
Abstract
Microtubule-based vesicle trafficking usually relies upon kinesin and dynein motors and few reports describe microtubule polymerisation driving directional vesicle trafficking. Here we show that Arabidopsis END BINDING1b (EB1b), a microtubule plus-end binding protein, directly interacts with SYP121, a SNARE protein that mediates the trafficking of the K+ channel KAT1 and its distribution to the plasma membrane (PM) in Arabidopsis guard cells. Knockout of AtEB1b and its homologous proteins results in a modest but significant change in the distribution of KAT1 and SYP121 in guard cells and consequently delays light-induced stomatal opening. Live-cell imaging reveals that a portion of SYP121-associated endomembrane compartments co-localise with AtEB1b at the growing ends of microtubules, trafficking along with the growth of microtubules for targeting to the PM. Our study reveals a mechanism of vesicle trafficking driven by microtubule growth, which is involved in the redistribution of PM proteins to modulate guard cell movement.
Similar content being viewed by others


Introduction
Spatial positioning and motility of endomembrane compartments and organelles are fundamental to cellular functions in all eukaryotic cells and are primarily governed by the cytoskeleton. Microtubule-based vesicle trafficking and organelle positioning are commonplace in animal cells and are known to depend on kinesin and dynein motor proteins1,2,3. In plant cells, it has long been accepted that directional vesicle motility is predominantly dependent on actin-based myosin motors4,5. Later on, however, researchers reveal a critical role for microtubules in endoplasmic reticulum (ER) motility and morphology6, in insertion and tethering of cellulose synthase complexes to the plasma membrane (PM)7,8 and in endocytic recycling of the auxin efflux carrier PIN2 to the PM in plants9. It has been proposed that microtubules and their motors might anchor or slow down organelles/vesicles for their targeting to appropriate destinations. In addition, the plus-end binding protein, CLASP (CLIP-170 ASSOCIATED PROTEIN), was shown to mediate the association of sorting endosomes with microtubules10. Interestingly, intracellular small cellulose synthases-containing compartments were reported to tether with microtubules, and track with the depolymerising ends of microtubules8, which suggests a role for microtubule dynamics in organelle motility. Yet reports of microtubule growth-based directional vesicle trafficking in plant cells are lacking.
Stomata are surrounded by a pair of guard cells and are present on the epidermis of all aerial parts of almost all angiosperm. In response to various hormonal and environmental stimuli, such as abscisic acid, light and atmospheric CO2 levels, as well as abiotic and biotic stresses, guard cells precisely regulate photosynthetic gas exchange and water transpiration, and restrict pathogen invasion by regulating stomatal opening and closing11,12,13,14,15,16,17. During stomatal movement, the volume of each guard cell changes rapidly, driven by the transport of K+, Cl- and other solutes across the PM and guard cell tonoplast, with consequent effects on the osmotic content of the cell and its turgor15,18. In this process, vesicle trafficking of ion channels in guard cells contributes to stomatal movement19,20. It has been demonstrated that the regulation of the recycling of the K+ channel KAT1 to the PM is important for stomatal movement. Furthermore, delivery and positional anchoring of KAT1 to the PM has been found to be dependent on SNARE (soluble N-ethylmaleimide- sensitive factor adaptor protein receptor) protein SYP121 (SYNTAXIN OF PLANTS 121) in tobacco and Arabidopsis19,20. Interestingly, it has also been reported that SYP121 binding to KAT1 promotes KAT1 channel activity21.
In guard cells of opening stomata, both actin filaments (F-actin) and microtubules are arranged in a pattern radiating from the ventral side toward the dorsal side. In early studies, by observing the reorganisation during stomatal movement and based on pharmacological analyses, F-actin was believed to be involved in stomatal movement22,23; however, reports on the function of microtubules in the stomatal movement are controversial. Some studies have suggested that microtubules are required for opening but not for closing of the stomata; whereas, some proposed that microtubules are not required for the function of guard cells24,25,26. Eventually, emerging evidence supported the fact that microtubule dynamics in guard cells were involved in stomatal movement27,28,29. Eisinger et al. reported that guard cells of closed stomata displayed fewer microtubules, whereas an increase in microtubules was observed in guard cells during stomatal opening30,31. Furthermore, pharmacological analyses implied that the disruption of microtubules prevented stomatal opening and that the stabilisation of microtubules caused a delay in stomatal closure30. The expression of the microtubule-binding domains of microtubule-associated proteins (MAPs), such as MAP4 or elongation factor-1α (EF-1α), led to a failure in light-induced stomatal opening27,28. All these evidence suggest an important role for microtubule organisation and dynamics in the proper function of guard cells. In addition, a hypothetical role of microtubules in modulating the activity of K+ and Ca2+ channels was proposed32. Nevertheless, the underlying mechanism remains unclear.
The End-binding 1 (EB1) family members are well-known microtubule plus-end-tracking proteins (+TIPs) conserved in yeasts, animals and plants29,33. These proteins directly regulate the dynamics of microtubule plus ends or recruit other +TIPs, and act as master integrators of the +TIP interaction network34. The Arabidopsis genome encodes three EB1 orthologues, AtEB1a, AtEB1b and AtEB1c, which are grouped into two subclades35. AtEB1a and AtEB1b exhibit relatively high similarity at the amino-acid level with mammalian EB1 and present as comet-like structures to track growing plus ends of microtubules (a pattern conserved among EB1 proteins in different species)34,35. Interestingly, AtEB1a and AtEB1b can interact with each other35, but not with AtEB1c. AtEB1c is more divergent, with a C-terminal tail containing a functional nuclear localisation signal (NLS) and a likely extra functional motif(s) absent in AtEB1a/b. The distinct features of the tail region discriminate it from other EB1 proteins35. Genetic analyses have indicated that AtEB1 proteins function during root growth and responses to touch and gravity signals36,37. Lindeboom et al. reported the absence of AtEB1 proteins reduced growth velocities of microtubule plus ends but increased shrinkage velocities38, suggesting AtEB1 proteins regulate microtubule dynamics by inhibiting the shrinkage of microtubule plus ends. Their study also hinted that AtEB1 proteins are involved in the blue light-stimulated reorientation of cortical microtubule arrays in etiolated hypocotyl cells38.
Interestingly, the relatively strong expression of all three AtEB1 genes in guard cells was revealed35, but the role of associated AtEB1 proteins in stomatal movements remains unclear. In the present study, we found that light-induced stomata opening was significantly delayed in eb1a 1b 1c triple mutant as was also the redistribution of KAT1 and SYP121 to the PM of guard cells. A direct interaction between SYP121 and AtEB1a/b was detected. Using AtEB1b-GFP as a fluorescent probe, we performed live cell imaging and revealed that SYP121 was associated with the trans-Golgi network/early endosome (TGN/EE) compartments and recruited to microtubule plus-ends by AtEB1b, trafficking with the growth of microtubule for targeting to the PM. Our study reveals microtubule plus-end-directed vesicle trafficking in plants, establishing a novel mechanism for redistribution of PM proteins in guard cells during light-induced stomatal opening.
Results
AtEB1 functions in stomatal opening
Previous studies demonstrated that all AtEB1 genes were expressed in various tissues in Arabidopsis, although relatively stronger in guard cells35. A role for AtEB1 proteins in light-stimulated cortical microtubule reorientation in hypocotyl epidermal cells was also suggested38. To determine if AtEB1 proteins participate in light-induced stomatal opening, we measured the stomatal apertures of the wild type (Col-0) and the eb1a 1b 1c triple mutant. Arabidopsis leaves were maintained in the dark for 2 h to completely close stomata, followed by a 1 h illumination before measurements. We observed that stomatal apertures in the wild type were significantly larger than in the eb1a 1b 1c mutant. A similar result was consistently obtained after 2 h of light induction (Fig. 1a, b). Furthermore, infrared thermal imaging analysis39 revealed warmer leaves in eb1a 1b 1c plants when compared with the wild type after approximately 2 h light illumination, indicating a reduced transpiration rate in eb1a 1b 1c plants (Fig. 1c). In addition, Arabidopsis whole-rosette gas-exchange measurements40 exhibited defects in light-induced changes in stomatal conductance in eb1a 1b 1c plants when compared with the wild type (Supplementary Fig. 1). These results demonstrate that stomatal opening was delayed in the eb1a 1b 1c mutant, suggesting AtEB1 proteins are important for stomatal opening. To confirm this, we introduced native promoter-driven AtEB1a, AtEB1b, and AtEB1c into eb1a 1b 1c mutant, respectively, and obtained the complementary lines, namely COM-EB1a, COM-EB1b, and COM-EB1c (Fig. 1a, b and Supplementary Fig. 2a, b). Light-induced stomatal openings in COM-EB1a and COM-EB1b lines were similar to the wild type (Fig. 1a, b), indicating a redundant function for AtEB1a and AtEB1b in this process. AtEB1c has been shown to accumulate in the interphase nucleus and plays a role in spindle pole positioning and chromosome segregation during mitosis35. Interestingly, in this study, we found a partial recovery of stomatal-opening phenotype in eb1a 1b 1c triple mutant by expressing AtEB1c, although the eb1c single mutant had no defect in stomatal opening (Supplementary Fig. 2c). We then generated the eb1a 1b double mutant and found that the stomatal aperture in eb1a 1b double mutant was significantly smaller than that in the wild type, but larger than that in the eb1a 1b 1c triple mutant (Supplementary Fig. 2d). Taken together, the above genetic results indicated a major role of functional redundant AtEB1a and AtEB1b in stomatal opening, whereas AtEB1c might play a minor role or compensate when AtEB1a and AtEB1b were absent. Therefore, we mostly used AtEB1b for subsequent experiments.

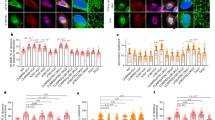
a Representative images of stomatal aperture in wild-type (Col-0), eb1a 1b 1c mutant and complementary lines (COM-EB1a and COM-EB1b) before and after light treatment. A total of three independent experiments were performed and obtained similar results. Bars = 10 μm. b Quantification of stomatal aperture size in wild type, eb1a 1b 1c mutant and complementary lines. Abaxial epidermal strips were peeled and exposed under white light. The image on the left illustrates how the size of the stomatal aperture was measured. c Representative false-colour infrared images show that the leaf temperature of eb1a 1b 1c plants is higher than for the wild type after 2 h light illumination. A quantitative analysis of leaf temperature is shown on the right. A total of three independent experiments were performed and obtained similar results. d More AtEB1b-mCherry comet-like dots were detected in guard cells with opened stomata under light, in comparison to guard cells with closed stomata in the dark. Consistently, more microtubules (visualised by GFP-TUA6) were noted in guard cells under light compared with dark conditions. The max-intensity projection of Z-stack images is shown. Bar = 5 µm. e Quantification of AtEB1b-mCherry density in guard cells as shown in d. f Quantification of microtubule density (indicated by the occupancy) in guard cells expressing GFP-TUA6 as shown in d. g Quantification of cortical microtubule orientation in guard cells expressing GFP-TUA6 as shown in d. Data in b, c, and e–g are presented as box plots. The whiskers indicate the maximum/minimum values and the centre line within the box reflects the median value and the boundary lines of box indicate the 1st quartile (upper) and the 3rd quartile (lower) value of the dataset. The data in b were analysed using RStudio software with the agricolae package. Different letters indicate significant differences analysed by one-way ANOVA with Duncan’s new multiple range test, P < 0.01. In c, e–g, ***P < 0.001, two-tailed Student’s t-test. The “n” below each box indicates the number of analysed guard cells.
AtEB1a and AtEB1b associate with the plus ends of growing microtubules and exhibit comet-like structures35,41. To investigate their role in stomata opening, we used AtEB1b-mCherry (driven by the native promoter) as a fluorescent probe to follow cellular processes. Co-localisation analysis of AtEB1b-mCherry with GFP-tagged α-tubulin 6 (TUA6) clearly showed that comet-like structures of AtEB1b-mCherry were anchored to the growing ends of microtubules in guard cells (Fig. 1d and Supplementary Movie 1). In the dark, the density of AtEB1b-mCherry dots in guard cells with closed stoma was 6.9 ± 2.2 per 10 µm2 (± indicates s.d., n = 218); the density increased to 9.0 ± 2.4 per 10 µm2 (± indicates s.d., n = 219) in guard cells in the 1-h light-induced open stomata (Fig. 1d, e). We then used the occupancy index to indicate microtubule density in guard cells with opened or closed stomata, respectively, as previously described with brief modification42,43. The leaves expressing GFP-TUA6 were kept in the dark for 2 h to close the stomata. The occupancy of microtubules in one guard cell with closed stomata was 15.9% (n = 218). After 1-h light treatment, the microtubule occupancy significantly increased to 26.2% along with the stomata opening (n = 219) (Fig. 1d, f). The above evidence indicates that growing microtubules increased in guard cells upon light treatment, since AtEB1b associates with the growing plus-end of microtubules35. Moreover, we evaluated microtubule orientation as previously described42,44, the results suggested a significant change in microtubule orientation during the stomatal opening process (Fig. 1g). This is consistent with a previous report showing the orientation of cortical microtubule changes during stomatal closure43.
AtEB1 proteins involved in KAT1 redistribution during stomatal opening
It has been suggested that plant cells also respond to environmental conditions by regulating the abundance and distribution of PM proteins that comprise the functional interface with the environment, through endocytosis and recycling processes as occurs in animals and yeast45,46. This regulation provides a prompt and efficient means of response to external signals in addition to altering transcriptional output. Stomatal opening requires K+ uptake through K+ channels in the PM14. After being kept in the dark, guard cell re-opening in intact plants can be very fast, whereas transcripts of the K+ channel, KAT1, do not change much in studies of guard cell gene expression47. Sutter et al. reported that abscisic acid (ABA) triggered KAT1 endocytosis and stomatal closure, whereas endosomal-localised KAT1 recycled back to the PM during stomatal reopening after ABA washout20, suggesting an important role for KAT1 endocytotic recycling in modulating its levels at the PM in the regulation of stomatal movement20,48,49. To test whether AtEB1 proteins modulate light-induced stomatal opening by affecting the PM distribution of KAT1, we generated transgenic plants expressing ProKAT1::KAT1-mNeonGreen and compared KAT1 trafficking during stomatal opening in wild-type and eb1a 1b 1c guard cells. Z-stack images (0.5-μm step size) of guard cells were acquired, and the projection image was shown in Fig. 2a, after maintenance in the dark for 2 h, the KAT1-mNeonGreen signal at the PM was dim in guard cells surrounding closed stoma in both wild-type and mutant cells. Consistent with a previous report48, a 1 h light illumination induced stomatal opening and the redistribution of KAT1-mNeonGreen fluorescence to the PM in wild-type guard cells; however, an obvious delay in stomatal opening and the redistribution of KAT1-mNeonGreen to the PM was observed in eb1a 1b 1c triple mutant guard cells (Fig. 2a). To quantitatively analyse the PM redistribution of KAT1-mNeonGreen in guard cells upon light exposure, we compared the PM/cytosol ratio of mean KAT1-mNeonGreen fluorescence intensity in the wild type and eb1a 1b 1c mutant. As shown (Fig. 2b), a significantly higher ratio was observed in the wild type when compared with eb1a 1b 1c mutant after 1 h or 2 h light treatment. Moreover, expression of AtEB1a or AtEB1b rescued the PM localisation and dynamics of KAT1 in COM-EB1a and COM-EB1b lines (Supplementary Fig. 3a). These results suggested a role for AtEB1 proteins in KAT1 redistribution to the PM during light-induced stomatal opening. Furthermore, we applied 10 µM amiprofos-methyl (APM, a microtubule-disrupting drug) along with the 1-h light treatment to the wild-type guard cells. We found that depolymerisation of microtubules significantly reduced the light-induced PM-distribution of KAT1-mNeonGreen compared with the untreated control (Supplementary Fig. 3b, c), which further supported the important role of microtubules in KAT1 localisation.

a Distribution of mNeonGreen-tagged KAT1 (KAT1-mNeonGreen) in guard cells from wild-type (Col-0) and eb1a 1b 1c mutant plants before and after light treatment. Light stimulus increased the redistribution of KAT1-mNeonGreen to the PM in wild-type guard cells, whereas this process was dramatically delayed in eb1a 1b 1c cells. Z-stack images (0.5-μm step size) were acquired, and the max-intensity projection is shown. Total four independent experiments were performed and obtained similar results. Bars = 5 µm. b The ratio of mean KAT1-mNeonGreen fluorescence intensity between the PM and cytosol (excluding chloroplast auto-fluorescence) was measured in guard cells from wild-type and eb1a 1b 1c mutant plants as shown in a. c Representative pseudo-coloured images of intracellular K+ levels in guard cells expressing lc-LysM GEPII1.0 during a light-induced stomatal opening process. Total of five independent experiments were performed and obtained similar results. Bars = 5 µm. d Quantitative analysis of light-induced intracellular K+ dynamics from wild-type and eb1a 1b 1c mutant guard cells based on FRET ratios (FRET/CFP). Data in b and d are presented as boxplots. The whiskers indicate the maximum/minimum values and the centre line within the box reflects the median value and the boundary lines of box indicate the 1st quartile (upper) and the 3rd quartile (lower) value of the dataset. The data were analysed using RStudio software with the agricolae package. Different letters indicate significant differences analysed by one-way ANOVA with Duncan’s new multiple range test, P < 0.05. The “n” below each box indicates the number of analysed guard cells.
During stomatal opening, K+ is pumped into guard cells through inward potassium channels on the PM (such as KAT1), resulting in increased turgor pressure14. Therefore, we suspected that reduction of the KAT1 channel at the PM (Fig. 2a, b) may perturb K+ influx and be at least partially responsible for delayed light-induced stomatal opening in the eb1a 1b 1c mutant. We used a ratiometric GEPII-based K+ biosensor which fuses K+ binding domains of the bacterial K+-binding protein with CFP and YFP to quantify cellular K+ concentrations ([K+]c) using FRET analysis50. As shown (Fig. 2c, d), after 2 h of darkness, [K+]c as indicated by the FRET ratio (FRET/CFP) in wild-type guard cells was similar to eb1a 1b 1c cells. Upon light illumination, [K+]c was gradually increased along with the stomata opening, consistent with a previous report14, however, the increase in [K+]c in eb1a 1b 1c guard cells was significantly less than in wild-type cells (Fig. 2c, d). This result was consistent with less KAT1-mNeonGreen on the PM in eb1a 1b 1c guard cells when compared with wild-type cells (Fig. 2a, b).
From this evidence, we propose that AtEB1 proteins function in the distribution/redistribution of the KAT1 potassium channel on the PM, and consequently, are involved in light-induced stomatal opening.
AtEB1b mediates SYP121 redistribution during stomatal opening
Arabidopsis SYP121 protein belongs to the Qa-SNARE family. PM-localised SYP121 is highly dynamic and is continuously recycled from and to the PM51. A previous study indicated that SYP121 facilitates stomatal reopening, and the trafficking and recycling of K+ channels to the PM48. We, therefore, tested if AtEB1 interacted with SYP121 to regulate stomatal opening. We used AtEB1b and a SYP121△C fragment containing the essential N-terminal residues, a Habc domain and a H3 Qa-SNARE motif (Supplementary Fig. 4a, b) as described previously52, for the following analyses: Firstly, an in vitro pull-down assay was performed using glutathione S-transferase fused SYP121△C (GST-SYP121△C) and His-AtEB1b; His-AtEB1b was found to directly interact with GST-SYP121△C (Fig. 3a and Supplementary Fig. 4c). Secondly, a luciferase complementation imaging (LCI) analysis also confirmed the direct interaction between AtEB1b and SYP121 (Fig. 3b). We then conducted a co-immunoprecipitation (Co-IP) assay using Arabidopsis transgenic plants expressing AtEB1b-GFP or GFP alone. AtEB1b-GFP or GFP alone was precipitated, respectively, from plant extracts with anti-GFP antibody; the SYP121 protein co-precipitated with AtEB1b-GFP, but not with GFP alone, as identified by an anti-SYP121 antibody (Fig. 3c). This further indicates that AtEB1b and SYP121 may co-exist within the same complex in vivo. Lastly, we performed a bimolecular fluorescence complementation (BiFC) assay in tobacco leaves and observed YFP signals with a typical mobile comet-like pattern (similar to AtEB1b-GFP) in leaf epidermal cells co-expressing YN-SYP121 and AtEB1b-YC (Fig. 3d and Supplementary Movie 2). This result hinted that SYP121 interacted with AtEB1b at microtubule plus ends. However, the AtEB1b truncated fragment, AtEB1bCH35, which lacks the coiled-coil domain and L region for microtubule-binding, and also misses the tail region for interaction with other proteins, did not interact with SYP121 (Fig. 3d).

a An in vitro pull-down assay was performed with His-AtEB1b protein and GST-SYP121△C. His-AtEB1b pulled down by GST-SYP121△C was detected by anti-His antibody. However, His-AtEB1b could not be pulled down by GST alone. One representative result from three independent analyses is presented here. GST-SYP121△C is indicated with an asterisk. b Luciferase complementation imaging assay showing the interaction between SYP121 and AtEB1b. Co-expression of cLuc-SYP121/ECAP-nLuc was used as a negative control. A representative result from three independent analyses is shown in the left panel, and the pseudo-colour bar indicates the range of luminescence. The expression levels of cLuc-SYP121, AtEB1b-nLuc, and ECAP-nLuc were indicated by the qRT-PCR analysis (right panel). c AtEB1b co-immunoprecipitated with SYP121. Proteins extracted from transgenic plants expressing AtEB1b-GFP were immunoprecipitated using anti-GFP antibody and analysed by western blot with anti-SYP121 antibody. Transgenic plants expressing GFP alone were used as negative controls. One representative result from three independent analyses is presented here. AtEB1b-GFP is indicated with asterisks. d BiFC assay confirming the interaction between AtEB1b and SYP121. A truncated AtEB1CH fragment was used as a negative control. The max-intensity projection of Z-stack images is shown in the upper panel. The lower-left panel indicates the conserved domains of AtEB1b or AtEB1bCH. CH, calponin-homologous domain; L, linker region; CC, coiled-coiled domain; T, tail region. The expression levels of YN-SYP121, AtEB1b-YC, and YC-AtEB1bCH are indicated by the qRT-PCR analysis (lower-right panel). A representative result from three independent analyses is shown. Bars = 6 µm. In b and d, Nicotiana benthamiana ACTIN (NbACTIN) was used as an internal control, data are presented as mean ± s.d. **P < 0.01, ***P < 0.001, two-tailed Student’s t-test. ns, not significant.
To further address the functional relationship between AtEB1 and SYP121, we compared the SYP121 localisation pattern in wild-type and eb1a 1b 1c mutant guard cells. In wild-type guard cells with open stoma, most GFP-SYP121 signals were detected in the PM; only a few GFP-SYP121 punctates with weak fluorescence were seen in the cytosol (Fig. 4a). As shown in Supplementary Fig. 5 and Supplementary Table 1, fluorescent protein-tagged SYP121 partially co-localised with HAP13g-RFP and GFP-VAMP722 (markers for the TGN/EE compartment). This indicates that those cytosolic fluorescent punctates in Fig. 4a were GFP-SYP121-associated TGN/EE. Considerably more GFP-SYP121-associated TGN/EE existed in the cytosol, with reduced signal at the PM, in eb1a 1b 1c mutant guard cells than in the wild-type cells (Fig. 4a). Quantification analysis revealed a significantly higher average PM/cytosol ratio of GFP-SYP121 fluorescence intensity in the wild type compared with eb1a 1b 1c guard cells (Fig. 4b). The average number of GFP-SYP121-associated TGN/EE in the cytosol of eb1a 1b 1c guard cells was significantly greater than in wild-type guard cells (Fig. 4c). Consistently, the percentage of GFP-SYP121 fluorescence intensity on TGN/EE versus cytosol was significantly greater in the eb1a 1b 1c mutant than the wild-type guard cells (Fig. 4d).

a GFP-SYP121 localisation pattern in wild-type (Col-0) (left) and eb1a 1b 1c mutant (right) guard cells under normal growing condition (The max-intensity projection of Z-stack images is shown). Reduced PM localisation and increased fluorescent punctates in the cytosol were detected in eb1a 1b 1c mutant guard cells. Arrowheads indicate GFP-SYP121-associated TGN/EE. A total three independent experiments were performed and obtained similar results. Bars = 5 µm. b The ratio of the mean GFP-SYP121 fluorescence intensity (PM vs. cytosol) was measured in wild-type and eb1a 1b 1c mutant guard cells. c Quantification of GFP-SYP121-associated TGN/EE in wild-type and eb1a 1b 1c mutant guard cells. d The percentage of GFP-SYP121 fluorescence intensity (endosomal-associated vs. cytosol). e AtEB1 proteins promote the light-induced redistribution of SYP121 to the PM. Wild-type and eb1a 1b 1c mutant seedlings expressing GFP-SYP121 were kept in the dark for 2 h and then transferred to light for 0.5 h before observation. In wild-type guard cells, light efficiently induced the accumulation of GFP-SYP121 at the PM, whereas GFP-SYP121-associated TGN/EE dramatically decreased in the cytosol. However, this process was significantly delayed in eb1a 1b 1c guard cells. The max-intensity projection of Z-stack images is shown. Total six independent experiments were performed and obtained similar results. Bars = 5 µm. f Quantification of GFP-SYP121-associated TGN/EE in wild-type and eb1a 1b 1c mutant guard cells before and after light treatment. g Quantitative analysis of the ratio of GFP-SYP121 mean fluorescence intensity (PM vs. cytosol) in wild-type and eb1a 1b 1c mutant guard cells, before and after light illumination. Data in b–d, f, and g are presented as boxplots. The whiskers indicate the maximum/minimum values and the centre line within the box reflects the median value and the boundary lines of the box indicate the 1st quartile (upper) and the 3rd quartile (lower) value of the dataset. Data in b–d, ***P < 0.001, two-tailed Student’s t-test. Data in f and g were analysed using RStudio software with the agricolae package. Different letters indicate significant differences analysed by one-way ANOVA with Duncan’s new multiple range test, P < 0.05. The “n” above each box indicates the number of analysed guard cells.
Our investigation strongly suggested that the delay in stomatal opening in the eb1a 1b 1c mutant could be attributed to the decreased localisation of SYP121 at the PM of guard cells. We, therefore, tracked any changes in GFP-SYP121 localisation in wild-type and eb1a 1b 1c guard cells before and after 0.5 h of light treatment. After 2 h in darkness, most of the GFP-SYP121 signal associated with cytosolic TGN/EE in guard cell bearing closed stoma and the average TGN/EE number per cell was similar between wild-type and eb1a 1b 1c guard cells (Fig. 4e, f). Upon light illumination, the GFP-SYP121 signal quickly increased at the PM, and the number of GFP-SYP121-associated TGN/EE dramatically decreased in the cytosol of wild-type cells at 0.5 h after light exposure (Fig. 4e–g). In light-treated eb1a 1b 1c cells, the average number of GFP-SYP121-associated TGN/EE per 100 µm2 was significantly higher than wild-type cells (Fig. 4e, f), and the 0.5 h light-induced increase in the PM/cyto ratio of GFP-SYP121 signal was significantly less than that in wild-type cells (Fig. 4g), suggesting a delay in SYP121 redistribution to the PM in eb1a 1b 1c cells. Moreover, the light-induced PM-distribution of GFP-SYP121 was recovered in COM-EB1a and COM-EB1b (Supplementary Fig. 6a, b). These indicated that AtEB1 proteins are important for SYP121 redistribution to the PM during stomatal opening.
To further test this, 50 μM Brefeldin A (BFA) and 50 μM CHX (cycloheximide) was applied and this was then followed by a 3 h washout of BFA. BFA inhibits exocytosis but not endocytosis, and CHX is a protein biosynthesis inhibitor. BFA treatment induced a similar aggregation of GFP-SYP121 in BFA compartments in wild-type and eb1a 1b 1c cells, implying little difference in the internalisation of SYP121 (Supplementary Fig. 7a, b). Once BFA was washed out, SYP121-containing BFA bodies disappeared in nearly 62.4% of wild-type cells, but could still be detected in about 79.6% of eb1a 1b 1c cells (Supplementary Fig. 7b). This indicated that AtEB1 proteins are important for the redistribution of internalised SYP121 to the PM, but may not be required for SYP121 internalisation.
To further confirm that AtEB1 proteins are conducive to SYP121 redistribution on the PM during light-induced stomatal opening, we performed a fluorescence recovery after photo-bleaching (FRAP) analysis. We drew a region of interest (ROI) at the dorsal side of a guard cell with an open stoma expressing GFP-SYP121 (Supplementary Fig. 7c), and then measured the average GFP-SYP121 fluorescence intensity in the ROI before and after bleaching. We found a significantly slower recovery rate of GFP-SYP121 fluorescence at the PM in eb1a 1b 1c guard cells when compared with wild-type cells. Moreover, APM treatment interfered with the recovery of GFP-SYP121 fluorescence at the PM (Supplementary Fig. 7c–e). These evidences further suggested a role for AtEB1 proteins in the redistribution of SYP121 to the PM region in guard cells during light-induced stomatal opening.
Microtubule growth drives endomembrane trafficking of SYP121
We next asked if such AtEB1-mediated SYP121 redistribution to the PM was dependent on microtubules. We first applied 10 µM microtubule-disrupting drug APM for 30 min to wild-type guard cells kept in the dark. Compared with the untreated control, light-induced stomatal opening was dramatically inhibited (Supplementary Fig. 8a). Further exploration of the PM/cytosol ratio for GFP-SYP121 fluorescence intensity indicated that the disruption of microtubules significantly reduced the light-induced redistribution of GFP-SYP121 to the PM (Supplementary Fig. 8b, c). In addition, the movement of GFP-SYP121-associated TGN/EE was severely affected by APM treatment, about 69.2% of them were nearly immobile or moving at a speed <0.1 μm s−1, which was a higher proportion than that for cells without APM treatment (48.5%; Supplementary Fig. 8d). Taken together, above observations imply that the light-induced redistribution of SYP121 to the PM is at least partially dependent on microtubules.
As we observed SYP121 directly interacted with AtEB1b in vitro and in vivo (Fig.3) and moved together (Supplementary Movie 2), we predicted that SYP121-associated TGN/EE could be recruited by AtEB1 proteins and anchor at the plus ends of growing microtubules during light-induced stomatal opening. First, we observed the dynamics of AtEB1b-mCherry and GFP-SYP121 in guard cells by spinning disc confocal microscopy. AtEB1b-mCherry dots were observed moving constantly along a path, indicating the growing ends of microtubules (Supplementary Movie 1). Interestingly, some SYP121-associated TGN/EE displayed random motion (Supplementary Movie 3), whereas other SYP121-associated TGN/EE travelled a certain distance and then disappeared (Supplementary Movie 4). A change in direction for moving SYP121-associated TGN/EE was also observed (Supplementary Movie 5). Notably, the velocity of GFP-SYP121-associated TGN/EE was seemingly reduced in eb1a 1b 1c guard cells. About 70.4% of GFP-SYP121-associated TGN/EE (a total of 345 TGN/EE from 40 cells) in eb1a 1b 1c cells moved at a speed of <0.1 μm s−1, more than that in wild-type cells (48.5%; a total of 332 TGN/EE from 39 cells). Next, we generated transgenic plants co-expressing mCherrry-SYP121 and AtEB1b-GFP to investigate their co-localisation and behaviour. The expression of mCherrry-SYP121 in syp121-6 rescued the mutant phenotype, indicating its proper functionality (Supplementary Fig. 9a, b). During light-induced stomatal opening, co-localisation between mCherry-SYP121-labeled compartment and AtEB1b-GFP was indeed detected (Supplementary Movie 6). A representative co-localisation event is shown in Fig. 5a, Supplementary Fig. 9c and Supplementary Movie 6, where a moving SYP121-associated TGN/EE instantaneously changed its path after associating with AtEB1b, and moved in the same direction as AtEB1b; this indicates that some SYP121 punctates (34 out of total 198 labelled TGN/EE from 11 cells) were moved by growing microtubules. However, not all SYP121-associated TGN/EE co-localised with AtEB1b-GFP dots, suggesting that only a proportion of SYP121-associated TGN/EE are transported by the growth of microtubules.

a Left: A representative time series of partial region in a guard cell (the image of intact cells was shown in Supplementary Fig. 9c) expressing AtEB1b-GFP and mCherry-SYP121. Spinning disc confocal microscopy was used and the focal plane was the interior of the cell. A SYP121-associated TGN/EE (magenta) moved away from the cell periphery (marked by a white dashed line). At 20 s, an AtEB1b-GFP dot (green) suddenly appeared and a co-localisation between AtEB1b-GFP and the mCherry-SYP121-associated TGN/EE was detected (white arrowheads). The SYP121-associated TGN/EE then changed its direction and moved toward the cell periphery. Better visualisation required increasing contrast (post-acquisition). White arrows indicate the moving direction of a mCherry-SYP121-associated TGN/EE. Bar = 2 µm. Right: The graphic depicts the moving tracks of AtEB1b-GFP particle and mCherry-SYP121-associated TGN/EE shown in the left panel. The whole tracking path with AtEB1b in green and SYP121 tracks in white (when co-localised with AtEB1b) or magenta (when no co-localisation was observed). b Left: Variable-angle epifluorescence microscopy was used to observe the co-localisation of mCherry-SYP121 with AtEB1b-GFP and their approach to the cell surface. A representative time series was shown. A mCherry-SYP121-associated TGN/EE (white arrowheads) appears and is stabilised at the cell surface at 0 s, and remained stationary for several seconds before its fluorescence faded at 8 s. Bar = 1.5 µm. *indicated the AtEB1b-GFP dots being tracked. Right: Kymograph of the region indicated in the left panel (white dash arrow), showing the stationary phase of mCherry-SYP121-associated TGN/EE co-localised with AtEB1b-GFP. The green and magenta arrows represent the AtEB1b and SYP121 trajectory, respectively.
We monitored a total of 34 pairs of AtEB1b-GFP dots and mCherry-SYP121-labeled TGN/EE that exhibited associated movements over a distance > 0.5 µm. Of these, 14 pairs clearly demonstrated that the moving direction of SYP121-associated TGN/EE changed after interacting with AtEB1b-GFP and being orientated by microtubule growth direction. For another 20 pairs, the SYP121-associated TGN/EE and AtEB1b-GFP dot interacted and moved together at the beginning of the observation (Supplementary Movie 7); although it was hard to tell if the direction of SYP121-associated TGN/EE had changed, it still clearly indicated a vesicle trafficking of SYP121 driven by microtubule growth. We propose that AtEB1 proteins recruit SYP121-associated TGN/EE to the plus end of microtubules via binding with SYP121. However, we cannot exclude the possibility that SYP121-associated compartments might also be transported by motor proteins such as myosin or kinesin along microfilaments or microtubules.
Interestingly, shortly after the SYP121-associated TGN/EE reached the periphery of the cell, the fluorescence of the mCherry-SYP121 weakened and disappeared, which was likely the result of its fusion with the PM (Fig. 5a and Supplementary Movie 6). We then focused on the periclinal cell surface using variable-angle epifluorescence microscopy (VAEM), a technology suitable for observing the dynamics of proteins near the PM in plants53. We observed total 58 mCherry-SYP121-associated TGN/EE co-localised with AtEB1b-GFP and tracked their approach to the cell surface (Fig. 5b). After their first appearance at the cell periphery (near the PM), 14 mCherry-SYP121/AtEB1b-GFP-colocalised TGN/EE remained stationary for at least 2 s before the fluorescence faded (Fig. 5b). We suspect that these compartments were delivered to the cell periphery and fused with the PM.
To verify this assumption, we used VAEM to monitor and analyse the dynamics of SYP121-associated TGN/EE at the cell surface in wild-type guard cells expressing GFP-SYP121. As shown (Fig. 6a–c), GFP-SYP121 displayed as discrete punctate at the periclinal cell surface. The appearance, stationary state (>2 s), and disappearance of some GFP-SYP121 punctates at distinct sites were clearly revealed by kymograph analysis (Fig. 6b). This type of behaviour was similar to the exocytosis event described previously54,55. Similar behaviour of GFP-SYP121-associated TGN/EE was also found in eb1a 1b 1c cells, although the loss function of AtEB1 proteins led to significantly reduced occurrence (3.0 ± 0.3 events/100 μm2/10 s) than in wild-type cells (5.0 ± 0.5 events/100 μm2/10 s). Moreover, we found that the appearance of GFP-SYP121 punctates was significantly interfered by the APM treatment (Fig. 6d), indicating a role of microtubules in the delivery of GFP-SYP121-associated TGN/EE to the cell periphery.

a A wild-type (Col-0) guard cell expressing GFP-SYP121 visualised using variable-angle epifluorescence microscopy, showing discrete GFP-SYP121 punctates near the periclinal cell surface of the guard cell. The yellow box indicates the ROI region used to calculate the frequency of the SYP121-associated punctates approached and distributed to the cell surface, and represents the location where the kymograph plot was generated in b. Bar = 10 µm. b Kymograph of a region of interest indicated by the yellow box in a, showing the appearance, stationary (>2 s), and disappearance of each distinct GFP-SYP121 punctate at the cell surface/PM (a behaviour similar to the previously reported exocytosis events). The span of each white straight line in the kymograph represents the stationary duration of distinct GFP-SYP121 punctate. c A GFP-SYP121-associated punctate (yellow arrows) was enlarged from a, showing its appearance at the cell surface at 0 s. It kept stationary about 4 s before the fading of its fluorescence, indicating an exocytosis event. Bar = 0.35 µm. d The rate of GFP-SYP121-associated punctates with exocytosis-like dynamic behaviour (appeared, stationed, then disappeared) at the cell surface were calculated in wild-type (without or with APM treatment) and eb1a 1b 1c guard cells. Data were presented as mean ± s.e. (n = 17 ROI from 12 wild-type guard cells without APM; n = 23 ROI from 14 eb1a 1b 1c mutant guard cells; n = 24 ROI from 13 APM-treatment wild-type guard cells). **P < 0.01, ***P < 0.001, two-tailed Student’s t-test. e Duration time of stationary GFP-SYP121-associated punctate near the cell surface in wild-type and eb1a 1b 1c guard cells. Data were presented as mean ± s.e. (n = 3). A total of 127 and 145 events were measured from three independent biological experiments in wild type and eb1a 1b 1c mutant, respectively. *P < 0.05, **P < 0.01, two-tailed Student’s t-test. ns, not significant.
We also analysed the resident time of stationary GFP-SYP121 punctates at the cell periphery before the disappearance of their fluorescence (Fig. 6e). We found that most GFP-SYP121-associated TGN/EE (about 70%) in wild-type guard cells displayed resident time less than 5 s (Fig. 6e), significantly more than that in eb1a 1b 1c cells. The average resident time of GFP-SYP121 punctates in wild-type cells was 5.0 ± 0.4 s (mean ± s.e.), whereas it was extended to 7.1 ± 0.9 s (mean ± s.e.) in eb1a 1b 1c cells. We suspected that the reduction of PM-localised SYP121 in eb1a 1b 1c guard cells (Fig. 4 and Supplementary Fig. 7) caused less efficiency in membrane fusion during exocytosis, thereby leading to extension of the residency time of exocytic vesicles.
In conclusion, a hypothetic model of microtubule growth-driven endomembrane trafficking during light-induced stomatal opening is shown in Fig. 7. SYP121-associated TGN/EE directly interact with AtEB1 +TIP proteins, and, through microtubule growth, are propelled and delivered to the cell periphery for redistribution to the PM. It has been shown that PM-localised SYP121 facilitates the recycling of KAT1 to the PM, and the subsequently increased K+ channels on the PM are important for light-induced stomatal opening19,46 (Fig. 7). Further analyses of light-induced stomatal opening were performed using kat1 (SALK_127506C), syp121-6 single mutants56, and quadruple mutants eb1a 1b 1c kat1 and eb1a 1b 1c syp121-6 (Supplementary Fig. 10). We found similar phenotypes in these mutants as that in the eb1a 1b 1c triple mutant, which strongly supports our hypothetical model.

1 During stomata closing, PM-localised KAT1 and SYP121 proteins were internalised into the cytosol; 2 During stomata opening, co-localisation between SYP121 and AtEB1 proteins recruits the SYP121-associated TGN/EE to the growing end of microtubules; 3 The SYP121-associated TGN/EE is delivered to the cortex, close to the PM for exocytosis; 4 PM-associated SYP121 subsequently facilitates the redistribution of KAT1 K+ channel and SYP121 to the PM48,56. Moreover, PM-associated SYP121 also binds to KAT1 and promotes KAT1 channel activity21.
Discussion
Previous studies and our present work reported two approaches for regulation of KAT1 function during stomatal opening. By single guard cell recordings in intact plants, Roelfsema et al.57 demonstrated that light induces hyperpolarisation of the PM to activate the activity of inward rectifying K+ channels. A recent report by Lefoulon et al. indicated that PM-anchored SYP121 binding to KAT1 promoted KAT1 channel activity21. In addition to channel-activity regulation, microscopy analysis revealed that dynamic population-control of KAT1 channels at the PM also plays an important role in the regulation of stomatal movements20,48. KAT1 was reported to be internalised into endomembrane compartments when guard cells were kept in darkness or treated with abscisic acid, whereas be redistributed to the PM during stomatal opening/reopening20,48 (present work). Interestingly, in single intact guard cells impaled with microelectrodes, Roelfsema et al.57 found that K+ currents did not disappear when guard cells were kept in darkness. This could be due to the fact that not all KAT1 channels are internalised into the cytosol, and the remaining PM-resided KAT1 is activated by the hyperpolarsation of the PM (Roelfsema et al.57 also proposed that factors other than light can induce hyperpolarised membrane potentials as well). Indeed, further investigation is needed for this hypothesis. In addition, it was shown that multiple inward-rectifying K+ channels exist in guard cells and KAT1 was not essential for stomatal action58. It is worthy of study in the future to testify whether AtEB1 proteins-mediated microtubule-growth-driven endomembrane trafficking is also involved in regulation of other K+ channels in Arabidopsis. Besides, aquaporin PIP2;7 (which was also implicated in stomatal movement) has been shown to directly interact with SYP121, and its delivery to the PM depends on SYP12159,60. Because the PM-localised SYP121 participates in driving membrane fusion during exocytosis, we reckon that AtEB1-mediated microtubule growth-based mechanism might not be specific for KAT1. More likely, it is also adopted for some other transmembrane proteins to distribute to the PM via SYP121-mediated exocytosis. This hypothesis needs further investigation in future studies.
Our investigation suggests an important role for microtubule growth in vesicle trafficking during light-induced stomatal opening. Although it is well known that the power produced by microtubule elongation and shrinkage is essential for chromosome movements61, it is of interest to investigate how this is involved in vesicle trafficking. In plant cells, little evidence supports the involvement of microtubules in the long-distance trafficking of organelles or endomembrane compartments previously. However, there is mounting evidence to suggest that microtubules and their motors are involved in targeting secretory organelles to functionally appropriate destinations, by anchoring or slowing down these organelles4,5,10,62. Our observations agree with this hypothesis: the association of SYP121-associated TGN/EE with AtEB1 at the plus end of microtubules allows the delivery of vesicles to the cell periphery, which would facilitate the subsequent docking and fusion of SYP121-associated TGN/EE to the PM. Since not all SYP121-resident TGN/EE were associated with AtEB1b (Fig. 5, Supplementary Fig. 9c and Supplementary Movie 6), other mechanisms must exist to deliver SYP121 to the PM, which is of interest for future studies. In addition, another microtubule plus end-binding protein, CLASP, was found to directly interact with the retromer SNX1 and also has a role in controlling SNX1 vesicle morphology and behaviour9. It was proposed that the recruitment of SNX1 vesicles to the plus end of microtubules increases vesicle residency time close to the PM9. Do different plus end-binding proteins recognise distinct vesicles and/or play different roles in microtubule-dependent vesicle trafficking? What specifies those vesicles that are recognisable to various microtubule plus end-binding proteins? These are interesting questions worthy of further investigation.
A previous report demonstrated that vacuole remodelling in guard cells during stomatal movement requires SNARE VTI11 and intact microtubules63. This raises an interesting question concerning whether microtubule growth is also a driving force in VTI11 positioning and hence in membrane fusion at the vacuole during stomatal opening. Therefore, whether microtubule growth-driven endomembrane trafficking is involved in more than one SNARE protein-mediated vesicle trafficking is worthy of further investigation.
EB1 proteins are evolutionarily conserved microtubule plus end-binding proteins in yeasts, animals and plants29,33. They directly regulate microtubule dynamics, or indirectly by recruiting other microtubule plus end-binding proteins and acting as master integrators organising the +TIP interaction network34,35,38,64. In Arabidopsis, Lindeboom et al.38 reported that the absence of AtEB1 proteins reduces the growth rate of microtubule plus ends, but leads to an accelerated shrinkage velocity, although they have no obvious effects on the catastrophe or rescue rates of microtubules. In addition, Molines et al.65 suggested a function for AtEB1a and AtEB1b in the interaction of adjacent microtubules, contributing to microtubule bundling and organisation and root growth in Arabidopsis. Therefore, besides the recruitment of SYP121-associated TGN/EE to microtubule plus ends, AtEB1 proteins may also facilitate microtubule growth-based vesicle trafficking by modulating the dynamics/behaviour of plus ends of growing microtubules, and by regulating the organisation of microtubule array. Nevertheless, in plants, the actin-based myosin system has long been accepted to direct vesicle transport4,5. It was reported previously that the localisation of SYP123, SYP124, and SYP125 in tip growing pollen tubes and root hairs is closely related to F-actin66,67,68. Interestingly, animal EB1 can bind to F-actin69,70, and EB1 proteins are evolutionarily conserved in yeasts, animals, and plants. However, to date there is no such interaction reported in plants, and whether AtEB1 proteins also mediate vesicle transport through possible interaction with F-actin is an interesting question that merits further investigation.
In Arabidopsis, AtEB1a and AtEB1b belong to the same subclade, whereas AtEB1c constitutes another subgroup35. Different from AtEB1a and AtEB1b, AtEB1c-GFP lacks the characteristic tail and accumulates in the nucleus of the intermitotic phase35,71. Interesting, we found that all three AtEB1s could interact with SYP121 (Supplementary Fig. 11). However, based on a series of genetic analyses (Supplementary Fig. 2), we proposed that functional redundant AtEB1a and AtEB1b play major roles in stomatal opening, whereas AtEB1c might be compensatory when AtEB1a and AtEB1b were absent. It has been shown that AtEB1c is enriched in the spindle and the middle region of the phragmoplast, where the positive end of microtubules are more concentrated during mitosis36. After the rupture of the nuclear membrane, AtEB1c was found associated with the ends of growing microtubules in a comet-like pattern35,72,73. Ho et al.74 reported that robust microtubule polymerisation (indicated by fluorescently tagged AtEB1c) took place in the phragmoplast midline. This raises an interesting question of whether AtEB1c mediates phragmoplast microtubule trapping and guides vesicle fusion with the cell plate and is worthy of further study.
Notably, when we use fusicoccin (FC, 5 µM) to by-pass the light signal to induce stomatal opening in the dark, we also detected defects in stomatal opening and K+ accumulation in eb1a1b1c guard cells (Supplementary Fig. 12). This evidence suggests that AtEB1-mediated regulation of stomatal opening is not specific to the light.
The dynamics of the microtubule plus end have been proposed as a mechanism in searching for cortical destination subdomains in Drosophila and mammalian cells75,76. Gu et al.77 reported that both KIF3/kinesin II and EB1 participate in the axonal targeting of Kv1 voltage-gated K+ channels. An association with both EB1 and KIF3 enhanced the ability of Kv1-containing vesicles to stay associated with microtubules. The association with EB1 was also speculated to facilitate the delivery of Kv1-containing vesicles to the axonal membrane—if the microtubule plus end is close enough to the axonal membrane. This implies microtubule plus-end-directed vesicle trafficking in animal cells, although direct evidence is lacking. Microtubule plus-end-directed vesicle trafficking has rarely been reported to date and yet our findings raise the important question of whether this could be a widespread mechanism underlying critical cellular activity at the cortex of all eukaryotic cells. Further studies in animal/fungal systems are needed to demonstrate this; such investigations will broaden our understanding of directional endomembrane trafficking in all organisms.
Methods
Plant materials
Arabidopsis thaliana ecotype Columbia (Col-0) was used as the wild type. The eb1a 1b 1c triple mutant was described previously by Komaki et al.35 The eb1a 1b double mutant was obtained by hybridising the eb1a 1b 1c triple mutant with the wild-type plant. The complementary lines COM-EB1a, COM-EB1b, and COM-EB1c were obtained by introducing ProAtEB1a::AtEB1a, ProAtEB1b::AtEB1b, or ProAtEB1c::AtEB1c into the eb1a 1b 1c triple mutant, respectively.
The syp121-6 mutant and syp121-6 ProSYP121::GFP-SYP121 (named as COM 18) transgenic lines were documented in our previously published article56. The eb1a 1b 1c GFP-SYP121 line was obtained by crossing COM 18 with the eb1a 1b 1c mutant. Then, eb1a 1b 1c GFP-SYP121 plants were crossed with COM-EB1a or COM-EB1b to generate AtEB1 complementary lines expressing GFP-SYP121. To verify the functionality of mCherry-SYP121, ProSYP121::mCherry-SYP121 was introduced into syp121-6 plants to generate the complementary line—COM9. A 35S::AtEB1b-GFP transgenic line was kindly provided by Prof. Shanjin Huang (Tsinghua University, China). The 35S::AtEB1b-GFP line was crossed with ProSYP121::mCherry-SYP121 to generate dual-labelled transgenic plants. ProAtEB1b::AtEB1b-mCherry was transformed into 35S::GFP-TUA6 transgenic lines for dual labelling of AtEB1b and microtubules.
Arabidopsis transgenic marker lines HAP13-RFP and WAVE22R were kindly provided by Prof. Yan Zhang (Shandong Agricultural University, China); mCherry-MEMB12 and mRFP-ARA7 were kindly provided by Prof. Ruixi Li (Southern University of Science and Technology, China); GFP-VAMP722 line was provided by Prof. Jinxing Lin (Beijing Forestry University, China). Above marker lines were crossed with ProSYP121::GFP-SYP121 or ProSYP121::mCherry-SYP121 to obtain dual labelling lines.
ProKAT1::KAT1-mNeonGreen was transformed into eb1a 1b 1c and wild-type plants, respectively. Then, the eb1a 1b 1c KAT1-mNeonGreen plant was crossed with COM-EB1a or COM-EB1b to obtain complementary lines expressing KAT1-mNeonGreen. An Arabidopsis transgenic line expressing K+ biosensor lc-LysMGEPII1.050 was kindly provided by Prof. Yi Wang (China Agricultural University, China), and an eb1a 1b 1c line expressing lc-LysMGEPII1.0 was obtained by crossing. A T-DNA insertion mutant of KAT1 (SALK_127506C) was ordered from the ABRC.
Agrobacterium-mediated transformation was performed by using the floral dip method.
Plasmid construction
To generate the ProKAT1::KAT1-mNeonGreen construct, the native KAT1 promoter (3-kb fragment upstream of the start codon) was amplified from the wild-type genomic DNA, and the coding sequence of KAT1 was amplified from a cDNA library using specific primers. Resulting products were then cloned into a pCAMBIA1300 vector harbouring a mNeonGreen tag coding sequence.
To generate the ProSYP121::mCherry-SYP121 construct, a 1.8-kb fragment before the start codon was amplified from wild-type genomic DNA as the native promoter. The coding sequence of SYP121 was amplified from a cDNA library. The corresponding products were subsequently cloned into a pCAMBIA1300 vector harbouring the coding sequence of a mCherry tag.
A native promoter (2-kb fragment before the start codon) and the coding sequence of AtEB1a, AtEB1b, or AtEB1c were amplified respectively, and then cloned into a pCAMBIA1300 vector to generate ProAtEB1a:: AtEB1a, ProAtEB1b:: AtEB1b, or ProAtEB1c::AtEB1c. The amplification primers used for cloning are shown in Supplementary Table 2.
Stomatal aperture bioassay
To analyse light-induced stomatal opening, leaves from 2-3-week-old plants were incubated in buffer containing 10 mM 2-(N-morpholino) ethanesulfonic acid (MES)-Tris buffer, pH 6.1, and kept in the dark for 2 h to ensure stomata were completely closed. Then, abaxial epidermal strips were peeled from leaves and transferred to buffer containing 50 mM KCl, 10 mM MES-Tris (pH 6.1) and illuminated with white light. At different time points, abaxial epidermal strips were mounted on a glass slide and immediately observed under a microscope (Olympus BX51) equipped with a digital camera (DP72). Three independent experiments were conducted. Stomatal aperture size was measured using ImageJ software.
Leaf temperature measurement
Images of 2-3-week-old wild-type and eb1a 1b 1c triple mutant plants were acquired using a ThermaCAM SC1000 infrared camera (FLIR Systems Inc., Wilsonville, OR, USA), and false-colour infrared images were processed by IRWIN REPORTER software (version 5.31). Three independent experiments were performed.
Stomatal conductance measurement
The whole plant stomatal conductance40 was detected using an Ecotek LI-6400 system (LI-COR, http://www.licor.com). The mature leaves of 4–5-week-old Arabidopsis plants were exposed to 400 ppm CO2 and 47% relative humidity (24 °C). One hour light illumination (100 μmol m−2 s−1) was applied to measure the changes of stomatal conductance in response to light. All measurements were taken once per 2 min, and four independent experiments were performed.
Pull down assay
The SYP121 ∆C fragment was amplified, and then cloned into a pGEX4T-1 vector. The GST-tagged SYP121∆C (GST- SYP121∆C) was expressed in BL21 cells after induction by 1 mM IPTG for 4 h at 28 °C and then purified using glutathione Sepharose 4B beads (GE Healthcare, Uppsala, Sweden) according to the manufacturers’ instructions. The coding sequence of AtEB1b was amplified and then cloned into a pET30a (+) vector with a 6 × His tag. The recombinant His-AtEB1b protein was expressed in BL21 cells, and purified using nickel-nitrilotriacetic acid column (Qiagen, Hilden, Germany) according to the manufacturers’ instructions.
For the in vitro pull-down assay, GST and GST- SYP121△C recombinant proteins were pre-incubated with glutathione Sepharose 4B beads in PBS buffer (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4) and then added 50 μg His-AtEB1b protein at 4 °C for 2 h. Unbound proteins were removed by PBS buffer with at least three times. Eluted pull-down mixtures were analysed by 10% SDS-PAGE. His-AtEB1b protein was detected using a monoclonal anti-His antibody (diluted 1:5000, Sigma, MO, USA, H1029,) and GST-tagged proteins were detected using a monoclonal anti-GST antibody (diluted 1:5,000, Sigma, MO, USA, G1160) with secondary antibody (diluted 1:10,000; Abmart, Shanghai, China, M21001S). The uncropped original blot image is shown in Supplementary Fig. 13.
Co-immunoprecipitation Analysis
Arabidopsis transgenic plants expressing 35S::GFP or 35S::AtEB1b-GFP were used for Co-IP analysis. Total protein was extracted from two-week-old plants using extraction buffer (10 mM DTT, 5 mM EGTA, 5 mM EDTA, 10 mM NaF, 10 mM Na3VO4, 5% glycerol, 1 mM phenylmethylsulfonyl fluoride, 1 x protease inhibitor cocktail (Roche, Mannheim, Germany), and 100 mM HEPES), and then centrifuged at 14,000 × g for 30 min at 4 °C. Protein extracts were incubated with anti-GFP conjugated mAb-magnetic agarose beads (MBL, Japan, D153-10) at 4 °C for 2 h. The beads were washed five times with wash buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.05% NP-40). Bound proteins were analysed by 10% SDS-PAGE and western blotted using purified rabbit-derived anti-SYP121 antibody56 (diluted 1:1,000). The uncropped original blot image is shown in Supplementary Fig. 13.
BiFC assay
To construct AtEB1s-YC and YN-SYP121, the coding sequence of AtEB1s and SYP121 was amplified from a cDNA library and cloned into the pSPYCE(M) and pSPYNE (R) 173 plasmid, respectively56. The coding sequence of AtEB1b CH domain was amplified and YC-AtEB1bCH was used as the negative control. Nicotiana benthamiana leaves were transformed by Agrobacterium-mediated transformation78. After 48 h, the leaves were observed using laser confocal microscopy (Carl Zeiss, LSM880, Germany) and a custom-built microscope equipped with Yokagawa W1 spinning disk (Yokagawa) and a Primer 95B sCOMS camera (Teledyne Photometrics, Arizona, USA).
LCI assay
The SYP121 coding sequence was cloned and fused with the C-terminal of cLuc in the pCAMBIA-35S-cLuc vector. The AtEB1b coding sequence was cloned and fused with the N-terminal of nLuc in the pCAMBIA-35S-nLuc vector. Recombinant plasmids were transformed into A. tumefaciens (GV3101) using electroporation and introduced into N. benthamiana via Agrobacterium-mediated infection56. After 48 h, luminescence signals were imaged using a cool charge-coupled device (CCD) imaging system (Berthold, LB985, Germany).
FRAP assay
To perform analyses, a region of interest (ROI) was drawn on the dorsal side of a guard cell. Fluorescence within the ROI was bleached by a 488 nm laser with 100% laser power. The fluorescence recovery of GFP-SYP121 was recorded with 5% laser power at 15 s intervals for 6.5 min. The mean GFP intensity on the guard cell dorsal side PM was measured using ImageJ software. All experimental data were normalised using the following formula79:
(Fn = normalised grey value of fluorescence, Ft = grey value of fluorescence at a selected time point after photobleaching, Fmax = maximal grey value of fluorescence before photobleaching and Fmin = minimal grey value of fluorescence after photobleaching). Normalised data were used to perform curve fitting in OriginPro software (9.1) and the resulting curves were fitted with the following equation:
(Y = grey value after exponential fitting, A1, t1 and y0 represent parameters in the curve and t = time).
qRT-PCR Analysis
For qRT-PCR analysis, the total RNA of 10-day-old Arabidopsis seedlings was extracted with TRIzol reagent (Invitrogen, Scotland, UK) according to the manufacturer’s instructions. RNA was reverse transcribed with oligo d(T)18 primers using the TaKaRa (Shiga, Japan) reverse transcription system. Primers used for qRT-PCR are shown in Supplementary Table 2.
Spinning disc confocal microscopy
An Olympus IX81 inverted microscope equipped with a Yokogawa spinning-disc confocal head (Yokogawa Electric, Tokyo, Japan) and Primer 95B sCOMS camera (Teledyne Photometrics) or iXon 897 EMCCD camera (Andor Technology, Belfast, UK) was used for time-course imaging. Guard cells expressing GFP (or mNeonGreen)- and/or mCherry-tagged proteins were excited by 488 and 561 nm, respectively, and emission signals were observed using an Olympus objective. Images were acquired using VisiView software 6D (Visitron Systems GmbH, Gutenbergstr, Germany) and processed using ImageJ and Photoshop software (Adobe Systems, San Jose, USA).
To observe protein subcellular localisation patterns in guard cell during light-induced stomatal opening, 10-d-old Arabidopsis seedlings expressing fluorescence protein-tagged proteins were kept in the dark for 2 h to allow stomata to close completely. The seedlings were then exposed to light to induce stomatal opening. For projection, Z-stack optical sections of guard cells were taken with 0.5 μm step sizes.
To quantitative analysis of AtEB1b dots and microtubule density, maximum-intensity projections of Z-stack optical sections were used. The average number of AtEB1b-mCherry dots per 10 µm2 was taken as the density of AtEB1b-mCherry dots in one guard cell. To analyse microtubule density, Tubeness and threshold plugin were applied to produce binary images. The microtubule density was indicated by the occupancy of the GFP signal per guard cells42. To evaluate microtubule orientation, ImageJ software was used to measure the angle of cortical microtubule to the inner surface of the stomata42,44. More than 218 cells from three independent biological experiments were analysed.
For pharmacological analysis, guard cells expressing GFP-SYP121 were treated with 50 µM BFA for 30 min and observed under the microscope, either immediately or after a 3 h wash out. CHX (50 µM) was added in the reagents for BFA treatment and washout. To determine the role of microtubules, 10-day-old seedlings expressing GFP-SYP121 (or KAT1-mNeonGreen) were kept in the dark for 120 min before treatment with 10 μM APM in the dark for another 30 min. Seedlings were then exposed to light for 30 min (60 min, KAT1-mNeonGreen) before being observed under the microscope.
To quantify fluorescence intensity of GFP-SYP121 on the PM or in the cytosol80,81: we first used FM4-64, a membrane-impermeable fluorescent styryl dye to label the PM of guard cells expressing GFP-SYP121 to determine the PM and cytoplasm regions. Subsequently, a 0.5-μm-wide band was created along all the PM of each cell and the average fluorescence intensity was calculated by ImageJ software and determined as PM signals. Another region was created immediately underneath the band, including the whole cytoplasmic area, and the average fluorescence intensity was determined as cytosol signals. The fluorescence ratio of the PM/cytosol was then determined.
To analyse the co-localisation between GFP-SYP121 and different endomembrane compartments, ImageJ plugin Coloc 2 was used to measure Pearson’s coefficients82.
To measure endosome velocity, a time series of images was collected and trajectories and velocities of GFP-SYP121-associated TGN/EE were tracked using ImageJ plugin Manual Tracking.
To quantify the proportion of ProKAT1::KAT1-mNeonGreen at the dorsal periphery/PM relative to the whole cell20,48, Z-stack images (0.5-μm step size) of guard cells were acquired, and the projection image was used to calculate the PM/cytosol ratio of KAT1-mNeonGreen fluorescence intensity. We drew a 0.5-μm-wide band along the PM at the dorsal side of a guard cell and calculated the mean grey values in this band using ImageJ software. The mean grey values of cytoplasm (chloroplast autofluorescence was excluded) was also measured by ImageJ software. The fluorescence ratio of PM versus cytoplasm was counted.
To record K+ concentrations in guard cells, Arabidopsis seedlings expressing lc-LysM GEPII1.0 (combines K+-binding domains of the bacterial K+ binding protein (KBP) with CFP and YFP variants) to enable FRET ratio-based recordings were used, and FRET-based K+ imaging was acquired using a Zeiss 880 microscope, with an excitation wavelength of 440 nm and emission wavelengths of 450–480 nm (CFP) and 520–570 nm (FRET)50. The median section of the guard cell was captured and the average fluorescence intensity of the cytosol (both CFP and FRET images) calculated using Image J software. The fluorescence ratio images for the FRET/CFP were then acquired by ImageJ plugin Calculator Plus.
To visualise the distribution of fluorescence-tagged proteins to the PM, time-lapse images were collected after focusing on the cell surface using VAEM equipped with a 150 × 1.45–NA UAPON TIRF objective (Olympus) and Primer 95B sCOMS camera (Teledyne Photometrics). For GFP-SYP121, time lapse images were acquired with 0.5-s intervals for 1 min. For analysis of the delivery events of GFP-SYP121, the Stack Reslice function were applied to trace the motion trajectory of each particle and generate kymograph images. A successful delivery event was defined as a straight line in the kymograph54,55 that endured at least 2 s. The span of straight line was defined as the lifetime of an individual particle.
To observe the co-localisation of AtEB1b-mCherry and GFP-tubulin, time series imaging was performed with 1-s intervals over a 20-s period using spinning-disc confocal microscopes equipped with Primer 95B sCOMS camera (Teledyne Photometrics). To observe the co-localisation of AtEB1b-GFP and mCherry-SYP121 in the interior of the guard cell, time series imaging was performed over a 55-s period with 5-s intervals from 7-day-old seedlings by using spinning-disc confocal microscope equipped with iXon 897 EMCCD camera (Andor Technology). To observe the co-localisation of AtEB1b-GFP and mCherry-SYP121 on the cell surface/PM of guard cells, time series imaging was performed over a 300-s period with 1-s intervals from 4-day-old seedlings by using VAEM equipped with a 150×1.45–NA UAPON TIRF objective (Olympus) and Primer 95B sCOMS camera (Teledyne Photometrics).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Biological materials can be obtained upon request. Source data are provided with this paper.
References
Schliwa, M. & Woehlke, G. Molecular motors. Nature 422, 759–765 (2003).
Vale, R. D. The molecular motor toolbox for intracellular transport. Cell 112, 467–480 (2003).
Hirokawa, N., Noda, Y., Tanaka, Y. & Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 10, 682–696 (2009).
Buchnik, L., Abu-Abied, M. & Sadot, E. Role of plant myosins in motile organelles: is a direct interaction required? J. Int. Plant Biol. 57, 23–30 (2015).
Perico, C. & Sparkes, I. Plant organelle dynamics: cytoskeletal control and membrane contact sites. N. Phytol. 220, 381–394 (2018).
Foissner, I., Menzel, D. & Wasteneys, G. O. Microtubule-dependent motility and orientation of the cortical endoplasmic reticulum in elongating characean internodal cells. Cell Motil. Cytoskeleton 66, 142–155 (2009).
Crowell, E. F. et al. Pausing of Golgi bodies on microtubules regulates secretion of cellulose synthase complexes in Arabidopsis. Plant Cell 21, 1141–1154 (2009).
Gutierrez, R., Lindeboom, J. J., Paredez, A. R., Emons, A. M. & Ehrhardt, D. W. Arabidopsis cortical microtubules position cellulose synthase delivery to the plasma membrane and interact with cellulose synthase trafficking compartments. Nat. Cell Biol. 11, 797–806 (2009).
Ambrose, C. et al. CLASP interacts with sorting nexin 1 to link microtubules and auxin transport via PIN2 recycling in Arabidopsis thaliana. Dev. Cell 24, 649–659 (2013).
Brandizzi, F. & Wasteneys, G. O. Cytoskeleton-dependent endomembrane organization in plant cells: an emerging role for microtubules. Plant J. 75, 339–349 (2013).
Acharya, B. R. & Assmann, S. M. Hormone interactions in stomatal function. Plant Mol. Biol. 69, 451–462 (2009).
Hubbard, K. E., Siegel, R. S., Valerio, G., Brandt, B. & Schroeder, J. I. Abscisic acid and CO2 signalling via calcium sensitivity priming in guard cells, new CDPK mutant phenotypes and a method for improved resolution of stomatal stimulus-response analyses. Ann. Bot. 109, 5–17 (2012).
Kim, T. H., Bohmer, M., Hu, H., Nishimura, N. & Schroeder, J. I. Guard cell signal transduction network: advances in understanding abscisic acid, CO2, and Ca2+ signaling. Annu. Rev. Plant Biol. 61, 561–591 (2010).
Shimazaki, K., Doi, M., Assmann, S. M. & Kinoshita, T. Light regulation of stomatal movement. Annu. Rev. Plant Biol. 58, 219–247 (2007).
Yang, J., Li, C. L., Kong, D. X., Guo, F. Y. & Wei, H. B. Light-mediated signaling and metabolic changes coordinate stomatal opening and closure. Front. Plant Sci. 11, 601478 (2020).
Kollist, H., Nuhkat, M. & Roelfsema, M. R. Closing gaps: linking elements that control stomatal movement. N. Phytol. 203, 44–62 (2014).
Lawson, T. & Blatt, M. R. Stomatal size, speed, and responsiveness impact on photosynthesis and water use efficiency. Plant Physiol. 164, 1556–1570 (2014).
Hedrich, R. Ion channels in plants. Physiol. Rev. 92, 1777–1811 (2012).
Sutter, J. U., Campanoni, P., Tyrrell, M. & Blatt, M. R. Selective mobility and sensitivity to SNAREs is exhibited by the Arabidopsis KAT1 K+ channel at the plasma membrane. Plant Cell 18, 935–954 (2006).
Sutter, J. U. et al. Abscisic acid triggers the endocytosis of the Arabidopsis KAT1 K+ channel and its recycling to the plasma membrane. Curr. Biol. 17, 1396–1402 (2007).
Lefoulon, C., Waghmare, S., Karnik, R. & Blatt, M. R. Gating control and K+ uptake by the KAT1 K+ channel leaveraged through membrane anchoring of the trafficking protein SYP121. Plant Cell Environ. 41, 2668–2677 (2018).
Kim, M., Hepler, P. K., Eun, S. O., Ha, K. S. & Lee, Y. Actin filaments in mature guard cells are radially distributed and involved in stomatal movement. Plant Physiol. 109, 1077–1084 (1995).
Eun, S. O. & Lee, Y. Actin filaments of guard cells are reorganized in response to light and abscisic acid. Plant Physiol. 115, 1491–1498 (1997).
Couot-Gastelier, L. & Louguet, P. Effet de la colchicine sur les mouvements des stomates et l’ultrastructure des cellules stomatiques deTradescantia virginiana. Bull. Soc. Bot. Fr. Lett. Bot. 139, 345–356 (1992).
Jiang, C. J., Nakajim, N. & Kondo, N. Disruption of microtubules by abscisic acid in guard cells of Vicia faba L. Plant Cell Physiol. 37, 697–701 (1996).
Assmann, S. M. & Baskin, T. I. The function of guard cells does not require an intact array of cortical microtubules. J. Exp. Bot. 49, 163–170 (1998).
Marcus, A. I., Moore, R. C. & Cyr, R. J. The role of microtubules in guard cell function. Plant Physiol. 125, 387–395 (2001).
Moore, R. C. & Cyr, R. J. Association btween elongation factor-1alpha and microtubules in vivo is domain dependent and conditional. Cell Motil. Cytoskeleton 45, 279–292 (2000).
Gardiner, J. & Marc, J. Arabidopsis thaliana, a plant model organism for the neuronal microtubule cytoskeleton? J. Exp. Bot. 62, 89–97 (2011).
Eisinger, W., Ehrhardt, D. & Briggs, W. Microtubules are essential for guard-cell function in Vicia and Arabidopsis. Mol. Plant 5, 601–610 (2012).
Eisinger, W. R., Kirik, V., Lewis, C., Ehrhardt, D. W. & Briggs, W. R. Quantitative changes in microtubule distribution correlate with guard cell function in Arabidopsis. Mol. Plant 5, 716–725 (2012).
Galatis, B. & Apostolakos, P. The role of the cytoskeleton in the morphogenesis and function of stomatal complexes. N. Phytol. 161, 613–639 (2004).
Nehlig, A., Molina, A., Rodrigues‑Ferreira, S., Honoré, S. & Nahmias, C. Regulation of end-binding protein EB1 in the control of microtubule dynamics. Cell Mol. Life Sci. 74, 2381–2393 (2017).
Akhmanova, A. & Steinmetz, M. O. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 9, 309–322 (2008).
Komaki, S. et al. Nuclear-localized subtype of end-binding 1 protein regulates spindle organization in Arabidopsis. J. Cell Sci. 123, 451–459 (2010).
Bisgrove, S. R., Lee, Y. R., Liu, B., Peters, N. T. & Kropf, D. L. The microtubule plus-end binding protein EB1 functions in root responses to touch and gravity signals in Arabidopsis. Plant Cell 20, 396–410 (2008).
Galva, C. et al. The microtubule plus-end tracking proteins SPR1 and EB1b interact to maintain polar cell elongation and directional organ growth in Arabidopsis. Plant Cell 26, 4409–4425 (2014).
Lindeboom, J. J. et al. CLASP stabilization of plus ends created by severing promotes microtubule creation and reorientation. J. Cell Biol. 218, 190–205 (2019).
Zheng, W. N. et al. AP3M harbors actin filament binding activity that is crucial for vacuole morphology and stomatal closure in Arabidopsis. Proc. Natl Acad. Sci. USA 116, 18132–18141 (2019).
Negi, J. et al. A Dof transcription factor, SCAP1, is essential for the development of functional stomata in Arabidopsis. Curr. Biol. 23, 479–484 (2013).
Chan, J., Calder, G. M., Doonan, J. H. & Lloyd, C. W. EB1 reveals mobile microtubule nucleation sites in Arabidopsis. Nat. Cell Biol. 5, 967–971 (2003).
Higaki, T., Kutsuna, N., Sano, T., Kondo, N. & Hasezawa, S. Quantification and cluster analysis of actin cytoskeletal structures in plant cells: role of actin bundling in stomatal movement during diurnal cycles in Arabidopsis guard cells. Plant J. 61, 156–165 (2010).
Fukuda, M., Hasezawa, S., Asai, N., Nakajima, N. & Kondo, N. Dynamic organization of microtubules in guard cells of Vicia faba L. with diunral cycle. Plant Cell Physiol. 39, 80–86 (1998).
Dou, L., He, K., Peng, J., Wang, X. & Mao, T. The E3 ligase MREL57 modulates microtubule stability and stomatal closure in response to ABA. Nat. Commun. 12, 2181 (2021).
Murphy, A. S., Bandyopadhyay, A., Holstein, S. E. & Peer, W. A. Endocytotic cycling of PM proteins. Annu. Rev. Plant Biol. 56, 221–251 (2005).
Peer, W. A. Plasma membrane protein trafficking. In The Plant Plasma Membrane, Plant Cell Monographs (Springer, Berlin). 19, 31–56 (2011).
Philippar, K. et al. Auxin activates KAT1 and KAT2, two K+-channel genes expressed in seedlings of Arabidopsis thaliana. Plant J. 37, 815–827 (2004).
Eisenach, C., Chen, Z. H., Grefen, C. & Blatt, M. R. The trafficking protein SYP121 of Arabidopsis connects programmed stomatal closure and K+ channel activity with vegetative growth. Plant J. 69, 241–251 (2012).
Meckel, T., Hurst, A. C., Thiel, G. & Homann, U. Endocytosis against high turgor: intact guard cells of Vicia faba constitutively endocytose fluorescently labelled plasma membrane and GFP-tagged K-channel KAT1. Plant J. 39, 182–193 (2004).
Wang, F. L. et al. A potassium-sensing niche in Arabidopsis roots orchestrates signaling and adaptation responses to maintain nutrient homeostasis. Dev. Cell 56, 781–794 (2021).
Nielsen, M. E. & Thordal-Christensen, H. Recycling of Arabidopsis plasma membrane PEN1 syntaxin. Plant Signal Behav. 7, 1541–1543 (2012).
Karnik, R. et al. Arabidopsis Sec1/Munc18 protein SEC11 is a competitive and dynamic modulator of SNARE binding and SYP121-dependent vesicle traffic. Plant Cell 25, 1368–1382 (2013).
Wan, Y. L. et al. Variable-angle total internal reflection fluorescence microscopy of intact cells of Arabidopsis thaliana. Plant Methods 7, 27 (2011).
Fendrych, M. et al. Visualization of the exocyst complex dynamics at the plasma membrane of Arabidopsis thaliana. Mol. Biol. Cell 24, 510–520 (2013).
Zhang, W. W., Huang, L., Zhang, C. H. & Staiger, C. J. Arabidopsis myosin XIK interacts with the exocyst complex to facilitate vesicle tethering during exocytosis. Plant Cell 33, 2454–2478 (2021).
Cui, X. K. et al. Arabidopsis SYP121 acts as an ROP2 effector in the regulation of root hair tip growth. Mol. Plant 15, 1008–1023 (2022).
Roelfsema, M. R., Steinmeyer, R., Staal, M. & Hedrich, R. Single guard cell recordings in intact plants: light-induced hyperpolarization of the plasma membrane. Plant J. 26, 1–13 (2001).
Szyroki, A. et al. KAT1 is not essential for stomatal opening. Proc. Natl Acad. Sci. USA 98, 2917–2921 (2001).
Laloux, T., Matyjaszczyk, I., Beaudelot, S., Hachez, C. & Chaumont, F. Interaction between the SNARE SYP121 and the plasma membrane aquaporin PIP2;7 involves different protein domains. Front. Plant Sci. 11, 631643 (2021).
Hachez et al. Arabidopsis SNAREs SYP61 and SYP121 coordinate the trafficking of plasma membrane aquaporin PIP2;7 to modulate the cell membrane water permeability. Plant Cell 26, 3132–3147 (2014).
Cheerambathur, D. K. & Desai, A. Linked in: formation and regulation of microtubule attachments during chromosome segregation. Curr. Opin. Cell Biol. 26, 113–122 (2014).
Liu, Z. Y., Persson, S. & Zhang, Y. The connection of cytoskeletal network with plasma membrane and the cell wall. J. Integr. Plant Biol. 57, 330–340 (2015).
Zheng, J. M., Han, S. W., Rodriguez-Welsh, M. F. & Rojas-Pierce, M. Homotypic vacuole fusion requires VTI11 and is regulated by phosphoinositides. Mol. Plant 7, 1026–1040 (2014).
Galjart, N. Plus-end-tracking proteins and their interactions at microtubule ends. Curr. Biol. 20, R528–R537 (2010).
Molines, A. T. et al. EB1 contributes to microtubule bundling and organization, along with root growth, in Arabidopsis thaliana. Biol. Open 7, bio030510 (2018).
Silva, P. A., Ul-Rehman, R., Rato, C., Sansebastiano, G. P. D. & Malho, R. Asymmetric localization of Arabidopsis SYP124 syntaxin at the pollen tube apical and sub-apical zones is involved in tip growth. BMC Plant Biol. 10, 179 (2010).
Ichikawa et al. Syntaxin of plant proteins SYP123 and SYP132 mediate root hair tip growth in Arabidopsis thaliana. Plant Cell Physiol. 55, 790–800 (2014).
Ichikawa, M., Iwano, M. & Sato, M. H. Nuclear membrane localization during pollen development and apex-focused polarity establishment of SYP124/125 during pollen germination in Arabidopsis thaliana. Plant Reprod. 28, 143–151 (2015).
Alberico, E. O. et al. Interactions between the microtubule binding protein EB1 and F-Actin. J. Mol. Biol. 428, 1304–1314 (2016).
Henty-Ridilla, J. L., Rankova, A., Eskin, J. A., Kenny, K. & Goode, B. L. Accelerated actin filament polymerization from microtubule plus ends. Science 352, 1004–1009 (2016).
Dixit, R., Chang, E. & Cyr, R. Establishment of polarity during organization of the acentrosomal plant cortical microtubule array. Mol. Biol. Cell 17, 1298–1305 (2006).
Novak, D. et al. Analysis of formin functions during cytokinesis using specific inhibitor SMIFH2. Plant Physiol. 186, 945–963 (2021).
Zhang, L. N. et al. Analysis of formin functions during cytokinesis using specific inhibitor SMIFH2. Plant Physiol. 186, 945–963 (2021).
Ho, C. M. et al. Interaction of antiparallel microtubules in the phragmoplast is mediated by the microtubule-associated protein MAP65-3 in Arabidopsis. Plant Cell 23, 2909–2923 (2011).
Rogers, S. L., Wiedemann, U., Hacker, U., Turck, C. & Vale, R. D. Drosophila RhoGEF2 associates with microtubule plus ends in an EB1-dependent manner. Curr. Biol. 14, 1827–1833 (2004).
Barth, A. I., Caro-Gonzalez, H. Y. & Nelson, W. J. Role of adenomatous polyposis coli (APC) and microtubules in directional cell migration and neuronal polarization. Semin. Cell Dev. Biol. 19, 245–251 (2008).
Gu, C. et al. The microtubule plus-end tracking protein EB1 is required for Kv1 voltage-gated K+ channel axonal targeting. Neuron 52, 803–816 (2006).
Waadt, R. et al. Multicolor bimolecular fluorescence complementation reveals simultaneous formation of alternative CBL/CIPK complexes in planta. Plant J. 56, 505–516 (2008).
Chang, M. & Huang, S. J. Arabidopsis ACT11 modifies actin turnover to promote pollen germination and maintain the normal rate of tube growth. Plant J. 83, 515–527 (2015).
Xu, T. D. et al. Cell surface ABP1-TMK auxin-sensing complex activates ROPGTPase signaling. Science 343, 1025–1028 (2014).
Baster, P. et al. SCF(TIR1/AFB)-auxin signalling regulates PIN vacuolar trafficking and auxin fluxes during root gravitropism. EMBO J. 32, 260–274 (2013).
Liu, L. et al. Actomyosin and CSI1/POM2 cooperate to deliver cellulose synthase from Golgi to cortical microtubules in Arabidopsis. Nat. Commun. 14, 7442 (2023).
Acknowledgements
We thank Dr. Zhenbiao Yang (University of California, Riverside) and Dr. Takashi Hashimoto (Nara Institute of Science and Technology) for critical reading and comments on the article. We thank Dr. Liwen Jiang (The Chinese University of Hong Kong) for advice on the observation of exocytosis events. We thank Dr. Yi Wang (China Agricultural University) for providing Arabidopsis seeds that expressing lc-LysM GEPII1.0. We thank Dr. Tonglin Mao (China Agricultural University) for providing pCAMBIA-35S-AtEB1a-nLuc vector. We thank Dr. Xiaohan Wang, Dr. Legong Li (Capital Normal University) for the help and advice on gas exchange measurement. We thank Dr. Jinglong Qiu (Institute of Microbiology, Chinese Academy of Sciences) and Dr. Dingzhong Tang (Fujian Agriculture and Forestry University) for providing Arabidopsis seeds that expressing 35S::GFP-SYP121. We thank Dr. Bo Liu (University of California, Davis) and Dr. Shanjin Huang (Tsinghua University) for providing Arabidopsis seeds that expressing 35S::AtEB1b-GFP. We thank Dr. Jinxing Lin and Dr. Xi Zhang (Beijing Forestry University) for helpful advice on VAEM image analysis. This research was supported by the National Key R&D Program of China (2022YFE0108200) and the Natural Science Foundation of China (32230030, 32061143018) to Y.F.
Author information
Authors and Affiliations
Contributions
All authors designed experiments. H.Z., S.W., Y.H., X.C. performed the experiments, H.Z., S.W. analysed the results. X.D. and L.Z. helped to interpret and discuss the results. M.Y. and Y.F. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Takumi Higaki and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhong, H., Wang, S., Huang, Y. et al. Endomembrane trafficking driven by microtubule growth regulates stomatal movement in Arabidopsis. Nat Commun 15, 7967 (2024). https://doi.org/10.1038/s41467-024-52338-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52338-x
