
Abstract
Ribosomal DNA (rDNA) encodes the ribosomal RNA genes and represents an intrinsically unstable genomic region. However, the underlying mechanisms and implications for genome integrity remain elusive. Here, we use Bloom syndrome (BS), a rare genetic disease characterized by DNA repair defects and hyper-unstable rDNA, as a model to investigate the mechanisms leading to rDNA instability. We find that in Bloom helicase (BLM) proficient cells, the homologous recombination (HR) pathway in rDNA resembles that in nuclear chromatin; it is initiated by resection, replication protein A (RPA) loading and BRCA2-dependent RAD51 filament formation. However, BLM deficiency compromises RPA-loading and BRCA1/2 recruitment to rDNA, but not RAD51 accumulation. RAD51 accumulates at rDNA despite depletion of long-range resection nucleases and rDNA damage results in micronuclei when BLM is absent. In summary, our findings indicate that rDNA is permissive to RAD51 accumulation in the absence of BLM, leading to micronucleation and potentially global genomic instability.
Similar content being viewed by others


Introduction
The nucleolus is the most distinct feature of the mammalian nucleus and plays a critical role in ribosome biogenesis. This process is driven by RNA polymerase I (RNA pol I) mediated transcription of the ribosomal RNA genes, collectively referred to as the rDNA, which takes place in the nucleolus1. In human cells rDNA is composed of hundreds of repeats that are clustered on the short arms of the five acrocentric chromosomes (13, 14, 15, 21 and 22) and form the nucleolar organizer regions (NORs)2,3. One or more NORs can contribute rDNA to the same nucleolus4.
The formation of the nucleolus upon rDNA transcription creates a physical environment displaying liquid-like properties5, distinct from the rest of the nucleus, with selective protein retention influencing the factors associated with rDNA6. Furthermore, the actively transcribed rRNA genes are densely packed with RNA pol I and largely depleted of nucleosomes, resulting in a highly specialized chromatin environment7.
The rDNA is intrinsically unstable and was found to be one of the most frequently rearranged chromosomal regions in solid tumors8. Several features of the rDNA may contribute to its instability, including the presence of identical sequences in multiple genomic locations, the concentration of rDNA repeats in the nucleolus, high transcriptional activity and the open chromatin structure resulting from the dense RNA pol I occupancy9,10. Currently, however, our understanding of rDNA instability and its impact on the rest of the genome in mammalian cells is limited.
An accumulating number of studies have described how the nucleolus responds to DNA lesions. Many of these studies have focused on DNA double-strand breaks (DSBs), a deleterious type of lesion that can result in genomic rearrangements or loss of genetic material. To minimize the consequences of DSBs, cells activate the DNA damage response (DDR) that serves to prevent duplication or segregation of the DNA until the damage is repaired11. The DDR in the nucleolus (the nucleolar DDR or n-DDR) is distinct from that elsewhere in the nucleus, with specific adaptations to the nucleolar compartment. DSBs in the rDNA induce ATM-dependent phosphorylation of the nucleolar protein Treacle that recruits the MRE11-RAD50-NBS1 (MRN) complex and TOPBP112,13. Treacle, the MRN complex, and TOPBP1 then recruit and activate ATR, leading to transcriptional shut-down and nucleolar restructuring14,15. Nucleolar reorganization results in the translocation of the damaged rDNA to the nucleolar periphery into so-called nucleolar caps, where each cap represents a single NOR4,16,17. After separation of NORs into individual caps, the rDNA becomes accessible to factors promoting the homologous recombination (HR) repair pathway. This repair pathway utilizes a homologous sequence as template for DNA repair and HR factors cannot be detected within the nucleolar interior space, presumably to prevent recombination between rDNA originating from different chromosomes14,15,18,19,20. Generally, HR is activated in G2 and uses a sister chromatid as the template for repair21.
Bloom synd rome (BS) is a rare disease characterized by DNA repair defects and early onset of multiple cancers. By the age of 40, 80% of BS patients have developed cancer, which is the leading cause of death in BS22. At the cellular level, BS is characterized by high levels of genome instability with elevated sister chromatid exchange, chromosomal rearrangements, deletions, and micronuclei23,24. Of particular relevance to this study, rDNA was found to be hyper-unstable in BS cell lines, even when compared to other repair deficient syndromes25,26. These findings suggest that BS cells can be used as a model to study mechanisms underlying rDNA instability.
BS is caused by mutations in the gene encoding the Bloom helicase (BLM), a member of the RecQ helicases family, exerting 3′−5′ helicase activity27. BLM regulates multiple steps of the HR repair pathway activated upon DSBs, overall favoring anti-recombinogenic outcomes23. Initially, BLM facilitates long-range resection to generate single-stranded DNA (ssDNA) that is rapidly coated by replication protein A (RPA)28,29,30,31. After resection, RPA is replaced by RAD51, which is required for homology search and strand invasion of the sister chromatid. The loading of RAD51 is promoted by BRCA1 and dependent on BRCA232. BLM has both pro-and anti-recombinogenic roles at this intermediate step, by first promoting, then negatively regulating RAD51 activity and D-loop formation33,34,35,36. In complex with RMI1, RMI2, and Topoisomerase IIIα (TOP3A) (known as the BTRR complex), BLM is responsible for the dissolution of repair intermediates such as double Holliday junctions (dHJ) and promotes a non-crossover repair outcome. Furthermore, BLM plays a role in mitosis in the segregation of chromosomes by resolving ultra-fine bridges that can arise from unprocessed HR intermediates24,37,38,39.
In this study, we characterize the role of BLM in rDNA repair and use BS patient-derived and BLM-deficient cells to study the mechanisms resulting in destabilization of rDNA. In BLM-deficient cells we find that RAD51 accumulation occurs despite compromised recruitment of upstream HR factors (RPA, BRCA1 and BRCA2) to rDNA DSBs. Depletion of nucleases promoting long-range resection also does not prevent RAD51 recruitment to rDNA in the absence of BLM. However, depletion of the short-range resection factor MRE11 moderately decreases RAD51 accumulation and an increased dependency on BRCA1 is observed in BLM-deficient cells. Our data further indicate that RAD51 accumulation in rDNA in the absence of BLM drive micronuclei formation as cells go through mitosis. Collectively, our data suggest that rDNA allows RAD51 accumulation in the absence of BLM resulting in micronuclei formation and potentially global genome instability.
Results
BLM is recruited to rDNA DSBs and promotes cellular survival
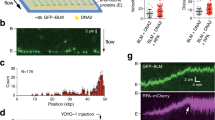
To understand the mechanisms underlying rDNA instability in BLM-deficient cells26, we started out by characterizing the role of BLM in the n-DDR. First, we examined BLM localization after rDNA damage, induced in Cas9-expressing U2OS cells (U2OS-Cas9) transfected with guide RNA plasmids (gRNAs) targeting the rDNA (Supplementary Fig. 1a, b)14, over a time-course of 6 h after damage. During this time, rDNA translocates from the nucleolar interior to the periphery of the nucleolus where it forms nucleolar caps. GFP-tagged NBS1 was used as a damage marker as it rapidly associates with rDNA DSBs and is transferred with rDNA to nucleolar caps14. Prior to damage (0 h), BLM presents a nucleoplasmic and dispersed nucleolar signal. Three hours after rDNA damage, when interior nucleolar foci form, no BLM association is detected with nucleolar foci. After 6 h, however, BLM co-localizes with NBS1 in nucleolar caps (Fig. 1a). Automated quantification by Quantitative Image Based Cytometry (QIBC40), also confirmed accumulation of BLM in nucleolar caps (Fig. 1b). While NBS1 is recruited independently of the cell cycle (Supplementary Fig. 1c), we found increased BLM recruitment to nucleolar caps in S/G2 phases (Fig. 1c). BLM recruitment to nucleolar caps was further confirmed in HT1080 cells expressing the I-PpoI nuclease (Fig. 1d) and in HeLa cells transfected with Cas9 and rDNA targeting gRNAs (Cas9-gRNA) as ribonucleoproteins complexes (RNPs) (Fig. 1e). In both cases, BLM accumulates in nucleolar caps after translocation of rDNA DSBs to the nucleolar periphery. To determine if BLM is recruited specifically to rDNA DSBs we performed chromatin immunoprecipitation (ChIP) (Fig. 1f). After gRNA treatment, BLM was detected near the break (primer A), with no enrichment measured further away from the break site (primers B-E). RNA Pol I was included as a reference localizing to the transcribed region of rDNA (primers A, B, and C) (Fig. 1f)18. In summary, BLM accumulates at rDNA DSBs in nucleolar caps with an increased proportion in S/G2.

a Time course of BLM recruitment. U2OS-Cas9 NBS1-GFP cells treated with rDNA gRNA plasmids and stained with BLM antibody 0-3-6 h after gRNA transfection. Scale bar 10 μm. b QIBC quantification of BLM cap foci number per cell 6 h after transfection with EV or gRNA plasmids. The graph depicts one representative out of three biological replicates (n = 341,162 cells/condition/experiment). Box limits indicate the central 50% with the horizontal line marking the mean value. Points more than 1.5 times the inter-quartile range outside the box are plotted as outliers (dots), while whiskers cover the remaining points. For statistical analysis a binomial distribution model was applied. c Cell cycle distribution of cells with BLM cap foci. Cells were treated as in b, and the cell cycle distribution of cells with at least one BLM cap foci was analyzed by QIBC. The graph depicts mean values +/−SEM. (n = 113, 44, 63 (G1); 46, 14, 31 (S); 61, 28, 68 (G2) cells/condition/experiment). Statistical analysis was done using a Poisson distribution model. d BLM recruitment to rDNA breaks upon I-PpoI nuclease induction in HT1080 cells. Scale bar 10 μm. e BLM recruitment to rDNA breaks upon Cas9-Scaffold/gRNA RNP transfection in Hela cells. Scale bar 10 μm. f BLM recruitment to the rDNA repeat analyzed by ChIP in U2OS-Cas9 cells. The gRNA1 cut site and the position of the amplified regions are indicated. RNA pol I ChIP was used as a control. The graph depicts mean values +/−SEM. g Percentage of cells displaying at least one NBS1 cap foci. U2OS-Cas9 cells were treated with siRNA for 24 h and subsequently transfected with EV or gRNA plasmids for 6 h. The graph depicts mean values +/−SEM (n = 291, 266 (EV,siCon); 351, 324 (EV, siBLM1); 251, 323 (EV, siBLM2); 210, 198, 315 (gRNA, siCon); 193, 193, 211 (gRNA, siBLM1); 195, 207, 272 (gRNA, siBLM2) cells/condition/experiment). Two biological replicates for EV samples. For statistical analysis of the gRNA treated samples, one-way Anova was applied. h Quantification of colony survival assay of U2OS WT and BLM−/− cells. Values are normalized to the Cas9-Scaffold RNP treated controls. The graph depicts mean values +/−SEM. Red dots indicate technical replicates from three biological replicates (n = 361, 347, 320 (WT, EV); 238, 180, 297 (BLM−/−, EV); 395, 239, 374(WT, gRNA); 111, 81, 151(BLM−/−, gRNA) colonies/condition/experiment). For statistical analysis, one-way Anova was applied. Source data are provided as a Source Data file for panels b, c, f, g and h. Statistical significance is depicted with p-values. Three biological replicates unless specifically stated (QIBC quantitative image-based cytometry, EV empty vector).
We next investigated how the absence of BLM affects the response to rDNA DSBs. BLM knockdown was conducted using two independent siRNAs (Supplementary Fig. 1d) that did not alter cell cycle distribution (Supplementary Fig. 1e). We found that nucleolar reorganization and cap formation appear functional in BLM depleted cells based on the translocation of the two rDNA-bound proteins, UBF and Treacle14, from the nucleolar interior to the periphery (Supplementary Fig. 1f, g). We also assessed if BLM depletion affects DDR proteins known to associate with damaged rDNA after translocation to the nuclear periphery14,19. We found that γH2AX and phosphorylated ATM levels, as well as the recruitment of MDC1 and 53BP1 to the nucleolar periphery, were independent of BLM (Supplementary Fig. 2a–d). Next, to determine if the n-DDR is enhanced in the absence of BLM, we quantified cells with NBS1 cap foci. We observed an increased number of cells with NBS1 cap foci in BLM depleted samples (Fig. 1g), suggesting that the absence of BLM may either increase the accumulation of repair proteins or impair the clearance kinetics of nucleolar caps, given that NBS1 protein levels remained unchanged (Supplementary Fig. 2e). Furthermore, BLM-deficient cells were found to be more sensitive to rDNA damage. Survival analysis revealed a 50% reduction in colonies in U2OS BLM−/− cells after Cas9-gRNA RNP treatment compared to the control Cas9-Scaffold RNP, while BLM WT cells survived rDNA damage (Fig. 1h). Together these data show proficient nucleolar restructuring and accumulation of initial n-DDR factors in nucleolar caps, but an increased sensitivity to rDNA DSBs in the absence of BLM.
BLM promotes RPA, BRCA1 and BRCA2 recruitment to rDNA DSBs
The repair mechanism in rDNA is confined to individual nucleolar sub-compartments with HR taking place in the nucleolar caps10,19. Recruitment of BLM to nucleolar caps, therefore, indicates a role for BLM in HR in rDNA. The role of BLM in promoting resection and regulating HR is widely recognized29,41. Resection generates ssDNA stretches that are rapidly coated with RPA to enhance their stability. Resection initiation is dependent on MRE11 and CtIP followed by long-range resection by BLM together with either DNA2 or EXO128,30. Consequently, in the absence of BLM resection was found to be impaired in nuclear chromatin29,30.
To determine if rDNA resection is compromised in BLM-deficient cells, we examined the DSB-associated resection marker RPA32. RPA was recruited to the nucleolar caps in control cells, indicative of ongoing resection14,15 (Fig. 2a). In BLM depleted cells, however, RPA accumulation in the caps was largely diminished (Fig. 2a) and the number of cells with RPA positive caps was significantly decreased (Fig. 2b). Total cellular levels of RPA, however, were unchanged (Supplementary Fig. 3a). Upon depletion of CtIP, EXO1 and DNA2 we found either a pronounced or partial loss of RPA32 recruitment to rDNA (Fig. 2a, b and Supplementary Fig. 3b) correlating with the expected impairment of resection reported in nuclear chromatin28,29,30,42.

a RPA32 recruitment to nucleolar caps. U2OS-Cas9 cells were treated with siRNAs for 24 h and subsequently transfected with gRNA plasmids for 6 h, and immunostained with antibody against RPA32. b Quantification of cells with at least one RPA32 cap focus after depletion of resection factors. Cells were treated as in a. The graph depicts mean values +/−SEM. Three biological replicates (two in case of EXO1) (n-values in source data file). For statistical analysis, data was square root transformed and analyzed with one-way Anova. c BRCA1 recruitment to nucleolar caps. Cells were treated as in a and immunostained with antibody against BRCA1. d Quantification of cells treated with gRNA from c. Left panel: quantification of BRCA1 cap foci number per cell (n = 120, 151, 191 cells/condition). Right panel: quantification of BRCA1 cap foci intensity (n = 287, 276, 271 cells/condition). The graphs show one representative experiment. Box limits indicate the central 50% with the horizontal line marking the mean value. Points more than 1.5 times the inter-quartile range outside the box are plotted as outliers (dots), while whiskers cover the remaining points. For statistical analysis a Poisson distribution model was used (left panel) or data were square root transformed (right panel) and analyzed with one-way Anova. e BRCA2 recruitment to nucleolar caps. U2OS-Cas9 cells were treated as in a, and immunostained with antibody against BRCA2. f Quantification of cells with at least one BRCA2 positive cap focus. Cells were treated as in e. The graph depicts mean values +/−SEM (n = 90, 105, 104 (siCon); 88, 102, 101 (siBLM1); 99, 111, 116 (siBLM2) cells/condition/experiment). For statistical analysis, one-way Anova was applied. Scale bars 10 μm. Source data are provided as a Source Data file for panels b, d and f. Three biological replicates unless specifically stated. Statistical significance is depicted with p-values. (EV: empty vector).
In the following steps of HR, BRCA1 promotes DSB processing, resection regulation, and replacement of RPA with RAD51 on ssDNA32,43,44,45. We therefore investigated if BLM depletion influenced BRCA1 recruitment to rDNA DSBs14,15,19,20. In BLM depleted cells, BRCA1 foci formation could still be detected (Fig. 2c), but foci count and intensity were significantly reduced (Fig. 2d), while protein levels of BRCA1 were not affected (Supplementary Fig. 3c). BRCA2, an HR factor primarily engaged with RAD51 loading onto ssDNA stretches32,46, follows a similar pattern to RPA and BRCA1 with reduced accumulation in rDNA in BLM depleted cells (Fig. 2e, f).
Collectively, these data indicate that the processing of DSBs is compromised and that BRCA1 and BRCA2 accumulation at rDNA is diminished in BLM-deficient cells.
RAD51 accumulation is independent of long-range resection factors after rDNA DSBs
BRCA1 and BRCA2 promote the displacement of RPA on ssDNA by RAD51, a recombinase initiating homology search and strand invasion of the sister chromatid32,44,45,46. Lower accumulation of BRCA1 and BRCA2 might be an indication of compromised HR. We therefore assessed if RAD51 accumulation is also impaired as it would be anticipated after rDNA damage in BLM depleted cells. Interestingly, however, the number and intensity of RAD51 cap foci were not decreased by BLM depletion (Fig. 3a, b), suggesting that RAD51 can still accumulate at rDNA regardless of the impaired upstream processing. RAD51 protein levels remained unchanged in BLM-deficient cells (Supplementary Fig. 3d).

a RAD51 recruitment to nucleolar caps. U2OS-Cas9 cells were treated with siRNA for 24 h, transfected with EV or gRNA plasmids for 6 h, and immunostained with antibody against RAD51. Scale bar 10 μm. b Left panel: quantification of RAD51 cap foci number per cell (n = 102, 86, 117 cells/condition). Right panel: quantification of RAD51 cap foci intensity (n = 167, 116, 154/condition). Cells were treated as in a. The graphs show one representative biological replicate. Box limits indicate the central 50% with the horizontal line marking the mean value. Points more than 1.5 times the inter-quartile range outside the box are plotted as outliers (dots), while whiskers cover the remaining points. For statistical analysis a Poisson distribution model was used (left panel) or data were square root transformed (right panel) and analyzed with one-way Anova. c Recruitment of RPA32 (upper panel) and RAD51 (lower panel) in gRNA treated BLM−/− cells reconstituted with GFP-BLM WT or mutants. Cells were transfected with plasmids (48 h) and rDNA damage was induced by Cas9-gRNA RNP transfection for 6 h. The graphs depict mean values +/−SEM (n-values in source data file). d Quantification of RPA32 (upper panel) and RAD51 (lower panel) recruitment after depletions of resection factors. Cells were treated as in a and immunostained with antibodies against RPA32 and RAD51. The graphs depict mean values +/−SEM (n-values in source data file). For statistical analysis data were log transformed and analyzed with one-way Anova. e Recruitment of RAD51 in Cas9-gRNA treated BLM−/− cells after MRE11 depletion (60 h), Cas9-gRNAs (8 h), and immunostaining with antibodies against RAD51. The graphs depict RAD51 positive nucleolar caps (n = 129 cells/condition/experiment). For statistical analysis, one-way Anova was applied. f Measurement of ssDNA in nucleolar caps by BrdU detection. U2OS WT or BLM−/− cells were grown in 10 µM BrdU for 24 h prior to Cas9-gRNA treatment for 8 h (n = 1074, 1613, 2007 (WT, gRNA); 911, 1274, 1202 (BLM−/−, gRNA) foci/condition/experiment). Data are presented as individual values with a horizontal line marking the mean. The graphs show one representative biological replicate. For statistical analysis data were log transformed and one-way Anova was applied. g Recruitment of RAD51 in gRNA treated WT or BLM−/− cells after depletion with indicated siRNAs. Cells were treated with siRNA for 24 h and subsequently transfected with Cas9-gRNA for 6 h, and immunostained with antibodies against RAD51. The graphs depict mean values +/−SEM (n-values in source data file). For statistical analysis data were log transformed to get normal distribution and were analyzed with one-way Anova. Source data are provided as a Source Data file for panels b–g. Three biological replicates unless stated. Statistical significance is depicted with p-values.
To further understand if impaired recruitment of upstream repair factors is linked to defective resection of rDNA, we performed reconstitution assays with a panel of GFP-tagged BLM mutants: I841T (ATPase domain, helicase-dead47), K1270V (HRDC domain, no dHJ dissolution48), S1342D-S1345D (nucleolar localization mutant49) and the BLM-Ash mutant (a dysfunctional truncated protein, commonly mutated in BS patients)50 (Supplementary Fig. 3e). The mutants were introduced in BLM−/− cells (Supplementary Fig. 3f, g) in combination with RNP complexes to induce rDNA DSBs. As expected, no RPA foci formation was observed in U2OS BLM−/− cells. RPA foci were rescued by ectopic expression of BLM WT, K1270V and the S1342D-S1345D mutants, but not by the helicase-dead I841T or the blm-Ash mutants (Fig. 3c, Supplementary Fig 3g), in agreement with BLM’s role in resection, which is dependent on its helicase activity30. In the case of RAD51, foci formation was proficient after rDNA DSB induction, regardless of BLM status (Fig. 3c, Supplementary Fig 3g), supporting our finding that RAD51 recruitment is at least partially uncoupled from the upstream processing steps of the HR cascade in rDNA.
As mentioned above, the resection process is initiated by MRE11 and CtIP, conducting initial short-range resection, followed by long-range resection by BLM together with either DNA2 or EXO128,30. To determine if BLM-independent contributions of DNA2 or EXO1 are facilitating RAD51 accumulation, we performed co-depletion of multiple resection factors and measured RPA32 and RAD51 recruitment. RPA32 accumulation in nucleolar caps was significantly reduced upon depletion of CtIP or BLM, as well as co-depletion of BLM/DNA2, BLM/EXO1, DNA2/EXO1 and BLM/DNA2/EXO1 (Figs. 2b and 3d). However, accumulation of RAD51 was generally unaffected apart from a 15% reduction observed after co-depletion of BLM/DNA2 (Fig. 3d). In summary, based on measurements of RPA accumulation our results strongly indicate that RAD51 recruitment to rDNA is independent of long-range resection factors.
The MRN nuclease complex initiates resection and we therefore examined if short-range resection, mediated by MRE11, is required for RAD51 accumulation. We depleted MRE11 in BLM-deficient cells (Supplementary Fig. 4a) and scored the number of nucleolar caps positive for RAD51. MRE11 depletion reduced the number of RAD51 containing caps by 30 percent (Fig. 3e), suggesting that MRE11 promotes RAD51 accumulation in nucleolar caps in the absence of BLM. In addition, we examined if WRN, an alternative helicase to BLM that can facilitate DNA2 resection51, is required for RAD51 accumulation in the absence of BLM, but our findings did not support this requirement (Supplementary Fig. 4b).
Although RPA is commonly used as a proxy for resection, the accumulation of RAD51 indicates the presence of single-stranded DNA (ssDNA) in response to rDNA damage. We therefore set out to measure ssDNA by BrdU-detection under native conditions52 in BLM-deficient cells. Under these conditions BrdU is not accessible to the BrdU-antibody in double-stranded DNA but can only be recognized in ssDNA. We found that BLM-deficient cells have lower levels of BrdU in nucleolar foci after rDNA damage compared to control cells consistent with defective long-range resection (Fig. 3f).
Since RAD51 loading onto ssDNA is dependent on BRCA253 we examined the dependency on both BRCA1 and BRCA2 in BLM-deficient cells. BRCA2 depletion fully abrogated RAD51 recruitment after rDNA DSBs in both BLM pro- and deficient cells (Fig. 3g). BRCA1 depletion, however, had only a mild impact on RAD51 loading in BLM proficient cells but strongly impaired RAD51 accumulation in BLM-deficient cells (Fig. 3g). These findings support BRCA2 mediated accumulation of RAD51 onto ssDNA and an increased need for BRCA1 when upstream processing is impaired.
We then wanted to examine if RAD51 loading occurs genome-wide in the absence of BLM. We therefore treated cells with 5 Gy of ionizing radiation (IR) and followed RPA and RAD51 accumulation. We found that in U2OS BLM−/− cells both RPA and RAD51 recruitment to radiation induced foci is impaired, as previously reported29,30,41,54 (Supplementary Fig. 4c, d). These data indicate that the RAD51 loading observed in rDNA does not occur genome-wide after exposure to IR.
BLM depletion induces rDNA-containing micronuclei
To understand the response of BLM-deficient cells to rDNA damage over longer periods of time we followed cell cycle progression, repair kinetics and senescence over a prolonged timeframe. Cells were treated with Cas9 and gRNA for 6 h and followed for an additional 24 h (Supplementary Fig. 5a). The Cas9 protein level peaked after 6 h and gradually degraded throughout the time course (Supplementary Fig. 5b). The level of endogenous nucleolar DSBs was elevated in BLM-deficient cells compared to the control, whereas RNP treatment induced an increase in nucleolar DSBs in both BLM−/− and control cells (Supplementary Fig. 5c). We also quantified nucleolar repair foci and found an increased accumulation until 12 h post transfection followed by a plateau and a subsequent decline after 24 h, with BLM-deficient cells having increased levels of DNA damage throughout the time course (Supplementary Fig. 5d) consistent with previous findings55. The fraction of senescent cells was increased in the BLM-deficient population, however the induction of senescence after rDNA damage was minor in both control and BLM-deficient cells (Supplementary Fig. 5e). Our cell cycle analysis revealed that after 30 h a substantial fraction of cells was still going through mitosis aligning with previously published observations14, however, the proportion of cells in S-phase was decreased, particularly in BLM−/− cells after rDNA damage (Supplementary Fig. 5f). To determine if rDNA damage in BLM-deficient cells is potentially harmful to daughter cells, we followed U2OS WT and BLM−/− cells expressing an endogenously tagged version of the nucleolar protein Treacle through mitosis. U2OS BLM proficient cells resolve rDNA DSBs and proceed normally through mitosis (Fig. 4a). In BLM-deficient U2OS cells, we observed chromosome segregation errors, with rDNA present on the tips of lagging chromosomes or chromatin connecting the condensed DNA in anaphase. As cells go through mitosis, the unsegregated rDNA results in the formation of micronuclei (Fig. 4b).

a, b Snapshots from live cell imaging of mitotic U2OS WT and BLM−/− cells endogenously expressing GFP-Treacle used as a marker of rDNA and transfected with H2B-RFP adenovirus plasmid to visualize DNA. Cells were treated with Cas9-gRNA RNPs. Scale bar 10 μm. c Analysis of micronuclei formation. U2OS-Cas9 cells were treated with siRNA for 24 h and subsequently transfected with EV or gRNA plasmids. Cells were then treated with cytochalasin B for 24 h, stained with DAPI and imaged with confocal microscopy. Arrows indicate micronuclei. Scale bar 60 μm. d Quantification of micronuclei in bi-nucleated cells from c. The graph depicts mean values +/−SEM (n-values in source data file). For statistical analysis, one-way Anova was applied. e Quantification of UBF positive micronuclei. Cells were treated as in c, and immunostained with UBF antibody to visualize rDNA. The graph depicts mean values +/−SEM (n-values in source data file). For statistical analysis data were square root transformed to get normal distribution and were analyzed with one-way Anova. f Representative images of UBF positive and negative micronuclei from cells in e. Scale bar 10 μm. g Quantification of micronuclei (left panel) and rDNA (visualized by UBF) containing micronuclei (right panel) in U2OS WT and BLM−/− cells 6 h after rDNA damage was induced by Cas9-gRNA RNP transfection. The graphs depict mean values +/− SEM (n-values in source data file). For statistical analysis data were square root transformed (left panel) or not transformed (right panel) to get normal distribution and were analyzed with one-way Anova. h Representative images of micronuclei in BS patient cells (GM08505, GM03402, GM02932, GM02548) and control fibroblast (GM08402) during normal proliferation. Cells were immunostained with Treacle antibody to visualize rDNA. Scale bar 10 μm. i Quantification of micronuclei (left panel) and rDNA (Treacle) containing micronuclei (right panel) in BS patient cells upon normal cell proliferation. The graphs depict mean values +/−SEM (n-values in source data file). Five biological replicates. For statistical analysis data were log transformed (left panel) and square root transformed (right panel) and were analyzed with one-way Anova. Source data are provided as a Source Data file for panels d, e, g and i. Three biological replicates unless stated. Statistical significance is depicted with p-values. (EV: empty vector).
Micronuclei can affect genome stability severely as chromosomes or parts of chromosomes are separated from the rest of the genome56. We therefore set out to quantify micronucleation in BLM-deficient cells exposed to rDNA DSBs. We found that transient BLM depletion or rDNA DSBs alone induced either no or a minor increase in micronuclei formation (Fig. 4c, d). However, when BLM depletion and rDNA DSBs were combined, cells showed a three-fold induction of micronuclei compared to control cells (Fig. 4c, d).
To examine how often rDNA can be found in these micronuclei, we stained for rDNA, using UBF as a marker. When BLM depletion and rDNA DSBs are combined, UBF positive micronuclei occur in ~25% of the cells (Fig. 4e, f), corresponding to the total increase in micronuclei (Fig. 4d). This finding suggests that in the absence of BLM, rDNA DSBs drive micronuclei formation. Similarly, when loss of BLM was permanent (U2OS BLM−/− cells), we also observed a higher frequency of rDNA containing micronuclei compared to U2OS WT cells after rDNA DSBs (Fig. 4g).
To assess the physiological relevance of rDNA damage in relation to BS, we analyzed micronucleation in a panel of fibroblast cell lines from BS patients (GM08505, GM03402, GM02932, GM02548). All cell lines showed elevated levels of micronuclei compared to the control fibroblast cell line, GM08402. In the BS cell lines, rDNA containing micronuclei, marked by Treacle, accounted for 20–50% of all micronuclei (Fig. 4h, i), indicating that rDNA instability could potentially contribute to the genomic instability and pathogenesis of BS.
RAD51 induces mitotic errors leading to micronuclei
We speculated that micronucleation could be driven by unscheduled RAD51 accumulation and the formation of joint molecules, causing mitotic mis-segregation. To address the role of RAD51 in micronuclei formation in BLM-deficient cells, we quantified micronuclei after RAD51 depletion (Supplementary Fig. 5g). Depletion of BLM and RAD51, alone and in combination, increased the total number of micronuclei upon rDNA DSBs compared to control cells (Fig. 5a), whereas BLM depletion alone resulted in the highest number of rDNAcontaining micronuclei (Fig. 5b). Moreover, the increase in rDNA-positive micronuclei caused by BLM depletion could be rescued by co-depletion of RAD51 (Fig. 5b), suggesting that RAD51 accumulation is a major contributor to rDNA instability in BLM-deficient cells, either through the formation of joint molecules or by preventing the use of BLM independent repair pathways.

a Quantification of micronuclei in bi-nucleated cells upon depletion of BLM, RAD51, or BLM/RAD51. U2OS-Cas9 cells were treated with siRNA for 24 h and with EV/gRNA plasmids (6 h). Cells were then treated with cytochalasin B for 24 h and stained with DAPI. The graph depicts mean values +/−SEM (n-values in source data file). For statistical analysis data were square root transformed and were analyzed with one-way Anova. b Quantification of rDNA positive micronuclei as in a combined with UBF staining. The graph depicts mean values +/−SEM (n-values in source data file). For statistical analysis data were square root transformed and were analyzed with one-way Anova. c Quantification of micronuclei in bi-nucleated cells upon depletion of the BTRR complex and transfected with gRNA plasmids. Cells were treated as in a. The graph depicts mean values +/−SEM (n-values in source data file). Four biological replicates. For statistical analysis data were log transformed and analyzed with one-way Anova. d Quantification of rDNA (UBF) positive micronuclei in cells from c. The graph depicts mean values +/−SEM (n-values in source data file). For statistical analysis data were square root transformed and analyzed with one-way Anova. e rDNA copy number quantified in U2OS WT and BLM−/− cells using ddPCR. Cells were treated with control or rDNA gRNA for 6 h followed by 72 h of recovery prior to DNA extraction. The graphs depict mean values +/−SEM (n = 3). For statistical analysis data were log transformed and analyzed with one-way Anova. f Proposed model for the role of BLM in rDNA instability. Left panel: in BLM proficient conditions, BLM promoted resection takes place after DSB induction (1 + 2). Rad51 is recruited to resected DNA and HR can take place (3). The BTRR complex promotes dissolution (4), and normal cell division takes place (5). Middle panel: resection is impaired in BLM-deficient conditions in nuclear chromatin (1 + 2), HR is impeded (3) and genomic instability occurs (4). Right panel: resection is impaired in rDNA in BLM-deficient conditions (1 + 2), RAD51 accumulation still occurs (3), and joint rDNA molecules give mitotic problems in the absence of the BTRR complex (4), leading to rDNA driven micronuclei formation after cell division (5). Created with BioRender.com released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license. Source data are provided as a Source Data file for panels a–e. Three biological replicates unless specifically stated. Statistical significance is depicted with p-values.
BLM carries out several of its functions as part of the BTRR complex, including resection and dissolution of repair intermediates31,37. Faulty dissolution of repair intermediates can compromise chromosome segregation and lead to micronucleation24. To address if BLM functions in the context of this BTRR dissolvasome to repair rDNA, we depleted individual components of the complex (Supplementary Fig. 5h). Consistent with previous findings57, depletion of RMI1 affected the stability of RMI2. Depletion of TOP3A, RMI1 and RMI2 increased the total number of micronuclei to a lesser extent than BLM depletion (Fig. 5c, Supplementary Fig. 5i). However, co-depletion of RMI1, RMI2, and TOP3A together with BLM showed no additive effect compared to individual knock-down, indicative of the components functioning in the same pathway (Fig. 5c). We then specifically examined rDNA containing micronuclei and found that rDNA-containing micronuclei were induced by depletion of individual BTRR complex components (Fig. 5d). However, upon co-depletion no additive effect could be measured, suggesting that BLM functions in the BTRR complex to maintain rDNA stability.
HR repair has been connected to loss of rDNA repeats20. We therefore examined rDNA copy number in BLM pro- and deficient cells. We found that control cells had approximately twice as many rDNA copies as BLM-deficient cells (Fig. 5e). Interestingly, rDNA copies were lost upon rDNA damage in control cells, whereas this was not the case in BLM-deficient cells (Fig. 5e).
Discussion
The human genome contains several types of repetitive sequences that must be preserved through successive cell divisions. Recent studies have documented specialized repair pathways operating within these domains to ensure their stability10,58. However, rDNA recombines with a rate of more than 10% in meiosis59 and represents one of the most commonly rearranged genomic regions in solid tumors8, supposedly due to unscheduled recombination. The mechanisms underlying rDNA recombination are therefore both important for the plasticity of the genome in healthy cells and likely to contribute to the genomic instability associated with human diseases, especially cancer. To gain insights into the mechanisms underlying rDNA hyper-recombination, we used BS as a model due to its hyper-unstable rDNA25,26 and combined it with targeted rDNA DSBs induced by CRISPR-Cas914. Our data uncovered a long-range resection-independent mechanism for RAD51 accumulation at rDNA, leading to rDNA hyper-instability in BLM-deficient cells.
When DSBs are inflicted in rDNA, the special n-DDR, adapted to the nucleolar environment, is activated. rDNA transcription is rapidly silenced by the ATM-Treacle-MRN signaling axis, and rDNA moves to the periphery of the nucleolus where it is condensed in nucleolar caps13,14,15,17,18,19,20,60. In nucleolar caps the homologous recombination pathway can be activated to facilitate rDNA repair16,19,60,61. Our data suggest a function of BLM in this stage of rDNA DSB repair as nucleolar restructuring remains proficient in BLM depleted cells (Supplementary Fig. 1f, g) and BLM recruitment occurs in nucleolar caps at the periphery (Fig. 1a, d, e). The augmented DSB-associated nucleolar cap occurrence and increased formation of micronuclei in BLM depleted cells (Figs. 1g and 4a–g) document the importance of BLM in rDNA repair. Defective resection of rDNA in BLM-deficient cells (Figs. 2a, b and 3f) suggests that BLM functions to promote the HR pathway, in agreement with previous results in nuclear chromatin29. Participation of the BLM helicase in the nucleolar DNA damage response therefore overlaps with its function in the HR pathway in nuclear chromatin.
BLM has previously been shown to localize to the nucleolus and promote rDNA transcription62. To address if the function of BLM in rDNA repair is distinct from its role in rDNA transcription, we performed functional reconstitution assays and found that a BLM mutant (S1342D-S1345D), excluded from the nucleolar interior where rDNA transcription takes place, can still accumulate at the nucleolar periphery and rescue the impaired resection observed in BLM-deficient cells (Fig. 3c and Supplementary Fig. 3g). These findings support a role of BLM in repair of rDNA distinct from its role in rDNA transcription.
The HR repair pathway in nuclear chromatin is commonly presented as a hierarchical process with several sequential steps starting with short-range resection, long-range resection, and formation of the RAD51 filament followed by invasion of the sister chromatid63. Our study of DSB repair in rDNA reveals that within this genomic domain, depletion of long-range resection factors does not abrogate RAD51 accumulation (Fig. 3a, b, d and g). When the dependency on BLM for RAD51 loading was assessed broadly across nuclear chromatin after IR, we found an impairment of RAD51 accumulation in the absence of BLM (Supplementary Fig. 4c, d). The accumulation of RAD51 may therefore be dependent on the chromatin context and be more complex than it is generally depicted.
Our data show that BRCA1 and BRCA2 accumulation is reduced at rDNA in BLM-deficient cells, but nevertheless is still required for RAD51 loading (Fig. 3g). The dependency on BRCA1 for RAD51 accumulation was increased in BLM-deficient cells, possibly to reach sufficient BRCA2 accumulation, required for RAD51 loading64.
rDNA appears to be less dependent on resection factors and does not require the same level of processing to facilitate RAD51 accumulation as previously reported29,30. The nature of the substrate for RAD51 accumulation needs to be further investigated, however the dependence on BRCA2 and the detection of ssDNA in nucleolar caps after rDNA damage, although reduced in BLM-deficient cells, suggest that ssDNA is involved. Furthermore, MRE11 promotes RAD51 accumulation supporting that ssDNA contributes to this process (Fig. 3e). Interestingly, in BLM-deficient cells RAD51 was shown to accumulate at stalled replication forks65 and further examinations of the mechanism associated with accumulation of RAD51 in rDNA are needed to determine if similarities between these two processes exist. Alternatively, the open chromatin conformation and low density of nucleosomes in the transcribed regions of rDNA could expose ssDNA serving as a platform for RAD51 filament formation7. A recent study found a key role of BLM in the creation of ssDNA loops during DNA end processing and suggested that loop formation may facilitate RAD51 filament formation66. It is not unreasonable to envision that ssDNA loops may arise as rDNA is condensed in nucleolar caps at the nucleolar periphery making BLM dispensable for RAD51 loading. Other studies have found that RAD51 filaments may be stabilized by the absence of BLM due to the reported role of BLM in displacing RAD5134. BLM depletion was shown to stabilize RAD51 filaments even in shorter resected tracts35 and hyper-accumulation of RAD51 has been observed in BS cells67,68. These findings suggest that the permissiveness of rDNA to RAD51 accumulation in the absence of BLM could be driven by multiple mechanisms and will require further investigations to be fully elucidated.
Our data support previous reports documenting a dual role of BLM in HR, initially promoting resection and later dissolution of repair intermediates (Fig. 5f, left panel). In the absence of BLM, resection is impeded in nuclear chromatin and the subsequent steps of the HR pathway are impaired (Fig. 5f, middle panel). In contrast, impaired resection in rDNA does not hinder the recruitment of the RAD51 recombinase, likely leaving BLM-deficient cells with joined DNA molecules (Fig. 5f, right panel). Such structures may require the dissolution activity of the BTRR complex for faithful chromosome segregation, however, since the BTRR complex is also implicated in the regulation of resection31, impairment of this step may further contribute to rDNA instability.
Examination of rDNA copy number, in BLM-deficient cells, confirms the previously reported importance of BLM for rDNA stability26. In these cells, we observed a significant reduction in the rDNA copy number. Notably, upon induction of DSBs BLM-deficient cells did not lose rDNA copies as seen in control cells (Fig. 5e). Both the reduced number of rDNA repeats and the lack of rDNA loss likely contribute to the sensitivity of BLM-deficient cells to rDNA damage. The mechanistic details of the process leading to loss of rDNA repeats remain unclear and will need further investigations to be fully understood.
The repetitive nature and intrinsic instability raise a conceptual question of whether rDNA damage is of importance to broader destabilization of the genome and tumorigenesis. Our data shows how rDNA can drive micronucleation when DNA repair is compromised (Fig. 4a–g), a feature of many cancers, and rDNAcontaining micronuclei were detected in BS patient cell lines (Fig. 4h, i). Micronuclei can result in large numbers of rearrangements in entire chromosomes, leading to gene dosage imbalances, and are viewed as one of the key causes of tumor evolution56. It will therefore be interesting to further investigate how rDNA-driven genomic instability may be relevant to normal cells, tumorigenesis and the pathology of BS. Furthermore, the importance of BLM in repair of rDNA could open therapeutic possibilities for BS patients and cancer patients with aberrant BLM expression.
Methods
Cell culture
The following cell lines were used in the study: U2OS-Cas9 generated in the lab and previously published in ref. 14, U2OS WT purchased from ATCC, U2OS endogenously tagged with GFP-Treacle generated from WT U2OS, BLM−/− and parental U2OS gifted by Dr. Eros Lazzerini Denchi, and U2OS BLM−/− endogenously tagged with GFP-Treacle derived from this cell line. Hela was purchased from the ATCC and Bloom syndrome patient cell lines and control fibroblasts (GM08505, GM03402, GM02932, GM02548 and GM08402) were purchased from the Coriell Institute. HT1080 I-PpoI cell line was published by Oka et al., 2011. U2OS-Cas9 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Gibco D5030) containing 10% fetal bovine serum (FBS) (Invitrogen), 100 U penicillin, 100 ug/ml streptomycin (P/S), 1 ug/ml puromycin and 400 ug/ml neomycin (G418). U2OS WT, BLM−/−, Hela and HT1080 cells were grown in DMEM (10% FBS + P/S). Bloom syndrome patient cell lines and control fibroblasts were grown in Minimal Essential Medium (α-MEM, Gibco 12571063) complemented with 15% inactivated FBS and P/S For GM02458, 20% FBS was added. To induce I-PpoI endonuclease activity in the HT1080 cell line, 4-hydroxytamoxifen (4OHT) was added to a final concentration of 300 nM.
Generation of cell lines
BLM proficient and deficient U2OS cell lines expressing endogenously tagged GFP-Treacle were generated using CRISPR/Cas9. Two plasmids were used to express 1) Cas9 nuclease and a scaffold/gRNA (pX330-NTCOF1 gRNA #1) and 2) the donor DNA for homology directed repair (pBluescript-HDR-GFP-NTCOF1). To introduce a GFP at the N-terminus of Treacle, we designed sgRNAs close to TCOF1 start codon using the Benchling CRISPR Design tool (https://benchling.com.). The sgRNAs were subcloned into the Cas9/gRNA-expressing plasmid using the general protocol from Zhang lab69. Briefly, the primers NTCOF1 gRNA#1 FWD and NTCOF1 gRNA#1 REV were phosphorylated and annealed prior to being introduced into BbsI-digested pX330 (pX330-U6-Chimeric_BB-CBh-hSpCas9 was a gift from Feng Zhang, Addgene plasmid #42230). For the donor DNA plasmid, we used approx. 900 bp long homology arms around TCOF1 start codon. The left and right homology arms were amplified by PCR from genomic DNA obtained from U2OS using the primers LHA-AvrII FWD + LHA-AgeI REV and RHA-XhoI-FWD + RHA-EcoRI REV, respectively. The left homology arm was digested with AvrII and AgeI and cloned into NheI+AgeI digested pEGFP-C1 vector (Clontech). Subsequently, the right homology arm was introduced using EcoRI and XhoI. Finally, the fragment containing both homology arms separated by the GFP sequence was PCR amplified using the primers HindIII FWD and BamHI REV and cloned into pBluescript (Agilent technologies) non-expressing vector using HindIII+BamHI.
BLM proficient and deficient U2OS cells were transfected with both plasmids in a 1:2 ratio (pX330: pHDR) using LTX Lipofectamine (Thermo), according to manufacturer’s protocol. The total amount of DNA ranged from 1 to 3 ug. 24 h after transfection, cells were retransfected with the donor plasmid only. GFP-expressing cells were enriched using FACS Melody (BD). In the case of U2OS BLM proficient cells, individual clones were isolated. U2OS BLM deficient cells did not allow us to obtain individual clones and we therefore performed the experiments with a pool. GFP insertion in the TCOF1 gene was checked by PCR. GFP-Treacle expression was confirmed by fluorescence microscopy and western blot. Primer sequences are listed in Supplementary Table 1.
gRNA plasmid transfections
Transient transfections were carried out using Lipofectamine LTX with plus reagent (Invitrogen) according to the manufacturer’s protocol and collected 0–6 h post-transfection unless otherwise stated. The four gRNA plasmids used for transfection were ampicillin resistant pMA plasmids (generated by the Luo laboratory) expressing either rDNA14 (Supplementary Table 2) or a control pMA vector expressing the gRNA scaffold but no target sequence (EV). Three rDNA gRNA plasmids were used to induce Cas9 mediated nucleolar DSBs, pooled in a ratio 1:1:1 of each.
Ribonucleoprotein (RNP) complex transfection
In cells that do not have a stable expression of Cas9, DSBs in the rDNA were induced by directly transfecting ribonucleprotein complexes consisting of purified recombinant Cas9 protein (TrueCut Cas9 Protein v2, Invitrogen) and synthetic guide RNA (TrueGuide, Invitrogen) using Lipofectamine CRISPRMAX (Invitrogen) according to the manufacturer´s protocol but using only 25% of the recommended amount of protein and gRNA, always keeping the stoichiometric ratio as recommended. We used a combination of gRNA1 and gRNA3 (molar ratio 1:1) in these experiments, and used the TrueguideTM sgRNA Negative Control, non-targetting 1 (Thermo Scientific, #A35526) as a control.
RNA interference
siRNA-mediated depletions were performed with a final concentration of 15 nM, 20 nM, or 50 nM of siRNA duplexes using Lipofectamine RNAiMAX (Invitrogen). 6 h after transfection, the medium was removed and replenished with fresh medium and cells were incubated for 24 h, unless otherwise stated in the figure legend. The siRNA oligonucleotides were obtained from Eurofins Genomics. The sequences of the siRNA used in this study are listed in Supplementary Table 3.
Immuno-fluorescence
Cells were grown on glass coverslips and fixed in 4% paraformaldehyde at room temperature (RT) for 10 min and subsequently permeabilized with 0.5% Triton X-100 for 10 min at RT. Samples used for phospho-RPAS4/8 staining were pre-extracted with 0.1% Triton X-100 in phosphate-buffered saline (PBS) on ice for 2 min, washed once with PBS and fixed as described above. Samples were incubated with primary antibody for 60 min at RT, 30 min with secondary antibody and 5 mg/ml DAPI (Invitrogen # D1306) in a moist chamber. Cells were washed three times in PBS between stainings, rinsed with water, mounted with Vectashield Mounting medium (Vector Laboritories), and sealed with nail polish. The antibodies used for immunofluorescence in this study are listed in Supplementary Table 4.
For quantitative analysis using the ImageXpress microscope (Molecular Devices), 5000 cell/well were plated in 96-well plates suitable for fluorescence (Vision Plate 4ti-0221, 4titude-). After treatment cells were fixed and stained as described above, and eventually wells were filled up with 200 ul PBS for imaging.
Microscopy and image analysis
Confocal microscopy
Qualitative image analysis of fluorescence was carried out using the point scanning confocal microscope LSM800 (Zeiss), using the 63x oil immersion objective and ZEN Software (Zeiss). The files were processed afterwards using Adobe Photoshop. The scoring of NBS1, RPA32, BRCA2 nucleolar foci containing cells was performed manually.
High throughput confocal imaging
Quantitative Image Based Cytometry (QIBC) was performed with the ImageXpress Micro Confocal High-Content Imaging System (Molecular Devices) using a 60x air objective. Images were taken in an automated fashion and experiments were repeated three times.
Image segmentation and analysis were performed using the MetaXpress high-content image acquisition and analysis software version 6.7.2.290 (Molecular Devices). The following pipeline was generated for image segmentation:
DAPI was used to detect the nuclei using the “identify blobs” command. For nucleolar segmentation, RNA Polymerase II signal was used due to its low intensity in the nucleolus. First, an inversion mask “H-basin” was applied, then nucleoli were recognized by “identify blobs” command. The initial nucleolar mask was further separated into nucleolar rim and nucleolar interior. NBS1 and DDR foci were enhanced by background correction “Top Hat”, foci were then recognized by “identify round objects” in the case of NBS1 and “identify blobs” in the case of DDR factors. Based on the two nucleolar segments and the central pixels of the foci, they were associated with one of the nucleolar compartments by using the “Keep marked object” function, resulting in “cap foci” or “interior foci”.
High throughput imaging of irradiated samples was performed using ScanR automated imaging platform (Olympus) with a 40x air objective, and analyzed with the ScanR analysis software (Olympus, v3.2.0). For image segmentation, DAPI was used to define cell nuclei. RPA and RAD51 foci were detected using the “Spot detector” function, counted and their intensity was measured per cell.
Live-cell imaging
For time-lapse imaging, U2OS Treacle-GFP cells were infected with H2B-RFP containing adenoviral vector (gift from Dr. Marin Barisic) 24 h before imaging. 6 h after virus infection, cells were treated with Cas9-Scaffold or gRNA RNPs to induce rDNA DSBs. Imaging was performed in a heated incubation chamber at 37 °C with controlled humidity and 5% CO2, using Plan-Apochromat 63×/1.4NA oil objective on a Zeiss Axio Observer Z1 microscope with a CSU-X1 spinning disk confocal head (Yokogawa Corporation of America). Images were acquired using an iXon Ultra 888 EM-CCD camera (Andor Technology) and the Slidebook 6.0 software. Images were processed with ImageJ Jiji (1.53t) package. Cells in promethaphase were located, and twelve 1,2 μm-separated z-planes were collected every 2 min until mitosis was completed.
Repair kinetics
For analyzing repair kinetics, 200.000 U2OS parental and BLM−/− cells were seeded in a well of a 6-well plate. The next day cells were transfected with gRNA RNPs as described above. The transfection reagent was washed off after 6 h. Samples for immunofluorescence and Western blot were collected at the indicated timepoints (0-6-12-24-30 h).
The quantification of 53BP1 foci indicative of DNA repair kinetics was done using the CellProfilerTM (version 4.2.6) cell image analysis software (Broad Institute). For detection of nuclei, DAPI was identified through the “IdentifyPrimaryObjects” module. For segmentation of nucleoli, the RNA polymerase II signal was used due to its low intensity in the nucleolus. This was done by enhancing “dark holes” using the “EnhanceOrSuppressFeatures” module, and nucleoli were recognized by the “IdentifyPrimaryObjects” module. Nucleoli were expanded and shrunk by a specified number of pixels to include foci located only at the nucleolar periphery using the “ExpandOrShrinkObjects” module. Further, 53BP1 caps masked in the nucleolar periphery “ring” were enhanced using the “EnhanceOrSuppressFeatures” module and identified using the “IdentifyPrimaryObjects” module. The percentage of cap positive nuclei were measured using the “RelateObjects” module.
High-throughput data filtering and visualization
Data filtering was performed based on NBS1 cap foci counts. After gRNA transfection, only cells with at least one NBS1 positive cap foci were considered, thereby categorized as “damaged”. Cell cycle phases were defined by the DAPI signal. Boxplots depict one representative experiment, with the corresponding significance values calculated on all the experimental repetitions performed (n = 3).
Senescence-associated β-galactosidase detection
Senescence was determined using the Senescence Cells Histochemical Staining Kit (Sigma-Aldrich, CS0030). Briefly, the cells were incubated with 1x Fixation Buffer for 7 min at RT. Next, the cells were washed and incubated with staining mixture (containing X-gal) at 37 °C without CO2 for 8 h. The experiments were repeated three times, and the cells were photographed using an inverted Leica ICC50 HD microscope at 10× magnification for qualitative detection of SA-β-Gal activity. The numbers of positive (blue) and negative cells were counted, and the percentage of SA-β-Gal-positive cells was calculated as the number of positive cells divided by the total number of cells counted.
Cell cycle analysis by EdU incorporation
Cells were grown on coverslips and incubated 1 h with 10 uM EdU before fixing. Samples were fixed in 4% paraformaldehyde at room temperature for 10 min and permeabilized in 0.5% Triton X-100 for 10 min at room temperature. Click-iT chemistry was used for detecting EdU (Thermo Fischer Scientific, #C10340) and subsequently, samples were incubated with 5 mg/ml DAPI (Invitrogen # D1306) in PBS for 2 min. EdU intensity was measured using the ScanR automated microscope (Olympus). Images were acquired with a 20x air objective, and image segmentation and analysis were performed with the ScanR software (Olympus) using standard algorithms for detection of nuclei based on DAPI staining. After segmentation of nuclei, the desired parameters for the different nuclei were quantified, with single parameters (mean and total intensities for DAPI and EdU). These values were exported and analyzed with TIBCO Software Analyst, version 12.0.3. This software was used to quantify absolute, median, and average values in cell populations and to generate the graph in Supplementary Fig. 5f. Where indicated cells were treated with 3.3 µM Nocodazole (Sigma-Aldrich M1404) for 12 h.
Western blotting
Cell lysates were prepared in clear denaturing lysis buffer (200 mM Tris–HCL (pH 6,8), 40% glycerol (99.5%), 8% SDS pure (95%)). Cell lysates were boiled for 5 min at 95 °C and sonicated to homogenize the samples. Protein concentration was measured by Qbit fluorimeter (Invitrogen), according to the manufacturer´s protocol. Before loading the samples on a gel, DTT was added to a final concentration of 100 mM as well as 1 ul bromophenol blue per sample and denatured by heating the samples at 95 °C for 5 min. Proteins were separated using NuPAGE 3–8% Tris-Acetate or 4–12% Bis-Tris NuPAGE gels (Invitrogen) and transferred to membranes using iBlot Dry Blotting System (Invitrogen) with 20 V for 10 min. For antibodies and concentrations used for Western blot, see Supplementary Table 4.
Micronuclei quantification
Cells were pre-treated with corresponding siRNAs and gRNAs as described above. 3 h after gRNA transfection, Cytochalasin B (Sigma C6762) was added to the cells to a final concentration of 5 µg/ml, diluted in fresh medium and incubated for 24 h at 37 °C, 5% CO2. Cells were fixed and processed for microscopy as described. Micronuclei present around bi-nucleated cells and their UBF content was quantified manually.
Site-directed mutagenesis and reconstitution experiments
The BLM mutant plasmids were generated by site-directed mutagenesis using the QuikChange Site-Directed Mutagenesis kit (Agilent Technologies), according to the manufacturer’s protocol. BLM mutations were inserted into a pEGFP-BLM-WT plasmid (gift from Andrew Blackford)65, confirmed by Sanger sequencing. For primer sequences, see Supplementary Table 5.
The BLM mutant expression plasmids generated for this study, along with the BLM WT expression plasmid, were transiently transfected into U2OS BLM−/− cells using the Lipofectamine LTX and Plus reagent according to the manufacturer’s protocol. Cells were incubated for 48 h with the plasmids for proper protein expression and localization. Following plasmid transfection, rDNA damage was induced by RNP transfection, as described above. 6 h after damage induction, cells were fixed and processed for microscopy as described.
Quantitative image analysis was performed manually. Images were obtained using a 40x oil objective on LSM800 confocal microscope (Zeiss). In the reconstituted samples only GFP positive cells were counted. Upon gRNA treatment only GFP-positive cells with UBF/Treacle caps were considered. Scoring was based on whether RPA32 or RAD51 nucleolar cap foci were present in the reconstituted cells. Experiments were repeated three times.
Quantitative DNA-end resection assay
End resection was analyzed using BrdU detection under native conditions and was performed as previously described52 with some alterations. Cells were grown on coverslips and labeled with 10 µM BrdU for 24 h prior to RNP transfections for induction of rDNA damage. Following RNP transfection, the cells were incubated for 8 h before fixing in 70% ethanol overnight at −20 °C. Cells were blocked in 5% FBS in PBS for 1 h at room temperature and subsequently incubated with primary antibodies diluted in 5% FBS in PBS for 2 h at room temperature. Cells were then stained with secondary antibodies diluted in 5% FBS and PBS for 1 h at room temperature, washed and mounted using Fluoromount-GTM mounting medium (Invitrogen, #00-4958-02) and sealed with nail polish. Washes between incubations were done in 0.1% Tween 20 in PBS. Coverslips were kept at 4 °C before imaging on LSM800 confocal microscope (Zeiss).
Quantitative image analysis was performed on at least 100 cells for each condition. Images were obtained using a 40x oil objective on LSM800 confocal microscope (Zeiss). Automated analysis measuring intensity of BrdU in caps was done in CellProfilerTM (version 4.2.6) cell image analysis software and the following pipeline was developed for image segmentation:
For detection of nuclei, DAPI was identified through the “IdentifyPrimaryObjects” module. For segmentation of nucleoli, the RNA polymerase II signal was used due to its low intensity in the nucleolus. This was done by enhancing “dark holes” using the “EnhanceOrSuppressFeatures” module, and nucleoli were recognized by the “IdentifyPrimaryObjects” module. Nucleoli were expanded and shrunk by a specified number of pixels to include foci located only at the nucleolar periphery using the “ExpandOrShrinkObjects” module. Further, TCOF1 caps masked in the nucleolar periphery “ring” were enhanced using the “EnhanceOrSuppressFeatures” module and identified using the “IdentifyPrimaryObjects” module. Lastly, the intensity of BrdU was measured using the “MeasureObjectIntensity” modules within the nucleolar caps.
Droplet digital PCR (ddPCR) to measure rDNA copy number
U2OS WT and BLM−/− cells were grown in a 12-well plate (3 samples per condition, 2 technical replicates per sample). Cells were transfected with ribonucleoprotein (RNP) complexes (Cas9-gRNA/Cas9-negative control gRNA). The media was changed 6 h after transfection and genomic DNA was extracted 3 days post-transfection using QIAmp DNA mini kit (#51306, Qiagen).
The rDNA copy number was quantified by droplet digital PCR (ddPCR) as described before70 using the QX200 system (BioRad) according to manufacturer’s instructions. The PCR mixture comprised of 1x ddPCR Supermix for Probes without dUTPs (Bio-Rad #186304), 2 Units of Hae III, 0.9 µM of each primer, 0.25 µM of the probe and approximately 1 ng of genomic DNA, with a final volume of 20 µl. Prior to droplet generation, samples were incubated at room temperature for 15 min to facilitate DNA digestion by the restriction enzyme. Data analysis was conducted using Quantasoft (version1.7.4.0917 Bio-Rad). Normalization of rDNA copy numbers was performed relative to TBP copy number, serving as the internal control. Primer/probe sequences are listed in Supplementary Table 5.
ChIP
ChIP experiments were performed as in Larsen et al. 13. Briefly, 100 ug of chromatin were used for each IP, incubated with 3 ul IgG antibody, 3 ul of RNA pol I antibody or 4 ul BLM antibody (Supplementary Table 4). BLM ChIP samples were washed with low salt concentration buffer 1B (10 mM Tris-HCL pH8, 300 mM NaCl, 0.1% SDS, 1% TritonX-100). Purified DNA was used for quantitative PCR (qPCR) with the SYBR Green detection system. Five primer pairs spanning the ribosomal DNA were used in addition to non-specific, GAPDH primers18. ChIP efficiency was calculated as a percentage of DNA input precipitated. Each primer pair in RNA pol I ChIP was normalized for primer D in undamaged condition (EV), while for BLM ChIP, it was normalized for primer A in undamaged condition. Each ChIP was performed in three biological replicates and the mean of the normalized efficiency and SEM is depicted on the graph. For primer sequences see Supplementary Table 5.
Quantification of double-strand breaks (dSTRIDE)
Direct detection of DSBs was performed by intoDNA using the dSTRIDE procedure, as previously described (Kordon et al., 2020), followed by counter-staining with anti-RNA polymerase II antibody (1:500, abcam, ab5408) for 90 minutes, anti-mouse IgG Fab2 AF488 antibody (1:800, Cell Signaling, 4408S) for 60 min and HCS CellMask Deep Red Stain (1:10000, Invitorgen, H32721A) for 15 min. All of the counter-stainings were performed at room temperature. The coverslips with fixed cells were then mounted with Mounting Medium With DAPI (Fluoroshield, Abcam, AB104139) and stored at 4 °C until imaging. Fields of view (FOVs) for imaging (at least 6 FOVs with a total of 200–300 cells per sample) were chosen randomly over the surface of the coverslip. The images were then collected as 3D confocal stacks using Cell Discoverer 7 LSM 900 microscope, Zeiss. Polymerase RNA II and CellMask levels were measured as integrated fluorescence from the nuclear region. Identification of STRIDE foci was based on 3D local maxima of fluorescence. Only the foci localized inside the 3D masks were included in further data analysis. Nuclei were recognized based on DAPI staining. 3D nucleus masks were created for each recognized nucleus and its projection. Nucleoli analysis was performed inside the 3D nucleoli masks identified based on CellMask staining. Areas with the highest fluorescence inside the 3D nucleus masks were defined as the nucleoli area.
Experimental repetitions and statistics
All experiments were performed three times unless otherwise stated. Error bars represent the standard error of the mean in all figures. Statistical analysis was performed using analysis of variance with the experimental repetitions used as the blocking factor in R-studio 2023.06.0 Build 421 (ggplot2 package). The Tukey post hoc test was applied. Datasets with continuous values (fluorescent intensities, count averages) were tested and transformed to get normal distribution (see figure legends) and no transformation was applied when distribution was already normal. For binomial data (foci positive/negative cells), a binomial distribution model was applied, and for discreet values (counts), a Poisson distribution model was used. Statistical significance in the Supplementary Figures is depicted with stars (*p < 0.05, **p < 0.01, ***p < 0.001), and no legend: not significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information. Source data are provided with this paper.
References
Raska, I., Shaw, P. J. & Cmarko, D. Structure and function of the nucleolus in the spotlight. Curr. Opin. Cell Biol. 18, 325–334 (2006).
Henderson, A. S., Warburton, D. & Atwood, K. C. Location of ribosomal DNA in the human chromosome complement. Proc. Natl Acad. Sci. USA 69, 3394–3398 (1972).
Schmickel, R. D. Quantitation of human ribosomal DNA: hybridization of human DNA with ribosomal RNA for quantitation and fractionation. Pediatr. Res. 7, 5–12 (1973).
Floutsakou, I. et al. The shared genomic architecture of human nucleolar organizer regions. Genome Res. 23, 2003–2012 (2013).
Feric, M. et al. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 165, 1686–1697 (2016).
Emmott, E. & Hiscox, J. A. Nucleolar targeting: the hub of the matter. EMBO Rep. 10, 231–238 (2009).
Schachner, C. et al. Establishment and Maintenance of Open Ribosomal RNA Gene Chromatin States in Eukaryotes. Methods Mol. Biol. 2533, 25–38 (2022).
Stults, D. M. et al. Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 69, 9096–9104 (2009).
Potapova, T. A. et al. Superresolution microscopy reveals linkages between ribosomal DNA on heterologous chromosomes. J. Cell Biol. 218, 2492–2513 (2019).
Gal, Z., Nieto, B., Boukoura, S., Rasmussen, A. V. & Larsen, D. H. Treacle Sticks the Nucleolar Responses to DNA Damage Together. Front. Cell Dev. Biol. 10, 892006 (2022).
Jackson, S. P. & Bartek, J. The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009).
Ciccia, A. et al. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc. Natl Acad. Sci. USA 111, 18631–18636 (2014).
Larsen, D. H. et al. The NBS1-Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat. Cell Biol. 16, 792–803 (2014).
Korsholm, L. M. et al. Double-strand breaks in ribosomal RNA genes activate a distinct signaling and chromatin response to facilitate nucleolar restructuring and repair. Nucleic Acids Res. 47, 8019–8035 (2019).
Mooser, C. et al. Treacle controls the nucleolar response to rDNA breaks via TOPBP1 recruitment and ATR activation. Nat. Commun. 11, 123 (2020).
van Sluis, M. & McStay, B. Nucleolar reorganization in response to rDNA damage. Curr. Opin. Cell Biol. 46, 81–86 (2017).
Harding, S. M., Boiarsky, J. A. & Greenberg, R. A. ATM Dependent Silencing Links Nucleolar Chromatin Reorganization to DNA Damage Recognition. Cell Rep. 13, 251–259 (2015).
Marnef, A. et al. A cohesin/HUSH- and LINC-dependent pathway controls ribosomal DNA double-strand break repair. Genes Dev. 33, 1175–1190 (2019).
van Sluis, M. & McStay, B. A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 29, 1151–1163 (2015).
Warmerdam, D. O., van den Berg, J. & Medema, R. H. Breaks in the 45S rDNA Lead to Recombination-Mediated Loss of Repeats. Cell Rep. 14, 2519–2527 (2016).
Scully, R., Panday, A., Elango, R. & Willis, N. A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 20, 698–714 (2019).
Cunniff, C. et al. Health supervision for people with Bloom syndrome. Am. J. Med. Genet A 176, 1872–1881 (2018).
Croteau, D. L., Popuri, V., Opresko, P. L. & Bohr, V. A. Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 83, 519–552 (2014).
Chan, K. L., North, P. S. & Hickson, I. D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 26, 3397–3409 (2007).
Therman, E., Otto, P. G. & Shahidi, N. T. Mitotic recombination and segregation of satellites in Bloom’s syndrome. Chromosoma 82, 627–636 (1981).
Killen, M. W., Stults, D. M., Adachi, N., Hanakahi, L. & Pierce, A. J. Loss of Bloom syndrome protein destabilizes human gene cluster architecture. Hum. Mol. Genet 18, 3417–3428 (2009).
Mohaghegh, P., Karow, J. K., Brosh, R. M. Jr, Bohr, V. A. & Hickson, I. D. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 29, 2843–2849 (2001).
Daley, J. M. et al. Enhancement of BLM-DNA2-Mediated Long-Range DNA End Resection by CtIP. Cell Rep. 21, 324–332 (2017).
Gravel, S., Chapman, J. R., Magill, C. & Jackson, S. P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22, 2767–2772 (2008).
Nimonkar, A. V. et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362 (2011).
Soniat, M. M., Nguyen, G., Kuo, H. C. & Finkelstein, I. J. The MRN complex and topoisomerase IIIa-RMI1/2 synchronize DNA resection motor proteins. J. Biol. Chem. 299, 102802 (2023).
Roy, R., Chun, J. & Powell, S. N. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat. Rev. Cancer 12, 68–78 (2011).
Bugreev, D. V., Mazina, O. M. & Mazin, A. V. Bloom syndrome helicase stimulates RAD51 DNA strand exchange activity through a novel mechanism. J. Biol. Chem. 284, 26349–26359 (2009).
Bugreev, D. V., Yu, X., Egelman, E. H. & Mazin, A. V. Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev. 21, 3085–3094 (2007).
Patel, D. S., Misenko, S. M., Her, J. & Bunting, S. F. BLM helicase regulates DNA repair by counteracting RAD51 loading at DNA double-strand break sites. J. Cell Biol. 216, 3521–3534 (2017).
Bachrati, C. Z., Borts, R. H. & Hickson, I. D. Mobile D-loops are a preferred substrate for the Bloom’s syndrome helicase. Nucleic Acids Res. 34, 2269–2279 (2006).
Manthei, K. A. & Keck, J. L. The BLM dissolvasome in DNA replication and repair. Cell Mol. Life Sci. 70, 4067–4084 (2013).
Chan, Y. W., Fugger, K. & West, S. C. Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat. Cell Biol. 20, 92–103 (2018).
Wu, L. & Hickson, I. D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 426, 870–874 (2003).
Toledo, L. I. et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103 (2013).
Grabarz, A. et al. A role for BLM in double-strand break repair pathway choice: prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 5, 21–28 (2013).
Sartori, A. A. et al. Human CtIP promotes DNA end resection. Nature 450, 509–514 (2007).
Yun, M. H. & Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 459, 460–463 (2009).
San Filippo, J., Sung, P. & Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 77, 229–257 (2008).
Zhao, W. et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 550, 360–365 (2017).
Tarsounas, M., Davies, D. & West, S. C. BRCA2-dependent and independent formation of RAD51 nuclear foci. Oncogene 22, 1115–1123 (2003).
Guo, R. B. et al. Structural and functional analyses of disease-causing missense mutations in Bloom syndrome protein. Nucleic Acids Res. 35, 6297–6310 (2007).
Wu, L. et al. The HRDC domain of BLM is required for the dissolution of double Holliday junctions. EMBO J. 24, 2679–2687 (2005).
Tangeman, L., McIlhatton, M. A., Grierson, P., Groden, J. & Acharya, S. Regulation of BLM Nucleolar Localization. Genes 7, 69 (2016).
Li, L., Eng, C., Desnick, R. J., German, J. & Ellis, N. A. Carrier frequency of the Bloom syndrome blmAsh mutation in the Ashkenazi Jewish population. Mol. Genet Metab. 64, 286–290 (1998).
Sturzenegger, A. et al. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J. Biol. Chem. 289, 27314–27326 (2014).
Nishi, R. et al. Systematic characterization of deubiquitylating enzymes for roles in maintaining genome integrity. Nat. Cell Biol. 16, 1016–1026 (2014).
Belan, O. et al. Visualization of direct and diffusion-assisted RAD51 nucleation by full-length human BRCA2 protein. Mol. Cell 83, 2925–2940.e2928 (2023).
Cohen, S. et al. A POLD3/BLM dependent pathway handles DSBs in transcribed chromatin upon excessive RNA:DNA hybrid accumulation. Nat. Commun. 13, 2012 (2022).
Rassool, F. V., North, P. S., Mufti, G. J. & Hickson, I. D. Constitutive DNA damage is linked to DNA replication abnormalities in Bloom’s syndrome cells. Oncogene 22, 8749–8757 (2003).
Crasta, K. et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58 (2012).
Xu, D. et al. RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev. 22, 2843–2855 (2008).
Kalousi, A. & Soutoglou, E. Nuclear compartmentalization of DNA repair. Curr. Opin. Genet Dev. 37, 148–157 (2016).
Stults, D. M., Killen, M. W., Pierce, H. H. & Pierce, A. J. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 18, 13–18 (2008).
Tsaridou, S. et al. 53BP1-mediated recruitment of RASSF1A to ribosomal DNA breaks promotes local ATM signaling. EMBO Rep. 23, e54483 (2022).
Korsholm, L. M. et al. Recent advances in the nucleolar responses to DNA double-strand breaks. Nucleic Acids Res. 48, 9449–9461 (2020).
Grierson, P. M. et al. BLM helicase facilitates RNA polymerase I-mediated ribosomal RNA transcription. Hum. Mol. Genet 21, 1172–1183 (2012).
Bythell-Douglas, R. & Deans, A. J. A Structural Guide to the Bloom Syndrome Complex. Structure 29, 99–113 (2021).
Zhang, F., Fan, Q., Ren, K. & Andreassen, P. R. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol. Cancer Res. 7, 1110–1118 (2009).
Shorrocks, A. K. et al. The Bloom syndrome complex senses RPA-coated single-stranded DNA to restart stalled replication forks. Nat. Commun. 12, 585 (2021).
Xue, C. et al. Bloom helicase mediates formation of large single-stranded DNA loops during DNA end processing. Nat. Commun. 13, 2248 (2022).
Bischof, O. et al. Regulation and localization of the Bloom syndrome protein in response to DNA damage. J. Cell Biol. 153, 367–380 (2001).
Wu, L., Davies, S. L., Levitt, N. C. & Hickson, I. D. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem. 276, 19375–19381 (2001).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Xu, B. et al. Ribosomal DNA copy number loss and sequence variation in cancer. PLoS Genet 13, e1006771 (2017).
Acknowledgements
We thank Dr. Andrew Blackford for the GFP-BLM construct and Dr. Eros Lazzerini Denchi for the U2OS parental and BLM−/− cells. We thank all members of the Nucleolar Stress and Disease Group and the Genome Integrity Group from the Danish Cancer Institute, and the Hickson Group from Copenhagen University for critical discussion and input to the project. We thank Dr Susana Eibes González and Dr. Marin Barisic from the Cell Division and Cytoskeleton group, for assistance with live cell imaging of mitotic cells. We thank Dr. Niels Mailand for allowing us to use his lab space for experiments. We thank Tiina Dietrich and Dr. Christoffel Dinant, head of the DCI core facility for bioimaging, for technical support. Funding: Independent Research Fund Denmark [6110-00506B] (D.H.L.) Independent Research Fund Denmark [8045-00057A] (D.H.L.) Independent Research Fund Denmark [8048-00072A] (L.L.) Independent Research Fund Denmark [DFF 1026-00241B] (J.B.) Novo Nordisk Foundation [NNF18OC0052647] (D.H.L.) Novo Nordisk Foundation [NNF21OC0068988, NNF21OC0072031] (Y.L.) Novo Nordisk Foundation [NNF22OC0078971] (D.H.L.) Danish Cancer Society-Knæk Cancer [R311-A18224] (D.H.L.) Danish Cancer Society-Knæk Cancer [R167-A11068] (P.Galanos) Danish Cancer Society-Knæk Cancer [R364-A21079] (P.Galanos) Danish Cancer Society-Knæk Cancer [R302-A17506] (D.H.L.) Danish Cancer Society [A10965] (D.H.L.) Danish Cancer Society [R322-A17482] (J.B.) Lundbeck Foundation [R347-2020-2219] (B.N.) Lundbeck Foundation [R322-2019-2577, R445-2023-788] (PGalanos) European Union’s Horizon 2020 research and innovation programme [899417] (Y.L.) Swedish Research Council [VR-MH 2014-46602-117891-30] (J.B.) Danish Cancer Institute, Danish Cancer Society (E.D., P.Guldberg, C.D., D.H.L.)
Author information
Authors and Affiliations
Contributions
Conceptualization: D.H.L., Z.G. Methodology: Z.G., SBoukoura, K.C.O., S.Badawi, B.N., L.M.K., S.B.G., E.D., A.V.R., C.D., W.L., H.X., X.P., S.A., D.-E.S., P.Galanos, L.L., P.Guldberg, Y.L., D.H.L. Investigation: Z.G., S.Boukoura, K.C.O., S.Badawi, B.N., L.M.K., S.B.G., E.D., A.V.R., C.D., W.L., H.X., X.P., S.A., D.-E.S., P.Galanos, L.L., Supervision: D.H.L., Y.L., J.B., L.L., P.Guldberg, P.Galanos. Writing—original draft: Z.G., D.H.L. Writing—review & editing: Z.G., S.Boukoura, K.C.O., S.Badawi, B.N., L.M.K., S.B.G., A.V.R., C.D., PGalanos, L.L., PGuldberg, J.B., Y.L., D.H.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gál, Z., Boukoura, S., Oxe, K.C. et al. Hyper-recombination in ribosomal DNA is driven by long-range resection-independent RAD51 accumulation. Nat Commun 15, 7797 (2024). https://doi.org/10.1038/s41467-024-52189-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52189-6
This article is cited by
-
C8orf33 dictates DNA double-strand break repair choice by modulating KAT8-mediated H4K16 acetylation
Cell Death & Disease (2025)
