
Abstract
The 5´–3´ exoribonuclease Rat1/Xrn2 is responsible for the termination of eukaryotic mRNA transcription by RNAPII. Rat1 forms a complex with its partner proteins, Rai1 and Rtt103, and acts as a “torpedo” to bind transcribing RNAPII and dissociate DNA/RNA from it. Here we report the cryo-electron microscopy structures of the Rat1-Rai1-Rtt103 complex and three Rat1-Rai1-associated RNAPII complexes (type-1, type-1b, and type-2) from the yeast, Komagataella phaffii. The Rat1-Rai1-Rtt103 structure revealed that Rat1 and Rai1 form a heterotetramer with a single Rtt103 bound between two Rai1 molecules. In the type-1 complex, Rat1-Rai1 forms a heterodimer and binds to the RNA exit site of RNAPII to extract RNA into the Rat1 exonuclease active site. This interaction changes the RNA path in favor of termination (the “pre-termination” state). The type-1b and type-2 complexes have no bound DNA/RNA, likely representing the “post-termination” states. These structures illustrate the termination mechanism of eukaryotic mRNA transcription.
Similar content being viewed by others


Introduction
DNA-dependent RNA polymerase (RNAP) performs gene transcription in three steps—initiation, elongation, and termination—regulated by specific initiation, elongation, and termination factors1,2. During mRNA transcription by eukaryotic RNA polymerase II (RNAPII), transcribed pre-mRNA undergoes 5´-capping, splicing, and 3´-polyadenylation, and these processes occur co-transcriptionally3. Assembly of the transcription machinery and its association with processing factors are regulated by post-translational modifications, particularly the phosphorylation of the heptad repeat sequence within the C-terminal domain (CTD) of the RNAPII Rpb1 subunit4,5,6,7,8,9.
Following transcription initiation from the promoter, RNAPII travels along the gene body as a transcription elongation complex (EC) to transcribe the gene10,11. In the final step of transcription, the EC terminates transcription at a termination site on the gene by dissociating the DNA and RNA. Precise termination control is essential to maintain and regulate the quality of RNA products, transcriptomes, and genome integrity. Despite its importance, detailed molecular mechanisms underlying transcription termination remain largely unknown, especially in eukaryotes. Eukaryotes use multiple mechanisms for transcription termination depending on the type of gene: protein-coding mRNA genes, replication-dependent histone genes, and non-coding RNA genes12,13. Termination of mRNA transcription is a complex process coupled with mRNA processing. Efficient termination of mRNA transcription requires co-transcriptional cleavage and polyadenylation of the pre-mRNA at the polyadenylation site, followed by 5´–3´ degradation of the downstream nascent RNA transcript to dislodge RNAPII from DNA12,14,15,16,17,18,19,20,21.
The yeast Rat1 protein (Xrn2 in humans) is an essential 5´–3´-exoribonuclease responsible for this final step22,23,24,25. Rat1 forms a binary complex with Rai1, a partner protein that stimulates the Rat1 exoribonuclease activity26,27. Rtt103 is a CTD-interaction domain protein that recruits Rat1-Rai1 to the 3´-end of genes to facilitate EC disassembly28,29. Rat1 is recruited to the monophosphorylated RNA 5´-end newly generated by the pre-mRNA cleavage and is proposed to act as a “torpedo” to terminate transcription; it digests the RNA emerging from EC in the 5´–3´-direction, catches up with the EC, and dissociates DNA/RNA from RNAPII15,22,28,30,31. EC disassembly may be accompanied by an allosteric change in the RNAPII structure13,32,33. However, the molecular mechanisms by which Rat1 interacts with EC and mediates termination remain unclear.
In the present study, we analyzed the structures of the Rat1-Rai1-Rtt103 complex and the Rat1-Rai1-bound RNAPII complexes using cryo-electron microscopy (cryo-EM). These structures provide insights into the Rat1-Rai1-mediated termination of mRNA transcription by RNAPII.
Results
Cryo-EM structure of the Rat1-Rai1-Rtt103 complex
We prepared the recombinant proteins of Rat1, Rai1, and Rtt103 from Komagataella phaffii (K. pastoris). Purified Rat1 efficiently degrades a 5´-monophosphorylated 31 nt RNA (P-31RNA-Cy5) either in the presence or absence of Rai1 (Supplementary Fig. 1). Mutations in two or five acidic residues in the Rat1 active site (Supplementary Fig. 2) (D233A/D235A (Rat1 2A); E203A/E205A/D233A/D235A/D330A (Rat1 5A); and E203Q/E205Q/D233N/D235N/D330N (Rat1 2Q3N)) abolished the nuclease activity of Rat1 (Supplementary Fig. 1b, lanes 6–8). Rai1 or its catalytically inactive mutant (E213A/D215A (Rai1 2A), Supplementary Fig. 3) showed no nuclease activity, consistent with the fact that the substrate for Rai1 is a 5´-triphosphorylated RNA or a 5´-capped RNA (Supplementary Fig. 1b, lanes 9 and 10). In the following structural analyses, we used mutant Rat1 and Rai1 proteins to avoid RNA digestion.
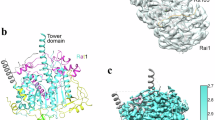
We prepared the Rat1-Rai1-Rtt103 complex by mixing Rat1 (2A), Rai1(2A), and Rtt103, and determined the cryo-EM structure of the complex at 3.5 Å (Fig. 1, Supplementary Figs. 5–8, Supplementary Table 1). Although the sample contained a 17 nt RNA, its density was not observed. In this complex, two Rat1-Rai1 heterodimers (dimers 1 and 2) form a tetramer ((Rat1-Rai1)2) (Fig. 1 and Supplementary Movie 1). Two Rat1 molecules form an interface between dimers 1 and 2. The kinked α12 helix (residues 370–407) of Rat1 in dimers 1 and 2 interacts with the α20 helix (residues 812–819) of Rat1 in dimers 2 and 1 to form a pseudo-two-fold symmetric arrangement (Fig. 1a–c). We found that the N-terminal domain of Rtt103 (Supplementary Fig. 4) was asymmetrically bound to the two Rai1 molecules (Fig. 1a, b, d). Three helices of Rtt103 form extensive interactions with one face of Rai1 in dimer 1, whereas the N-terminal helix of Rtt103 is close to the same face of Rai1 in dimer 2.

a, b The K. phaffii Rat1-Rai1 heterotetramer complexed with Rtt103 is represented as a surface and ribbon model. Rat1 is colored green and grass green, Rai1 is colored marine and purple blue and the N-terminal domain of Rtt103 (Rtt103N) is colored magenta. Bottom, view after a 90° rotation about the horizontal axis from the upper panel. c A close-up view of the Rat1 dimer interface. The Rat1 residues involved in dimerization are shown by green stick models. Hydrogen bonding interactions are represented by dotted lines. d A close-up view of the interface between Rai1 and Rtt103N. The Rai1 and Rtt103 residues are shown by blue and magenta stick models, respectively. A transparent ribbon model of the Rat1-Rai1 (blue and green) complexed with Rtt103 (magenta) is shown in the background. Hydrogen bonds and ionic interactions between Rai1 and Rtt103 are represented by dotted lines.
Cryo-EM analysis of the Rat1-Rai1-associated RNAPII complexes
Subsequently, we analyzed the structure of the RNAPII EC associated with the Rat1-Rai1 exonuclease complex. The EC was assembled by mixing K. phaffii RNAPII with a DNA/RNA scaffold. To reconstitute the pre-termination complex, the EC was mixed with Rat1(2Q3N), Rai1(2A), and Rtt103. We examined five RNAs of different lengths (31, 26, 22, 20, and 17 nt) (Fig. 2a, Supplementary Fig. 9). We also examined the conditions without DNA/RNA. The complex species were partially purified by glycerol gradient sedimentation (Supplementary Figs. 10–15) and crosslinked with BS3 before the cryogrids were prepared; 2000 to 12,000 cryo-EM images were collected for each sample for single-particle analysis (Supplementary Figs. 16, 17 and Supplementary Tables 1, 2). Classification of the particles yielded two distinct classes of RNAPII-Rat1-Rai1 complexes (referred to as types 1 and 2), which differ in the Rat1-Rai1 binding mode to RNAPII (Fig. 2b). In type 1, the Rat1-Rai1 complex docked around the RNA exit site of the EC. In contrast, in the type-2 complex, the Rat1-Rai1 complex occupies the main channel of RNAPII. The type-1 complex was observed when RNA lengths were 20, 22, 26, and 31 nt (Fig. 2a and Supplementary Fig. 17). In contrast, only the type-2 complex was observed in the presence of the 17 nt RNA or in the absence of DNA/RNA (Fig. 2a and Supplementary Fig. 17). Thus, in the range we investigated, the type-1 complex formed when the RNA length was 20 nt or longer. As the Rat1-Rai1 density was best resolved in the type-1 complex yielded by the 22nt RNA, we collected a large dataset for this complex and performed a detailed structural analysis (Supplementary Figs. 18, 19 and Supplementary Table 1). The cryo-EM structures of the type-1 and type-2 complexes were determined at 3.3 Å and 3.4 Å resolution, respectively (Supplementary Fig. 19). The structures are described in detail below.

a Schematic representation of the EC-Rat1-Rai1 complex and the RNA sequences used in this study. Y, the indicated complex was observed in cryo-EM analysis; ND, not detected (Supplementary Fig. 17). b Structures of the type-1 and type-2 RNAPII-Rat1-Rai1 complexes. The composite maps were generated from the overall-masked maps and the Rat1-Rai-masked focused maps by Phenix64. The maps are colored to match the structural models (gray, RNAP; green, Rat1, blue, Rai1; yellow, DNA; red, RNA). c Structure of the elongation complex (EC) engaged with Rat1-Rai1 at the RNA exit (the type-1 complex). The structural model is displayed in two orientations. RNAPII, template DNA, non-template DNA, RNA, Rat1, and Rai1 are colored gray (Rpb7 in brown), yellow, orange, green, and blue, respectively. Transparent surfaces of DNA/RNA, Rat1, and Rai1 are overlaid. d Close-up view of the Rpb7-Rat1 interface. e The Rat1-binding site overlaps with the binding sites of the elongation factors Spt5 and Spt6. The structural model of the type-1 complex is shown in transparent surface, colored as in (c). The structural model of RNAPII EC (PDB 7XN7) is superimposed with the type-1 complex by the Rpb1 subunit. Elongation factors Spt4, Spt5 and Spt6 are shown in cartoon representation and colored purple, blue, and pink, respectively. f The Rat1-binding site does not overlap with the binding sites of the Paf1 complex (Paf1, Leo1, Ctr9, Cdc73, and Rtf1), Elf1, and Spn1. The structural model of the type-1 complex is shown in transparent surface, colored as in (c). The structural model of RNAPII EC (PDB 7XN7) is superimposed with the type-1 complex by the Rpb1 subunit, and the elongation factors are shown in cartoon representation colored in light blue. g A close-up view of the interaction between the Rpb1 dock domain and the Rat1 α20 helix. Cryo-EM map (transparent surfaces) is overlaid on the structural model. h A close-up view of the interaction between the Rpb2 wall domain and the Rat1 α12 helix. The cryo-EM map (transparent surfaces) is overlaid on the structural model.
Structure of the RNAPII pre-termination complex engaged with Rat1-Rai1 (type 1)
The structural analysis of the type-1 complex revealed that the Rat1-Rai1 exonuclease complex was engaged at the RNA exit site of EC, likely representing the state just prior to transcription termination (Fig. 2c and Supplementary Movie 2). Although Rat1-Rai1 forms a heterotetramer ((Rat1-Rai1)2) in its free form, it forms a heterodimer (Rat1-Rai1) upon binding to RNAPII. Rtt103 was not observed. While Rat1 in the Rat1-Rai1 complex directly interacts with RNAPII, Rai1 is oriented away from RNAPII (Fig. 2c). Rat1 orients its exonuclease active site toward RNAPII to capture the 5´-end of RNA emerging from the RNA exit tunnel of RNAPII. The stalk of RNAPII forms a major interface with Rat1 (Fig. 2d). One side of Rat1 extensively interacts with the Rpb7 subunit of the stalk. While the Rat1 N-terminal loop (residues 28–39) fits well with the hydrophobic face of the Rpb7 β-barrel, the C-terminal segment containing Arg715 and Asp 780 contacts the most distal part of the stalk. The segment connecting α5 and β4 (residues 184–186) is also involved in this interaction. The orientation of the stalk was different from that in the EC, likely due to this interaction (Supplementary Fig. 20)34. The Rat1-binding site overlaps with the binding sites of the transcription elongation factors Spt6 and Spt5 (the KOW3-4 domains)34,35, suggesting that Rat1 can access RNAPII after these factors or domains are dissociated from RNAPII (Fig. 2e). On the other hand, there is no overlap between Rat1 and other elongation factors such as the Paf1 complex (Paf1, Leo1, Ctr9, Cdc73, and Rtf1), Spn1, and Elf1 (Fig. 2f).
In addition to the stalk interaction, the α20 helix (residues 810–819) of Rat1 contacts the dock domain of the Rpb1 subunit of RNAPII (Fig. 2g). The kinked α12 helix (residues 385–407) also contacts the wall domain36 of the Rpb2 subunit of RNAPII (Fig. 2h). The tip helix of the wall domain is displaced and disordered, likely due to this interaction. These interactions form a continuous basic channel connecting the RNAPII RNA exit to the Rat1 active site (Fig. 3a, b, Supplementary Fig. 21). The RNA segment emerging from the RNA exit tunnel assumes an extended conformation and is accommodated within the channel. Consequently, the RNA path is largely different from that in the EC (Fig. 3c, d). The 5´-terminal three nucleobases (A1-U2-A3) are stacked to each other and the 5´-terminal nucleobase (A1) is stacked with His59 at the bottom of the active site (Fig. 3e, f, Supplementary Fig. 21b). The 5´-phosphate group of A1 is specifically recognized by Lys109, Gln113, Arg116, and Arg117 on the tower helix (α4) at the bottom of the active site37. The scissile phosphate bond also contacts Lys109 and Arg116 and is close to the putative catalytic residues Glu205, Asp233, and Asp330, which are mutated to Gln or Asn to avoid RNA cleavage. Thus, Rat1 docked to the RNA exit of RNAPII to extract RNA into its active site, largely changing the RNA path.

a Cryo-EM map of the RNA in the type-1 complex. The cryo-EM map was segmented by ChimeraX65, and regions around the RNA and DNA/RNA hybrid are shown as transparent surfaces overlaid on the structural model. b RNA channel in the type-1 complex. A transparent surface of RNAPII and Rat1 (colored by electrostatic surface potentials) is overlaid on the cartoon model (colored as in Fig. 2c). c, d Comparing the RNA path at the RNAPII RNA exit. The structure of RNAPII EC (PDB 7XN7, purple) is superposed on the type-1 complex (colored as in Fig. 2c) by the Rpb1 subunit. e, f Structure of the Rat1 active site with the RNA 5´-end incorporated. e The cryo-EM map (transparent surfaces) is overlaid on the structural model (cartoon and stick models). f The side chains of the Rat1 residues interacting with the RNA are shown as stick models. Putative hydrogen bonds between Rat1 and RNA are shown as yellow dashed lines. The wild-type amino acids are shown in parentheses for the residues mutated in the Rat1 active site (residues 205, 233, and 330).
Particle classification identified another structural class similar to the type-1 complex, referred to as type-1b (Fig. 4). In this complex, the RNAPII conformation and the position of the bound Rat1-Rai1 complex are similar to those in the type-1 complex. However, the type-1b complex lacks clear nucleic acid densities within RNAPII, leaving the RNAPII main and RNA-exit channels empty. Despite the lack of DNA/RNA, the RNAPII clamp remained in a closed conformation, similar to that in the EC. We presume that the type-1b complex was formed after the release of DNA/RNA from the type-1 complex via a transient conformational change in RNAPII, such as the opening of the clamp, due to the binding of Rat1-Rai1.

a, b Structures of the type-1 and type-1b complexes. Cryo-EM maps (white surfaces) are overlaid on the structural models (cartoon representation, colored as in Fig. 2). c Comparison of the type-1 and type-1b complexes. The structural models for the type-1 and type-1b complexes are superimposed by the Rpb1 subunit.
Structure of the Rat1-Rai1 complex bound within the RNAPII cleft (type 2)
We also identified a different structural class (type 2) wherein the Rat1-Rai1 complex is bound to RNAPII in a manner different from that of the type-1 complex. In the type-2 complex, the Rat1-Rai1 complex occupies the main cleft of RNAPII instead of DNA/RNA (Fig. 5a–c and Supplementary Movie 3). Rat1 is sandwiched between the Rpb1 clamp head and Rpb2 lobe domains, whereas, Rai1 is sandwiched between the Rpb1 clamp and Rpb2 protrusion domains (Fig. 5a, d, e). Rat1 uses the same face to interact with RNAPII in the type-1 and type-2 complexes (Fig. 5f, g). The face that interacts with the RNAPII stalk in type 1 interacts with the RNAPII clamp and Rpb5 subunit in type 2. The Rat1 α20 helix that binds to the RNAPII dock in type 1 interacts with the basic surface of the Rpb2 lobe in type 2 (Fig. 5d).

a Structure of Rat1-Rai1 bound within the RNAPII cleft (the type-2 complex). The structural model is displayed in two orientations, colored as in Fig. 2. The transparent surfaces of Rat1 and Rai1 are overlaid. b Comparison of the type-1 and type-2 complexes. The structural models for the type-1 (light orange) and type-2 complexes (colored as in (a)) are superimposed by the Rpb2 subunit. Structural models for RNAPII are shown in cartoon representation. The RNAPII clamp of the type-1 complex is colored red. c Comparison of the Rat1-Rai-1 binding sites in the type-1 and type-2 complexes. The structural models are superimposed by the Rpb2 subunit. d A close-up view of the interaction between the Rpb2 lobe and the Rat1 α20 helix. Cryo-EM map is overlaid on the structural model. e A close-up view of the interaction between the RNAPII clamp and the Rai1. Cryo-EM map is overlaid on the structural model. f, g Comparison of the Rat1 and RNAPII interfaces in the type-1 and type-2 complexes. RNAPII is shown as a cartoon representation, and Rat1 is shown as a surface representation. The Rat1 regions interacting with RNAPII in the type-1 complex are colored salmon (Rat1-Rpb7 interface, Fig. 2d), yellow (the α20 helix), and orange (the α12 helix), respectively. h Overlapping binding sites for Rat1-Rai1 and double-stranded DNA in the transcription pre-initiation complex (PIC). The structural model of the type-2 complex is shown in cartoon representation, colored as in (a). The structural model of RNAPII PIC (PDB 5FZ5) is superimposed on the type-2 complex by the Rpb2 subunit. The template DNA (yellow) and the non-template DNA (orange) of the PIC are shown in surface representation. i Overlapping binding sites for Rat1-Rai1 and elongation factors Spt4, Spt5, and Elf1. The structural model of the type-2 complex is shown in cartoon representation, colored as in (a). The structural model of RNAPII EC (PDB 7XN7) is superposed on the type-2 complex by the Rpb2 subunit. Elongation factors Spt4, Spt5, and Elf1 are shown in surface representation.
The Rat1-Rai1 binding site overlaps with the binding site of double-stranded DNA in the transcription pre-initiation complex (PIC)38,39 (Fig. 5h) and the binding sites of the elongation factors Elf1, Spt5 (the NGN and KOW1 domains), and Spt4 in EC40,41 (Fig. 5i). The type-2 complex preferentially formed in the absence of DNA/RNA (Supplementary Fig. 17). This suggests that when RNAPII has no bound DNA/RNA, Rat1-Rai1 preferentially binds to the empty main cleft of RNAPII, rather than to the RNA-exit site. Therefore, Rat1-Rai1 may occlude the RNAPII cleft when RNAPII releases DNA/RNA from the cleft during termination. The type-2 Rat1-Rai1 binding may also prevent non-specific DNA binding by RNAPII until it is recycled into a PIC to initiate another round of transcription.
Discussion
In this study, we determined the cryo-EM structure of the termination factor complex Rat1-Rai1-Rtt103 from the yeast K. phaffii. Two Rat1-Rai1 dimers form a tetramer ((Rat1-Rai1)2), mediated by the Rat1-Rat1 interaction, and Rtt103 is bound between the two Rai1 molecules. We also determined the structures of RNAPII associated with the Rat1-Rai1 heterodimer complex. Rat1-Rai1 revealed two alternative RNAPII-binding modes (types 1 and 2). In the type-1 complex, Rat1-Rai1 binds to the RNAPII stalk, orienting the exonuclease active site of Rat1 toward the RNA exit site of EC. Rat1 forms direct contacts with the RNAPII dock and wall domains, and incorporates the RNA 5´-end emerging from the RNA exit. This Rat1 interaction changes the direction of RNA, as compared with that in EC. As the type-1 complex was observed with RNAs longer than 20 nt, this state likely represents the “pre-termination” state. Consistently, we also identified the type-1b complex, in which DNA/RNA is dissociated from RNAPII (the “post-termination” state). In contrast, Rat1-Rai1 was bound within the widely open RNAPII main cleft in the type-2 complex. As the type-2 complex was preferentially formed in the absence of DNA/RNA (Supplementary Fig. 17), this complex could represent another post-termination state after the DNA/RNA release from the RNAPII cleft.
Although Rat1, Rai1, and Rtt103 form a pentamer, Rat1-Rai1 was associated with RNAPII as a Rat1-Rai1 heterodimer. Superimposition of the Rat1-Rai-Rtt103 pentamer structure with the Rat1-Rai-associated RNAPII structures (either type 1 or 2) resulted in severe steric clash of the second Rat1 molecule with RNAPII (Supplementary Fig. 22). This suggests that Rat1-Rai1 must become a dimer to bind RNAPII. Rtt103 was only observed in the Rat1-Rai1-Rtt103 pentamer structure and not in the Rat1-Rai1-associated RNAPII structures (type 1 or 2). In the Rat1-Rai1-Rtt103 pentamer, the N-terminal domain of Rtt103 is held between the two Rai1 molecules in the Rat1-Rai1 heterotetramer. When bound to RNAPII, Rat1-Rai1 forms a heterodimer, containing only a single Rai1 molecule to interact with Rtt103, presumably with a weaker affinity for Rtt103 than the Rat1-Rai1 tetramer. Rtt103 consistently dissociated during the sample preparation of the Rat1-Rai1-bound EC (Supplementary Figs. 10–15). This suggests that Rtt103 may dissociate from Rat1-Rai1 once Rat1-Rai1 is recruited to the RNAPII EC.
These structures outline the mechanism of eukaryotic mRNA transcription termination as follows (Fig. 6). (i) Rat1-Rai1 exists as a tetramer but dissociates into a dimer when bound to RNAPII. When cleavage of mRNA precursor occurs at the polyadenylation site, Rat1 captures the monophosphorylated RNA 5´-end and starts trimming of the RNA to track RNAPII. (ii) When Rat1 trimmed the RNA to its length of 20–22 nt, Rat1-Rai1 forms a close contact with RNAPII. Rat1 binding to the EC requires the prior dissociation of Spt6 and Spt4/5 from the EC. Spt4/5 can remain bound to RNAPII through tight binding by the Spt5 KOW5 domain, even when the other Spt5 domains are dissociated from RNAPII40,42,43. Further RNA trimming increases the tension of RNA between the Rat1 and RNAPII active sites, leading to perturbation/destabilization of the DNA/RNA hybrid within the transcription bubble or in the RNAPII-DNA/RNA contacts. (iii) This triggers bubble collapse and DNA/RNA release. (iv) After termination, Rat1-Rai1 is relocated to the empty DNA-binding site of RNAPII for occlusion. Alternatively, Rat1-Rai1 could move from the RNA exit site to the RNAPII cleft while digesting the RNA to actively dislodge DNA/RNA from the RNAPII cleft. Rat1-Rai1 bound to the RNAPII cleft would protect RNAPII until it is recycled into a PIC for another round of transcription.

(i) Rat1-Rai1 exists as a tetramer in free form, and the tetramer holds one Rtt103 molecule. When recruited to transcribing RNAPII, the Rat1-Rai1 tetramer dissociates into a dimer. The Rat1-Rai1 binding to RNAPII requires the prior dissociation of Spt6 and Spt4/5 from RNAPII (except for the KOW5 domain of Spt5). (ii) Rat1 captures the monophosphorylated RNA 5´-end, which is generated by cleavage of mRNA precursor at the polyadenylation site and starts trimming of the RNA. When Rat1 trimmed the RNA to its length of 20–22 nt, Rat1-Rai1 forms a close contact with RNAPII. (iii) Further RNA trimming triggers bubble collapse and DNA/RNA release. (iv) After termination, Rat1-Rai1 is relocated to the empty DNA-binding site of RNAPII for occlusion. Alternatively, Rat1-Rai1 could move from the RNA-exit site (ii) to the RNAPII cleft (iv), to actively dislodge DNA/RNA from the RNAPII cleft. Rat1-Rai1 bound to the RNAPII cleft would protect RNAPII until it is recycled into a PIC for another round of transcription.
Rat1 is involved not only in mRNA transcription by RNAPII, but also in rRNA transcription by RNA polymerase I (RNAPI)44,45. Superimposition of the type-1 complex structure with the RNAPI EC structure shows no apparent steric clash between Rat1 and RNAPI, except for the RNAPI stalk (Supplementary Fig. 23). Rat1 can bind RNAPI in the same manner as the type-1 complex if the orientation of the RNAPI stalk changes similar to that of RNAPII. In contrast, the stalk of RNA polymerase III (RNAPIII) contains specific insertions, which could make Rat1 binding incompatible.
Rat1 binds to the RNA exit site of RNAPII to incorporate the RNA into its active site. This view is analogous to the manner in which the bacterial termination factor Rho interacts with transcribing RNAP46,47 (Supplementary Fig. 24). Although Rho is an ATP-dependent RNA helicase/translocase, it binds the similar part of RNAP (the β-flap and zinc-binding domains) to extract nascent RNA. Although these enzymes differ in their molecular structures, enzymatic activities, and detailed RNAP interactions, they may share similarities in that they are dedicated to binding nascent RNA and RNAP to destabilize the EC, suggesting a mechanistic generality of transcription termination.
Rat1/Xrn2 action in mRNA transcription termination is tightly coupled with the prior cleavage of the mRNA precursor and polyadenylation by the cleavage and polyadenylation (CPA) complex12. Further studies are needed to clarify how Rat1/Xrn2 cooperates with the CPA complex at appropriate timing to complete transcription termination coupled with mRNA 3´-processing.
Methods
Materials
Biochemical and molecular biology procedures were performed using commercially available materials, enzymes, and chemicals. The synthesized DNAs and RNAs were purchased from Eurofins Genomics (Tokyo, Japan).
Cloning, expression, and purification of kpRat1 proteins
The DNA fragment encoding the Komagataella phaffii Rat1 (kpRat1) gene was polymerase chain reaction (PCR) amplified from a plasmid containing the codon-optimized kpRat1 gene as a template. The kpRat1 gene, a maltose-binding protein (MBP) gene with a human rhinovirus 3 C (HRV-3C) cleavage site at the N-terminus, and a hexahistidine (6×His) tag at the C-terminus were cloned into the pET-47 vector. Escherichia coli Rosetta 2 (DE3) cells (Novagen, 71397) were transformed with the plasmid and selected on an LB agar plate supplemented with 50 μg mL−1 kanamycin. Single colonies were used to inoculate 10 mL of Plusgrow II culture media (Nacalai, 08202-75), supplemented with 50 μg mL−1 kanamycin, and grown at 37 °C for 4 h. The starter culture was transferred to two-liter flasks of the Plusgrow II culture and then grown at 37 °C. When the OD600 reached 0.6, the temperature was lowered to 20 °C. Protein expression was induced with 1 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG) and the cells were grown at 20 °C for 21 h. The E. coli cells were collected by centrifugation and stored at −80 °C. The cells were resuspended in 50 mM potassium phosphate buffer (pH 7.4) containing 500 mM NaCl, 25 mM imidazole, 5 mM β-mercaptoethanol, and a protease inhibitor cocktail (cOmplete ULTRA, EDTA-free, Roche, 05892953001) (buffer A), then lysed by sonication on ice. The cell lysate was centrifuged at 15,000×g for 15 min at 4 °C, and the supernatant was applied to a HisTrap column (Cytiva, 17524802), which was equilibrated with buffer A. The kpRat1-MBP-6×His fusion protein was eluted with buffer A containing 400 mM imidazole instead of 25 mM imidazole and collected. The eluted fraction was collected and dialyzed against 40 mM potassium phosphate buffer (pH 7.4), containing 50 mM NaCl and 1 mM dithiothreitol (DTT) (buffer B). The dialyzed fraction was then applied to a Resource Q column (Cytiva, 17117901), washed with buffer B, and the bound proteins were eluted using a linear gradient of 50–537.5 mM NaCl. The C-terminal MBP-6×His-tag was cleaved using HRV-3C protease at 4 °C overnight. The dialyzed fraction was then loaded onto a HiTrap Heparin column (Cytiva, 17040703), washed with buffer B, and the bound kpRat1 proteins were eluted using a linear gradient of 50–1025 mM NaCl. The eluted fractions were collected, dialyzed against 20 mM HEPES-NaOH buffer (pH 7.5) containing 200 mM NaCl and 10 mM β-mercaptoethanol (buffer C) and concentrated by ultracentrifugation. Aliquots of the kpRat1 protein were flash-cooled in liquid nitrogen and stored at −80 °C. The catalytically inactive mutant kpRat1 proteins, kpRat1(E203Q/E205Q/D233N/D235N/D330N)[kpRat1(2Q3N)], kpRat1(E203A/E205 A/D233A/D235A/D330A)[kpRat1(5A)], and kpRat1(D213A/D215A)[kpRat1(2A)] were expressed and purified in the same manner as the wild-type kpRat1 protein. The initially constructed kpRat1(2A) and kpRat1(5A) mutants had protein yields less than one-tenth of wild-type kpRat1. As the Ala substitutions that increase hydrophobicity in the active site were thought to be the cause of the low protein yields, we created a kpRat1(2Q3N) mutant with two substitutions from Glu to Gln and three substitutions from Asp to Asn in the active site. The yield of kpRat1(2Q3N) was comparable to that of wild-type kpRat1. The yields of the kpRat1(2A), kpRat1(5A), and kpRat1(2Q3N) proteins were 0.3 mg, 0.24 mg, and 2.67 mg per 1 L medium, respectively. The kpRat1(2A) protein was used to prepare the Rat1-Rai1-Rtt103 complex, whereas the kpRat1(2Q3N) protein was used to prepare the RNAPII-Rat1-Rai1 complexes.
Cloning, expression, and purification of kpRai1 proteins
The DNA fragment encoding K. phaffii Rai1 (kpRai1) gene was PCR-amplified from K. phaffii genomic DNA and cloned into a modified pCR2.1 vector possessing a 6×His-tag and an HRV-3C cleavage site attached at its N-terminus. E. coli KRX cells (Promega, L3001) were transformed with the plasmid and selected on an LB agar plate supplemented with 50 μg mL−1 kanamycin. Single colonies were inoculated into 10 mL of Plusgrow II culture media (Nacalai), supplemented with 50 μg mL−1 kanamycin, and grown at 37 °C for 4 h. The starter culture was transferred to one-liter flasks containing Plusgrow II culture and then grown at 37 °C. When the OD600 reached 0.5, the temperature was lowered to 20 °C. The protein expression was induced with 0.2% rhamnose, and the cells were grown at 20 °C for 21 h. The E. coli cells were collected by centrifugation and stored at −80 °C. Subsequently, the cells were resuspended in buffer A and lysed by sonication on ice. The cell lysate was centrifuged at 15,000×g for 15 min at 4 °C, and the supernatant was applied to a HisTrap column (Cytiva), which was equilibrated with buffer C. The proteins were eluted with buffer A containing 400 mM imidazole instead of 25 mM imidazole and collected. The eluted fraction was collected and dialyzed against buffer B. The dialyzed fraction was then applied to a Resource Q column (Cytiva), washed with buffer B, and the bound proteins were eluted using a linear gradient of 50–537.5 mM NaCl. The N-terminal 6×His-tag was cleaved with HRV-3C protease at 4 °C overnight. The dialyzed fraction was then loaded onto a HiTrap Heparin column (Cytiva), washed with buffer B, and the bound proteins were eluted using a linear gradient of 50–1025 mM NaCl. The eluted fractions were collected, dialyzed against buffer C, and concentrated using ultracentrifugation. Aliquots of the purified kpRai1 protein were flash-cooled in liquid nitrogen and stored at −80 °C. The catalytically inactive mutant kpRai1 protein, kpRai1(E213A/D215A)[kpRai1(2A)], was expressed and purified in the same manner as wild-type kpRai1.
Cloning, expression, and purification of kpRtt103 protein
The DNA fragment encoding K. phaffii Rtt103 (kpRtt103) gene was PCR-amplified from K. phaffii genomic DNA and cloned into modified pCR2.1, vector with an HRV-3C cleavage site and a 6×His-tag attached at its C-terminus. E. coli KRX cells were transformed with the plasmid and selected on an LB agar plate supplemented with 50 μg mL−1 kanamycin. Single colonies were inoculated into 10 mL of Plusgrow II culture media (Nacalai), supplemented with 50 μg mL−1 kanamycin, and grown at 37 °C for 4 h. The starter culture was transferred to one-liter flasks containing Plusgrow II culture and grown at 37 °C. When the OD600 reached 0.6, the temperature was lowered to 20 °C. Protein expression was induced with 0.2% rhamnose, and the cells were grown at 20 °C overnight. E. coli cells were collected by centrifugation and stored at −80 °C. The cells were then resuspended in buffer A and lysed by sonication on ice. The cell lysate was centrifuged at 15,000×g for 15 min at 4 °C, and the supernatant was applied to a HisTrap column (Cytiva) that was equilibrated with buffer A. The proteins were eluted with buffer A containing 400 mM imidazole instead of 25 mM imidazole and collected. The eluted fraction was collected and dialyzed against buffer B. The dialyzed fraction was then applied to a Resource Q column (Cytiva), washed with buffer B, and the bound proteins were eluted using a linear gradient of 50–537.5 mM NaCl. The N-terminal 6×His-tag was cleaved with HRV-3C protease at 4 °C overnight. The dialyzed fraction was then loaded onto a HiTrap Heparin column (Cytiva), washed with buffer B, and the bound proteins were eluted using a linear gradient of 50–1025 mM NaCl. The eluted fractions were pooled, concentrated, and applied to a HiLoad 16/60 Superdex 200 column (Cytiva, 28989335) equilibrated with 30 mM potassium phosphate buffer (pH 7.4) containing 200 mM NaCl and 1 mM DTT. The eluted fractions were collected, dialyzed against buffer C, and concentrated using ultracentrifugation. Aliquots of purified kpRtt103 protein were flash-cooled in liquid nitrogen and stored at −80 °C.
Purification of RNA polymerase II from K. phaffii
kpRNAPII containing the TAP-tagged Rpb2 subunit was purified from a genetically modified strain of K. phaffii48,49. Approximately 400 g of K. phaffii cells were resuspended in 1.25 times volume of lysis buffer (40 mM Tris–HCl buffer (pH 7.5), containing 100 mM potassium acetate, 1 mM EDTA, 5 mM DTT, 0.1 % Triton X-100, 1 mM PMSF, and cOmplete EDTA-free protease inhibitor (Roche, 46931320)). After disruption, the cell lysate was centrifuged to remove cell debris, and the supernatant was loaded onto a Q Sepharose Fast Flow column (Cytiva, 17051001) equilibrated with lysis buffer. After washing the column with wash buffer (20 mM Tris–HCl buffer (pH 7.5), containing 150 mM potassium acetate, 1 mM EDTA, and 0.1 % Triton X-100), the protein was eluted with buffer K1000 (20 mM Tris–HCl buffer (pH 7.5), containing 1000 mM potassium acetate, and 5% glycerol). The eluted fractions were then loaded onto an anti-FLAG M2 affinity gel column (Sigma-Aldrich, A2220-25ML). The column was washed with buffer K500 (20 mM Tris–HCl buffer (pH 7.5), containing 500 mM potassium acetate and 5% glycerol), and the fraction containing RNAPII was eluted with buffer K500 containing 3×FLAG peptide (0.25 mg/mL). Finally, the protein was purified by Resource Q column (Cytiva, 17117901), eluted by a linear gradient of potassium acetate from 0 to 1 M in the buffer T (20 mM Tris–HCl buffer (pH 7.5), containing 1 μM Zn(OAc)2, 0.1 mM Tris(2-carboxyethyl)phosphine [TCEP], and 5% glycerol). The RNAPII-containing fractions were dialyzed against RNAPII buffer (20 mM Tris-acetate (pH 8.0), 150 mM potassium acetate, 1 μM zinc acetate, 0.1 mM TCEP, and 5% glycerol) before concentration.
In vitro assay of the 5’→3’-exoribonuclease activity
kpRat1/Rai1 activity assays were conducted in 10-μL reaction mixtures containing 20 mM HEPES-NaOH (pH 7.5), 100 mM NaCl, 5 mM MgCl2, 5 mM DTT, and 0.75 μM 5´-phosphorylated/3´-Cy5-labeled 31 nt RNA substrate (5´-P-UAAUCCCAUAUAUAUGCAUAAAGACCAGGCU-Cy5-3´)(P-31RNA-Cy5). The purified kpRat1 (1.5 μM) and kpRai1 (1.5 μM) proteins were mixed, and incubated at 0 °C for 1 h. The reaction was terminated by the addition of 1 volume of 2 x sodium dodecyl sulfate (SDS) loading buffer (0.125 M Tris–HCl (pH 6.8), 4% SDS, 10% glycerol, 10% β-mercaptoethanol, and 0.03% Orange G) and heat treatment (95 °C for 3 min). Reaction products were resolved by 10–20% SDS-polyacrylamide gel electrophoresis (PAGE), visualized using LAS4000 (GE Healthcare), and stained with SimplyBlue SafeStain (Thermo Fisher Scientific, LC6060).
Preparation of the Rat1-Rai1-Rtt103-RNA complex for cryo-EM analysis
5´-Cy5-labeled 17-nt RNA (5´-Cy5-CTAGTAATGACCAGGCU-3´) (Cy5-17RNA), kpRat1(2A), kpRai1(2A), and kpRtt103 were mixed in a 2:1:2:2 molar ratio (15 μM, 7.5 μM, 15 μM, and 15 μM, respectively) in 20 mM HEPES-NaOH buffer (pH 7.5), containing 100 mM NaCl, 5 mM MgCl2, and 2 mM DTT, and incubated on ice for 30 min to allow for association. After filtration, the samples were loaded and purified on a Superose 6 Increase 10/300 GL column (Cytiva, 29091596) in 20 mM HEPES-NaOH buffer (pH 7.5) containing 100 mM NaCl, 1 mM MgCl2, and 1 mM DTT. The peak corresponding to the complex was pooled, concentrated by centrifugal filtration (Amicon), and exchanged with 20 mM HEPES-NaOH buffer (pH 7.5) containing 100 mM NaCl, 1 mM MgCl2, and 1 mM TCEP. Protein-nucleic acid complexes (5–10 μL) were crosslinked with 5 mM bissulfosuccinimidyl suberate (BS3) (Toronto Research Chemicals Inc, S777500) on ice for 30 min and quenched with 100 mM Tris–HCl (pH 7.5).
Preparation of RNAPII bound with Rat1-Rai1-Rtt103 for cryo-EM analysis
To assemble the EC bound to the Rat1-Rai1-Rtt103 complex for cryo-EM studies, purified kpRat1(2Q3N), kpRai1(2A), and kpRtt103 were incubated with the pre-assembled kpRNAPII-DNA/RNA-scaffold complex in a buffer containing 20 mM HEPES-NaOH (pH 7.5), 100 mM NaCl, 10 mM KCl, 1 mM MgCl2, and 1 mM DTT. The DNA/RNA scaffold consists of a non-template DNA (5´-GTCAAGGCAGTACTAGTAATTTAGCAATCCAACTACTTTATCTTTTAATCAATCTACAATAACTGGGGGGCTACCGACGCTAGGGATCCT-3´), a template DNA (5´-AGGATCCCTAGCGTCGGTAGCCCCCCAGTTATTGTAGATTGATTAAAAGATAAAGTAGTTGAGCCTGGTCATTACTAGTACTGCCTTGAC-3´), and a 5′-phosphorylated/3´-Cy5-labeled 22 nt RNA (5´-P-AUAUAUGCAUAAAGACCAGGCU-Cy5-3´) (P-22RNA-Cy5) (Supplementary Fig. 9). The template and non-template DNAs were designed to have a 9 bp mismatch, so the scaffold contains a stable DNA-RNA hybrid in the transcription bubble. The P-22RNA-Cy5, template DNA, and non-template DNA were mixed and annealed at 95 °C for 2 min in annealing buffer (10 mM HEPES-NaOH (pH 7.5), 50 mM NaCl), and the temperature gradually decreased from 95 °C to room temperature. kpRNAPII (1.4 μM) was mixed with the scaffold (1.6 μM), and incubated on ice for 1 h in RNAPII complex buffer (20 mM HEPES-NaOH buffer (pH 7.5), containing 100 mM NaCl, 10 mM KCl, 1 mM MgCl2, and 5 mM DTT). Subsequently, kpRat1(2Q3N) (8 μM), kpRai1(2A) (8 μM), and kpRtt103 (8 μM) were added, and incubated on ice for 6 h to allow for association.
The protein-nucleic acid complexes were then fractionated by ultracentrifugation sedimentation on a glycerol gradient. The glycerol gradient (20 mM HEPES-NaOH buffer (pH 7.5) containing 100 mM NaCl, 10 mM KCl, 1 mM MgCl2, 5 mM DTT, 10–30% glycerol) was prepared by layering the lowest glycerol concentration solution to the highest glycerol concentration solution from the bottom of the tube with a syringe. The protein-nucleic acid complexes were applied on the top of the gradient solution and ultracentrifuged at 94,000×g at 4 °C for 17.75 h, using an S55S rotor (Eppendorf Himac Technologies). The fractionated samples containing the kpRNAPII/DNA-RNA scaffold/Rat1(2Q3N)/Rai1(2A)/Rtt103 complexes were pooled and concentrated using Amicon Ultra 50 K centrifugal filters (Millipore, UFC505024), and the buffer was exchanged with 20 mM HEPES-NaOH buffer (pH 7.5) containing 100 mM NaCl, 10 mM KCl, 1 mM MgCl2, and 1 mM TCEP. Crosslinking of the protein-nucleic acid complexes (5–10 μL) was performed with 5 mM BS3 on ice for 40 min and quenched with 100 mM Tris–HCl (pH 7.5).
Cryo-EM sample preparation and data collection
Crosslinked complexes (2.5–3.0 μL) were applied onto 300-mesh Quantifoil R1.2/1.3 Cu grids (Quantifoil Micro Tools GmbH, Germany), which were glow-discharged before use with a PIB-10 ION Bombarder (Vacuum Device Inc.). The grids were then blotted for 3.0 s at 10 °C and 75% humidity and plunge-frozen in liquid ethane using an EM GP2 (Leica, Germany). For the kpRat1(2A)/Rai1(2A)/Rtt103 complex and the kpRNAPII/DNA-RNA scaffold/Rat1(2Q3N)/Rai1(2A) complexes with 31 nt, 22 nt and 17 nt RNAs, cryo-EM data were collected with a 300 kV Krios G4 transmission electron microscope (Thermo Fisher Scientific, USA) equipped with a BioQuantum energy filter and K3 camera (Gatan, USA). Images were recorded at ×105,000 magnification, with a calibrated pixel size of 0.83 Å/pixel. The exposure times were set to 2.2–2.4 s with a total accumulated dose of 53–62 electrons per Å2. All images were automatically recorded using EPU software (Thermo Fisher Scientific, USA). For the kpRat1(2A)/Rai1(2A)/Rtt103 complex, a total of 11,000 images were collected with a defocus range from −2.4 μm to −1.6 μm. For the kpRNAPII/DNA-RNA scaffold/Rat1(2Q3N)/Rai1(2A) complexes with 31 nt, 22 nt and 17 nt RNAs, 12,304, 21,135 and 6828 images were collected, respectively, with a defocus range from −2.4 μm to −1.4 μm. For the kpRNAPII/DNA-RNA scaffold/Rat1(2Q3N)/Rai1(2A) complexes with 26 nt and 20 nt RNAs and the kpRNAPII/ Rat1(2Q3N)/Rai1(2A) complex (with no DNA/RNA), cryo-EM data were collected using a 200 kV Tecnai Arctica transmission electron microscope (Thermo Fisher Scientific, USA) equipped with a K2 camera (Gatan, USA). For each sample, approximately 2300 images were recorded at ×23,500 magnification with a calibrated pixel size of 1.47 Å/pixel, using SerialEM software50. The exposure times were set to 14–16 s with a total accumulated dose of 50 electrons per Å2. The statistics for data collection and refinement are presented in Supplementary Tables 1 and 2.
Image processing and 3D reconstruction of the Rat1-Rai1-Rtt103 complex
Image processing of kpRat1(2A)/Rai1(2A)/Rtt103 data was performed using Relion3.1, unless otherwise specified51,52. After motion correction, the CTF parameters were estimated using CTFFIND53, and the initial particles were picked using a Relion blob picker. After preliminary 2D and 3D classifications, a Topaz network was trained with good particles, and the particles were re-picked using Topaz54. Repeated cycles of 2D classification, 3D classification, 3D refinement, Bayesian polishing, and CTF refinement were performed to select good particles and improve the resolution (Supplementary Fig. 6). The C2 symmetry was applied in most of these steps. Finally, a 3.47 Å map of kpRat1/Rai1 was obtained from 526,250 particles, with C2 symmetry applied. To resolve the Rtt103N density between the dimers, the final steps of 3D classification were performed without symmetry using a broad mask around the whole molecule, and with local angular searches and the symmetry relaxation (C2) options. Subsequently, the final 3.78 Å map of kpRat1/Rai1/Rtt103N was obtained from 77,924 particles. The resolution numbers were estimated based on the Gold-standard Fourier shell correlation (FSC) cutoff of 0.143.
Image processing and preliminary single-particle analysis of the RNAPII-Rat1-Rai1 complexes
Motion correction was performed using Relion3.151,52 and CTF parameters were estimated using CTFFIND53. Particle picking was performed using Topaz54. Particles containing RNAPII were selected by 2D classification and subjected to 3D classification using Relion3.151,52 (Supplementary Fig. 17). The sample containing the 22-nt RNA yielded the complex wherein the Rat1-Rai1 density was best resolved and was subjected to further data collection and analysis.
3D reconstruction of the RNAPII-Rat1-Rai1 complex
Particle picking was performed using Warp55 and Topaz54, and the particle coordinates from each program were merged after 2D classification. Approximately 1,500,000 particles containing RNAPII were selected by 2D and 3D classification and subjected to 3D refinement, followed by local CTF estimation, beam tilt refinement, and Bayesian polishing to improve resolution. The particles were further sorted by 3D classification with blush regularization using relion556 into apo RNAPII, EC, and RNAPII-Rat1-Rai1 complexes (type 1 and type 2) (Supplementary Fig. 18). The 3D classes of EC, which showed a weak density of Rat1-Rai1 at the type-1 binding site, were subjected to focused 3D classification using a mask around Rat1-Rai1 and the RNAPII stalk. Particles with a clear Rat1-Rai1 density were merged with type-1 particles from the preceding 3D classification. The particle-picking network of Topaz was trained on the particle coordinates of the type-1 and type-2 complexes, and the particles were re-picked using the trained network. Approximately 1,000,000 type-1-like particles were refined with an overall mask and subtracted with a mask around Rat1-Rai1 and the RNAPII stalk. 3D classifications of the subtracted particles were performed to exclude particles with poor Rat1-Rai1 density. Approximately 113,000 particles with a clear Rat1-Rai1 density were reverted to the original particles and subjected to 3D classification using an overall mask. The particles were sorted into classes with and without a clear DNA/RNA density. Approximately 64,000 particles with clear DNA/RNA density (type 1) were further classified with a Rat1-Rai1-stalk mask, and 44,794 particles were selected for the final reconstruction of a 3.3 Å resolution map, based on the Gold-standard FSC 0.143 cutoff (Supplementary Fig. 19). Approximately 57,000 particles without clear DNA/RNA density (type 1b) were further classified with a Rat1-Rai1-stalk mask, and 31,706 particles were selected for the final reconstruction of a 3.4 Å resolution map, based on the Gold-standard FSC 0.143 cutoff (Supplementary Fig. 19). For the type-2 complex, approximately 140,000 particles were classified as apo-RNAPII and RNAPII-Rat1-Rai1 complexes with different RNAPII clamp orientations. The class with the most particles (~53,000) and the best map quality was selected for further refinement. The particles were initially refined with an overall mask and then subtracted with a mask around Rat1-Rai1. 3D classifications of the subtracted particles were performed to exclude particles with poor Rat1-Rai1 density, and 49,931 particles with clear Rat1-Rai1 density were selected for the final reconstruction of a 3.4 Å resolution map based on the Gold-standard FSC 0.143 cutoff (Supplementary Fig. 19). For each complex, local reconstructions using masks around Rat1-Rai1 (and the RNAPII stalk for type-1) were performed and used for model building (Supplementary Fig. 19).
Model building and refinement
To build the five structures, the structures of kpRat1(2Q3N), kpRai1(2A), and kpRtt103, which were generated by Alphafold257, and kpRNAPII (PDB: 5XOG) were placed and rigid-body fitted into the cryo-EM map using UCSF Chimera58. The models were manually built using WinCoot 0.9.8.759 and ISOLDE60 under the guidance of the cryo-EM map, and overall real-space refinement was performed using Phenix 1.961. The data validation statistics are presented in Supplementary Table 1. The final model was validated using Molprobity62 and Procheck63. Figures were prepared using UCSF Chimera 1.17, ChimeraX, and PyMOL2.4.0 [http://pymol.sourceforge.net/].
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM maps and coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) and Protein Data Bank (PDB), respectively, with accession codes: EMD-39221 and 8YFE(Rat1-Rai1), EMD-39211 and 8YF5(Rat1-Rai1-Rtt103N), EMD-39226 and 8YFQ(EC-Rat1-Rai1 (type 1)), and EMD-39227 and 8YFR(RNAPII-Rat1-Rai1 (type 2)). The atomic models used in this study are available in the Protein Data Bank under accession codes 2KM4, 3FQD, 5FZ5, 5XOG, and 7XN7. The source data underlying Supplementary Figs. 1b, 1c, 5a, 5b, 10a, 10b, 11a, 11b, 12a, 12b, 13a, 13b, 14a, 14b, and 15, are provided as a Source Data file. Source data are provided with this paper.
References
Osman, S. & Cramer, P. Structural biology of RNA polymerase II transcription: 20 years on. Annu. Rev. Cell Dev. Biol. 36, 1–34 (2020).
Kwak, H. & Lis, J. T. Control of transcriptional elongation. Annu. Rev. Genet. 47, 483–508 (2013).
Bentley, D. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 15, 163–175 (2014).
Komarnitsky, P., Cho, E. & Buratowski, S. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14, 2452–2460 (2000).
Buratowski, S. The CTD code. Nat. Struct. Biol. 10, 679–680 (2003).
Eick, D. & Geyer, M. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem. Rev. 113, 8456–8490 (2013).
Hsin, J. P. & Manley, J. L. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 26, 2119–2137 (2012).
Jeronimo, C., Collin, P. & Robert, F. The RNA polymerase II CTD: the increasing complexity of a low-complexity protein domain. J. Mol. Biol. 428, 2607–2622 (2016).
Harlen, K. & Churchman, L. The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat. Rev. Mol. Cell Biol. 18, 263–273 (2017).
Mohamed, A. A., Vazquez Nunez, R. & Vos, S. M. Structural advances in transcription elongation. Curr. Opin. Struct. Biol. 75, 102422 (2022).
Sekine, S., Ehara, H., Kujirai, T. & Kurumizaka, H. Structural perspectives on transcription in chromatin. Trends Cell Biol. 34, 211–224 (2023).
Rodríguez-Molina, J., West, S. & Passmore, L. Knowing when to stop: transcription termination on protein-coding genes by eukaryotic RNAPII. Mol. Cell 83, 404–415 (2023).
Eaton, J. & West, S. Termination of transcription by RNA Polymerase II: BOOM! Trends Genet. 36, 664–675 (2020).
Logan, J., Falck-Pedersen, E., Darnell, J. & Shenk, T. A poly(A) addition site and a downstream termination region are required for efficient cessation of transcription by RNA polymerase II in the mouse βmaj-globin gene. Proc. Natl Acad. Sci. USA 84, 8306–8310 (1987).
Proudfoot, N. How RNA polymerase II terminates transcription in higher eukaryotes. Trends Biochem. Sci. 14, 105–110 (1989).
Connelly, S. & Manley, J. A functional mRNA polyadenylation signal is required for transcription termination by RNA polymerase II. Genes Dev. 2, 440–452 (1988).
Birse, C., Minvielle-Sebastia, L., Lee, B., Keller, W. & Proudfoot, N. Coupling termination of transcription to messenger RNA maturation in yeast. Science 280, 298–301 (1998).
Bentley, D. Rules of engagement: co-transcriptional recruitment of pre-mRNA processing factors. Curr. Opin. Cell Biol. 17, 251–256 (2005).
Proudfoot, N. Ending the message: poly(A) signals then and now. Genes Dev. 25, 1770–1782 (2011).
Porrua, O. & Libri, D. Transcription termination and the control of the transcriptome: why, where and how to stop. Nat. Rev. Mol. Cell Biol. 16, 190–202 (2015).
Proudfoot, N. Transcriptional termination in mammals: stopping the RNA polymerase II juggernaut. Science 352, aad9926 (2016).
West, S., Gromak, N. & Proudfoot, N. Human 5’→3’ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature 432, 522–525 (2004).
Kim, M. et al. Distinct pathways for snoRNA and mRNA termination. Mol. Cell 24, 723–734 (2006).
Kilchert, C. & Vasiljeva, L. mRNA quality control goes transcriptional. Biochem. Soc. Trans. 41, 1666–1672 (2013).
Nagarajan, V., Jones, C., Newbury, S. & Green, P. XRN 5’→3’ exoribonucleases: structure, mechanisms and functions. Biochim. Biophys. Acta 1829, 590–603 (2013).
Stevens, A. & Poole, T. 5’-exonuclease-2 of Saccharomyces cerevisiae. Purification and features of ribonuclease activity with comparison to 5’-exonuclease-1. J. Biol. Chem. 270, 16063–16069 (1995).
Xue, Y. et al. Saccharomyces cerevisiae RAI1 (YGL246c) is homologous to human DOM3Z and encodes a protein that binds the nuclear exoribonuclease Rat1p. Mol. Cell Biol. 20, 4006–4015 (2000).
Kim, M. et al. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432, 517–522 (2004).
Lunde, B. et al. Cooperative interaction of transcription termination factors with the RNA polymerase II C-terminal domain. Nat. Struct. Mol. Biol. 17, 1195–1201 (2010).
Connelly, S. & Manley, J. A CCAAT box sequence in the adenovirus major late promoter functions as part of an RNA polymerase II termination signal. Cell 57, 561–571 (1989).
Han, Z. et al. DNA-directed termination of RNA polymerase II transcription. Mol. Cell 83, 3253–3267 (2023).
Luo, W., Johnson, A. & Bentley, D. The role of Rat1 in coupling mRNA 3’-end processing to transcription termination: implications for a unified allosteric-torpedo model. Genes Dev. 20, 954–965 (2006).
Eaton, J., Francis, L., Davidson, L. & West, S. A unified allosteric/torpedo mechanism for transcriptional termination on human protein-coding genes. Genes Dev. 34, 132–145 (2020).
Ehara, H., Kujirai, T., Shirouzu, M., Kurumizaka, H. & Sekine, S. Structural basis of nucleosome disassembly and reassembly by RNAPII elongation complex with FACT. Science 377, eabp9466 (2022).
Vos, S. et al. Structure of activated transcription complex Pol II-DSIF-PAF-SPT6. Nature 560, 607–612 (2018).
Cramer, P., Bushnell, D. A. & Kornberg, R. D. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science 292, 1863–1876 (2001).
Xiang, S. et al. Structure and function of the 5’→3’ exoribonuclease Rat1 and its activating partner Rai1. Nature 458, 784–788 (2009).
He, Y. et al. Near-atomic resolution visualization of human transcription promoter opening. Nature 533, 359–365 (2016).
Plaschka, C. et al. Transcription initiation complex structures elucidate DNA opening. Nature 533, 353–358 (2016).
Ehara, H. et al. Structure of the complete elongation complex of RNA polymerase II with basal factors. Science 357, 921–924 (2017).
Ehara, H. et al. Structural insight into nucleosome transcription by RNA polymerase II with elongation factors. Science 363, 744–747 (2019).
Crickard, J. B., Fu, J. & Reese, J. C. Biochemical analysis of yeast suppressor of Ty 4/5 (Spt4/5) reveals the importance of nucleic acid interactions in the prevention of RNA Polymerase II arrest. J. Biol. Chem. 291, 9853–9870 (2016).
Zeng, Y., Zhang, H., Wu, X. & Zhang, Y. Structural basis of exoribonuclease-mediated mRNA transcription termination. Nature 628, 887–893 (2024).
Braglia, P., Kawauchi, J. & Proudfoot, N. Co-transcriptional RNA cleavage provides a failsafe termination mechanism for yeast RNA polymerase I. Nucleic Acids Res. 39, 1439–1448 (2011).
El Hage, A., Koper, M., Kufel, J. & Tollervey, D. Efficient termination of transcription by RNA polymerase I requires the 5’ exonuclease Rat1 in yeast. Genes Dev. 22, 1069–1081 (2008).
Murayama, Y. et al. Structural basis of the transcription termination factor Rho engagement with transcribing RNA polymerase from Thermus thermophilus. Sci. Adv. 9, eade7093 (2023).
Molodtsov, V., Wang, C., Firlar, E., Kaelber, J. & Ebright, R. Structural basis of Rho-dependent transcription termination. Nature 614, 367–374 (2023).
Higo, T. et al. Development of a hexahistidine-3x FLAG-tandem affinity purification method for endogenous protein complexes in Pichia pastoris. J. Struct. Funct. Genomics 15, 191–199 (2014).
Ehara, H., Umehara, T., Sekine, S.-I. & Yokoyama, S. Crystal structure of RNA polymerase II from Komagataella pastoris. Biochem. Biophys. Res. Commun. 487, 230–235 (2017).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Scheres, S. H. Processing of structurally heterogeneous cryo-EM data in RELION. Methods Enzymol. 579, 125–157 (2016).
Scheres, S. A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 415, 406–418 (2012).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Tegunov, D. & Cramer, P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152 (2019).
Kimanius, D. et al. Data-driven regularisation lowers the size barrier of cryo-EM structure determination. Nat. Methods 21, 1216–1221 (2024).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Croll, T. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 74, 519–530 (2018).
Afonine, P. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012).
Davis, I. et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 (2007).
CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 (1994).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. D75, 861–877 (2019).
Goddard, T. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Acknowledgements
The cryo-EM experiments were performed with the Krios G4 microscope at the RIKEN Yokohama Cryo-EM Facility. This work was supported by the Japan Society for the Promotion of Science KAKENHI (grant numbers JP20H05690, JP20H05906, and JP24H00062 to S.S.).
Author information
Authors and Affiliations
Contributions
T.Y. and S.S. designed this study. T.Y., Y.M. and H.E. performed cryo-EM analyses. T.Y. performed protein preparations and biochemical analyses. M.G. prepared K. phaffii RNAPII. M.A. performed DNA cloning and mutagenesis. T.Y., Y.M., H.E. and S.S. wrote the manuscript. All of the authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dong Wang and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yanagisawa, T., Murayama, Y., Ehara, H. et al. Structural basis of eukaryotic transcription termination by the Rat1 exonuclease complex. Nat Commun 15, 7854 (2024). https://doi.org/10.1038/s41467-024-52157-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52157-0
This article is cited by
-
DSIF factor Spt5 coordinates transcription, maturation and exoribonucleolysis of RNA polymerase II transcripts
Nature Communications (2025)
-
Molecular basis for the interaction between Saccharomyces cerevisiae Rtt103 and the Rat1-Rai1 complex
Nature Communications (2025)
