP-NLRC5720-1866 K*R, or GFP-NLRC5K*R and endogenous NLRC5. In each condition, Flag-Ank5 would have still prevented endogenous NLRC5 nuclear transit and mediated its proteasomal degradation.
We extended this approach to Orientia infection. HeLa cells were infected for 24 h after which they were transfected to express GFP-NLRC5, -NLRC5720-1866 K*R, or -NLRC5K*R and then incubated with IFNγ or vehicle for another 24 h. The IFNγ-stimulated increase in surface MHC class I expression on GFP-positive cells was quantified. IFNγ treated cells expressing GFP-NLRC5 exhibited a reduction in MHC class I surface levels during infection (Fig. 7i). We attribute this to two likelihoods. First, the ability of Ank5 to mediate degradation of a sufficient portion of the GFP-NLRC5 pool would free it up to also target endogenous NLRC5. Second, based on Fig. 7a–c and Supplementary Fig. 10c–e, Ank5 blocks nuclear translocation of both endogenous and GFP-tagged NLRC5. While we cannot rule out the potential actions of other Orientia effectors during infection, results observed for cells expressing GFP-NLRC5720-1866 K*R and GFP-NLRC5K*R argue against this possibility. Indeed, IFNγ stimulated a 12-fold increase in surface MHC class I on infected cells expressing GFP-NLRC5720-1866 K*R or GFP-NLRC5K*R versus their uninfected counterparts (Fig. 7i). Because these increases would have been exclusively due to transactivation of MHC class I expression by endogenous NLRC5, we conclude that both degradation-resistant NLRC5 proteins bound Ank5 to competitively inhibit its access to endogenous NLRC5. If other Orientia effectors are critical for Ikeda to reduce surface MHC class I levels, then rescue of surface MHC class I expression would not have been observed.
Overall, the entirety of our data establish that Ank5 uses a two-pronged approach to negatively regulate NLRC5 transactivation of MHC class I surface expression. Ank5 binds NLRC5 to retain it in the cytoplasm, which alone is sufficient to prevent MHC class I gene and protein expression, and recruits the SCF complex to mediate ubiquitination of NLRC5 and its subsequent proteasomal degradation.
Discussion
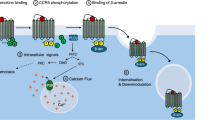
Like O. tsutsugamushi, many other intracellular microbes deploy AR-F-box proteins that counter eukaryotic immunity, benefit microbial fitness, and drive pathogenicity54,58,59,60,61,62,63. Yet nearly all these proteins’ modes of action are unknown. Here we elucidated the mechanism by which O. tsutsugamushi Ank5 binds NLRC5, blocks its nuclear translocation, and directs its ubiquitination and proteasomal degradation to impair MHC class I expression (Fig. 8). In doing so, we reveal NLRC5 as a target for microbial immunomodulation. Prior to this report, known strategies by which viruses and intracellular bacteria subvert the MHC class I pathway included altering subcellular transport of, inhibiting peptide loading on, and increasing degradation of MHC class I molecules2,3,4,6,7,117. Mycobacterium tuberculosis protein PPE38 modestly reduces MHC class I transcription when expressed in Mycobacterium smegmatis, but the underlying mechanism has not been examined5. O. tsutsugamushi sequesters NLRC5 in the cytoplasm, directs its ubiquitination, and promotes its degradation in a MG132-sensitive manner. Ectopically expressed Ank5 phenocopies all three phenomena. Ank5-mediated NLRC5 cytoplasmic retention and degradation translate to decreased HLA-A and β2 M transcription, which reduces MHC class I cellular and cell surface levels. The ability of Ank5 to sequester NLRC5 in the cytoplasm is F-box-independent, while its competency for directing NLRC5 ubiquitination and proteasomal degradation requires its F-box and host Skp1. These findings are consistent with the observation that Ank5 interactions with Skp1 and two other SCF complex proteins, Cul1 and RING box 1, are F-box-dependent56. Overexpression of dominant negative Ank5-F-boxAAAAA that can bind but not degrade NLRC5 protects it from Orientia-induced degradation by outcompeting bacterial-derived Ank5. Overall, Ank5 enables O. tsutsugamushi modulation of NLRC5-dependent MHC class I expression.

Ank5 utilizes its fourth AR to bind the N-terminal portion of NLRC5 and retain the transactivator in the cytoplasm. Ank5 also uses its F-box to direct SCF-mediated ubiquitination of the NLRC5 C-terminal leucine-rich repeat (LRR) domain, which promotes NLRC5 proteasomal degradation. This dual-action mechanism prevents MHC class I gene transactivation to ultimately lower MHC class I levels on the cell surface. Created with BioRender.com, released under a Creative Commons Attribution-Noncommercial-NoDerivs 4.0 International license.
Ank5 binds the N-terminal half of NLRC5. This interaction requires Ank5 AR4 and critically involves Y103 therein. The absolute sequence conservation of AR4 among Ikeda, UT76, and Wuj/2014 Ank5 proteins suggests that its functional essentiality has been maintained by selective pressure. This contention is further supported by the conserved abilities of Ikeda and UT76 to lower NLRC5 and MHC class I cellular levels during infection and for ectopically expressed Ikeda Ank5 and UT76 Ank5 to recapitulate these phenomena in an AR4- and F-box-dependent manner. NLRC5 and its differential splice variants are expressed in hematopoietic cells and tissues. Whereas isoform 3 ends at residue 720 and lacks the LRR domain, the other five splice variants extend beyond K1194 and have LRR regions41. Ank5 binds wt NLRC5 and isoform 3 but cannot promote degradation of isoform 3, which together with K1194 being the only LRR residue that is ubiquitinated in O. tsutsugamushi infected cells and cells ectopically expressing Ank5, reinforces the importance of K1194 over K200 for Ank5-mediated NLRC5 proteasomal degradation. Because the LRR region is critical for MHC class I transactivation107, this finding also supports that O. tsutsugamushi strains that express Ank5 would be able to inhibit the nuclear translocation of and rid their host cells of all NLRC5 isoforms competent for transactivating MHC class I expression. O. tsutsugamushi promotes a more complex NLRC5 interactome than in uninfected cells. Aside from those involved in protein degradation, the roles of proteins in this interactome in mediating NLRC5 degradation or other modulatory functions remain to be determined.
NLRC5 deficiency has profound impacts on adaptive immunity. In nlrc5−/− mice, MHC class I surface expression is severely reduced in CD4+ T cells, CD8+ T cells, and B cells118,119,120. APCs from these mice are impaired for inducing MHC class I-dependent antigen-specific stimulation of co-cultured CD8+ T cells for proliferation, IFNγ production, and cytolytic activity85,118,119. Consequently, nlrc5-/- mice are more susceptible to intracellular pathogens as they exhibit higher Listeria monocytogenes loads and are less effective at clearing influenza A and rotavirus infections85,119,120,121. NLRC5 elevated expression in cancer cells is associated with MHC class I gene expression, susceptibility to CD8+ T-cell-mediated killing, and tumor growth inhibition122,123,124,125. Conversely, its suppression correlates with reduced MHC class I expression and lower cancer patient survival rates. Immune escape of cancer cells due to NLRC5 suppression can result from multiple mechanisms122. However, the mode of action by which the proto-oncogene BMI1 impairs the NLRC5-MHC class I axis is of notable mention because it is reminiscent of Ank5. BMI1, an E3 ubiquitin ligase component of polycomb repressive complex 1, binds NLRC5 to induce its ubiquitination and degradation, which reduces MHC class I surface expression and CD8+ T cell activation126.
Of more than 1900 bacterial genomes, the Ikeda chromosome encodes the fifth-largest number of Anks, accounting for 2.4% of its content. By comparison, ank genomic content for Homo sapiens, Mus musculus, and the average of all obligate intracellular bacteria except Chlamydia species, which do not have any ank genes, is 3.1%, 1.7%, and 0.8%, respectively52. Retention of such a high ank content over its reductive evolution suggests the importance of Anks to O. tsutsugamushi pathobiology. However, very few have been studied and their functions are poorly characterized65,66,67,68,70. The significance of Ank5 to O. tsutsugamushi pathogenesis is apparent. By upregulating Ank5 during cytoplasmic replication in endothelial cells, the bacterium can orchestrate NLRC5 degradation so that it can grow to high numbers in host cells rendered incapable of eliciting adaptive immunity via MHC class I antigen presentation. The Leptotromidium mites that vector O. tsutsugamushi are considered its primary reservoirs11. However, laboratory-conducted studies demonstrated that naïve chiggers could acquire the bacterium from infected mice and in rare instances their offspring could infect new hosts127,128. Suppressing the NLRC5-MHC class I axis could augment Orientia reaching a sufficient load in rodent hosts that maximizes its chance for reacquisition via mite feeding before immune responses capable of clearing the infection are elicited. Given that mites lack adaptive immunity, the Ank5 mechanism uncovered here is exclusively relevant to Orientia infection in mammals and argues that mammals might play a role in shaping the immunomodulatory/virulence capacity of O. tsutsugamushi strains. Ank5-mediated immunosuppression would also potentially contribute to disease outcomes associated with infection of endothelial cells in accidental human hosts. High O. tsutsugamushi burdens are observed during late disease, and severe manifestations in patients and experimentally infected mice at least partially stem from endothelial cell colonization32,129,130,131,132,133,134,135.
Differences in genomic carriage and expression of Ank5 could influence scrub typhus pathogenesis. Indeed, while NLRC5 and MHC class I levels are pronouncedly reduced in cells infected with Ikeda or UT76, NLRC5 expression is markedly increased and MHC class I levels unaltered during Karp infection. Therefore, Ank5 affords strains that carry and express it the advantage of downregulating NLRC5-dependent anti-oriential immune responses. Notably, the studies that concluded that MHC class I and CD8+ T cells are vital for preventing fatal O. tsutsugamushi infections and CD8+ T cells contribute to dysregulated Th1 immunopathologies in murine models were conducted using Karp33,34,129,132. Yet, despite being unable to degrade NLRC5 and block HLA-ABC and β2 M expression, Karp still impairs MHC class I cell surface presentation. Although the mechanism is undefined, Karp might do so using Ank6 or Ank9. Of the four Anks besides Ank5 shown herein to reduce MHC class I surface expression when ectopically expressed, Karp carries these two53. Ank6 and Ank9 dysregulate NF-κB and protein secretion, respectively56,65,66, both of which positively regulate the MHC class I pathway1,136,137. Theoretically, Ank4, Ank6, Ank9, and Ank13 could contribute to Ikeda subversion of MHC class I surface expression. However, counter to this possibility, IFNγ-stimulates robust increases in surface MHC class I on Ikeda-infected cells expressing GFP-NLRC5720-1866 K*R or GFP-NLRC5K*R. In these cells, MHC class I expression would be exclusively attributable to endogenous NLRC5 because GFP-NLRC5720-1866 K*R or GFP-NLRC5K*R would competitively inhibit oriential Ank5. If Ank4, Ank9, Ank6, and Ank13 – none of which target NLRC5 and hence would not be antagonized by either K-to-R degradation-resistant NLRC5 protein – were key for Ikeda to reduce surface MHC class I levels, then GFP-NLRC5720-1866 K*R or GFP-NLRC5K*R would not rescue surface MHC class I expression during infection. Because they do, we argue that Ank5 is the primary effector that Ikeda uses to counter the MHC class I pathway.
NLRC5 has been implicated in signal transduction, Type I interferon responses, selective autophagy, and possibly inflammasome activation83,138,139. O. tsutsugamushi strains that carry/express Ank5 or other Anks could contribute to strain-specific variations in scrub typhus immunopathologies independent of MHC class I. In support of this premise, a dual RNA-seq comparison of endothelial cells infected with Karp or O. tsutsugamushi strain UT176 showed that UT176 induced a stronger proinflammatory response and was cleared in mice while Karp stimulated IL-33 alarmin-based immunopathologies and was pathogenic in mice. Although neither strain carries ank5, they differentially express putative virulence factors during infection, including several anks25. Given that O. tsutsugamushi Anks varies strain-to-strain43,49,50,53, and their functions are mostly unknown, future work must define additional modes of action for these effectors and determine their roles in scrub typhus pathogenesis.
In conclusion, our study establishes Ank5 as an O. tsutsugamushi virulence factor that promotes NLRC5 degradation using two eukaryotic-like motifs that simultaneously bind NLRC5 and nucleate the SCF ubiquitin ligase complex. Ank5 binding to the NLRC5 N-terminus prevents the transactivator from trafficking into the nucleus. As an adapter protein in this complex scaffold, Ank5 also directs selective ubiquitination of NLRC5 most likely on K1194 to target it to the proteasome. Though each mechanism is sufficient to independently prevent MHC class I gene expression, Ank5 synergistically employs both to maximally rid the host cell of NLRC5 and significantly reduce MHC class I surface levels. The similarity between Ank5 and BMI1 mechanisms of action is a striking example of convergent evolution in that both an intracellular pathogen and oncogene co-opt ubiquitination/proteasomal degradation to subvert the NLRC5-MHC class I axis. As exemplified for BMI1126, this strategy likely contributes to the abilities of Ikeda and other O. tsutsugamushi strains that express Ank5 to evade CD8 + T cell-mediated immunity and additional NLRC5-dependent immune responses. By unveiling NLRC5 as a target for microbial immunomodulation, this study advances understanding of how O. tsutsugamushi and potentially other intracellular pathogens maximize survival in their intracellular niches.
Methods
Cultivation of cell lines and O. tsutsugamushi infections
Uninfected HeLa cells (CCL-2; American Type Culture Collection [ATCC], Manassas, VA) and HeLa cells infected with O. tsutsugamushi str. Ikeda (NC_010793.1) were maintained as previously described66. EA.hy926 endothelial cells (CRL-2922; ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with l‐glutamine, 4.5 g d‐glucose and 100 mg sodium pyruvate (Gibco, Gaithersburg, MD) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS) (Gemini Bioproducts, West Sacramento, CA), 1X minimal essential medium containing non‐essential amino acids (Gibco) and 15 mM HEPES (Gibco) in a humidified incubator at 37 °C with 5% atmospheric CO2. The Ikeda, UT76, and Karp O. tsutsugamushi strains were used in this study. For routine propagation, bacteria were grown in HeLa cells. For experimental use, infected HeLa cells (≥90%) were mechanically disrupted using glass beads followed by differential centrifugation to recover host cell-free bacteria as described66 and then subsequently incubated with naïve HeLa or EA.hy926 cells. Cells undergoing mock infections were incubated with an equivalent volume of bead-lysed HeLa or EA.hy926 cells. Synchronous infections were achieved by replacing inoculum with fresh media at 2 to 4 h post infection (p.i.) Experiments were verified for achieving a multiplicity of infection (MOI) of 10 in at least 80% of the host cells by assessing coverslips of infected cells by immunofluorescence as described below. Samples in which less than 80% of the cells were infected were not analyzed further.
Plasmid constructs
Empty pEGFP-C1 was included as a control51. pFlag-Ank4, -Ank5, -Ank6, -Ank9, -Ank13, -Ank5∆F-box, and -Ank5-F-boxAAAAA as well as pGFP-Ank4, -Ank5, -Ank6, -Ank9, and -Ank13 were generated previously51,56. Primers used to introduce the following mutations (Supplementary Table 4) were designed using the In-Fusion Cloning Primer Design Tool v1.0 (www.takarabio.com). pFlag-Ank5∆AR1-F-boxAAAAA (lacks residues 3-33), -Ank5∆AR2-F-boxAAAAA (lacks residues 34-66), -Ank5∆AR3-F-boxAAAAA (lacks residues 67-100), -Ank5∆AR4-F-boxAAAAA (lacks residues 101-133), -Ank5L68A-F-boxAAAAA, -Ank5Y69A-F-boxAAAAA, -Ank5Y103A-F-boxAAAAA, and -Ank5Y105A-F-boxAAAAA were generated using TaKaRa Bio USA (San Francisco, CA) In-Fusion Mutagenesis protocol and pFlag-Ank5-F-boxAAAAA as template. pFlag-Ank5∆AR4 and pGFP-Ank5∆AR4 were generated using In-Fusion Mutagenesis and pFlag-Ank5 or pGFP-Ank5 as template, respectively. pGFP-Ank5-F-boxAAAAA and pGFP-Ank5∆AR4-F-boxAAAAA were generated by digesting pFlag-Ank5-F-boxAAAAA and pFlag-Ank5∆AR4-F-boxAAAAA with EcoRI-HF and BamHI and subcloning the released fragments into the multicloning site in pEGFP-C1. Mammalian codon optimized UT76 ank5 was synthesized and cloned into p3xFlag-CMV-7.1 downstream and in-frame with a 3xFlag tag coding sequence by Genewiz (Burlington, MA) to generate pFlag-UT76-Ank5. pFlag-UT76-Ank5 served as template for the generation of pFlag-UT76-Ank5∆F-box and pFlag-UT76-Ank5∆AR4 using TaKaRa Bio USA (San Francisco, CA) In-Fusion Mutagenesis protocol and previously described Ank5∆F-box56 or Ank5∆AR4 primers listed in Supplementary Table 4. pCMV-Tag2b-Flag-NLRC5 (Addgene plasmid #37521; http://n2t.net/addgene:37521; RRID:Addgene_37521), pcDNA3.1-3xmyc-B-NLRC5 (Addgene plasmid #37509; http://n2t.net/addgene:37509; RRID:Addgene_37509), and pcDNA3.1-3xmyc-B-NLRC5 iso3 (Addgene plasmid #37510; http://n2t.net/addgene:37510; RRID:Addgene_37510) were gifts from Thomas Kufer. pcDNA3.1-HA-Ubiquitin (Ub) was a gift from Edward Yeh (Addgene plasmid #18712; http://n2t.net/addgene:18712; RRID:Addgene_18712). pmyc-NLRC5720-1866 K*R and pmyc-NLRC5K*R were generated (GenScript; Piscataway, NJ) using pcDNA3.1-3xmyc-B-NLRC5 as template and replacing lysine residues with arginine in the region C-terminal of AA720 or in the entirety of the protein, respectively. myc-NLRC5720-1866 K*R and myc-NLRC5K*R coding sequences were PCR amplified and subcloned into pEGFP-C1 using primers listed in Supplementary Table 4. Full-length tsa56 (OTT_0945) was amplified from O. tsutsugamushi str. Ikeda genomic DNA with tsa56-Full-F and tsa56-Full-R primers. The subsequent amplicon was cloned into TOPO vector pCR4.0 to generate pCR4.0::Ot_tsa56. All plasmid constructs were confirmed by sequence analysis (Genewiz).
Transfection
HeLa or EA.hy926 cells grown to 90% confluency were transfected with plasmid DNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and incubated at 37 °C in a humidified incubator at 5% atmospheric CO2 for 18 to 24 h. The amount of DNA used for transfections was modified from that recommended by the Lipofectamine 2000 protocol per plasmid to accommodate varying levels of transfection efficiency, as determined by western blot densitometric signal. After incubation, spent media was removed. The cells were washed once with PBS before being processed for further applications.
siRNA knockdown
HeLa cells grown to 90% confluency were transfected with ON-TARGETplus human Skp1 SMARTpool siRNA or non-targeting siRNA (GE Healthcare Dharmacon, Inc., Lafayette, CO) at a final concentration appropriate to the vessel size. After 48 h, the media was replaced and knocked down cells were transfected to express either Flag-Ank5 or Flag-Ank5∆F-box. After 24 h, lysates were collected and western-blotted as described below.
Pharmacologic and IFNγ treatments
To prevent eukaryotic protein synthesis or proteasome degradation during infection, O. tsutsugamushi- or mock-infected HeLa cells were treated with or without cycloheximide (50 μg ml−1 in ethanol; Sigma-Aldrich) or MG132 (5 μM in DMSO; Sigma-Aldrich), respectively, at 2 h p.i. and whole cell lysates were collected at 18 h p.i. To inhibit the proteasome in transfected cells, cells were first transfected for 18 to 24 h and then treated with 5 μM MG132 or DMSO for 18 to 24 h prior to collection. To stimulate expression of endogenous NLRC5, spent media was replaced with media containing 40 ng ml−1 human IFNγ (PeproTech, Rock Hill, NJ) or vehicle control (0.1% bovine serum albumin [BSA] in H2O) at 24 h after transfection or 2 h p.i. and kept on until collection.
Immunofluorescence
For MOI and flow cytometry coverslips, cells were fixed and permeabilized with −20 °C methanol. The cells were blocked in 5% (vol/vol) bovine serum albumin (BSA) in phosphate-buffered saline (PBS; 1.05 mM KH2PO4, 155 mM NaCl, 2.96 mM Na2HPO4, pH 7.4) and incubated with rabbit anti-TSA56 (65; 1:1000) followed by incubation with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen [A-11034], 1:10,000) or Alexa Fluor 594-conjugated goat anti-rabbit IgG (Invitrogen [A-11012], 1:10,000) in 5% BSA. Blocking and antibody incubations were performed for 1 h at room temperature with three PBS washes between each step. Coverslips were incubated with 300 nM 4′ 6-diamidino-2-phenylindole (DAPI; [D1306] Invitrogen) in PBS for 5 min, washed three times, and mounted using ProLong Gold Anti-fade reagent (Invitrogen). Coverslips were imaged with an Olympus BX51 spinning disc confocal microscope (Olympus, Shinjuku City, Tokyo, Japan) or a Zeiss LSM 700 laser scanning confocal microscope (Zeiss). The percentage of 100 infected cells per condition was calculated by examining the images. For assessing GFP-NLRC5 nuclear translocation, HeLa cells were co-transfected to express one of the three GFP-NLRC5 fusions and Flag-BAP or Flag-Ank5. After 24 h, the cells were stimulated with IFNγ for 24 h. Alternatively, HeLa cells infected with O. tsutsugamushi Ikeda or mock-infected controls were transfected to express one of the three GFP-NLRC5 fusions and treated with IFNγ at 24 h p.i. After 24 h, the cells were processed using the same protocol except that the cells were fixed in 2% (vol/vol) paraformaldehyde (Electron Microscopy Sciences, Hatfield PA) in PBS for 10 min followed by permeabilization in 1% (vol/vol) nonidet P-40 in PBS for 15 min with single PBS washes between steps. Cells expressing Flag-tagged protein were incubated with mouse anti-Flag (Sigma-Aldrich [F1804], 1:1000) and rabbit anti-GFP (ThermoFisher Scientific, Rockford, IL [A-11122], 1:1000) followed by Alexa Fluor 594-conjugated chicken anti-mouse IgG (Invitrogen [A-21201], 1:10,000) and Alexa Fluor 488-conjugated chicken anti-rabbit IgG (Invitrogen [A-21441], 1:10,000). Infected cells were incubated with rabbit anti-TSA5665 (1:1000) and mouse anti-GFP (Santa Cruz, Dallas, TX [SC-9996], 1:1000) followed by Alexa Fluor 594-conjugated chicken anti-rabbit IgG (Invitrogen [A-21442], 1:10,000) and Alex Fluor 488-conjugated chicken anti-mouse IgG (Invitrogen [A-21200], 1:10,000). Micrographs were obtained using a Leica DMi8 inverted microscope affixed with the following Leica package: Leica EL6000 lamp at 460 nm and 630 nm and band-pass filters at 420/30 nm and 570/20 nm. Image acquisition was performed with an Andor iXon Ultra 888 EMCCD camera (Oxford Instruments) and a 63X water-immersion objective with 1.2 numeric aperture.
Immunoprecipitation
For Fab-trap immunoprecipitations, HeLa cells expressing HA-Ub and Flag-NLRC5 for 18 h were synchronously infected with O. tsutsugamushi str. Ikeda. At 24 h p.i., cells were incubated with or without MG132 for 24 h followed by lysis in high saline Tris buffer (50 mM Tris HCl, 400 mM NaCl, 1 mM EDTA, pH 7.4) with 1.0% Triton x-100 (TBHS-T) spiked with Halt Protease and Phosphatase Inhibitor Cocktail (100X; Thermo). ChromoTek DYKDDDDK Fab-trap Agarose resin (Proteintech Group, Inc., Rosemont, IL) was washed with TBHS-T buffer two times, centrifuged at 2500×g for two min, and added to normalized cell lysates in a final volume of 400 μl. Samples were incubated with beads rotating at 4 °C for 1 h followed by centrifugation at 2500×g for 5 min and washing with TBHS-T four times. Proteins were eluted with Laemmli buffer containing 4% βME and a fraction of the eluate was resolved by SDS-PAGE next to input lysates and immunoblotted as described below. For anti-Flag affinity agarose immunoprecipitations, transfected HeLa cells were harvested and lysed in TBHS-T spiked with Halt Protease and Phosphatase Inhibitor Cocktail. Protein A/G agarose beads (ThermoFisher Scientific) were washed with TBHS-T buffer three times, centrifuged at 8600×g for 30 s, and added to normalized cell lysates in a final volume of 400 μl. The samples were rotated with beads at 4 °C for 4 h followed by centrifugation at 8600×g for 30 s. Recovered supernatants were added to Anti-Flag M2 Affinity Gel (MilliporeSigma) that had been washed three times with TBHS-T buffer. Samples rotated with beads at 4 °C overnight followed by centrifugation at 8600×g for 30 s and washed with TBHS-T six to ten times. Washed beads were resuspended in Laemmli buffer containing 4% βME and incubated at 100 °C for 5 min to elute bound proteins. Inputs (10–30 μg) and eluates were resolved by SDS-PAGE and screened by western blot.
Western blotting
Western blotting was performed as previously described68. Briefly, normalized amounts of eluates were resolved by SDS-PAGE in 4 to 15% TGX polyacrylamide gels (Bio-Rad) at 110 V for 15 min followed by 200 V for 25 min. Proteins were transferred onto nitrocellulose membrane in Towbin buffer at 100 V for 30 min. Blots were blocked in either 5% (vol/vol) non-fat dry milk or 5% (vol/vol) BSA in tris-buffered saline plus 0.05% Tween 20 (TBS-T) after which they were screened with rat anti-NLRC5 (MilliporeSigma [MABF260], 1:1000), rabbit anti-TSA5665 (1:1000), rabbit or mouse anti-Flag (Sigma-Aldrich [F7425 or F1804], 1:1000), rabbit anti-GFP (ThermoFisher Scientific, Rockford, IL [A-6455], 1:1000), mouse anti-GAPDH (Santa Cruz, Dallas, TX [sc-365062], 1:750), rabbit or mouse anti-myc (Abcam, Cambridge, United Kingdom [ab9106 and ab32], 1:1000), rabbit anti-HA (Abcam [ab9110], 1:4000), mouse anti-HLA-ABC heavy chain (Abcam [ab70328], 1:1000), rabbit anti-β2 M (Life Technologies, Carlsbad, CA [4H5L5], 1:1000), rabbit anti-Skp1 (Cell Signaling Technology, Danvers, MA [2156S], 1:750), rabbit anti-Nrf1 (Cell Signaling Technology [D5B10], 1:1000), rabbit anti-lamin A/C (Cell Signaling [2032S]; 1:1000), or mouse anti-Hsp90 (Santa Cruz [13119]; 1:250). Bound primary antibodies were detected using horseradish peroxidase (HRP)-conjugated horse anti-mouse IgG, anti-rabbit IgG, and anti-rat IgG (Cell Signaling Technology, [7076S, 7074S, 7077S], 1:10,000). All blots were incubated with either SuperSignal West Pico PLUS, SuperSignal West Dura, or SuperSignal West Femto chemiluminescent substrate (ThermoFisher Scientific) prior to imaging in a ChemiDoc Touch Imaging System (Bio-Rad). Bio-Rad Image Lab 6.0 software was used to obtain densitometric values.
Mass spectrometry-based immunoprecipitation proteomics
HeLa cells were transfected to express Flag-NLRC5 for 18 h and then synchronously infected with O. tsutsugamushi str. Ikeda. At 24 h p.i., cells were incubated with 5 μM MG132 for 24 h. In parallel, HeLa cells were transfected to express Flag-NLRC5 with either GFP-Ank5 or GFP-Ank5-F-boxAAAAA for 18 h after which point all samples were incubated with MG132 for 24 h. Populations of GFP-positive cells were isolated using FACS as described below and lysed with TBHS-T spiked with Halt Protease and Phosphatase Inhibitor Cocktail. Flag-NLRC5 was immunoprecipitated using Fab-trap agarose resin. The beads were washed, resuspended in elution buffer (65 mM Tris HCl, 2% SDS, with 50 mM dithiothreitol (DTT)), and incubated at 100 °C for 10 min to elute bound proteins. Inputs (10-30 μg) were resolved by SDS-PAGE and screened by western blot. Eluates were stored at −80 °C until processing for mass spectrometry. Eluates were load balanced (20 µg) and surfactant depleted with Pierce Detergent Removal Spin Columns (ThermoFisher Scientific). Samples were reduced with TCEP (10 mM), carboxymethylated with iodoacetamide (15 mM), and quenched with DTT (10 mM). In solution digests were performed overnight at pH 8 and 37 °C with mass spectrometry grade trypsin (Promega, Madison, WI) at a 1:33 enzyme-to-protein ratio and quenched with formic acid (0.15%). Peptide digests were analyzed as described previously140. Samples were loaded onto a Symmetry C18 trap column on a NanoAcquity (Waters, Milford, MA) ultra-performance liquid chromatography system and gradient eluted from 6% to 44% acetonitrile in water (0.1% formic acid) over 90 min at 400 nL min−1 using a 150 mm × 75 µm HSS T3 column maintained at 55 °C. Eluting peptides were electrosprayed into a Synapt G2-Si tandem mass spectrometer (Waters). Data were acquired in data-independent acquisition mode with ion-mobility separation and collision energy optimization (UDMSe). Ions were resolved between 400 and 1800 m/z at a nominal resolution of 25,000. Spectra were peak picked, integrated and precursor-to-product aligned using PLGS 3.0.3 with data aligned across biological replicates by retention time (±0.8 min), mobility drift time (±2 bins) and precursor ion mass (MH+, ±8 ppm) with EndogeSeq. Data were filtered to ion features that replicated in over 50% of samples and searched against the tryptic peptides of the human NLRC5 protein (accession Q86WI3) and matched to within ± 4 ppm precursor ion mass and ± 6 ppm product ion mass while accounting for fixed carboxymethylation and variable ubiquitination and phosphorylation with results controlled to a 5% false discovery rate. Ion peak area counts for ubiquitinated peptides were median centered, imputed for the limit of quantification, log-2 transformed and assessed as fold-change from respective control conditions with a student’s t-test.
Yeast two-hybrid
ULTimate yeast two-hybrid analysis was performed by Hybrigenics Services (Paris, France). Mammalian codon optimized O. tsutsugamushi str. Ikeda ank5∆F-box was PCR amplified and cloned as an N-terminal LexA fusion into pB27 (N-lexA-ank5∆F-box-C). Sequence fidelity was confirmed, and the construct was introduced into yeast (bait) and screened through mating with yeast bearing a randomly primed human placental library (prey). Positively selected clones were isolated, and captured prey fragments were PCR amplified, sequenced, and identified using NCBI Protein Basic Local Alignment Search Tool (BLASTP) (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The probability of specificity for each interaction was calculated using a predicted biological score (PBS), described as very high (A), high (B), or good (C) confidence between the two proteins141.
Analysis of ank5 homologs
The National Center for Biotechnology Information (NCBI) Nucleotide Basic Local Alignment Search Tool (BLASTN) was used to identify homologs of the representative Ikeda ank5 copy (OTT_RS01000) present in the genomes of other O. tsutsugamushi strains in GenBank. BLASTN analyzed nucleotide sequence identity and BLASTP analyzed amino acid identity of Ikeda Ank5 homologs identified in UT76 (UT76HP_01926) and Wuj/2014 (CP044031).
qPCR
Total genomic DNA (gDNA) was isolated using the DNeasy Mini Kit (Qiagen [69504]) and eluted in 20 to 30 μl of AE buffer. Concentration and purity were determined using a spectrophotometer (NanoVue Plus; GE, Boston, MA). Serial dilutions were prepared using pCR4.0::Ot_tsa56 to generate a standard curve. qPCR of standards and normalized gDNA was performed with PerfeCTa SYBR Green FastMix (QuantaBio, Beverly, MA) and primer pairs against tsa56 (Supplementary Table 1). Thermal cycling conditions used were 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 54 °C for 15 s. O. tsutsugamushi genome equivalents (GE) were quantified based on the standard curve using the CFX Maestro for Mac 1.0 software package (Bio-Rad).
RT-qPCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen[74104]) and eluted in 25 μl of RNase-free water. Concentration and purity were determined. One μg of RNA was treated with amplification grade DNase I (Invitrogen) following the manufacturer’s protocol. cDNA was generated using the iScript Reverse Transcription Supermix protocol (Bio-Rad). Parallel reactions were performed in the absence of reverse transcriptase. To verify successful genomic DNA depletion, both samples were subjected to PCR with human GAPDH-specific primers42 and MyTaq polymerase (Bioline, Taunton, MA). The resulting PCR amplicons were visualized by agarose gel electrophoresis. qPCR using cDNA as template was performed with SsoFast EvaGreen supermix (Bio-Rad) and primer pairs against O. tsutsugamushi 16S rDNA (ott16S)67 or ank551. Primers against ank5 were designed to produce amplicons from any one of the three identical ank5 copies. Thermal cycling conditions used were 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 55 °C for 5 s. Relative expression was determined using the 2−ΔΔCT method142 as part of the CFX Maestro for Mac 1.0 software package (Bio-Rad).
FACS
HeLa cells were washed with PBS, trypsinized, and recovered by centrifugation at 500×g for 5 min. The resulting cell pellets were resuspended in 1 ml of pre-sort buffer (EDTA (Versene) Solution 0.526 mM (Irvine Scientific, Santa Ana, CA) with 2% (vol/vol) heat-inactivated FBS) and flushed through a 40 µm mesh cell strainer (Greiner Bio-One, Monroe, NC). GFP-expressing cells were sorted from non-transfected cells on a Becton Dickinson (BD) FACSMelody using BDFACSChorus 1.3.3 (BD Biosciences) in Purity Sort mode. The GFP population was gated on positivity compared to non-transfected HeLa cells. Sorted cells were collected in 10% (vol/vol) FBS in PBS and pelleted at 500×g for 5 min before progressing to additional assays.
Use of ectopically expressed Ank5 proteins to counter Orientia Ank5
HeLa cells were transfected to express GFP-Ank5, GFP-Ank5-F-boxAAAAA, GFP-Ank5∆AR4, GFP-Ank5∆AR4-F-box-AAAAA, or GFP. At 18 h, the cells were subjected to FACS to isolate GFP-positive cells. Recovered cells were centrifugated at 500×g for 5 min, resuspended in 1 ml RPMI containing 10% FBS, and halved into 6-well plates. Plates were spun at 1000×g for 3 min to optimize cell adherence and incubated in a humidified incubator at 35 °C with 5% atmospheric CO2 for 18 h. Next, the cells were infected with O. tsutsugamushi str. Ikeda as described above. At 2 h p.i., media was replaced with fresh RPMI containing 1% (vol/vol) FBS and 40 ng ml−1 human IFNγ for 24 h. At this point, cells were collected, washed, lysed, and western-blotted as described above.
Flow cytometry
Transfected or O. tsutsugamushi infected HeLa cells were washed with PBS and lifted from the flask by incubating in EDTA (Versene) Solution 0.526 mM for 10 min. Transfected cells were pelleted and processed for FACS as described above or gated on GFP positivity. Recovered cells were incubated with Fc block (Miltenyi Biotec, Bergusch Gladbach, Germany, 1:100), vortexed, then incubated for 5 min on ice. A portion of the Fc blocked-cells were separated to accommodate the unlabeled control while the remaining samples were incubated with mouse anti-human HLA-ABC W6/32 (Invitrogen [MA5-11723], 1:40) on ice for 30 min. Between each step, cells underwent centrifugation at 200×g for 5 min and washed with 1 ml pre-sort buffer or PBS. Cells were incubated with Alexa Fluor 594-conjugated goat anti-mouse IgG (Invitrogen [A-11032], 1:100) for 20 min on ice followed by fixation with 500 μl of 4% (vol/vol) paraformaldehyde (Electron Microscopy Services) on ice for 20 min. Cells were washed once more with 1 ml PBS and resuspended in 300 μl PBS before flow cytometry was performed on a BD FACSMelody using BDFACSChorus 1.3.3. Ten thousand cells were analyzed per sample using FlowJo (version 10.8.1) software (BD Biosciences).
Ank5-NLRC5 protein–protein interaction prediction
NovaFold (DNASTAR, Madison, WI), which is based on the I-TASSER algorithm143,144,145, was used to model the NLRC5 NACHT domain using iterative assembly simulations. NLRC5 is 1866 amino acids in length, which exceeds the allowable size limit for NovaDock interaction modeling. Because the LRR region was found to be dispensable for the Ank5-NLRC5 interaction herein, we utilized the NACHT domain in modeling studies. Our rationale was further guided by the shared protein architecture between NLRC5 and a related NLR protein, CIITA. CIITA and NLRC5 both bind the host cell ankyrin protein, RFXANK, as part of the transactivating enhanceosome36 and CIITA utilizes its NACHT domain to do so146,147, suggesting this region of NLRC5 could be bound by Ank5. NovaDock (DNASTAR) predicted atomic protein docking interactions between Ank5 and the NLRC5 NACHT domain using the SwarmDock algorithm148. Protean 3D (DNASTAR) was used to visually depict the NovaDock predictions.
Nuclear fractionation
Cells were washed once with 1X PBS, collected, and processed for FACS as described above. Recovered GFP-positive cell populations were lysed following the Nuclear Extraction kit (Abcam, [ab113474] Cambridge, United Kingdom) protocol. Protein concentrations were determined by Bradford assay and equivalent amounts of all samples were resolved by SDS-PAGE in 4 to 15% TGX polyacrylamide gels (Bio-Rad), blocked, immunolabelled, and imaged as described above.
Bioinformatic analysis
STRING76 was used to analyze interaction networks. AlphaFold149,150 was used to predict tertiary structures for GFP-Ank5, Ank5, Ank5-F-boxAAAAA, Ank5∆AR1-F-boxAAAAA, Ank5∆AR2-F-boxAAAAA, Ank5∆AR3-F-boxAAAAA, and Ank5∆AR4-F-boxAAAAA. Alignment and color coding of the resulting program database files were done using the PyMOL Molecular Graphic System, version 1.2r3pre, Schrödinger, LLC.
Statistical analysis
Statistical analyses were performed using the Prism 8.0 software package (GraphPad, San Diego, CA). An unpaired student t-test was used to assess differences between pairs. One-way ANOVA with Tukey’s post hoc test was used to test for a significant difference among group means. One-way ANOVA with Dunnett’s post hoc test was used to test for a significant difference among group means compared to a control group mean. Statistical significance was set at p-values of <0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information/Source Data files or from the corresponding author upon request. NLRC5 protein, accession number Q86WI3 is available [https://www.ncbi.nlm.nih.gov/protein/Q86WI3/]. Source data are provided with this paper.
References
Jongsma, M. L. M., Guarda, G. & Spaapen, R. M. The regulatory network behind MHC class I expression. Mol. Immunol. 113, 16–21 (2019).
Ibana, J. A. et al. Chlamydia trachomatis immune evasion via downregulation of MHC class I surface expression involves direct and indirect mechanisms. Infect. Dis. Obstet. Gynecol. 2011, 420905 (2011).
Vachiery, N., Trap, I., Totte, P., Martinez, D. & Bensaid, A. Inhibition of MHC class I and class II cell surface expression on bovine endothelial cells upon infection with Cowdria ruminantium. Vet. Immunol. Immunopathol. 61, 37–48 (1998).
Barrionuevo, P. & Giambartolomei, G. H. Inhibition of antigen presentation by Brucella: many more than many ways. Microbes Infect. 21, 136–142 (2019).
Meng, L. et al. PPE38 protein of mycobacterium tuberculosis inhibits macrophage MHC class I expression and dampens CD8(+) T cell responses. Front. Cell Infect. Microbiol. 7, 68 (2017).
Neumeister, B. et al. Legionella pneumophila down-regulates MHC class I expression of human monocytic host cells and thereby inhibits T cell activation. Cell Mol. Life Sci. 62, 578–588 (2005).
Schuren, A. B., Costa, A. I. & Wiertz, E. J. Recent advances in viral evasion of the MHC Class I processing pathway. Curr. Opin. Immunol. 40, 43–50 (2016).
Richards, A. L. & Jiang, J. Scrub typhus: historic perspective and current status of the worldwide presence of orientia species. Trop. Med. Infect. Dis. https://doi.org/10.3390/tropicalmed5020049 (2020).
Banerjee, A. & Kulkarni, S. Orientia tsutsugamushi: the dangerous yet neglected foe from the East. Int. J. Med. Microbiol. 311, 151467 (2021).
Chung, M. H., Kang, J. S. & Lee, J. S. Tick-borne rickettsiosis and tsutsugamushi disease recorded in 313. Infect. Chemother. https://doi.org/10.3947/ic.2023.0105 (2024).
Xu, G., Walker, D. H., Jupiter, D., Melby, P. C. & Arcari, C. M. A review of the global epidemiology of scrub typhus. PLoS Negl. Trop. Dis. 11, e0006062 (2017).
Weitzel, T. et al. Endemic scrub typhus in South America. N. Engl. J. Med. 375, 954–961 (2016).
Weitzel, T. et al. Scrub Typhus in continental Chile, 2016-2018(1). Emerg. Infect. Dis. 25, 1214–1217 (2019).
Izzard, L. et al. Isolation of a novel Orientia species (O. chuto sp. nov.) from a patient infected in Dubai. J. Clin. Microbiol. 48, 4404–4409 (2010).
Kocher, C. et al. Serologic evidence of scrub typhus in the peruvian amazon. Emerg. Infect. Dis. 23, 1389–1391 (2017).
Ghorbani, R. P., Ghorbani, A. J., Jain, M. K. & Walker, D. H. A case of scrub typhus probably acquired in Africa. Clin. Infect. Dis. 25, 1473–1474 (1997).
Osuga, K., Kimura, M., Goto, H., Shimada, K. & Suto, T. A case of Tsutsugamushi disease probably contracted in Africa. Eur. J. Clin. Microbiol. Infect. Dis. 10, 95–96 (1991).
Chen, K. et al. Detection of Orientia spp. bacteria in field-collected free-living eutrombicula chigger mites, United States. Emerg. Infect. Dis. 29, 1676–1679 (2023).
Paris, D. H. et al. Orientia tsutsugamushi in human scrub typhus eschars shows tropism for dendritic cells and monocytes rather than endothelium. PLoS Negl. Trop. Dis. 6, e1466 (2012).
Moron, C. G., Popov, V. L., Feng, H. M., Wear, D. & Walker, D. H. Identification of the target cells of Orientia tsutsugamushi in human cases of scrub typhus. Mod. Pathol. 14, 752–759 (2001).
Paris, D. H., Shelite, T. R., Day, N. P. & Walker, D. H. Unresolved problems related to scrub typhus: a seriously neglected life-threatening disease. Am. J. Trop. Med. Hyg. 89, 301–307 (2013).
Jiang, J. & Richards, A. L. Scrub typhus: no longer restricted to the tsutsugamushi triangle. Trop. Med. Infect. Dis. 3, 11 (2018).
Rajapakse, S., Weeratunga, P., Sivayoganathan, S. & Fernando, S. D. Clinical manifestations of scrub typhus. Trans. R. Soc. Trop. Med. Hyg. 111, 43–54 (2017).
Taylor, A. J., Paris, D. H. & Newton, P. N. A systematic review of mortality from untreated scrub Typhus (Orientia tsutsugamushi). PLoS Negl. Trop. Dis. 9, e0003971 (2015).
Mika-Gospodorz, B. et al. Dual RNA-seq of Orientia tsutsugamushi informs on host-pathogen interactions for this neglected intracellular human pathogen. Nat. Commun. 11, 3363 (2020).
Luce-Fedrow, A. et al. Comparison of lethal and nonlethal mouse models of orientia tsutsugamushi infection reveals T-cell population-associated cytokine signatures correlated with lethality and protection. Trop. Med. Infect. Dis. https://doi.org/10.3390/tropicalmed6030121 (2021).
Lu, M. et al. Genetic recombination of Orientia tsutsugamushi strains from scrub typhus patients in Guangxi, Southwest China, and the analysis of clinical features. Microbes Infect. https://doi.org/10.1016/j.micinf.2023.105098 (2023).
Kim, D. M. et al. Differences in clinical features according to Boryoung and Karp genotypes of Orientia tsutsugamushi. PLoS ONE 6, e22731 (2011).
Inthawong, M. et al. A time-course comparative clinical and immune response evaluation study between the human pathogenic Orientia tsutsugamushi strains: Karp and Gilliam in a rhesus macaque (Macaca mulatta) model. PLoS Negl. Trop. Dis. 16, e0010611 (2022).
Robinson, D. M., Gan, E., Chan, T. C. & Huxsoll, D. L. Patterns of rickettsemia and antibody response in Silvered Leaf Monkeys (Presbytis cristatus) after inoculation with virulent and avirulent strains of Rickettsia tsutsugamushi. J. Infect. Dis. 135, 664–668 (1977).
Soong, L. et al. An intradermal inoculation mouse model for immunological investigations of acute scrub typhus and persistent infection. PLoS Negl. Trop. Dis. 10, e0004884 (2016).
Shelite, T. R. et al. Hematogenously disseminated Orientia tsutsugamushi-infected murine model of scrub typhus [corrected]. PLoS Negl. Trop. Dis. 8, e2966 (2014).
Xu, G. et al. CD8+ T cells provide immune protection against murine disseminated endotheliotropic Orientia tsutsugamushi infection. PLoS Negl. Trop. Dis. 11, e0005763 (2017).
Hauptmann, M. et al. Protective and pathogenic roles of CD8+ T lymphocytes in murine Orientia tsutsugamushi infection. PLoS Negl. Trop. Dis. 10, e0004991 (2016).
Cho, B. A. et al. Phenotypic characterization of peripheral T cells and their dynamics in scrub typhus patients. PLoS Negl. Trop. Dis. 6, e1789 (2012).
Kobayashi, K. S. & van den Elsen, P. J. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat. Rev. Immunol. 12, 813–820 (2012).
Meissner, T. B. et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl Acad. Sci. USA 107, 13794–13799 (2010).
Yao, Y. & Qian, Y. Expression regulation and function of NLRC5. Protein Cell 4, 168–175 (2013).
Kuenzel, S. et al. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J. Immunol. 184, 1990–2000 (2010).
Williams, G. S. et al. Mice lacking the transcription factor CIITA–a second look. Int. Immunol. 10, 1957–1967 (1998).
Neerincx, A. et al. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J. Biol. Chem. 285, 26223–26232 (2010).
Rodino, K. G. et al. The obligate intracellular bacterium Orientia tsutsugamushi targets NLRC5 to modulate the major histocompatibility complex class I pathway. Infect. Immun. 87, 813–811 (2019).
Nakayama, K. et al. The Whole-genome sequencing of the obligate intracellular bacterium Orientia tsutsugamushi revealed massive gene amplification during reductive genome evolution. DNA Res. 15, 185–199 (2008).
Tamura, A. et al. Isolation of Rickettsia tsutsugamushi antigenically different from Kato, Karp, and Gilliam strains from patients. Microbiol. Immunol. 28, 873–882 (1984).
Kim, K. H. et al. Severe scrub typhus with enterocolitis by the ikeda strain of Orientia tsutsugamushi. Infect. Chemother. 44, 469 (2012).
Varghese, G. M. et al. Molecular epidemiology and genetic diversity of Orientia tsutsugamushi from patients with scrub typhus in 3 regions of India. Emerg. Infect. Dis. 21, 64–69 (2015).
Park, H. et al. Case report: fulminant myocarditis successfully treated with extracorporeal membrane oxygenation in ikeda strain Orientia tsutsugamushi infection. Front. Cardiovasc. Med. 8, 795249 (2021).
Ohashi, N. et al. Demonstration of antigenic and genotypic variation in Orientia tsutsugamushi which were isolated in Japan, and their classification into type and subtype. Microbiol. Immunol. 40, 627–638 (1996).
Batty, E. M. et al. Long-read whole genome sequencing and comparative analysis of six strains of the human pathogen Orientia tsutsugamushi. PLoS Negl. Trop. Dis. 12, e0006566 (2018).
Cho, N. H. et al. The Orientia tsutsugamushi genome reveals massive proliferation of conjugative type IV secretion system and host-cell interaction genes. Proc. Natl Acad. Sci. USA 104, 7981–7986 (2007).
VieBrock, L. et al. Orientia tsutsugamushi ankyrin repeat-containing protein family members are Type 1 secretion system substrates that traffic to the host cell endoplasmic reticulum. Front. Cell. Infect. Microbiol. 4, 186 (2015).
Jernigan, K. K. & Bordenstein, S. R. Ankyrin domains across the Tree of Life. PeerJ 2, e264 (2014).
Giengkam, S. et al. Orientia tsutsugamushi: comprehensive analysis of the mobilome of a highly fragmented and repetitive genome reveals the capacity for ongoing lateral gene transfer in an obligate intracellular bacterium. mSphere 8, e0026823 (2023).
Herbert, M., Squire, C. & Mercer, A. Poxviral ankyrin proteins. Viruses 7, 709–738 (2015).
Noroy, C. & Meyer, D. F. The super repertoire of type IV effectors in the pangenome of Ehrlichia spp. provides insights into host-specificity and pathogenesis. PLoS Comput. Biol. 17, e1008788 (2021).
Gupta, S. et al. Functional characterization of non-ankyrin repeat domains of orientia tsutsugamushi ank effectors reveals their importance for molecular pathogenesis. Infect. Immun. https://doi.org/10.1128/iai.00628-21 (2022).
Nguyen, K. M. & Busino, L. The biology of F-box proteins: the SCF Family of E3 ubiquitin ligases. Adv. Exp. Med. Biol. 1217, 111–122 (2020).
Price, C. T., Al-Quadan, T., Santic, M., Rosenshine, I. & Abu Kwaik, Y. Host proteasomal degradation generates amino acids essential for intracellular bacterial growth. Science 334, 1553–1557 (2011).
Price, C. T. D. & Abu Kwaik, Y. Evolution and adaptation of legionella pneumophila to manipulate the ubiquitination machinery of its amoebae and mammalian hosts. Biomolecules https://doi.org/10.3390/biom11010112 (2021).
Lomma, M. et al. The Legionella pneumophila F-box protein Lpp2082 (AnkB) modulates ubiquitination of the host protein parvin B and promotes intracellular replication. Cell Microbiol. 12, 1272–1291 (2010).
Rubio, D. et al. Crosstalk between the type 1 interferon and nuclear factor kappa B pathways confers resistance to a lethal virus infection. Cell Host Microbe 13, 701–710 (2013).
Liu, Z. et al. A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity 54, 247–258 e247 (2021).
Mohamed, M. R. et al. Proteomic screening of variola virus reveals a unique NF-kappaB inhibitor that is highly conserved among pathogenic orthopoxviruses. Proc. Natl Acad. Sci. USA 106, 9045–9050 (2009).
Benko, S., Magalhaes, J. G., Philpott, D. J. & Girardin, S. E. NLRC5 limits the activation of inflammatory pathways. J. Immunol. 185, 1681–1691 (2010).
Beyer, A. R. et al. Orientia tsutsugamushi Ank9 is a multifunctional effector that utilizes a novel GRIP-like Golgi localization domain for Golgi-to-endoplasmic reticulum trafficking and interacts with host COPB2. Cell. Microbiol. https://doi.org/10.1111/cmi.12727 (2017).
Evans, S. M., Rodino, K. G., Adcox, H. E. & Carlyon, J. A. Orientia tsutsugamushi uses two Ank effectors to modulate NF-κB p65 nuclear transport and inhibit NF-κB transcriptional activation. PLoS Pathog. 14, e1007023 (2018).
Rodino, K. G. et al. Orientia tsutsugamushi modulates endoplasmic reticulum-associated degradation to benefit its growth. Infect. Immun. 86, e00596–00517 (2017).
Adcox, H. E. et al. Orientia tsutsugamushi nucleomodulin Ank13 exploits the RaDAR nuclear import pathway to modulate host cell transcription. mBio https://doi.org/10.1128/mBio.01816-21 (2021).
Wangsanut, T., Brann, K. R., Adcox, H. E. & Carlyon, J. A. Orientia tsutsugamushi modulates cellular levels of NF-kappaB inhibitor p105. PLoS Negl. Trop. Dis. 15, e0009339 (2021).
Min, C. K. et al. Multiple Orientia tsutsugamushi ankyrin repeat proteins interact with SCF1 ubiquitin ligase complex and eukaryotic elongation factor 1 alpha. PLoS One 9, e105652 (2014).
Ko, Y. et al. Active escape of Orientia tsutsugamushi from cellular autophagy. Infect. Immun. 81, 552–559 (2013).
Ha, N. Y., Cho, N. H., Kim, Y. S., Choi, M. S. & Kim, I. S. An autotransporter protein from Orientia tsutsugamushi mediates adherence to nonphagocytic host cells. Infect. Immun. 79, 1718–1727 (2011).
Ko, Y., Cho, N. H., Cho, B. A., Kim, I. S. & Choi, M. S. Involvement of Ca(2)+ signaling in intracellular invasion of non-phagocytic host cells by Orientia tsutsugamushi. Micro Pathog. 50, 326–330 (2011).
Adcox, H. E., Berk, J. M., Hochstrasser, M. & Carlyon, J. A. Orientia tsutsugamushi OtDUB is expressed and interacts with adaptor protein complexes during infection. Infect. Immun. 90, e0046922 (2022).
Radhakrishnan, S. K. et al. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 38, 17–28 (2010).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Beyer, A. R. et al. Orientia tsutsugamushi strain Ikeda ankyrin repeat-containing proteins recruit SCF1 ubiquitin ligase machinery via poxvirus-like F-box motifs. J. Bacteriol. 197, 3097–3109 (2015).
Werren, J. H. et al. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327, 343–348 (2010).
McClure, E. E. et al. Engineering of obligate intracellular bacteria: progress, challenges and paradigms. Nat. Rev. Microbiol. 15, 544–558 (2017).
Kala, D. et al. Diagnosis of scrub typhus: recent advancements and challenges. 3 Biotech 10, 396 (2020).
Blacksell, S. D. et al. Genetic typing of the 56-kDa type-specific antigen gene of contemporary Orientia tsutsugamushi isolates causing human scrub typhus at two sites in north-eastern and western Thailand. FEMS Immunol. Med. Microbiol. 52, 335–342 (2008).
Drukker, M. et al. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc. Natl Acad. Sci. USA 99, 9864–9869 (2002).
Hao, J. et al. NLRC5 restricts dengue virus infection by promoting the autophagic degradation of viral NS3 through E3 ligase CUL2 (cullin 2). Autophagy 19, 1332–1347 (2023).
Mehraj, V. et al. Monocyte responses in the context of Q fever: from a static polarized model to a kinetic model of activation. J. Infect. Dis. 208, 942–951 (2013).
Yao, Y. et al. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res. 22, 836–847 (2012).
Li, J., Mahajan, A. & Tsai, M. D. Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 45, 15168–15178 (2006).
Al-Khodor, S., Price, C. T., Kalia, A. & Abu Kwaik, Y. Functional diversity of ankyrin repeats in microbial proteins. Trends Microbiol. 18, 132–139 (2010).
Islam, Z., Nagampalli, R. S. K., Fatima, M. T. & Ashraf, G. M. New paradigm in ankyrin repeats: beyond protein-protein interaction module. Int. J. Biol. Macromol. 109, 1164–1173 (2018).
Sklenovsky, P., Banas, P. & Otyepka, M. Two C-terminal ankyrin repeats form the minimal stable unit of the ankyrin repeat protein p18INK4c. J. Mol. Model. 14, 747–759 (2008).
Zhang, B. & Peng, Z. A minimum folding unit in the ankyrin repeat protein p16(INK4). J. Mol. Biol. 299, 1121–1132 (2000).
Whitaker, R. H. & Placzek, W. J. MCL1 binding to the reverse BH3 motif of P18INK4C couples cell survival to cell proliferation. Cell Death Dis. https://doi.org/10.1038/s41419-020-2351-1 (2020).
Sedgwick, S. G. & Smerdon, S. J. The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem. Sci. 24, 311–316 (1999).
Mosavi, L. K., Minor, D. L. & Peng, Z.-Y. Consensus-derived structural determinants of the ankyrin repeat motif. Proc. Natl Acad. Sci. USA 99, 16029–16034 (2002).
Zheng, N. et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 416, 703–709 (2002).
Wong, K. et al. Structural mimicry by a bacterial F box effector hijacks the host ubiquitin-proteasome system. Structure 25, 376–383 (2017).
Mosavi, L. K., Cammett, T. J., Desrosiers, D. C. & Peng, Z.-Y. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 13, 1435–1448 (2004).
Nagarajan, U. M. et al. RFX-B is the gene responsible for the most common cause of the bare lymphocyte syndrome, an MHC class II immunodeficiency. Immunity 10, 153–162 (1999).
Nekrep, N., Geyer, M., Jabrane-Ferrat, N. & Peterlin, B. M. Analysis of ankyrin repeats reveals how a single point mutation in RFXANK results in bare lymphocyte syndrome. Mol. Cell. Biol. 21, 5566–5576 (2001).
Russo, A. A., Tong, L., Lee, J. O., Jeffrey, P. D. & Pavletich, N. P. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 395, 237–243 (1998).
Byeon, I. J. et al. Tumor suppressor p16INK4A: determination of solution structure and analyses of its interaction with cyclin-dependent kinase 4. Mol. Cell 1, 421–431 (1998).
Inohara, N. & Nunez, G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 3, 371–382 (2003).
Proell, M., Riedl, S. J., Fritz, J. H., Rojas, A. M. & Schwarzenbacher, R. The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS ONE 3, e2119 (2008).
Mótyán, J. A., Bagossi, P., Benkő, S. & Tőzsér, J. A molecular model of the full-length human NOD-like receptor family CARD domain containing 5 (NLRC5) protein. BMC Bioinform. 14, 275 (2013).
Kobe, B. & Kajava, A. V. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 11, 725–732 (2001).
Choo, S. Y. The HLA system: genetics, immunology, clinical testing, and clinical implications. Yonsei Med. J. 48, 11–23 (2007).
Shiina, T., Hosomichi, K., Inoko, H. & Kulski, J. K. The HLA genomic loci map: expression, interaction, diversity and disease. J. Hum. Genet. 54, 15–39 (2009).
Neerincx, A., Rodriguez, G. M., Steimle, V. & Kufer, T. A. NLRC5 controls basal MHC class I gene expression in an MHC enhanceosome-dependent manner. J. Immunol. 188, 4940–4950 (2012).
Barbash, O., Egan, E., Pontano, L. L., Kosak, J. & Diehl, J. A. Lysine 269 is essential for cyclin D1 ubiquitylation by the SCF(Fbx4/alphaB-crystallin) ligase and subsequent proteasome-dependent degradation. Oncogene 28, 4317–4325 (2009).
Barrera, S. P. et al. PKC-dependent GlyT1 ubiquitination occurs independent of phosphorylation: inespecificity in lysine selection for ubiquitination. PLoS ONE 10, e0138897 (2015).
Feng, Q., Sekula, D., Muller, R., Freemantle, S. J. & Dmitrovsky, E. Uncovering residues that regulate cyclin D1 proteasomal degradation. Oncogene 26, 5098–5106 (2007).
Fung, T. K., Yam, C. H. & Poon, R. Y. The N-terminal regulatory domain of cyclin A contains redundant ubiquitination targeting sequences and acceptor sites. Cell Cycle 4, 1411–1420 (2005).
King, R. W., Glotzer, M. & Kirschner, M. W. Mutagenic analysis of the destruction signal of mitotic cyclins and structural characterization of ubiquitinated intermediates. Mol. Biol. Cell 7, 1343–1357 (1996).
Li, H. et al. Regulation of NF-kappaB activity by competition between RelA acetylation and ubiquitination. Oncogene 31, 611–623 (2012).
Miranda, M., Dionne, K. R., Sorkina, T. & Sorkin, A. Three ubiquitin conjugation sites in the amino terminus of the dopamine transporter mediate protein kinase C-dependent endocytosis of the transporter. Mol. Biol. Cell 18, 313–323 (2007).
Treier, M., Staszewski, L. M. & Bohmann, D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell 78, 787–798 (1994).
Liu, S. et al. Interplay between bacterial deubiquitinase and ubiquitin E3 ligase regulates ubiquitin dynamics on Legionella phagosomes. Elife https://doi.org/10.7554/eLife.58114 (2020).
Del Balzo, D., Capmany, A., Cebrian, I. & Damiani, M. T. Chlamydia trachomatis infection impairs MHC-I intracellular trafficking and antigen cross-presentation by dendritic cells. Front. Immunol. 12, 662096 (2021).
Staehli, F. et al. NLRC5 deficiency selectively impairs MHC class I- dependent lymphocyte killing by cytotoxic T cells. J. Immunol. 188, 3820–3828 (2012).
Biswas, A., Meissner, T. B., Kawai, T. & Kobayashi, K. S. Cutting edge: impaired MHC class I expression in mice deficient for Nlrc5/class I transactivator. J. Immunol. 189, 516–520 (2012).
Lupfer, C. R., Stokes, K. L., Kuriakose, T. & Kanneganti, T. D. Deficiency of the NOD-like receptor NLRC5 results in decreased CD8(+) T cell function and impaired viral clearance. J. Virol. https://doi.org/10.1128/JVI.00377-17 (2017).
Sun, T., Ferrero, R. L., Girardin, S. E., Gommerman, J. L. & Philpott, D. J. NLRC5 deficiency has a moderate impact on immunodominant CD8(+) T-cell responses during rotavirus infection of adult mice. Immunol. Cell Biol. 97, 552–562 (2019).
Yoshihama, S. et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl Acad. Sci. USA 113, 5999–6004 (2016).
Zebertavage, L. K., Alice, A., Crittenden, M. R. & Gough, M. J. Transcriptional upregulation of NLRC5 by radiation drives STING- and interferon-independent MHC-I expression on cancer cells and T cell cytotoxicity. Sci. Rep. 10, 7376 (2020).
Rodriguez, G. M. et al. NLRC5 elicits antitumor immunity by enhancing processing and presentation of tumor antigens to CD8(+) T lymphocytes. Oncoimmunology 5, e1151593 (2016).
Li, X. et al. NLRC5 expression in tumors and its role as a negative prognostic indicator in stage III non-small-cell lung cancer patients. Oncol. Lett. 10, 1533–1540 (2015).
Lu, Z. H. et al. BMI1 induces ubiquitination and protein degradation of Nod‐like receptor family CARD domain containing 5 and suppresses human leukocyte antigen class I expression to induce immune escape in non‐small cell lung cancer. Kaohsiung J. Med. Sci. https://doi.org/10.1002/kjm2.12602 (2022).
Traub, R. & Wisseman, C. L. Jr. The ecology of chigger-borne rickettsiosis (scrub typhus). J. Med. Entomol. 11, 237–303 (1974).
Walker, J. S., Chan, C. T., Manikumaran, C. & Elisberg, B. L. Attempts to infect and demonstrate transovarial transmission of R. tsutsugamushi in three species of Leptotrombidium mites. Ann. N. Y Acad. Sci. 266, 80–90 (1975).
Soong, L. Dysregulated Th1 immune and vascular responses in scrub Typhus pathogenesis. J. Immunol. 200, 1233–1240 (2018).
Valbuena, G. & Walker, D. H. Approaches to vaccines against Orientia tsutsugamushi. Front. Cell Infect. Microbiol. 2, 170 (2012).
Sonthayanon, P. et al. Association of high Orientia tsutsugamushi DNA loads with disease of greater severity in adults with scrub typhus. J. Clin. Microbiol. 47, 430–434 (2009).
Thiriot, J., Liang, Y., Fisher, J., Walker, D. H. & Soong, L. Host transcriptomic profiling of CD-1 outbred mice with severe clinical outcomes following infection with Orientia tsutsugamushi. PLoS Negl. Trop. Dis. 16, e0010459 (2022).
Trent, B. et al. Polarized lung inflammation and Tie2/angiopoietin-mediated endothelial dysfunction during severe Orientia tsutsugamushi infection. PLoS Negl. Trop. Dis. 14, e0007675 (2020).
Soong, L. et al. Strong type 1, but impaired type 2, immune responses contribute to Orientia tsutsugamushi-induced pathology in mice. PLoS Negl. Trop. Dis. 8, e3191 (2014).
Shelite, T. R. et al. IL-33-dependent endothelial activation contributes to apoptosis and renal injury in orientia tsutsugamushi-infected mice. PLoS Negl. Trop. Dis. 10, e0004467 (2016).
Cornel, A. M., Mimpen, I. L. & Nierkens, S. MHC class I downregulation in cancer: underlying mechanisms and potential targets for cancer immunotherapy. Cancers https://doi.org/10.3390/cancers12071760 (2020).
Gobin, S. J., Keijsers, V., van Zutphen, M. & van den Elsen, P. J. The role of enhancer A in the locus-specific transactivation of classical and nonclassical HLA class I genes by nuclear factor kappa B. J. Immunol. 161, 2276–2283 (1998).
Benko, S., Kovacs, E. G., Hezel, F. & Kufer, T. A. NLRC5 Functions beyond MHC I Regulation-What Do We Know So Far? Front Immunol. 8, 150 (2017).
Cho, S. X. et al. MHC class I transactivator NLRC5 in host immunity, cancer and beyond. Immunology 162, 252–261 (2021).
Mostovenko, E. et al. Carbon nanotube exposure triggers a cerebral peptidomic response: barrier compromise, neuroinflammation, and a hyperexcited state. Toxicol. Sci. 182, 107–119 (2021).
Rain, J. C. et al. The protein-protein interaction map of Helicobacter pylori. Nature 409, 211–215 (2001).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Yang, J. et al. The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015).
Yang, J. & Zhang, Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181 (2015).
Zheng, W. et al. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods https://doi.org/10.1016/j.crmeth.2021.100014 (2021).
Zhu, X. S. et al. Transcriptional scaffold: CIITA interacts with NF-Y, RFX, and CREB to cause stereospecific regulation of the class II major histocompatibility complex promoter. Mol. Cell Biol. 20, 6051–6061 (2000).
Krawczyk, M., Masternak, K., Zufferey, M., Barras, E. & Reith, W. New functions of the major histocompatibility complex class II-specific transcription factor RFXANK revealed by a high-resolution mutagenesis study. Mol. Cell Biol. 25, 8607–8618 (2005).
Torchala, M. et al. Enhanced sampling of protein conformational states for dynamic cross-docking within the protein-protein docking server SwarmDock. Proteins 88, 962–972 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Acknowledgements
We thank Dr. Rebecca Martin for assisting with flow cytometry data analysis and Dr. Travis J. Chiarelli, Dr. Erol Fikrig, Dr. Leigh A. Knodler, Dr. Curtis B. Read, Dr. Jeanne Salje, Dr. Savannah E. Sanchez, and Dr. Mary M. Weber for critical review of this manuscript. This work was supported by National Institutes of Health-National Institute of Allergy and Infectious Diseases (www.niaid.gov) grants 1R01 AI123346, 2R56 AI123346, and 1R01 AI167857 to J.A.C., 1F32 AI174656A1 to P.E.A., and American Heart Association grant 20PRE35210610 to H.E.A. Laser scanning confocal was performed at the VCU Massey Comprehensive Cancer Center Microscopy Shared Resource, which is supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Author information
Authors and Affiliations
Contributions
Conceptualization, H.E.A., J.R.H., P.E.A., and J.A.C.; investigation, H.E.A, J.R.H., P.E.A., T.E.S., A.K.O., and K.G.R.; writing – original draft, H.E.A. and J.A.C.; writing – review and editing, H.E.A., P.E.A., A.K.O., and J.A.C.; supervision, J.A.C.; acquisition of grant support, J.A.C., A.K.O., H.E.A., and P.E.A.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Andrea Lipińska and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Adcox, H.E., Hunt, J.R., Allen, P.E. et al. Orientia tsutsugamushi Ank5 promotes NLRC5 cytoplasmic retention and degradation to inhibit MHC class I expression. Nat Commun 15, 8069 (2024). https://doi.org/10.1038/s41467-024-52119-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52119-6
This article is cited by
-
New insights into the structure domain and function of NLR family CARD domain containing 5
Cell Communication and Signaling (2025)
-
Orientia tsutsugamushi modulates p53, the cell cycle, and genotoxicity to maintain its intracellular niche
Nature Communications (2025)
