
Abstract
The development of hydrogen technologies is at the heart of a green economy. As prerequisite for implementation of hydrogen storage, active and stable catalysts for (de)hydrogenation reactions are needed. So far, the use of precious metals associated with expensive costs dominates in this area. Herein, we present a new class of lower-cost Co-based catalysts (Co-SAs/NPs@NC) in which highly distributed single-metal sites are synergistically combined with small defined nanoparticles allowing efficient formic acid dehydrogenation. The optimal material with atomically dispersed CoN2C2 units and encapsulated 7-8 nm nanoparticles achieves an excellent gas yield of 1403.8 mL·g−1·h−1 using propylene carbonate as solvent, with no activity loss after 5 cycles, which is 15 times higher than that of the commercial Pd/C. In situ analytic experiments show that Co-SAs/NPs@NC enhances the adsorption and activation of the key intermediate monodentate HCOO*, thereby facilitating the following C-H bond breaking, compared to related single metal atom and nanoparticle catalysts. Theoretical calculations show that the integration of cobalt nanoparticles elevates the d-band center of the Co single atoms as the active center, which consequently enhances the coupling of the carbonyl O of the HCOO* intermediate to the Co centers, thereby lowering the energy barrier.
Similar content being viewed by others

Introduction
Hydrogen is recognized as an important energy carrier for the ongoing global energy transition and can be a major driver reaching carbon neutrality1. Due to its physical properties, e.g., flammability and low density, safe and efficient storage as well as transportation of hydrogen are crucial issues for the realization of a hydrogen economy2,3,4. As a solution, liquid organic hydrogen carriers (LOHCs), which store and release hydrogen through chemical reactions, have been proposed. Compared to molecular hydrogen, such materials (methanol, toluene, dibenzyltoluene, etc) can be easily and conveniently handled5,6,7. Among the different conventional LOHCs, formic acid (FA) has comparably lower toxicity (LD50: 1.8 g/kg) and H2-capacity of 53 g/L or 4.4 wt %. Notably, FA is the only LOHC that allows both the storage and release of hydrogen under mild conditions in the presence of suitable catalysts, so that no significant external energy input is required1,8,9. Indeed, many precious metal catalysts have been developed for formic acid dehydrogenation, with palladium-based catalysts, for example, exhibiting superior activity 50–200 times higher than low-cost metal catalysts10,11,12. However, if the cost of the active metal is taken into account, e.g., palladium is >1000 times more expensive than
cobalt, the search for highly active and stable non-noble metal heterogeneous catalysts continues to attract the interest of many researchers in academia and industry13,14,15.
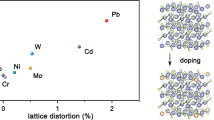
Although low-cost catalysts based on Mo and Co as well as noble/non-noble metal alloy nano-catalysts have been developed for FA dehydrogenation14,16, their gradual deactivation during the reaction is inevitable due to the occupation of metal active sites by protons, CO2, H2O, or formate anions (HCOO−), FA fouling, particle size aggregation, and possible CO poisoning17,18. More recently, it has been shown by us and others that single-atom catalysts (SACs) with highly dispersed CoIINx centers as active sites, have improved reactivity and acid resistance in formic acid dehydrogenation compared to nanoparticles17,19,20,21,22,23,24. In these Co–N–C materials, N atoms act as basic sites to promote FA deprotonation while enhancing structural stability by coordination to the central Co atom, whereas Co atoms provide H adsorption sites along with driving C–H rupture22,25,26. Unfortunately, the activity and stability of these catalysts are still far from present state-of-the-art homogeneous and heterogeneous noble metal catalysts (Fig. 1)13.

Surplus energy from renewable sources (such as solar or wind) can be produced by electrolyzing water. The hydrogen produced can be stored by LOHCs, a liquid in which hydrogenation and dehydrogenation can be reversible. In the dehydrogenation stage, hydrogen is the only product and the carrier liquid returns to its original state to be hydrogenated again. The hydrogen can be finally used in hydrogen refueling stations, batteries, and industrial constructions.
Recently, it has been reported that the intrinsic activity of defined SACs can be improved in the presence of other metal sites provided by different metal atoms or nanoparticles (NP) or nanoclusters (NC)27,28. This offers opportunities for further substrate adsorption and activation as well as regulation of the geometry and electronic structure of single-atom sites. Consequently, substrate adsorption/activation can be optimized, which provides better overall catalytic performance29,30. This gave us the idea of producing corresponding catalyst materials with hybrid active centers. While modified SACs have shown considerable potential in a wide range of catalytic applications31,32,33,34,35,36, to the best of our knowledge their role in the field hydrogen storage is unknown. In this respect, we report a general and reliable strategy to synthesize Co-based hybrid catalysts (Co-SAs/NPs@NC) consisting of defined nanoparticles and single-metal sites. The optimized Co-SAs/NPs@NC exhibits excellent formic acid dehydrogenation performance, superior to non-noble-based nano-structured catalysts (such as CoNx, cobalt single atom, cobalt@NC, and γ-Mo2N) and even noble metal catalysts. In situ characterization and DFT calculations of the active catalyst indicate that single-metal sites serve as active centers, and the present nanoparticles elevate the d-band center of Co atoms to promote HCOO* adsorption and activation, thereby reducing the reaction energy barrier.
Results and discussion
Synthesis and characterizations of Co-SAs/NPs@NC-T
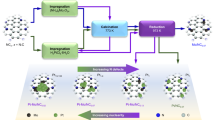
Zeolitic imidazolate frameworks (ZIFs) constitute well-defined three-dimensional precursors to provide catalysts with different kinds of metal species supported on nitrogen-doped carbonaceous materials (metal-NC catalysts)37,38. Thus, Co(NO3)2 and Zn(NO3)2 are combined with 2-methylimidazole in methanol to form the corresponding metal complexes in solution. After centrifugation and drying, the CoZn–ZIFs are pyrolyzed at different temperatures (750–950 °C) in an atmosphere of 6% H2 and 94% Ar. As shown below, the materials obtained are characterized by different active centers and are referred to as Co-SAs/NPs@NC-950, Co-SAs/NPs@NC-850, and Co-SAs/NPs@NC-750 (Fig. 2a). Specific experimental observations at some key steps in the synthesis process are detailed in Figs. S1–S3. Variable temperature powder X-ray diffraction (VTXRD) was performed to monitor the catalyst evolution. After the pyrolysis temperature reached 650 °C, the XRD patterns changed significantly due to the collapse of the ordered crystal structure of ZIFs (Fig. S4)39. As the temperature increased further, XRD patterns of Co-SAs/NPs@NC-850 and Co-SAs/NPs@NC-750 revealed two broad peaks at 20–30° and 40–50°, representing the amorphous carbon peaks (Fig. S5)40. Notably, three characteristic peaks at 44.2°, 51.5°, and 75.8°, belonging to metallic cobalt (JCPDS #15-0806), and the peak at 26.2° belonging to graphitic carbon (JCPDS #41-1487) were observed only in the XRD spectra of the Co-SAs/NPs@NC-950, suggesting that graphite-like encapsulated cobalt nanoparticles appeared on the catalyst41,42,43,44. Raman spectra shows that Co-SAs/NPs@NC-950 appeared to have stronger and narrower D and G peaks than the other samples, indicating a higher degree of graphitization (Fig. S6). Additionally, Co-SAs/NPs@NC-950 exhibits considerably higher Brunner-Emmett-Taylor (BET) surface area and pore volume (1261 m2·g−1 and 0.37 cm3·g−1) than the other samples and most of ZIF-derived NC materials (Fig. S7 and Table S1). Atomic absorption spectroscopy (AAS) shows cobalt contents of 2.69 wt%, 2.74 wt%, and 2.73 wt% in Co-SAs/NPs@NC-950, Co-SAs/NPs@NC-850, and Co-SAs/NPs@NC-750, respectively (Table S2). The zinc content of Co-SAs/NPs@NC-950, Co-SAs/NPs@NC-850, and Co-SAs/NPs@NC-750 increased progressively, which was attributed to the increased reduction and volatilization of Zn nodes with increasing pyrolysis temperatures (Zn, boiling point = 907 °C)45,46. Elemental analysis (EA) shows that as the pyrolysis temperature increases, the percentage of N decreases, the high content of O may be related to the adsorption of molecular O2 due to exposure to air. (Table S3). At certain cobalt contents, nanoparticles and isolated co-atoms coexist, which leads to a considerable increase in catalyst activity, as explained below.

a Schematic synthesis of Co-SAs/NPs@NC-T, where T represents the pyrolysis temperature (°C). b TEM images. c AC-HAADF-STEM image of Co-SAs/NPs@NC-950, the Co single atoms are marked with red circles. d EDS mapping of Co-SAs/NPs@NC-950.
Notably, transmission electron microscopy (TEM) proves the presence of distinct cobalt nanoparticles (NPs) with an average size of 7.5 ± 1.7 nm only in Co-SAs/NPs@NC-950 (Figs. 2b and S8). These nanoparticles are encapsulated by N-doped graphite-like carbon. The lattice stripe spacing of 0.361 and 0.201 nm is attributed to graphitic carbon (002) and metallic Co particles (111), respectively. Further, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) reveals Co NPs are surrounded by rich atomic cobalt in Co-SAs/NPs@NC-950 (Fig. 2c). However, only atomically dispersed cobalt atoms are observed on the support in the other two samples (Fig. S9). HAADF-STEM image with energy dispersive spectroscopy (EDS) shows a homogeneous distribution of C, N, Co, and segregated Co NPs in Co-SAs/NPs@NC-950 (Fig. 2d). All these results indicate that atomically dispersed Co sites and N-doped graphite-like carbon-encapsulated nanoparticles are successfully anchored on NC carriers in Co-SAs/NPs@NC-950, whereas only single-metal sites are present in the other samples.
Electronic and atomic structure analysis
The valence states and chemical compositions of the prepared materials are investigated using X-ray photoelectron spectroscopy (XPS). XPS survey spectra of the three catalysts shows the presence of the elements Co, N, C, and O, but Zn is present only in Co-SAs/NPs@NC-850 and Co-SAs/NPs@NC-750 (Fig. S10). As the pyrolysis temperature increases, the total N content decreases because N species are unstable and decompose into NH3 and NOx gases at higher temperatures (Table S4)47. Accordingly, the total C content gradually increases from Co-SAs/NPs@NC-750 to Co-SAs/NPs@NC-850 and Co-SAs/NPs@NC-950 (Figs. S11 and S12). The lower proportion of nitrogen atoms in the samples pyrolyzed at higher temperatures means that the number of NC carriers in Co-SAs/NPs@NC-950 should be lower than in other samples. This leads to a stronger sintering of the cobalt particles. The O 1s spectrum shows two peaks for C = O (531.6 eV) and C–O (533.5 eV), respectively (Fig. S13)48. As shown in Fig. 2a, the N 1s spectra can be deconvoluted into four characteristic nitrogen peaks for pyridinic N (398.4 eV), pyrrolic N (401.1 eV), graphitic N (402.3 eV), and Co–N (399.2 eV). The presence of Co–N bonds in all three samples indicates that a part of the N atoms is coordinated to single-metal sites, but the characteristics are quite different49. Applying a higher pyrolysis temperature significantly reduces the content of Co–N species from 43.7% in Co-SAs/NPs@NC-750 to 27.0% in Co-SAs/NPs@NC-850 and to 17.6% in Co-SAs/NPs@NC-950, which corresponds with an increase in the C content (Fig. 3a), suggesting a possible change in their Co–N coordination number and their partial replacement by C atoms50. The Zn 2p spectra revealed that this element is mainly present in the form of Zn2+. (Fig. S14)51. The Co 2p spectrum shows two prominent peaks at 780.8 and 796.1 eV, which are attributed to Co 2p3/2 and Co 2p1/2, respectively (Fig. 3b). The Co–N peaks are positively shifted in Co-SAs/NPs@NC-950 compared to Co-SAs/NPs@NC-850 and Co-SAs/NPs@NC-750, which indicates that the Co single atoms on the surface of Co-SAs/NPs@NC-950 are more electron depleted leading to higher oxidation state. It is noteworthy that only Co-SAs/NPs@NC-950 has a weak zero-valent cobalt (Co0) peak detected at 778.5 eV, evidencing the presence of nanoparticles originating from the aggregation of cobalt SAs at higher temperature52.

a N 1s and b Co 2p spectra of Co-SAs/NPs@NC-T. c XANES and d FT-EXAFS spectra at the Co K-edge of Co-SAs/NPs@NC-950, Co-SAs/NPs@NC-850 and Co-SAs/NPs@NC-750. e WT-EXAFS contour plots of Co-SAs/NPs@NC-950, Co-SAs/NPs@NC-850 and Co-SAs/NPs@NC-750. f the fitting curve of FT-EXAFS for Co-SAs/NPs@NC-950.
Next, synchronized X-ray absorption spectroscopy (XAS) is employed to analyze the electronic structure and coordination environment of Co species in the prepared samples. The cobalt valence states in Co-SAs/NPs@NC-950, Co-SAs/NPs@NC-850, and Co-SAs/NPs@NC-750 are detected by normalized Co-K-edge X-ray absorption near-edge structure (XANES) spectroscopy. As shown in Fig. 3c, the near-edge absorption of the three samples lies in between those of Co foil and CoO, demonstrating valence states of Co species between 0 and +253. In addition, a shift towards lower energies is observed from Co-SAs/NPs@NC-950 to Co-SAs/NPs@NC-850 and to Co-SAs/NPs@NC-750, indicating a lower oxidation state of Co in the opposite order. According to the linear combination fitting results, the Co valence state of Co-SAs/NPs@NC-950 is estimated to be +0.642, which is lower than that of Co-SAs/NPs@NC-850 (+1.376), and Co-SAs/NPs@NC-750 (+1.402). These results imply that the average oxidation state of the cobalt species in Co-SAs/NPs@NC-950 is significantly reduced, which in agreement with the results of XRD and HAADF-STEM and can be attributed to the coexistence of cobalt nanoparticles and single Co atoms41. The Co K-edge Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectroscopy shows a main peak at 1.32 Å attributed to the Co–N/Co–C shell, while the scattering path of metal Co–Co at 2.18 Å is only detected in Co-SAs/NPs@NC-950 (Fig. 3d). In addition, wavelet transform (WT) contour plots exhibit a maximum intensity at 6.7 Å−1 attributed to Co–N/Co–C, while only Co-SAs/NPs@NC-950 shows another maximum intensity at 8.8 Å−1 attributed to Co–Co bonds (Fig. 3e). In addition, the lease-squares EXAFS fitting analysis determined Co–N coordination numbers of 3.8, 3.2, and 2.3 and Co–C coordination numbers of 0, 0.9, and 1.8 for pyrolysis temperatures of 750, 850, and 950 °C, respectively (Figs. 3f, S15 and Table S1). More specifically, these latter results can be explained by the presence of both atomically dispersed CoN2C2 units and nanoparticles solely in Co-SAs/NPs@NC-950. In contrast, in Co-SAs/NPs@NC-850 and Co-SAs/NPs@NC-750 only CoN3C and CoN4 units are present. Apparently, with increasing pyrolysis temperature N atoms in CoN4 units are gradually replaced by C atoms, and cobalt SAs aggregate to form nanoparticles.
Formic acid dehydrogenation tests and active site exploration
To investigate the effect of preparation conditions on the performance of the different materials, reaction conditions from previous studies are used (Fig. S16)17,49. As shown in Fig. 4a, Co-SAs/NPs@NC-950 exhibits significantly higher activity than Co-SAs/NPs@NC-850 and Co-SAs/NPs@NC-750. Notably, all three prepared Co samples show superior behavior compared to standard commercial noble metal catalysts (Pd/C and Pt/C). Moreover, the Zn-ZIF-8 and Zn–N–C samples are no active for formic acid dehydrogenation, suggesting that Zn species are not active centers, but their effect on the activity can be ignored. In addition, Co-SAs/NPs@NC-850 and Co-SAs/NPs@NC-750 are pyrolyzed a second time at 950 °C for 1 h, which show an increase in activity but lower than Co-SAs/NPs@NC-950 (Fig. S17). Structural characterization of these materials shows the presence of Co nanoparticles in the re-pyrolyzed samples, but the low specific surface and the absence of graphite-like carbon result in lower activity compared to Co-SAs/NPs@NC-950 (Figs. S18–S20). The activities of the samples with different amounts of Co precursors are compared, too, and the highest activity is exhibited at an addition of 3.5 mol (Table S6 and Fig. S21). Obviously, the formation of different metal sites is influenced by the hydrogen content in the pyrolysis atmosphere and the pyrolysis time. Thus, the formic acid dehydrogenation activity of additional Co-SAs/NPs@NC-950 materials is evaluated. All the materials show medium to very good performance; however, none is better than Co-SAs/NPs@NC-950 (Figs. S22 and S23). The structural characterization of the materials shows that with the prolongation of pyrolysis time, the content of Co–N single-atomic sites gradually decreases due to the aggregation of metal atoms into nanoparticles, which explains the difference in activity between the samples with pyrolysis time of 0.5 h, 1 h and 2 h (Figs. S24–S28 and Table S7).

a Plot of volume of produced gas versus time for the dehydrogenation of FA applying different catalysts. Reaction conditions: FA (10 mmol, 377 μL), catalyst (30 mg), PC (6 mL), Tset: 110 °C, Tactual: 98 °C, 4 h. b Co-SAs/NPs@NC-950 (30 mg), different solvents. c Comparison of gas production rates of heterogeneous catalysts in organic solvents at 85−110 °C. d Recycling experiments with Co-SAs/NPs@NC-950. Reaction conditions: FA (10 mmol, 377 μL), Co-SAs/NPs@NC-950 (30 mg), solvent (6 mL), Tset: 110 °C, Tactual: 98 °C, each recycling reaction lasts one hour. Error bars are standard deviations, calculated from three activity testing.
In general, the performance of FA dehydrogenation catalysts is known to be highly dependent on the reaction conditions, especially the solvent used8,49. Utilizing water as a solvent, Co-SAs/NPs@NC-950 exhibits the highest initial reaction rate, but deactivation takes place, possibly due to the occupation of the active sites by protons or H2O18. Testing the catalysts in organic solvents such as 1,4-dioxane (DXA), n-butyl acetate (BAC), toluene (PhMe), triglyme, and cyclohexanone (CYC) show no improvement, too, and the best activity is observed in propylene carbonate (PC) (Fig. 4b and Table S8). Similarly, additives like triethylamine (NEt3) or sodium formate (HCOONa) have no further positive effect on the catalyst performance (Fig. S29). Under optimal reaction conditions, a gas production rate up to 1403.8 mL·g−1·h−1 is achieved (Fig. S30), which is significantly higher compared to all previously reported Co catalysts including SACs17,23,24. A dehydrogenation vs. dehydration selectivity of up to 99.96% is obtained in the respective experiments except for the reactions in water and with formate additive (Table S9). The activation energy is calculated to be 88.4 kJ/mol, which is comparable to noble metal catalysts (Fig. S31 and Table S10).
In addition, we compare a series of other heterogeneous catalysts for formic acid dehydrogenation under similar conditions (Fig. 4c, Table S11, and S12). As shown in Fig. 3c, the gas generation rate of Co-SAs/NPs@NC-950 surpasses most of the reported non-noble metal heterogeneous catalysts and is 15 and 10 times higher than that of commercial 5% Pd/C and 5% Pt/C catalysts.
An important feature for any practical application of (de)hydrogenation catalysts is their stability. Hence, a series of recycling experiments are done applying Co-SAs/NPs@NC-950. As shown in Fig. 4d, the initial activity and selectivity of the material are maintained for five consecutive runs (see also Table S13). Performing a long-term test, the gas production shows a linear increase over 72 h (Fig. S32). The cobalt content of used Co-SAs/NPs@NC-950 is 2.5 wt%, which is very close to that of the fresh catalyst, indicating no significant cobalt leaching (Table S14). No apparent color change or aggregation of the metal species is observed before or after the reaction (Fig. S33). AC-HAADF-STEM and EDS of the material applied in the long-term experiments indicate retention and homogeneous dispersion of atomically dispersed sites and no significant structural changes (Figs. S34 and S35). The characteristic peaks of Co0 and Co–N are still present in XPS, demonstrating the coexistence of Co NPs and single-metal sites, which also confirms the stability of the Co-SAs/NPs@NC-950 catalysts (Fig. S36).
To identify the most active sites responsible for formic acid dehydrogenation, separate materials with only single-metal sites (CoN2C2) or Co NPs are prepared according to previous studies17. The observed formic acid dehydrogenation activities under the same conditions are in the order of Co-SAs/NPs@NC-950 > Co SAs > Co NPs (Table S15), showing that the atomically dispersed CoN2C2 sites are more active than NPs. The kinetics of the reaction demonstrates that hydrogen release followed first-order reaction kinetics, but the slopes of several curves are not the same at different cobalt content, suggesting that the kinetics depend not only on formic acid but also on the active center (Fig. S37). Further kinetic studies show that the kinetic reaction order to cobalt content appears at 1.02 at low levels (less than 2.5%), considering the absence of cobalt metal peaks in XRD, which suggests that the almost uniformly distributed cobalt single-atom sites are the main active sites (Figs. S38 and S39). When the Co species content reaches 2.7%, a sudden increase in r occurs, indicating that the nanoparticles synergize well with the single atoms for higher activity. As the Co species content continues to increases, the curve becomes nonlinear, related to the increase of nanoparticles and the decrease of single-atomic sites. Therefore, the improved FA dehydrogenation performance of Co-SAs/NPs@NC-950 is a result of the synergistic behavior of single-metal sites and nanoparticles.
Study of HCOOH dehydrogenation mechanism
In situ diffuse reflectance infrared Fourier transform (in situ DRIFT) is carried out for an in-depth investigation to identify reaction intermediates in this process. After heating the sample to different reaction temperatures, two sets of frequencies are observed once formic acid was added (Fig. 5a). Three characteristic peaks of HCOOH* appear at 1089, 1217, and 1790 cm−1, which are attributed to the out-of-plane CH deformations π(CH), the C–O stretching vibration ν(C–O), and the C = O stretching vibration ν(C = O), respectively54,55. Another set of peaks at 1363 and 1592 cm−1 corresponds to the O–C–O symmetric vibrations νs(OCO) and O–C–O asymmetric stretching vibrations νas(OCO) of HCOO*, respectively33,56. As the reaction proceeds, the relative peaks of HCOOH* species and HCOO* species gradually attenuates. In general, formic acid decomposition comprises of three elementary steps: (I) formic acid adsorption at the active site, (II) H removal via formate or carboxylate pathway, and (III) combination of the two adsorbed H to generate hydrogen. HCOO* and COOH* have been recognized as key intermediates to determine the formate or carboxylate pathways, respectively57. Utilizing our catalytic system, only the characteristic peak of HCOO* appears, suggesting that formic acid decomposition follows exclusively the formate pathway58. Similar observations are made at lower temperatures of 78 °C and 88 °C (Fig. S40).

In situ DRIFT spectra of HCOOH dehydrogenation over a Co-SAs/NPs@NC-950 and b Co SAs. Legend represents in situ reaction time. c Time course of gas volumes produced using different isotopically labeled reagents. d Kinetic isotope effect data.
Analogous in situ DRIFT experiments are performed for the related Co NPs and Co SAs materials to explore the synergistic effects in the Co-SAs/NPs@NC-950 (Fig. 5b and S41). Both materials show a similar trend, but the characteristic peaks of HCOOH* and HCOO* slightly shift, indicating that the introduction of Co NPs changes the electronic structure of the single-atom centers. The characteristic νas(OCO) peak appears in Co-SAs/NPs@NC-950 and Co SAs but not in Co NPs, which further demonstrates that the intermediate formed by formic acid addition is a monodentate formate perpendicular to the surface plane and adsorbed on SAs as active sites59. Notably, significant vibrational enhancement of the π(CH) and ν(C = O) characteristic peaks is observed, which apparently leads to the distortion of HCOOH* and allows for easier reaction. As a result, the characteristic peaks of HCOOH* and HCOO* in Co-SAs/NPs@NC almost disappear after 2 min of reaction, which is much faster compared to the single-metal (6 min) and nanoparticle-based catalysts (12 min). All these results confirm that nanoparticle doping enhances the adsorption and activation of the intermediates and thus accelerates the reaction as proposed vide supra.
To further analyze the reaction pathway and identify the rate-determining step (RDS), KIE effects in the presence of Co-SAs/NPs@NC-950 are performed. Here, different formic acid isotopes such as HCOOH, HCOOD, DCOOH, and DCOOD, are used for the KIE studies. As shown in Fig. 5c, the rate of dehydrogenation decreases in the following order HCOOH > HCOOD > DCOOH > DCOOD. Further, the values of KHCOOH/KHCOOD, KHCOOH/KDCOOH, KHCOOD/KDCOOD, and KDCOOH/KDCOOD are computed as 1.14, 1.71, 2.16, and 1.44, respectively (Fig. 5d). Therewith, the C–H bond cleavage of HCOO* exhibits kH/kD values > 1.5 indicating a primary kinetic effect60,61, and seems to be the RDS in the dehydrogenation of HCOOH on Co-SAs/NPs@NC-950.
Furthermore, DFT calculations are performed to understand the effect of doped NPs to the intrinsic activity of Co-SAs. Models of Co-SAs/NPs@NC and Co-SAs are constructed (Figs. 6a and S42) based on the shown experiments and previous work52,62. After geometry optimization, small Co6 nanoparticles coexisting with single-atom units (CoN2C2) are identified, and the bond lengths of Co–C and Co–N in Co-SAs/NPs@NC are determined to be 1.87 Å and 1.90 Å, respectively, which is consistent with the XAFS results. The calculated partial density of states (PDOS) shows that the single Co metal atoms and nanoparticle composite (Co-SAs/NPs@NC) display higher hybridization near the Fermi level as compared to CoN2C2, leading to a more efficient electron transfer for HCOOH decomposition (Figs. 6b and S43). The corresponding d-band centers of Co-SAs/NPs@NC and Co-SAs are calculated to be −0.67 eV and −0.80 eV, respectively, with an elevation of 0.13 eV for Co-SAs/NPs@NC, which promotes the adsorption of HCOO* species by the tailored electronic structure of CoN2C2 after the introduction of NPs. Charge density difference shows a large electron cloud around both CoN2C2 units and nanoparticles, indicating strong interactions in between them due to electron exchange. Combined with Bader charge analysis, it is found that atomically dispersed Co loses 1.064e in Co-SAs/NPs@NC and 0.796e in Co SAs (Fig. S44). These results indicate that the integration of nanoparticles led to electron depletion at the Co site, resulting in an increase in the Co valence state, which is in agreement with the XPS results (Fig. 6c). The Co–O coupling properties for HCOO adsorption on Co-SAs/NPs@NC and Co SAs are analyzed by calculating the crystal orbital Hamiltonian group (COHP)63. As shown in Fig. 6d, the negative and positive values of –COHP correspond to the anti-bonding and bonding states, respectively. The Co–O (Co-carbonyl O of HCOO*) bonding strength for HCOO adsorption is evaluated by the integral of the –COHP value, which is 3.51 and 3.38 for Co-SAs/NPs@NC and Co-SAs, respectively. Similar results are shown for the adsorption of HCOOH, where the increase in –COHP integral value after nanoparticle doping indicates enhanced Co–O bonding, and thus promotes the activation of HCOO and HCOOH (Fig. S45).

a Lattice structure of Co-SAs/NPs@NC-950. b PDOS of Co-SAs/NPs@NC-950 and Co SAs. c 3D isosurfaces of charge density differences for HCOOH adsorption on Co-SAs/NPs@NC-950 and Co-SAs. d –pCOHP of the Co−O bond for HCOO adsorption on Co-SAs/NPs@NC-950 (left) and Co-SAs (right). e Reaction pathways of HCOOH dehydrogenation on Co-SAs/NPs@NC-950 and Co-SAs.
To further understand the excellent dehydrogenation performance of Co-SAs/NPs@NC, reaction pathways and energies are established. In detail, FA dehydrogenation involves five steps, including from HCOOH to HCOOH*, HCOOH* to HCOO* + H*, HCOO* + H* to 2H* + CO2*, 2H* + CO2* to 2H* + CO2, and 2H* to H2 (Fig. 6e). Formic acid molecular adsorption energy on the catalyst surface via carboxyl oxygen is lower than via hydroxyl oxygen (Figs. S46 and S47). Subsequently, the adsorbate preferentially undergoes O–H bond scission to form HCOO*, rather than C–H bond scission to form COOH* due to lower energy. Meanwhile, HCOO* adopts monodentate adsorption to promote bond breakage to produce CO2 and H2. These findings are consistent with the presence of νas(OCO) peaks in the in situ DRIFT, again indicating that in our studies FA decomposition follows the formate pathway. Importantly, consistent with KIE measurements, C–H dissociation has a much higher reaction energy barrier than the other reaction steps and constitutes the RDS. The optimal catalyst system Co-SAs/NPs@NC shows a lower energy barrier of 0.86 eV than Co-SAs (1.2 eV), leading to a significant improvement in the overall dehydrogenation performance. Notably, the presence of nanoparticles tailor the electronic structure of atomically dispersed co-active sites, which further enhances the adsorption and activation of the intermediates, thereby lowering the reaction barriers and facilitating hydrogen generation.
In summary, we demonstrate for the first time that the catalytic performance of hydrogen-generating catalysts can be significantly improved by using materials with both highly distributed single-metal sites as well as small defined nanoparticles. This concept has been confirmed by synthesizing Co-based nanoparticle-modified single-metal atom catalysts (Co-SAs/NPs@NC) as well as related materials with only single-metal sites (CoN2C2) or Co NPs. All materials are prepared by simple one-step pyrolysis methods. Structural analysis shows that the optimal catalyst (Co-SAs/NPs@NC-950) consists of atomically dispersed CoN2C2 units and N-doped graphite-like carbon-encapsulated small nanoparticles (7–8 nm). It exhibits an outstanding gas production rate up to 1403.8 mL·g−1·h−1 (H2: CO2 = 1.01:1) with a H2 vs CO selectivity of 99.96 % and maintains a constant activity for several days. The presented catalyst surpasses the activity of defined Co SAs and Pd/C catalysts by 4 and 15 times, respectively. In situ, DRIFT experiments show that Co-SAs/NPs@NC-950 exhibits stronger monodentate adsorption of HCOO* compared to Co-SAs, which is important in the formate pathway, and doped nanoparticles can promote HCOO* activation and accelerate C–H bond breaking which is identified to be the RDS. Theoretical calculations reveal that doping Co NPs elevate the d-band center of Co single atoms by 0.13 eV through interactions, enhancing the adsorption of intermediates HCOOH* and HCOO*, thus lowering the reaction barriers from 1.20 eV to 0.86 eV compared to Co SAs, which is responsible for the excellent activity.
In a broader sense, this study provides design ideas for novel single-metal atom catalysts and deepens the understanding how the catalytic performance can be enhanced by synergistic action of metal sites of different sizes. We believe this approach can be easily extended to many other catalytic systems, too.
Methods
Chemicals and materials
Co(NO3)2·6H2O (AR, 99%), Zn(NO3)2·6H2O (AR, 99%), 2-methylimidazole (98%), methanol (99.5%), propylene carbonate (PC, 99%), ethanol (AR, 99.7%) were purchased from Macklin, China. Formic acid (HCOOH, 98%) was purchased from Rhawn, China. All reagents were used directly without further purification, and the ultrapure water was prepared by the ultrapure purification system. Pt/C (5% mass loading) and Pd/C (5% mass loading) were purchased from Sigma–Aldrich.
Preparation of CoZn-ZIF
The synthesis of CoZn-ZIF nanocrystals was accomplished based on a previous procedure with some modifications23,64. Firstly, 30 mmol of Zn(NO3)2·6H2O (8.925 g) and 3.5 mmol of Co(NO3)2·6H2O (1.014 g) were mixed and dissolved in 300 mL of methanol. Then, 120 mmol of 2-methylimidazole (9.853 g) was dissolved in 100 mL of methanol and added to the above solution. The mixture was stirred at room temperature for 24 h. Finally, the product was separated by centrifugation at 6429 g for 10 min and washed thoroughly three times with methanol. The resulting powder was dried overnight at 60 °C under vacuum before use.
Preparation of Co-SAs/NPs@NC-T
For the synthesis of Co-SAs/NPs@NC-950, the dried powder of CoZn-ZIF was pyrolyzed at 950 °C for 1 h with a ramping rate of 5 °C min−1 under 6%H2 + 94%Ar flow. The sample was then cooled to room temperature to obtain Co-SAs/NPs@NC-950. For Co-SAs/NPs@NC-850 or Co-SAs/NPs@NC-750, the pyrolysis temperatures were changed to 850 and 750 °C, respectively. The prepared samples are ready for use without acid etching and other post-treatment.
Catalyst characterization
TEM (Transmission Electron Microscopy) measurements were performed on a Thermo Fisher Titan Themis, 60–300 “cubed” microscope fitted with aberration-correctors for the imaging and the probe forming lens at 300 kV. HAADF-STEM experiments were performed using FEI Titan G2 and FEI Titan Themis Z microscope equipped with a probe and image corrector, a four-segment DF4 detector. EDS elemental mapping images were also obtained on the FEI Titan Themis Z microscope. XPS analysis was performed on an X-ray photoelectron spectrometer (Thermo Fisher model ESCALAB 250Xi). The XANES and EXAFS of Co K-edge spectra are collected by Table XAFS-500 (Specreation Instruments Co., Ltd. China). The content of Co was determined by atomic absorption spectroscopy (AAS) (PinAAcle900T). X-ray diffraction spectroscopy (XRD) was recorded using an X-ray diffractometer (Bruker, Bruker D8 Advance, Germany). The nitrogen adsorption isotherm was performed on a physisorption instrument (Micromeritics, ASAP2020, USA).
HCOOH dehydrogenation procedure
The dehydrogenation reaction was carried out under argon atmosphere, using the standard Schlenk technique to remove air. The reaction vessel was evacuated and refilled with argon 6 times. The condenser water flow was turned on and the catalyst (30 mg) and solvent (6 mL) were added. The vessel was heated to the desired temperature by a thermostat and allow to equilibrate for 30 min. Then, formic acid (10 mmol, 377 µl) was added to the reaction vessel under a flow of argon. The three-way valve of the burettes was turned to release the pressure in the reactor, closed again, and then the measurement of the volume of gas produced was started using a manual burette (Fig. S16). After completing the reaction for the desired time, gas samples were taken for GC analysis using an argon-purged gas-tight syringe.
In situ DRIFT experiments
In situ DRIFT experiments were conducted on a Fourier transform infrared (FTIR) spectrometer (Thermo Fisher Scientific, Nicolet iS50) equipped with a mercury cadmium telluride (MCT) detector. The catalyst powder was put into a reaction cell (Harrick Scientific Products, Praying Mantis). After treatment of the catalysts with an Ar flow (50 mL/min) at room temperature, the samples were heated to the set temperature, followed by bubbling Ar (50 mL/min) in HCOOH solution and flowed into the in situ reaction cell for simulating the heterogeneous catalytic process. The IR spectra were recorded every 3.0 s intervals until 1 h.
Isotope-labeling experiments
HCOOH, DCOOH, HCOOD, and DCOOD were used as substrates in propylene carbonate. The rest of the conditions were consistent with the HCOOH dehydrogenation procedure.
First-principles calculations
First-principles calculations were performed using the density functional theory framework within the Vienna ab initio simulation package (VASP 5.4.4)65,66. A (5 × 5) supercell of the graphene surface with the lateral size of around 12.5 Å was employed as a substrate for the CoN2C2 and CoN2C2–Co6. A vacuum spacing of over 15 Å was added to avoid the interaction between neighboring substrate layers. The interaction between the ions and the electrons was described by projector augmented-wave (PAW) methods65,67. The generalized gradient approximation (GGA) functional of Perdew–Burke–Ernzerhof (PBE) with van der Waals correction proposed by Grimme was used68,69. The convergence criteria of total energies and forces were 10−6 eV/atom and 0.01 eV/Å. The energy cut-off value was set at 600 eV and Monkhorst-Pack 2 × 2 × 1 K-point mesh was adopted. The pseudopotentials used for the present models were constructed by the electron configurations as C 2s22p2 states, N 2s22p3 states, Co 3d74s2 states, H 1 s1 states, and O 2s22p4 states. The adsorption energies and electron density difference were calculated by subtracting the energy of the gas phase and surface species from the energy of the adsorbed system according to the adsorption or interfacial models70,71,72,73,74. The Gibbs free energy correction was used to convert DFT energies into the Gibbs free energy with considering vibration contribution to entropy and zero-point energy75. The climbing image nudge elastic band (CI-NEB) methods was employed to search the transition state for the reactions76.
Data availability
All data generated and analyzed in this study are included in this paper and their supplements or can be obtained from the corresponding authors upon reasonable request. Source data are provided with this paper.
Code availability
All code used in the simulations supporting this paper is available from the respective authors upon request.
References
Dutta, I. et al. Formic acid to power towards low-carbon economy. Adv. Energy Mater. 12, 2103799 (2022).
Sang, R. et al. Methyl formate as a hydrogen energy carrier. Nat. Catal. 6, 543–550 (2023).
Wei, D., Sang, R., Sponholz, P., Junge, H. & Beller, M. Reversible hydrogenation of carbon dioxide to formic acid using a Mn-pincer complex in the presence of lysine. Nat. Energy 7, 438–447 (2022).
Wei, D. et al. Toward a hydrogen economy: development of heterogeneous catalysts for chemical hydrogen storage and release reactions. ACS Energy Lett. 7, 3734–3752 (2022).
Modisha, P. M., Ouma, C. N. M., Garidzirai, R., Wasserscheid, P. & Bessarabov, D. The prospect of hydrogen storage using liquid organic hydrogen carriers. Energy Fuels 33, 2778–2796 (2019).
Niermann, M., Timmerberg, S., Drünert, S. & Kaltschmitt, M. Liquid organic hydrogen carriers and alternatives for international transport of renewable hydrogen. Renew. Sust. Energ. Rev. 135, 110171 (2021).
Preuster, P., Papp, C. & Wasserscheid, P. Liquid organic hydrogen carriers (LOHCs): toward a hydrogen-free hydrogen economy. Acc. Chem. Res. 50, 74–85 (2017).
Chen, Z. et al. Designing a robust palladium catalyst for formic acid dehydrogenation. ACS Catal. 13, 4835–4841 (2023).
Sun, Q., Wang, N., Xu, Q. & Yu, J. Nanopore-supported metal nanocatalysts for efficient hydrogen generation from liquid-phase chemical hydrogen storage materials. Adv. Mater. 32, 2001818 (2020).
Celaje, J. J. A. et al. A prolific catalyst for dehydrogenation of neat formic acid. Nat. Commun. 7, 11308 (2016).
Kar, S., Rauch, M., Leitus, G., Ben-David, Y. & Milstein, D. Highly efficient additive-free dehydrogenation of neat formic acid. Nat. Catal. 4, 193–201 (2021).
Li, S. et al. A simple and effective principle for a rational design of heterogeneous catalysts for dehydrogenation of formic acid. Adv. Mater. 31, 1806781 (2019).
Liu, M. et al. Heterogeneous catalysis for carbon dioxide mediated hydrogen storage technology based on formic acid. Adv. Energy Mater. 12, 2200817 (2022).
Yu, Z. et al. Selective dehydrogenation of aqueous formic acid over multifunctional γ-Mo2N catalysts at a temperature lower than 100 oC. Appl. Catal. B 313, 121445 (2022).
Wang, J., Xu, F., Jin, H., Chen, Y. & Wang, Y. Non-noble metal-based carbon composites in hydrogen evolution reaction: fundamentals to applications. Adv. Mater. 29, 1605838 (2017).
Sun, Q. et al. Zeolite-encaged Pd-Mn nanocatalysts for CO2 hydrogenation and formic acid dehydrogenation. Angew. Chem. Int. Ed. 59, 20183–20191 (2020).
Li, X. et al. Cobalt single-atom catalysts with high stability for selective dehydrogenation of formic acid. Angew. Chem. Int. Ed. 59, 15849–15854 (2020).
Martin, C., Quintanilla, A., Vega, G. & Casas, J. A. Formic acid-to-hydrogen on Pd/AC catalysts: kinetic study with catalytic deactivation. Appl. Catal. B 317, 121802 (2022).
Liu, L. & Corma, A. Metal Catalysts for Heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 118, 4981–5079 (2018).
Kaiser, S. K., Chen, Z., Akl, D. F., Mitchell, S. & Pérez-Ramírez, J. Single-atom catalysts across the periodic table. Chem. Rev. 120, 11703–11809 (2020).
Yang, Q., Jiang, Y., Zhuo, H., Mitchell, E. M. & Yu, Q. Recent progress of metal single-atom catalysts for energy applications. Nano Energy 111, 108404 (2023).
Shi, Z., Yang, W., Gu, Y., Liao, T. & Sun, Z. Metal-nitrogen-doped carbon materials as highly efficient catalysts: progress and rational design. Adv. Sci. 7, 2001069 (2020).
Tang, C. et al. A stable nanocobalt catalyst with highly dispersed CoNx active sites for the selective dehydrogenation of formic acid. Angew. Chem. Int. Ed. 56, 16616–16620 (2017).
Chernov, A. N., Astrakova, T. V., Sobolev, V. I. & Koltunov, K. Y. Liquid versus gas phase dehydrogenation of formic acid over Co@N-doped carbon materials. The role of single atomic sites. Mol. Catal. 504, 111457 (2021).
Wang, Q., Tsumori, N., Kitta, M. & Xu, Q. Fast dehydrogenation of formic acid over palladium nanoparticles immobilized in nitrogen-doped hierarchically porous carbon. ACS Catal. 8, 12041–12045 (2018).
He, L., Weniger, F., Neumann, H. & Beller, M. Synthesis, characterization, and application of metal nanoparticles supported on nitrogen-doped carbon: catalysis beyond electrochemistry. Angew. Chem. Int. Ed. 55, 12582–12594 (2016).
Gao, Y., Liu, B. & Wang, D. Microenvironment engineering of single/dual-atom catalysts for electrocatalytic application. Adv. Mater. 35, 2209654 (2023).
Zhao, C., Li, B., Liu, J. & Zhang, Q. Intrinsic electrocatalytic activity regulation of M-N-C single-atom catalysts for the oxygen reduction reaction. Angew. Chem. Int. Ed. 60, 4448–4463 (2021).
Wu, X. et al. Engineering the coordination sphere of isolated active sites to explore the intrinsic activity in single-atom catalysts. Nanomicro Lett. 13, 136 (2021).
Chen, R., Chen, S., Wang, L. & Wang, D. Nanoscale metal particle modified single-atom catalyst: synthesis, characterization, and application. Adv. Mater. 36, 2304713 (2024).
Zhang, M. et al. High-loading Co single atoms and clusters active sites toward enhanced electrocatalysis of oxygen reduction reaction for High-Performance Zn-Air Battery. Adv. Funct. Mater. 33, 2209726 (2023).
Huang, H. et al. Clusters induced electron redistribution to tune oxygen reduction activity of transition metal single-atom for metal-air batteries. Angew. Chem. Int. Ed. 61, e202116068 (2022).
Liu, H. et al. Asymmetric coordination of single-atom Co sites achieves efficient dehydrogenation catalysis. Adv. Funct. Mater. 32, 2207408 (2022).
Chen, S. et al. Unveiling the proton-feeding effect in sulfur-doped Fe-N-C single-atom catalyst for enhanced CO2 electroreduction. Angew. Chem. Int. Ed. 61, e202206233 (2022).
Zhou, Y. et al. Boosting oxygen electrocatalytic activity of Fe-N-C catalysts by phosphorus incorporation. J. Am. Chem. Soc. 145, 3647–3655 (2023).
Shang, H. et al. Engineering unsymmetrically coordinated Cu-S1N3 single atom sites with enhanced oxygen reduction activity. Nat. Commun. 11, 3049 (2020).
Wang, X. et al. Regulation of coordination number over single Co sites: triggering the efficient electroreduction of CO2. Angew. Chem. Int. Ed. 57, 1944–1948 (2018).
Yin, P. et al. Single cobalt atoms with precise N-coordination as superior oxygen reduction reaction catalysts. Angew. Chem. Int. Ed. 55, 10800–10805 (2016).
Wang, Q. et al. Evolution of Zn(II) single atom catalyst sites during the pyrolysis-induced transformation of ZIF-8 to N-doped carbons. Sci. Bull. 65, 1743–1751 (2020).
An, Z. et al. Highly active, ultra-low loading single-atom iron catalysts for catalytic transfer hydrogenation. Nat. Commun. 14, 6666 (2023).
Cheng, X. et al. Nano-geometric deformation and synergistic Co nanoparticles-Co-N4 composite sites for proton exchange membrane fuel cells. Energy Environ. Sci. 14, 5958–5967 (2021).
Tian, H. et al. High-power lithium–selenium batteries enabled by atomic cobalt electrocatalyst in hollow carbon cathode. Nat. Commun. 11, 5025 (2020).
Jin, H. et al. In situ cobalt–cobalt oxide/N-doped carbon hybrids as superior bifunctional electrocatalysts for hydrogen and oxygen evolution. J. Am. Chem. Soc. 137, 2688–2694 (2015).
Jiang, S. F. et al.High efficient removal of bisphenol A in a peroxymonosulfate/iron functionalized biochar system: mechanistic elucidation and quantification of the contributors. Chem. Eng. J. 359, 572–583 (2019).
Wang, Z. et al. Atomically dispersed Co2–N6 and Fe–N4 costructures boost oxygen reduction reaction in both alkaline and acidic media. Adv. Mater. 33, 2104718 (2021).
Xue, Y. et al. MOF-derived Co and Fe species loaded on N-doped carbon networks as efficient oxygen electrocatalysts for Zn-Air batteries. Nano. Micro. Lett. 14, 162 (2022).
Liang, J. et al. Regulating the coordination environment of mesopore-confined single atoms from metalloprotein-MOFs for highly efficient biocatalysis. Adv. Mater. 34, 2205674 (2022).
Tian, Q. et al. Mesoporous carbon spheres with programmable interiors as efficient nanoreactors for H2O2 electrosynthesis. Nat. Commun. 15, 983 (2024).
Shi, Y. et al. Atomically dispersed cobalt/copper dual-metal catalysts for synergistically boosting hydrogen generation from formic acid. Angew. Chem. Int. Ed. 62, e202313099 (2023).
Wu, Z. et al. Facilely tuning the first-shell coordination microenvironment in iron single-atom for fenton-like chemistry toward highly efficient wastewater purification. Environ. Sci. Technol. 57, 14046–14057 (2023).
Zhang, X. et al. Single zinc atom aggregates: synergetic interaction to boost fast polysulfide conversion in lithium-sulfur batteries. Adv. Mater. 35, 2208470 (2023).
Zhang, J. et al. Facile synthesis of cobalt cluster-CoNx composites: synergistic effect boosts electrochemical oxygen reduction. J. Mater. Chem. A 10, 16920–16927 (2022).
Wang, Z. et al. Understanding the synergistic effects of cobalt single atoms and small nanoparticles: enhancing oxygen reduction reaction catalytic activity and stability for zinc-air batteries. Adv. Funct. Mater. 31, 2104735 (2021).
Baber, A. E. et al. Assisted deprotonation of formic acid on Cu(111) and self-assembly of 1D chains. Phys. Chem. Chem. Phys. 15, 12291–12298 (2013).
Luo, Q., Feng, G., Beller, M. & Jiao, H. Formic acid dehydrogenation on Ni(111) and comparison with Pd(111) and Pt(111). J. Phys. Chem. C 116, 4149–4156 (2012).
Meunier, F. C., Dansette, I., Paredes-Nunez, A. & Schuurman, Y. Cu-bound formates are main reaction intermediates during CO2 hydrogenation to methanol over Cu/ZrO2. Angew. Chem. Int. Ed. 62, e202303939 (2023).
Herron, J. A., Scaranto, J., Ferrin, P., Li, S. & Mavrikakis, M. Trends in formic acid decomposition on model transition metal surfaces: a density functional theory study. ACS Catal. 4, 4434–4445 (2014).
Yu, Z. et al. Full spectrum decomposition of formic acid over γ-Mo2N-based catalysts: from dehydration to dehydrogenation. ACS Catal. 10, 5353–5361 (2020).
Karatok, M. et al. Achieving ultra-high selectivity to hydrogen production from formic acid on Pd–Ag alloys. J. Am. Chem. Soc. 145, 5114–5124 (2023).
Zhang, A., Xia, J., Yao, Q. & Lu, Z. H. Pd-WOx heterostructures immobilized by MOFs-derived carbon cage for formic acid dehydrogenation. Appl. Catal. B 309, 121278 (2022).
Hou, T. et al. Modulating oxygen coverage of Ti3C2Tx MXenes to boost catalytic activity for HCOOH dehydrogenation. Nat. Commun. 11, 4251 (2020).
Ma, Z. et al. Coexistence of Fe nanoclusters boosting Fe single atoms to generate singlet oxygen for efficient aerobic oxidation of primary amines to imines. ACS Catal. 12, 5595–5604 (2022).
Gong, Z. et al. Modulating metal-nitrogen coupling in anti-perovskite nitride via cation doping for efficient reduction of nitrate to ammonia. Angew. Chem. Int. Ed. 62, e202308775 (2023).
Hou, C. et al. A gas-steamed MOF route to P-doped open carbon cages with enhanced Zn-ion energy storage capability and ultrastability. Adv. Mater. 33, 2101698 (2021).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Peng, H., Yang, Z., Perdew, J. P. & Sun, J. Versatile van der waals density functional based on a meta-generalized gradient approximation. Phys. Rev. X 6, 041005 (2016).
Sun, J., Ruzsinszky, A. & Perdew, J. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 115, 36402 (2015).
Luo, B. et al. Interfacial electronic properties of ferroelectric nanocomposites for energy storage application. Mater. Today Energy 12, 136–145 (2019).
Luo, B. et al. Superhierarchical inorganic/organic nanocomposites exhibiting simultaneous ultrahigh dielectric energy density and high efficiency. Adv. Funct. Mater. 31, 2007994 (2020).
Zhang, Z. et al. Electronic structure and optical properties of SnO2/HC(NH2)2PbI3 interfaces from first-principles calculations. Surf. Interfaces 23, 100913 (2021).
Luo, B., Wang, X., Tian, E., Song, H. & Li, L. Interfacial electronic and structural properties of SiO2(010)/BaTiO3(001) from first-principles calculations. Ceram. Int. 43, 12988–12991 (2017).
Luo, B., Wang, X., Tian, E., Li, G. & Li, L. Structural and electronic properties of cubic KNbO3 (001) surfaces: A first-principles study. Appl. Surf. Sci. 351, 558–564 (2015).
Mok, D. H. et al. Data-driven discovery of electrocatalysts for CO2 reduction using active motifs-based machine learning. Nat. Commun. 14, 7303 (2023).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22176008 and 52202154, X.L.) and the National Key Research and Development Program (2023YFC3710700, X.L.). Bingcheng Luo acknowledges support from the High-performance Computing Platform of China Agricultural University. Bingcheng Luo also acknowledges support from the National Supercomputer Center in Tianjin, and the optimization calculations were performed on Tianhe new generation supercomputer. The authors thank Specreation Instruments Co., Ltd. China for XAFS measurements and Anhui University, China for HAADF-STEM images. We also thank the equipment support from the Analysis & Testing Center at Beihang University.
Author information
Authors and Affiliations
Contributions
M.B. and X.L. proposed and designed the research plan and experimental scheme. Y.Z.S., Y.S., and D.D.C. performed the characterization of the materials and data analysis. Y.Z.S. and R.Q.L. carried out the synthesis and the activity tests of the materials. X.L. and Y.Z.S. conducted mechanistic investigation experiments and analysis. B.C.L. and Z.L.Z. performed the first-principles calculations. Y.S. and Y.F.S. commented on the paper. M.B. H.J. and R.S. advised and supervised the work. X.L., Y.Z.S., and B.C.L. co-wrote the original manuscript. M.B., H.J., R.S., and X. L. reviewed and edited the manuscript. Y. Z. S. and B.C.L. contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jose Luis Santos, Chinnappan Sivasankar, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shi, Y., Luo, B., Sang, R. et al. Combination of nanoparticles with single-metal sites synergistically boosts co-catalyzed formic acid dehydrogenation. Nat Commun 15, 8189 (2024). https://doi.org/10.1038/s41467-024-52517-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52517-w
This article is cited by
-
Intrinsic nanoparticle-single-atom interplays steering radical versus nonradical pathways in catalytic ozonation
Nature Communications (2025)
-
Highly efficient heterogeneous thermal catalysis for noble-metal-free hydrogen production from formic acid
Nature Communications (2025)
-
Mechanistic insights into atomic-to-nanoscale synergistic electrocatalysis
Science China Materials (2025)
