
Abstract
Germline genetic context may play a significant role in the development and evolution of cancer, particularly in childhood cancers such as neuroblastoma. This study investigates the role of putatively functional germline variants in neuroblastoma, even if they do not directly increase disease risk. Our whole-exome sequencing analysis of 125 patients with neuroblastoma reveals a positive correlation between germline variant burden and somatic mutations. Moreover, patients with higher germline variant burden exhibit worse outcomes. Similar findings are observed in the independent neuroblastoma cohort where a higher germline variant burden correlates with a higher somatic mutational burden and a worse overall survival outcome. However, contrasting results emerge in adult-onset cancer, emphasizing the importance of germline genetics in neuroblastoma. The enrichment of putatively functional germline variants in cancer predisposition genes is borderline significant when compared to healthy populations (P = 0.077; Odds Ratio, 1.45; 95% confidence intervals, 0.94−2.21) and significantly more pronounced against adult-onset cancers (P = 0.016; Odds Ratio, 2.13; 95% confidence intervals, 1.10−3.91). Additionally, the presence of these variants proves to have prognostic significance in neuroblastoma (log-rank P < 0.001), and combining germline with clinical risk factors notably improves survival predictions.
Similar content being viewed by others
Introduction
As individuals age, the risk of accumulating mutations in their DNA increases significantly1,2. This accumulation of mutations is the primary reason that the risk of developing cancer increases over time3,4. However, the causes of childhood cancers such as neuroblastoma (the most common extracranial solid cancer in childhood5) may differ from those of adult-onset cancers6. Instead, inherited germline genetic variants may play a greater role in the development and presentation of tumors, as children have fewer opportunities to accumulate mutations.
Considerable research into germline genetics in neuroblastomas has revealed fundamental insights into predisposing germline variations. Genome-wide association studies (GWAS) have identified dozens of single-nucleotide polymorphisms associated with neuroblastoma risk7,8,9, while next-generation sequencing studies have reported several rare germline pathogenic variants in cancer predisposition genes (CPGs)10,11,12,13. However, variants identified from GWAS typically have modest effects14, and only a small proportion of patients with neuroblastoma exhibit known pathogenic germline variants in CPGs8,11,15,16. This indicates that most neuroblastomas occurred in the absence of highly penetrant germline variants. Additionally, previous studies have only focused on the predisposition to neuroblastoma associated with germline variants, but have overlooked the possibility that germline variants that do not possess a direct association with elevating the disease risk, could have a significant impact on neuroblastoma biology. Such variants could potentially contribute to the disease either collectively or by interacting with somatic mutations, as evidenced by recent research17,18,19,20.
We, therefore, hypothesized that putatively functional germline variants (pFGVs) that affect protein function or structure may influence tumor biology in neuroblastoma, even if they do not independently increase the risk of the disease. Furthermore, we anticipated that the degree of contribution to tumor biology would be greater in neuroblastoma than in adult-onset cancer. To explore these hypotheses, we conducted a germline whole-exome sequencing (WES) study of patients with neuroblastoma, focusing on two types of pFGVs: damaging missense and protein-truncating variants (PTVs). We investigated the role of pFGV burden as well as pFGVs in CPGs. To validate our findings in a separate neuroblastoma cohort, we replicated our analysis on WES data from the NCI-Therapeutically Applicable Research to Generate Effective Treatments (TARGET) neuroblastoma cohort6. We also analyzed pFGVs from The Cancer Genome Atlas (TCGA)21 and the Korean Genome Project (Korea1K)22 dataset to compare the roles of pFGVs in neuroblastoma patients to those in adult-onset cancer and healthy individuals.
Herein, we describe the role of germline variants in neuroblastoma beyond their role in the initiation of the disease. In addition, we show the differences in the impact of germline variants on neuroblastoma and adult-onset cancers. Overall, our study highlights the importance of considering the impact of germline variants in neuroblastoma and their potential implications for patient care and management.
Results
Germline variant burden of putative functional germline variants (pFGVs) is associated with somatic mutational burden in neuroblastoma
We performed WES of germline and somatic DNA from 125 Korean neuroblastoma patients at the Samsung Medical Center (SMC cohort), with 65 cases (52%) classified as high-risk and 60 cases (48%) classified as intermediate or low-risk (Supplementary Table 1). Median age at diagnosis was 3.12 and 49% of the patients were male. Following multiple filtering steps, as depicted in Fig. 1a, we identified a median burden of 41 (range, 27−58) pFGVs per patient, of which 24 were PTVs (Fig. 1b). We observed that patients with a higher germline variant burden (above the mean) harbored a higher somatic mutational burden (the total number of nonsynonymous mutations per coding area) (P = 0.018; Fig. 1c), indicating a positive association between the two factors. We also found a statistically significant but weak correlation (Pearson’s r = 0.18; P = 0.041; Fig. 1d) between the germline variant burden and log10-transformed somatic mutational burden across all protein-coding genes. This correlation maintained nominal statistical significance in patients without pFGVs in DNA damage repair (DDR) genes (Pearson’s r = 0.23; P = 0.032; Supplementary Fig. 1a). However, we found no significant correlation between the total number of rare synonymous germline variants and somatic mutational burden (Pearson’s r = −0.01; P = 0.942; Fig. 1e). These findings persisted when we implemented a down-sampling analysis, addressing potential biases due to the disproportionate volume of synonymous variants in relation to pFGVs (Supplementary Fig. 1b, c).

a Flow chart of the pipeline to identify pFGVs. b Box plot of all pFGVs, protein-truncating variants (PTV), and missense pFGVs in all patients (n = 125). c Box plot of somatic mutational burden compared with high (above mean, n = 66) vs. low germline variant burden (below mean, n = 59). d Correlation between germline variant burden and somatic mutational burden (log-transformed). e Correlation between number of rare synonymous germline variants and somatic mutational burden (log-transformed). Each box plot displays the median value as the center line, the upper and lower box boundaries at the first and third quartiles (25th and 75th percentiles) and the whiskers extend to points within 1.5 times the interquartile range. For all the scatter plots, the r represents Pearson’s correlation coefficient and the black line represents the fitted values from linear regressions, with 95% confidence intervals in gray. All P values are derived from two-sided test. Source data are provided as a Source Data file.
To validate our findings in an independent cohort, we analyzed germline and somatic exome data from the TARGET dataset. Consistent with our observations in the SMC cohort, we found that patients with a higher germline variant burden (above the mean) also had a significantly higher somatic mutational burden (P = 0.007; Fig. 2a). We also observed a significant correlation between germline variant burden and somatic mutational burden in all TARGET patients (Spearman’s ρ = 0.26; P = 0.0001; Fig. 2b). This correlation persisted in patients without pFGVs in DDR genes (Spearman’s ρ = 0.33; P = 0.0002; Supplementary Fig. 2a), as well as in analyses that excluded outliers identified using a Z-score threshold of 3 (Spearman’s ρ = 0.24; P = 0.0005; Supplementary Fig. 2b).

a Box plot of somatic mutational burden comparing high (above mean, n = 64) vs. low (n = 156) germline variant burden in the TARGET. Boxes represent interquartile ranges with the center line corresponding to the median. The whiskers extend to points within 1.5 times the interquartile range. The statistical analysis was performed using the Wilcoxon rank-sum test. b Correlation between germline variant burden and somatic mutational burden (log-transformed) in the TARGET. c Correlation between germline variant burden and somatic mutational burden (log-transformed) in the TCGA. d Trends in Spearman’s correlation coefficient and confidence intervals between germline variant burden and somatic mutational burden across age groups at diagnosis in the TARGET (white ethnicity, n = 160) and TCGA (n = 7482). For all the scatter plots, the ρ represents Spearman’s correlation coefficient and the black line represents the fitted values from linear regressions, with 95% confidence intervals in gray. Statistical analysis for (a–c) was performed using two-sided tests. The Jonckheere-Terpstra test for (d) was performed with a one-sided alternative hypothesis (less), indicating a decreasing trend. Source data are provided as a Source Data file.
To account for potential confounding factors, we employed a multivariable regression analysis. In analysis of the SMC cohort, after adjusting for median sequencing depth in both tumor and germline and the clinical risk, the somatic mutational burden continued to show a significant positive association with germline variant burden (β = 0.01, P = 0.045). This association was consistent among TARGET neuroblastoma patients, even when adjustments were made for race and median sequencing depth in both tumor and germline (β = 0.01, P = 0.016). We also investigated whether germline variants have a greater impact on neuroblastoma than adult-onset cancers. We analyzed the TCGA adult-onset solid cancer dataset and found a negligible negative correlation between germline variant burden and somatic mutational burden (Spearman’s ρ = −0.03; P = 0.024; Fig. 2c). However, we observed a different pattern of correlation when we analyzed the data across age groups. For patients with early-onset cancer (age at diagnosis <50 years), we found a very weak but positive association between germline variant burden and somatic mutational burden (Spearman’s ρ = 0.08; P = 0.002; Supplementary Fig. 2c), whereas for patients diagnosed at an older age (age at diagnosis ≥50 years), a very weak negative correlation was observed (Spearman’s ρ = −0.06; P < 0.00001; Supplementary Fig. 2d). Finally, we analyzed the association between germline variant burden and somatic mutational burden across all age groups at diagnosis. This analysis included patients with neuroblastoma from the white ethnicity subgroup within the TARGET cohort. Interestingly, we observed a decreasing trend in the correlation coefficient between germline variant burden and somatic mutational burden (Jonckheere-Terpstra test, P = 0.0006; Fig. 2d).
Germline variant burden of pFGVs have prognostic impact in neuroblastoma
While no significant association was found between germline variant burden and any known clinical risk factors for neuroblastoma (Supplementary Fig. 3), patients with a higher germline variant burden (above the mean) had a poorer progression-free survival (PFS) (Fig. 3a; log-rank P = 0.018) but not reduced overall survival (OS) (Fig. 3b; log-rank P = 0.455) in our SMC cohort. The clinical significance of germline variant burden persisted even after adjusting for age, stage, and MYCN status using a multivariable Cox model (adjusted HR, 2.78; 95% CI, 1.19−6.51; P = 0.018). In the TARGET cohort, patients with a higher germline variant burden (above the mean) had poorer OS than those with a lower germline variant burden (log-rank P = 0.005; Fig. 3c). After adjusting for ethnicity and MYCN status, the impact of a higher germline variant burden remained statistically significant (adjusted hazard ratio [HR], 1.70; 95% CI, 1.19−2.42; P = 0.003). However, there was no prognostic significance according to germline variant burden in the TCGA cohort (log-rank P = 0.643; Fig. 3d).

a Progression-free survival (PFS) for the patients in the SMC cohort. b Overall survival (OS) for the patients in the SMC cohort. c OS for patients in the TARGET cohort. d OS for patients in the TCGA cohort. Source data are provided as a Source Data file.
Enrichment analysis of pFGVs in CPGs of neuroblastoma
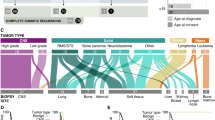
To prioritize genes with the greatest biological impact, we focused on 109 CPGs listed in the Cancer Gene Census (CGC) from the Catalogue Of Somatic Mutations In Cancer (COSMIC) database23 (Supplementary Data 1). In this analysis, we considered the role of each gene in cancer development, specifically targeting only missense variants of oncogenes, while examining all pFGVs in tumor suppressor genes (TSGs). In our SMC cohort, we identified 45 pFGVs and 31 affected CPGs in 39 of 125 patients (31%) (Fig. 4) (Supplementary Data 2). In the TARGET cohort, 97 pFGVs were identified in 79 patients (36%) (Supplementary Data 3). To determine whether pFGVs in CPGs were enriched in neuroblastoma compared with the general population and adult-onset cancers, we compared the prevalence of pFGVs in CPGs across cohorts while considering ethnicities. Our analysis revealed that pFGVs in CPGs were enriched in neuroblastoma in both the SMC and TARGET cohorts compared to healthy individuals (KOREA1K) and patients with adult-onset cancers (TCGA), respectively. In the SMC cohort, we observed a trend towards significance (P = 0.077; Odds Ratio, 1.45; 95% CI, 0.94−2.21) compared to KOREA1K, while in the TARGET cohort, we found a statistically significant difference (P = 0.027; Odds Ratio, 1.46; 95% CI, 1.04−2.05) compared to the TCGA cohort. Moreover, we observed a significant decreasing trend in the prevalence of pFGVs in CPGs across age in the TCGA cohort (Cochran–Armitage test for trend P = 0.00007; Supplementary Fig. 4). However, it was important to note that when refining our analysis based on the American College Medical Genetics (ACMG) guidelines for clinical interpretation24, which focuses sole on pathogenic or likely pathogenic (P/LP) variants in CPGs, there was a pronounced enrichment of these variants in neuroblastoma within the SMC cohort compared to the general population (P = 0.016; Odds Ratio, 2.13; 95% CI, 1.10−3.91).

Oncoprint of pFGVs in cancer predisposition genes (CPGs). The frequencies of pFGVs in CPGs are displayed as horizontal barplots (right). Source data are provided as a Source Data file.
pFGVs in CPGs are potential prognostic factors in neuroblastoma
Next, we investigated whether pFGVs in CPGs were associated with clinical factors and outcomes. pFGVs in CPGs were not associated with known clinical risk factors (Supplementary Table 2); however, we found that patients with pFGVs in CPGs had a higher incidence of family history of cancer in at least one second-degree relative (χ2(1) = 3.99; P = 0.046; Odds Ratio = 2.93; 95% CI, 1.00−8.61). Additionally, we found that pFGVs in CPGs may be potential risk factors, as they were associated with worse PFS (log-rank P = 0.00058; Fig. 5a) and OS (log-rank P = 0.025; Fig. 5b). We also analyzed the prognostic impact of other cancer-relevant genes (not CPGs), which are classified as either TSG or oncogenes in CGC (n = 565). However, no additional prognostic impact was observed for the other cancer-relevant genes (Supplementary Fig. 5). When we considered only P/LP variants according to the ACMG guidelines, a more pronounced distinction was observed in the family history of cancer between patients harboring P/LP variants in CPGs and those without such variants (χ2(1) = 5.18; P = 0.023; Odds Ratio = 5.42; 95% CI, 1.11−26.52). However, the survival differences in survival outcomes were significantly only for only OS (log-rank P = 0.009; Supplementary Fig. 6a), and not for PFS (log-rank P = 0.308; Supplementary Fig. 6b). Univariable Cox proportional-hazards analysis revealed that MYCN status, germline variant burden, and pFGVs in CPGs were associated with the risk of progression or relapse (Supplementary Fig. 7). Multivariable Cox proportional-hazards analysis showed that the presence of pFGVs in CPGs was independently associated with risk of progression or relapse, even after adjusting for age, stage, MYCN status, risk, and germline variant burden (adjusted HR, 2.91; 95% CI, 1.35−6.28; P = 0.006; Fig. 5c). Interestingly, even after excluding patients with pFGVs in CPGs, there was still a significant prognostic impact of the germline variant burden (PFS; log-rank P = 0.013; Fig. 5d). In the TARGET cohort, which consisted of patients aged >18 months, stage 4, and high-risk, a similar trend was observed, although the survival difference was not statistically significant when evaluating the presence of pFGVs in CPGs (OS, log-rank P = 0.191). However, in the subgroup analysis of the TARGET cohort, there was a significant difference in OS in patients without MYCN amplification based on the presence of pFGVs in CPGs (log-rank P = 0.016 and 0.414 for patients without and with MYCN amplification, respectively; Fig. 6a, b). These results were also consistent in the SMC cohort with nominal statistical significance (Fig. 6c, d). When assessing pathogenicity as per the ACMG guidelines, the TARGET cohort displayed significant differences in OS log-rank (P = 0.025). However, in the TCGA adult-onset cancer cohort, we observed the opposite trend, with no statistical significance in OS, as expected (log-rank P = 0.119).

a Kaplan–Meier survival curves for progression-free survival (PFS). b Kaplan–Meier survival curves for overall survival (OS). c Forest plot of Cox multivariable regression analysis for PFS. d Kaplan–Meier survival curves for PFS in patients without pFGVs in cancer predisposition genes (CPGs) according to germline variant burden. All P values are two-sided without correction for multiple comparisons. Source data are provided as a Source Data file.

a Overall survival (OS) for patients without MYCN amplification (TARGET cohort). b OS for patients with MYCN amplification (TARGET cohort). c OS for patients without MYCN amplification (SMC cohort). d OS for patients with MYCN amplification (SMC cohort). Source data are provided as a Source Data file.
The added predictive value of germline risk factors in neuroblastoma
As both the burden and presence of affected CPGs were independent risk factors for PFS, we investigated whether germline risk factors (high germline variant burden and presence of pFGVs in CPGs) provide additional benefits for stratifying patients with neuroblastoma. Therefore, we obtained the C-index distribution of clinical risk stratification in the development and internal validation groups using the bootstrap method. In the development group, the combination of germline risk factors and clinical risk factors (age, stage, MYCN status) demonstrated better discrimination power than clinical risk factors alone (mean C-index; 0.85 vs. 0.77, P < 0.00001 after Bonferroni correction; Fig. 7a). This finding was confirmed in the internal validation group (0.86 vs. 0.77; P < 0.00001 after Bonferroni correction; Fig. 7b).

a Training group. b Internal validation group. The box plots represent the C-index values calculated from 500 bootstrap samples, with a 6:4 training to validation split for each iteration. For all the box plots, the central line represents the median value, the top and bottom of the box represent the 25th and 75th percentiles, and the whiskers extend to points within 1.5 times the interquartile range. All P values are two-sided t-test results with Bonferroni correction for multiple comparisons. Source data are provided as a Source Data file.
Discussion
Cancer genomics research in pediatric patients has focused on the discovery of somatic drivers of tumorigenesis6,13,25,26, and has revealed that pediatric cancers, including neuroblastoma, have few somatic mutations. However, it is becoming increasingly clear that germline variants, inherent to each patient’s genetic makeup, can significantly shape tumor characteristics. Mounting evidence underscores the significance of germline variants, extending beyond cancer susceptibility to influence tumor progression and phenotype8,15,17,18,19,20,27,28,29,30,31,32. Our work builds on this foundation, focusing on the comprehensive analysis of rare germline variants and their broader implication in tumor biology and patient outcomes. In the present study, we comprehensively analyzed the role of rare, potentially harmful germline variants in patients to gain a holistic understanding of their influence.
It is widely recognized that the tumor mutational burden correlates with age at diagnosis33,34. This is because, as individuals age, environmental mutagens and mutations in DNA repair genes accumulate, which can lead to cancer development33,34. However, childhood cancers arise from different processes21. Our findings suggest that the burden of pFGVs affects somatic mutagenesis in neuroblastoma as well as in young adult-onset cancer patients. These results were consistent even in patients without pFGVs in DDR genes. In addition, we observed that the degree to which germline variants contributed to somatic mutation decreased over the course of their lifetimes. These findings are particularly noteworthy because previous studies on germline variant burden have only been conducted in adult-onset cancer and have not identified a reverse association between early-onset and late-onset cancer. Furthermore, while many studies have identified a personal germline variant burden in specific or manually curated gene lists17,19,30, we avoided potential biases by refraining from selecting specific genes for analysis. Finally, we also demonstrated that germline variant burden contributes not only to somatic mutations but also to neuroblastoma survival outcomes, which represents a significant finding.
Qing et al.19 have clearly described the association between germline variants and somatic mutations in adult-onset solid cancer, whereas our variant filtering process differed from theirs. In our analysis, we opted for the REVEL35 method, which has demonstrated superior performance in comparison to MetaSVM36, employed by Qing et al. Additionally, we further refined our selection by excluding variants that were present in more than 10% of each cohort, aiming to minimize false positives. Consequently, our findings present a narrower range of variants, with no more than 203 variants per patient in the TCGA cohort, in contrast to the 79−239 variant range reported in Qing’s study. Another difference is our study’s focus on pediatric patients and the inclusion of a wide array of genomic data, not limited to cancer-specific genes. This likely accounts for the observed weaker correlation between germline variants and somatic mutations compared to the associations reported by Qing et al.
We also investigated the prevalence and clinical relevance of germline variants in CPGs. The study by Kim et al., which includes analyses from the TARGET dataset that our research also examines, highlights the prevalence and potential prognostic implications of P/LP variants in CPGs29. However, it is important to recognize that our understanding of the role of these variants across a broader patient population remains limited. Additionally, most patients were predominantly of European ancestry, limiting their representativeness. Our study was designed to provide a comprehensive landscape of pFGVs in CPGs and explore the role of these variants in neuroblastoma, regardless of confirmed pathogenicity. We identified deleterious germline variants in CPGs of a substantial proportion of patients with neuroblastoma. We also demonstrated that using all pFGVs in CPGs was effective in predicting disease progression in a cohort of unselected patients (the SMC cohort). Importantly, we also observed that germline variant burden had a prognostic impact in patients without pFGVs in CPGs. This suggests that even pFGVs in nondefinitive CPGs may have a biological impact on neuroblastomas. Furthermore, we showed that pFGVs in CPGs serve as critical determinants of OS in patients without the strongest somatic driver alterations (MYCN amplification) in the both SMC and TARGET cohorts. Overall, our study may expand the definition of pathogenicity and highlights the significance of the identified pFGVs.
Neuroblastoma treatment strategies have considerably evolved over time, reflecting advances in medical research and clinical practice. It is essential to contextualize our findings within the treatment era of the patient cohorts studied. The TARGET cohort, comprising exclusively high-risk patients, experienced a wide variety of high-risk treatment protocols. These included different induction regimens37, the use of high-dose chemotherapy38, variations in both the chemotherapy regimens39 and the number of high-dose chemotherapy cycles40, adjustments in radiation therapy doses41, and the introduction of anti-GD2 maintenance therapy42,43. In contrast, the SMC cohort, which included patients from all clinical risk groups, could not utilize anti-GD2 therapy. Instead, for high-risk patients, it adopted the implementation of intensified tandem high-dose chemotherapy and high-dose MIBG treatment44,45. Despite these differences and changes in treatment paradigms, the prognostic value of germline variants remains evident.
However, our study has several limitations. First, we could not determine how the burden of germline variants affected somatic mutations. The association signals between germline variant burden and somatic mutation burden were weak, and it is important to note that it is unlikely that all the disruptions in protein-coding genes are equally important on somatic mutations. Furthermore, our control cohorts were not subjected to the same experimental conditions or variant calling processes as the case cohorts, as they relied on pre-processed variant data. This introduces a layer of complexity that might affect the comparability of our findings. The total count of germline variants and the identification of pFGVs in CPGs identified could have been affected by the specific experimental design and variant filtering processes, which varied across cohorts. Consequently, interpretations of the germline variant burden and the presence of pFGVs in CPGs should be approached with caution at an individual level, and this variance in methodology complicates direct comparisons between cohorts. Third, the lack of functional data on pFGVs hindered our ability to annotate and predict the effects of these variants on proteins, Additionally, the absence of analysis of parents’ data precluded us from determining the origin of the identified pFGVs. Finally, our pFGVs cannot supplant or diminish the importance of P/LP variants as defined by the ACMG. This is because a family history of cancer, enrichment, and some observed survival differences are more pronounced when adhering strictly to P/LP classifications compared to pFGVs.
Despite these limitations, this study has several important clinical implications. We showed that differences in somatic aberrations and outcomes between tumors could be partially explained by collectively considering a patient’s germline variants. This suggests that a larger number of germline variants may affect somatic mutations and outcomes in neuroblastoma, which differs from observations in adult-onset cancers. Additionally, we have broadened our understanding of pathogenic variants in CPGs, encompassing aspects beyond disease predisposition. Our study highlights the feasibility of incorporating germline risk factors into the clinical risk assessment of patients with neuroblastoma, as these germline risk factors were independent of known clinical risk factors and had an impact on patient outcomes. Our approach of using WES to investigate a broader range of pFGVs in patients with neuroblastoma may be generalizable to patients with other pediatric cancers and could have a broad impact.
Methods
Ethics statement
This study adheres to all applicable ethical regulations. The Samsung Medical Center Institutional Review Board approved the study (IRB No. 2015-11-053-014). Written informed consent for germline and tumor sequencing, as well as for the review of medical records for demographic, clinical, and pathological information, was obtained from the parents or legal guardians of all patients. Participants were not compensated, as their participation did not incur any additional costs. The study design did not take sex and gender into account.
Patient cohort and sequencing
We analyzed blood and tissue DNA from 125 neuroblastoma patients diagnosed between 2012 and 2021, initially identifying 145 patients with peripheral neuroblastoma tumors. After excluding ganglioneuroma cases (n = 6), tumors obtained post-relapse (n = 9), patients with unmatched DNA pairs confirmed by NGSCheckMate546 (n = 1), and non-primary site tumors (n = 4), our analysis focused on the remaining 125 cases. Most patients (82%) underwent prospective clinical sequencing. However, 23 patients (18%) were also included in this study using samples deposited at the SMC BioBank. This study included high-risk patients (n = 54) enrolled in the NB-2014 clinical trial (NCT02771743) designed to evaluate the potential benefits of response-adapted strategies in consolidation therapy. The trial design and clinical trial protocol have been previously documented47. The clinical outcomes were reported according to the trial’s objectives, including OS, PFS, and adverse events. In this analysis, we incorporated extended follow-up and survival data, which differs from the original trial that initiated follow-up after the induction period, as the original trial focused on the effects of consolidation treatment. Here, we calculated survival time starting from the time of diagnosis. Consequently, the outcome analysis used here includes non-prespecified exploratory outcomes for the NB-2014. These analyses, though not originally included in the NB-2014 trial specifications, were crucial for identifying the significance of germline variants. Genomic sequencing was performed on tumor and normal DNA extracted from fresh frozen (74%) or formalin-fixed paraffin-embedded tissues (26%) and mononuclear cells from peripheral blood, respectively, using a QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA). Tumor and matched DNA were enriched for exon regions using the SureSelectXT Human All Exon V5 kit (Agilent Technologies Inc., Santa Clara, CA, USA). All tumor specimens were reviewed by a pathologist to determine the percentage of viable tumors and their adequacy for sequencing. Patient samples were sequenced with pair-end 100-bp reads using the Illumina HiSeq 2500 platform (Illumina, Technologies Inc., San Diego, CA, USA). This study included only tumors obtained from the primary site.
Putatively functional germline variants
Illumina WES data were mapped to hg19 using Burrows-Wheeler Aligner (BWA)48 v0.7.17. Picard v2.17.5 (http://broadinstitute.github.io/picard/) and the genome analysis toolkit (GATK)49 v 4.0.2 was used for indel realignment, duplicate removal, and base- and quality-score recalibration. HaplotypeCaller in GATK was used for variant calling of SNVs and short indels. We removed variants with low quality (genotype quality-score <50) or inadequate read coverage (<10×), variants with a variant allele frequency (VAF) less than 30%, and variants commonly observed in healthy populations (>1% in ExAC50 or >1% KRGDB110051). The functional impact of missense germline variants was predicted using the REVEL score and annotated using the ClinVar52 (03-20-2022) database when available. We considered a missense variant to have a high functional impact if the REVEL score35 was ≥0.7 or listed as P/LP in ClinVar. PTVs, including frameshift indels, stop gain, stop loss, and splice-site variants, were also considered as pFGVs. Variants annotated as benign or likely benign in ClinVar were excluded. The deleterious variant burden in a sample was calculated as the total number of pFGVs in the coding regions. To remove platform-related artifacts, variants commonly observed (>10%) in the entire SMC cohort were also removed. Rare synonymous variants were defined as synonymous variants with an allele frequency <1% in the ExAC and KRGDB100 databases and as well as occurring in <10% of the cohort. The same QC filters were applied to both pFGVs and rare synonymous variants.
TARGET dataset
We obtained FASTQ files for germline and tumor samples from 222 patients using data from the Database of Genotypes and Phenotypes (dbGaP), accession phs000218.v24.p8 via the SRA Toolkit. We only retained individuals whose tumor sequencing was carried out on primary rather than metastatic tumors (220 patients). We then applied our variant calling pipeline to convert TARGET-NBL FASTQ files into a variant calling format (VCF) using the same methodology employed for our SMC cohort. The variants were then pre-filtered using previously reported QC steps29 for the TARGET data. We only retained variants with a read-depth coverage of at least 15 and a VAF of at least 0.2. Subsequently, we applied the same annotation steps and removed common variants within the TARGET cohort (>1% in ethnic-matched ExAC and >10% of the samples) to retrieve pFGVs, except KRGDB1100 (a variant resource specific to Koreans). Ethnicity was determined by using the R package EthSEQ53 (version 3.0.2) with a single-nucleotide polymorphism call rate threshold of 98% (Supplementary Fig. 8).
TCGA dataset
We downloaded the filtered variant calls (VCF) from 10,389 patients released by TCGA pan-cancer germline study21 (https://gdc.cancer.gov/about-data/publications/PanCancerAtlas-Germline-AWG). We acquired pFGVs from TCGA data using the same QC and annotation steps applied to our SMC cohort. We also removed common variants in ExAC (<1%) and variants found in >10% of the TCGA cohort. Children (age at diagnosis <18 years) and patients with hematological malignancies were excluded from the study. We also limited the analysis to individuals of self-reported white ethnicity in the TCGA pan-cancer cohort and compared it to the self-reported white ethnicity group in the TARGET cohort. As a result, we included 7482 patients with 31 types of solid cancers.
KOREA1K
The Korea1K22 dataset comprises the whole-genome data of 1094 healthy individuals. We downloaded the VCF files released by Korea1K and included 916 unrelated individuals. The variants were initially called on the hg38 genome assembly and lifted to the hg19 genome assembly using LiftoverVcf in the Picard package. We then extracted the pFGVs using our variant filtering pipeline.
Somatic mutational analysis
For the SMC and TARGET data, we called SNPs and small indels using Mutect254 and Manta55/Strelka256. We excluded common variants (VAF > 0.001 in gnomAD v.2.0), low variant allele fractions (VAF < 0.05), variants that did not have a minimum read-depth coverage of 30 reads, and those with fewer than three reads supporting the altered allele. Filtered variants were annotated using Variant Effector Predictor57 from the Ensembl database. In the TARGET data, we further filtered potential oxoG artifacts by removing G > T or C > A mutations with VAF < 0.15, as suggested for the TARGET data in previous reports6,58. The somatic mutational burden was calculated as the number of nonsynonymous variants. Somatic mutations in the TCGA were obtained from TCGA PanCancer Atlas MC3 set59, which is the result of applying an ensemble of seven mutation-calling algorithms, complete with scoring and artifact filtering60. Then we applied the same somatic mutation call pipeline used in the SMC cohort.
CPG and DDR gene sets
Among the 733 genes listed in the COSMIC database23 (CGC) (Supplementary Data 1), we compiled a list of 109 known CPGs according to their annotations. To account for their roles, we classified genes with only TSG annotations as TSG in our analysis. Eighty core DDR genes were obtained from Knijnenburg et al.61 (Supplementary Data 4).
MYCN amplification status
MYCN amplification status was determined primarily by fluorescence in situ hybridization (FISH). In cases where FISH data were unavailable, CNVKit (v.0.9.6)62 was used to determine MYCN amplification status. According to CNVKit analysis, MYCN was considered to be copy-gained when there were gains with log2 fold changes greater than 2.0 relative to the normal.
Statistical analysis
Normality of the data distribution was determined using the Shapiro–Wilk test. Differences in continuous traits between the two groups were determined using independent t-tests or non-parametric equivalent Wilcoxon rank-sum tests. Pearson or Spearman correlation coefficients were used to assess the relationship between germline variant burden and somatic mutational burden. We used the log10-transformed somatic mutational burden to increase normality. In the down-sampling analysis of synonymous variants, we incrementally reduced their count in 1% increments, starting from 10% and progressing to 100%. At each step of this process, the correlation between the number of rare synonymous germline variants and the total somatic mutation burden was recalculated. To assess trends across age groups in the TCGA, we used the Jonckheere-Terpstra trend test for continuous variables and the Cochran–Armitage trend test for categorical variables. Two-sided Fisher’s exact test was used to compare the enrichment of pFGVs in neuroblastomas to controls, and odds ratios with 95% intervals were reported. Kaplan–Meier analysis of PFS and OS were performed to compare the outcomes of patients with or without germline risk factors. The log-rank test was used to compare PFS and OS between groups. Univariable and multivariable Cox proportional-hazards regression models were used to assess whether a higher germline variant burden or the presence of pFGVs in CPGs were independently predictive of survival. A subgroup analysis between the presence of pFGVs in CPGs and MYCN status was also performed to evaluate any heterogeneous associations. Differences were considered statistically significant at P ≤ 0.05, and the tests were 2-tailed unless otherwise specified. For internal validation of our predictive model, we performed permutation testing over 500 iterations, randomly dividing the dataset into development (60%) and internal validation (40%) sets for each cycle. The model’s discriminatory power was quantitatively assessed using Harrell’s C-index, conducted with 500 bootstrap replicates to ensure robustness. All analyses were performed in R version 4.2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequencing data generated in this study are available in the NCBI Sequence Read Archive Repository under accession number PRJNA592880 and are publicly available. The TARGET neuroblastoma WES data files were downloaded from dbGaP under accession number phs000467. These data are available under dbGaP-controlled access for general research purposes. Approved users will receive access to the data for a period of 12 months, after which they will need to either renew their access or close out the project. The TCGA germline variants can be accessed at [https://gdc.cancer.gov/about-data/publications/PanCanAtlas-Germline-AWG], and these data also require dbGaP authorization (accession number phs000178.v11.p8). The publicly available TCGA somatic mutations are accessible at [https://gdc.cancer.gov/about-data/publications/mc3-2017]. KOREA1K data available through [http://koreangenome.org/], and access is subject to approval from the committee, with the process detailed at [http://1000genomes.kr/]. The remaining source data are available within the Article, Supplementary Information, or Source Data file. Source data are provided with this paper.
Code availability
All analyses were performed using standard publicly available software. Our custom code for analysis and figures is available at https://github.com/SGIlabes/NBL_Germline/ and a persistent copy of this repository is available via Zenodo (https://doi.org/10.5281/zenodo.13324781)63.
References
Mustjoki, S. & Young, N. S. Somatic mutations in “Benign” disease. N. Engl. J. Med. 384, 2039–2052 (2021).
Lee-Six, H. et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 574, 532–537 (2019).
Xie, M. et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472–1478 (2014).
Stratton, M. R., Campbell, P. J. & Futreal, P. A. The cancer genome. Nature 458, 719–724 (2009).
Maris, J. M. Recent advances in neuroblastoma. N. Engl. J. Med. 362, 2202–2211 (2010).
Pugh, T. J. et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 45, 279–284 (2013).
Maris, J. M. et al. Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N. Engl. J. Med. 358, 2585–2593 (2008).
Barr, E. K. & Applebaum, M. A. Genetic predisposition to neuroblastoma. Children 5, 119 (2018).
He, J. et al. Genetic variations of GWAS-identified genes and neuroblastoma susceptibility: a replication study in Southern Chinese children. Transl. Oncol. 10, 936–941 (2017).
Sausen, M. et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 45, 12–17 (2013).
Zhang, J. et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med. 373, 2336–2346 (2015).
Wang, Z. et al. Genetic risk for subsequent neoplasms among long-term survivors of childhood cancer. J. Clin. Oncol. 36, 2078–2087 (2018).
Fiala, E. M. et al. Prospective pan-cancer germline testing using MSK-IMPACT informs clinical translation in 751 patients with pediatric solid tumors. Nat. Cancer 2, 357–365 (2021).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484 (2019).
Akhavanfard, S., Padmanabhan, R., Yehia, L., Cheng, F. & Eng, C. Comprehensive germline genomic profiles of children, adolescents and young adults with solid tumors. Nat. Commun. 11, 2206 (2020).
Ritenour, L. E., Randall, M. P., Bosse, K. R. & Diskin, S. J. Genetic susceptibility to neuroblastoma: current knowledge and future directions. Cell Tissue Res. 372, 287–307 (2018).
Vosoughi, A. et al. Common germline-somatic variant interactions in advanced urothelial cancer. Nat. Commun. 11, 6195 (2020).
Srinivasan, P. et al. The context-specific role of germline pathogenicity in tumorigenesis. Nat. Genet. 53, 1577–1585 (2021).
Qing, T. et al. Germline variant burden in cancer genes correlates with age at diagnosis and somatic mutation burden. Nat. Commun. 11, 2438 (2020).
Vali-Pour, M., Lehner, B. & Supek, F. The impact of rare germline variants on human somatic mutation processes. Nat. Commun. 13, 3724 (2022).
Huang, K. L. et al. Pathogenic germline variants in 10,389 adult cancers. Cell 173, 355–370.e314 (2018).
Jeon, S. et al. Korean Genome Project: 1094 Korean personal genomes with clinical information. Sci. Adv. 6, eaaz7835 (2020).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–d947 (2019).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015).
Newman, S. et al. Genomes for Kids: the scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Discov. 11, 3008–3027 (2021).
Liu, Y. et al. Etiology of oncogenic fusions in 5,190 childhood cancers and its clinical and therapeutic implication. Nat. Commun. 14, 1739 (2023).
Diskin, S. J. et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat. Genet. 44, 1126–1130 (2012).
Cimmino, F. et al. Fine mapping of 2q35 high-risk neuroblastoma locus reveals independent functional risk variants and suggests full-length BARD1 as tumor-suppressor. Int. J. Cancer 143, 2828–2837 (2018).
Kim, J. et al. Germline pathogenic variants in neuroblastoma patients are enriched in BARD1 and predict worse survival. J. Natl Cancer Inst. 116, 149–159 (2024).
Esai Selvan, M., Onel, K., Gnjatic, S., Klein, R. J. & Gümüş, Z. H. Germline rare deleterious variant load alters cancer risk, age of onset and tumor characteristics. npj Precis. Oncol. 7, 13 (2023).
Mossé, Y. P. et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455, 930–935 (2008).
Wang, K. et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature 469, 216–220 (2011).
Robinson, P. S. et al. Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nat. Genet. 53, 1434–1442 (2021).
Blokzijl, F. et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 (2016).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885 (2016).
Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 24, 2125–2137 (2015).
Zage, P. E. et al. Outcomes of the POG 9340/9341/9342 trials for children with high-risk neuroblastoma: a report from the Children’s Oncology Group. Pediatr. Blood Cancer 51, 747–753 (2008).
Matthay, K. K. et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N. Engl. J. Med. 341, 1165–1173 (1999).
Berthold, F. et al. Myeloablative megatherapy with autologous stem-cell rescue versus oral maintenance chemotherapy as consolidation treatment in patients with high-risk neuroblastoma: a randomised controlled trial. Lancet Oncol. 6, 649–658 (2005).
Park, J. R. et al. Effect of tandem autologous stem cell transplant vs single transplant on event-free survival in patients with high-risk neuroblastoma: a randomized clinical trial. JAMA 322, 746–755 (2019).
Liu, K. X. et al. Prospective evaluation of radiation dose escalation in patients with high-risk neuroblastoma and gross residual disease after surgery: a report from the Children’s Oncology Group ANBL0532 study. J. Clin. Oncol. 38, 2741–2752 (2020).
Desai, A. V. et al. Outcomes following GD2-directed postconsolidation therapy for neuroblastoma after cessation of random assignment on ANBL0032: a report from the Children’s Oncology Group. J. Clin. Oncol. 40, 4107–4118 (2022).
Yu, A. L. et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 363, 1324–1334 (2010).
Sung, K. W. et al. Tandem high-dose chemotherapy and autologous stem cell transplantation in patients with high-risk neuroblastoma: results of SMC NB-2004 study. Bone Marrow Transpl. 48, 68–73 (2013).
Lee, J. W. et al. Incorporation of high-dose 131I-metaiodobenzylguanidine treatment into tandem high-dose chemotherapy and autologous stem cell transplantation for high-risk neuroblastoma: results of the SMC NB-2009 study. J. Hematol. Oncol. 10, 108 (2017).
Lee, S. et al. NGSCheckMate: software for validating sample identity in next-generation sequencing studies within and across data types. Nucleic Acids Res. 45, e103 (2017).
Seo, E. S. et al. Response-adapted consolidation therapy strategy for patients with metastatic high-risk neuroblastoma: Results of the SMC NB-2014 study. Pediatr. Blood Cancer 71, e31173 (2024).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 11.10.11–11.10.33 (2013).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Jung, K. S. et al. KRGDB: the large-scale variant database of 1722 Koreans based on whole genome sequencing. Database 2020, baz146 (2020).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985 (2014).
Romanel, A., Zhang, T., Elemento, O. & Demichelis, F. EthSEQ: ethnicity annotation from whole exome sequencing data. Bioinformatics 33, 2402–2404 (2017).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2015).
Saunders, C. T. et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Imielinski, M. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120 (2012).
Bailey, M. H. et al. Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385.e318 (2018).
Ellrott, K. et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281.e277 (2018).
Knijnenburg, T. A. et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 23, 239–254.e236 (2018).
Talevich, E., Shain, A. H., Botton, T. & Bastian, B. C. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 12, e1004873 (2016).
Seo, E. S. Germline functional variants contribute to somatic mutation and outcomes in neuroblastoma. Zenodo https://doi.org/10.5281/zenodo.13324781 (2024).
Acknowledgements
We thank all the subjects who participated in this research. K.W.S. was supported by grants from the National R&D Program for Cancer Control, Ministry of Health and Welfare, Republic of Korea (Grant No. 1520210). J.W.L. received support from the National Research Foundation of Korea (NRF), funded by the Korean government (Grant No. NRF-2020R1A2C1012723). Additionally, W.P. was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (Grant No. HR20C0025).
Author information
Authors and Affiliations
Contributions
W.P. and K.W.S. designed and supervised the study. E.S.S. and J.L. conducted the bioinformatics analysis. K.W.S., J.W.L., H.W.C., H.Y.J., and K.H.Y. contributed clinical samples. S.S. assisted with variant interpretation and data validation. J.W.L., H.W.C., H.Y.J., and K.H.Y. contributed clinical annotations and follow-up data. E.S.S. and J.W.L. wrote the manuscript and all authors contributed to the writing and provided comments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jinghui Zhang, Anders Valind, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Seo, E.S., Lee, J.W., Lim, J. et al. Germline functional variants contribute to somatic mutation and outcomes in neuroblastoma. Nat Commun 15, 8360 (2024). https://doi.org/10.1038/s41467-024-52128-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52128-5
