
Abstract
Membrane electrode assembly (MEA) cells incorporating Cu catalysts are effective for generating C2+ chemicals via the CO2 reduction reaction (CO2RR). However, the impact of MEA configuration on the inevitable reconstruction of Cu catalysts during CO2RR remains underexplored, despite its considerable potential to affect CO2RR efficacy. Herein, we demonstrate that MEA cells prompt a unique reconstruction of Cu, in contrast to H-type cells, which subsequently influences CO2RR outcomes. Utilizing three Cu-based catalysts, specifically engineered with different nanostructures, we identify contrasting selectivity trends in the production of C2+ chemicals between H-type and MEA cells. Operando X-ray absorption spectroscopy, alongside ex-situ analyses in both cell types, indicates that MEA cells facilitate the reduction of Cu2O, resulting in altered Cu surfaces compared to those in H-type cells. Time-resolved CO2RR studies, supported by Operando analysis, further highlight that significant Cu reconstruction within MEA cells is a primary factor leading to the deactivation of CO2RR into C2+ chemicals.
Similar content being viewed by others

Introduction
The electrochemical CO2 reduction reaction (CO2RR) has gained prominence as a leading technology for both mitigating atmospheric CO2 levels and transforming volatile electrical energy into storable chemical energy1,2,3,4. Among various chemicals from CO2RR, C2+ chemicals—those containing two or more carbon atoms, such as ethylene, ethanol, and n-propanol—stand out as key targets due to their high energy density and substantial market demand across a wide range of industrial applications5,6,7. To date, copper (Cu) catalysts have been distinguished for their ability to generate C2+ chemicals with significant Faradaic efficiency (FE), attributed to their ideal *CO binding strength in accordance with the Sabatier principle8,9,10,11. However, achieving precise selectivity towards C2+ chemicals amidst a range of potential products (H2, C1, and C2+) remains a formidable challenge in utilizing Cu catalysts.
The variation in product outcomes from the CO2RR is highly related to the surface morphologies of Cu catalysts12,13,14,15. There is a well-established consensus that under-coordinated sites of Cu, such as kinks, steps, and defects, play a pivotal role in stabilizing *OCCO intermediates. These intermediates are crucial for the kinetics of C2+ chemical production, facilitating a shift in product selectivity from C1 to C2+ compounds16,17,18,19,20,21. To create a high density of these under-coordinated sites, Cu oxides, or oxide-derived Cu, are commonly utilized. This approach leverages the natural tendency of Cu oxides to undergo reduction and reconstruction under CO2RR conditions, leading to the formation of defective Cu surfaces22,23,24. Several studies have empirically exploited the Cu defect sites, predominantly influenced by the initial Cu nanostructure, as a strategy to enhance FEC2+25,26,27,28,29.
The morphological transformations in Cu-based catalysts during CO2RR entail intricate processes such as the reduction of Cu oxides, fragmentation, dissolution cycling into Cu(OH)2- ions, and their subsequent reduction30,31,32. This simultaneous Cu reconstruction during CO2RR is highly interrelated to the reaction conditions18,27,31. In particular, the membrane electrode assembly (MEA) electrolyzer, which has recently been highlighted as a CO2RR reactor, operates under unconventional conditions, including gas-fed CO2 through a gas diffusion electrode (GDE) without a catholyte and a zero-gap configuration between the cathode, anion exchange membrane (AEM) and anode, comparing to a batch H-type cell reactor33,34,35,36. Due to these differences, the MEA cell exhibits significantly increased current density compared to the H-type cell, providing a distinct microenvironment for the catalyst. These distinctive features can significantly influence the reconstruction mechanisms of Cu-based catalysts during CO2RR, potentially altering the product distribution29,37. Despite its critical importance for advancing efficient and stable Cu catalysts in commercially viable devices, the investigation into Cu morphological changes within MEA environments remains sparse.
Herein, we employed three types of Cu-based catalysts: Cu2O nanocubes (Cu2O cube), Cu2O nanoparticle aggregate (Cu2O aggregate), and commercial Cu nanoparticles (cCu), as electrocatalysts to investigate the impact of initial morphologies and cell configurations on Cu reconstruction. Notably, the Cu2O aggregate exhibited the highest C2+/C1 ratio in the H-type cell, surpassing the performance of the other two catalysts, whereas the Cu2O cube demonstrated the most favorable C2+/C1 ratio in the MEA cell. Operando X-ray absorption spectroscopy (XAS) analysis indicated that the cell configurations markedly affect the reduction kinetics of Cu oxide during CO2RR. Moreover, ex-situ characterizations revealed that the accelerated reduction and reconstruction processes in MEA cells result in distinct Cu surface morphologies, which account for the divergent CO2RR performance trends observed in MEA versus H-type cells.
Results
Synthesis of Cu2O nanoparticles and preparation of catalyst electrodes
To understand the correlation between C2+ chemicals production and Cu state, we synthesized two Cu2O nanoparticles with different morphologies (i.e., Cu2O cube and Cu2O aggregate) and also prepared cCu (copper, 60–80 nm, Sigma Aldrich) as a control group to provide a contrasting morphology and oxidation state (detail synthesis in Methods) (Fig. 1a, b). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images showed that the Cu2O cube has monodisperse particles with an average size of 85.3 nm with a typical Cu2O lattice fringe of 0.31 and 0.24 nm, corresponding to Cu2O (110) and (111), respectively (Fig. 1c, d, Supplementary Fig. 1 and 2). Meanwhile, SEM images of Cu2O aggregate showed smooth spherical nanostructures with an average particle size of 92.9 nm, but TEM images represented it composed of a large number of small Cu2O domains, having specific Cu2O lattice fringes (Fig. 1e, f). X-ray diffraction (XRD) patterns of both the Cu2O cube and Cu2O aggregate confirmed their composition, displaying characteristic Cu2O peaks (Fig. 1g). However, the Cu2O aggregate displayed a smaller crystalline size in the XRD patterns, measuring 1.43 nm, compared to the Cu2O cube, which had a size of 4.58 nm (Supplementary Table 1). Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra of pristine Cu2O cube and Cu2O aggregate also clearly supported the Cu2O states (Fig. 1h). The cCu exhibited heterogeneity, yet their average particle size of 88.9 nm was comparable to that of the synthesized Cu2O catalysts. The XRD patterns and TEM images of cCu revealed both metallic Cu characteristic peaks and lattice fringes, as well as indications of partial oxidation (Supplementary Fig. 3). In the Fast Fourier Transform (FFT) image, the Cu2O cube displayed the distinct lattices of typical single crystalline Cu2O, whereas both the Cu2O aggregate and cCu showed ring patterns, indicative of polycrystalline nanostructures (Supplementary Fig. 4).

Synthetic protocols for the (a) Cu2O cube and (b) Cu2O aggregate. c, e TEM, d, f HRTEM, and lattice fringes of the (c, d) Cu2O cube and (e, f) Cu2O aggregate. g XRD patterns and (h) FT-EXAFS spectra of Cu2O cube and Cu2O aggregate.
Cell configuration-dependent CO2RR performance of Cu catalysts
As-prepared Cu catalysts were tested in both H-type and MEA cells to evaluate their CO2RR performances based on different cell configurations (Fig. 2 and Supplementary Fig. 5) and the catalysts’ initial nanostructures. The dependency of CO2RR performance on the applied potential for each type of Cu catalyst was analyzed using step-wise chronoamperometry. Within the H-type cell configuration, the FEC2+ of each Cu catalyst showed a volcano-like correlation as a function of applied potential (Fig. 2a–c and Supplementary Fig. 6). The maximum partial current density for C2+ chemicals production (jC2+) progressively rose as the applied potential neared −1.0 V (vs. reversible hydrogen electrode (RHE), with all potentials in the H-type cell referenced to RHE). Maximum FEC2+ were closely matched, recorded at 43.7, 44.4, and 43.2% for Cu2O cube, Cu2O aggregate, and cCu, respectively.

Cell configurations of the (a) H-type cell and (d) MEA cell. C2+ current densities of the Cu2O cube, Cu2O aggregate, and cCu in the (b) H-type cell and (e) MEA cell. C2+/C1 ratio of the Cu2O cube, Cu2O aggregate and cCu in the (c) H-type cell and (f) MEA cell. All error bars represent the standard deviation calculated from at least 3 data points. The applied potentials of the H-type cell were converted to RHE with 85% iR compensation, while the cell voltages for the MEA cell were presented as non-iR corrected values.
While the overall CO2RR efficiencies of the Cu-based catalysts showed similarities, the detailed performance metrics revealed distinct differences. The Cu2O cube demonstrated the lowest onset potential for converting CO2RR into C2+ chemicals. Conversely, the Cu2O aggregate achieved a significantly higher C2+/C1 ratio of 41.3 at −1.09 V, surpassing the ratios of 14.1 for the Cu2O cube and 25.2 for cCu. These findings confirm that the Cu2O cube and Cu2O aggregate provide CO2-to-C2+ favored reaction sites at low overpotential and C2+ selective active sites, respectively. cCu exhibited the highest onset potential to activate C2+ chemicals production, but more selective C2+ chemicals production compared to Cu2O cube at the optimum reduction potential.
To demonstrate the applicability of the relationship between Cu surface morphologies and CO2RR performances in different reaction conditions, MEA cell was utilized to evaluate these Cu-based catalysts with the identical electrode fabrication method to the H-type cell (Fig. 2d–f and Supplementary Fig. 7). The MEA cell remarkably improved FEC2+ and jC2+ even with the same catalysts. The FEC2+ of Cu2O cube, Cu2O aggregate, and cCu improved as increasing cell voltage, recording each maximum value as 63.3, 60.6, and 53.0%, respectively. The highest jC2+ of −227.7 mA cm-2 was achieved on the Cu2O cube among the Cu catalysts. The trend of C2+/C1 between Cu2O cube and Cu2O aggregate was intriguingly reversed, showing that Cu2O cube provided a higher C2+/C1 ratio than Cu2O aggregate. In particular, the Cu2O aggregate providing the highest C2+/C1 ratio in the H-type cell now represented the lowest C2+/C1 ratio in MEA. These observations provide evidence that the cell configurations can lead to different trends in CO2RR performances, even in the same Cu-based catalysts. Therefore, we postulate that the cell configuration may alter the Cu reconstruction process and result in different morphological features of Cu during CO2RR.
Operando XAS analysis during CO2RR in H-type and MEA cell
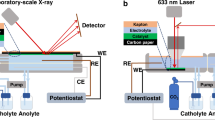
Real-time changes in the oxidation states and coordination numbers (CN) of each Cu-based catalyst were monitored in a custom-made H-type and MEA cell equipped with a transparent window capable of X-ray penetration (Fig. 3 and Supplementary Fig. 8). In the H-type cell, a step-wise potential application was employed, maintaining each voltage level for 15 min, with XAS spectra collected every 5 min (Fig.3a). In contrast, due to the accelerated Cu2O reduction, XAS spectra within the MEA cell were acquired at 3 V for 10 min and at 3.5 V for a duration of 120 min (Fig. 3e).

a Operando XAS spectra collection methods for the H-type cell. b Average oxidation states derived from the XANES spectra of the Cu2O cube and Cu2O aggregate samples in the H-type cell. FT-EXAFS spectra of the (c) Cu2O cube and (d) Cu2O aggregate samples in the H-type cell, respectively (Cu foil was scaled by 0.4 for better illustration). e Operando XAS spectra collection methods for the MEA cell. f Average oxidation states derived from the XANES spectra of the Cu2O cube and Cu2O aggregate samples in the MEA cell. Time-resolved FT-EXAFS spectra of the (g) Cu2O cube and (h) Cu2O aggregate samples in the MEA cell, respectively.
In the H-type cell, the Cu2O cube showed a gradual reduction with applied potential, whereas the Cu2O aggregate remained largely unchanged (Fig. 3b). This was evidenced by the decrease in white line intensity in the operando X-ray absorption near-edge structure (XANES) spectra with the application of reductive potentials, alongside a negative shift in edge energies (Supplementary Fig. 9). However, the kinetics of reduction as a function of potential and time varied significantly among the catalysts, dependent on their initial nanostructures. Among these samples, the Cu2O cube emerged as the most electrochemically active sample, demonstrating the most pronounced shift in its XANES spectra from open-circuit voltage (OCV) to −1.2 V. In the FT-EXAFS spectra of the Cu2O cube, Cu-O peaks (~ 1.5 Å) and Cu-Cu peaks associated with Cu2O (~ 2.7 Å) began diminishing from −0.6 V, with the Cu-Cu peaks indicative of metallic Cu (~2.2 Å) becoming significantly prominent from −1.0 V onward (Fig. 3c). Compared to the progressive reduction of the Cu2O cube during CO2RR over time, the Cu2O aggregate retained its Cu2O structure across all applied potentials, showing only minimal reduction after the initial phase (Fig. 3d). The cCu showed a notable metallic Cu phase after the initial reduction of residual surface Cu2O (Supplementary Fig. 10 and 11). Analysis of the FT-EXAFS spectra for cCu revealed a slight reduction of the CN for Cu-Cu within the error margin, from 11.4 ± 1.0 to 11.1 ± 1.0 (Supplementary Table 2).
Conversely, in the MEA cell, time-resolved operando XANES revealed that both the Cu2O cube and cCu were predominantly reduced into metallic Cu within the initial 5 min at 3 V (Fig. 3f and Supplementary Fig. 12). In contrast, while the Cu2O aggregate also experienced rapid reduction during the same timeframe, traces of residual Cu2O species were still distinct until 10 min at 3 V from its white line intensity. Simultaneously, FT-EXAFS spectra showed that Cu-O peaks (~ 1.5 Å) completely disappeared in Cu2O cube and cCu, and the Cu-Cu peaks of metallic Cu (~ 2.2 Å) emerged and grew within the first 10 min at 3 V (Fig. 3g and Supplementary Fig. 10), consistent with the XANES results. Cu2O aggregate showed a similar trend, albeit with the noticeable Cu-O peak at 3 V (Fig. 3h). Upon extending the operation to 3.5 V, all Cu-based catalysts predominantly displayed metallic Cu characteristics in their XANES spectra, with no substantial temporal variations. Nonetheless, changes in the local Cu geometries persisted throughout CO2RR, as evidenced by minor alterations in the CN derived from FT-EXAFS spectral fitting. The CN for Cu-Cu slightly decreased from 10.7 ± 0.9 to 10.4 ± 1.0 for the Cu2O cube, from 11.9 ± 1.6 to 11.6 ± 1.6 for cCu, and from 11.2 ± 0.7 to 11.6 ± 0.6 for the Cu2O aggregate (Fig. 3f and Supplementary Table 3).
Operando XAS showed that abrupt CO2RR conditions in the MEA cell trigger faster Cu2O reduction compared to the H-type cell. Therefore, we postulate that cell configurations can induce distinctive Cu oxidation states and surface morphologies during CO2RR.
Ex-situ characterizations of Cu and Cu2O catalysts post-CO2RR in H-type and MEA cells
To investigate how different reduction kinetics of Cu2O affect Cu state and surface morphologies, we characterized all Cu-based catalysts using ex-situ TEM and XRD after CO2RR in H-type and MEA cells (Fig. 4 and Supplementary Fig. 13 and 14). We additionally characterized Cu catalysts operated at 3 V for 10 min in the MEA cell because of the rapid Cu2O reduction in the MEA cell.

TEM images of (a) Cu2O cube, b Cu2O aggregate, c cCu, and (d) their XRD patterns after CO2RR in the H-type cell. TEM images of (e) Cu2O cube, f Cu2O aggregate, g cCu, and (h) their XRD patterns after CO2RR in the MEA cell for 10 min at 3 V. i–l Images show the same analyses after an additional 120 min at 3.5 V in the MEA cell.
In the H-type cell, all the Cu-based catalysts exhibited minimal structural changes after CO2RR (Fig. 4a–d and Supplementary Fig. 13a–c). The Cu2O cube largely preserved its cubic shape, showing only slight surface bumpiness. Both the Cu2O aggregate and cCu also retained their original nanostructures, though some small debris was observed near the nanoparticles. XAS showed that the Cu2O cube exhibited characteristic peaks of both Cu2O and metallic Cu, with the corresponding lattice fringes observed in XRD patterns and high resolution TEM (HRTEM) images. FFT images revealed polycrystalline ring patterns indicative of multi-domains of Cu and Cu2O formed during CO2RR, suggesting a mix of phases in the Cu2O cube (Supplementary Fig. 14d). The Cu2O aggregate, in contrast, displayed only Cu2O characteristic peaks and maintained polycrystalline ring patterns similar to those of the pristine material (Supplementary Fig. 14e). The post-CO2RR cCu showed exclusive metallic Cu XRD patterns along with Cu (111) and (200) lattice fringes and ring patterns (Supplementary Fig. 14f). From XRD patterns, the crystal size of Cu2O cube and cCu decreased from 4.58 nm and 9.17 nm to 3.55 nm and 8.53 nm, respectively, while that of Cu2O aggregate represented only minor changes (Supplementary Table 1).
In the MEA cell environment, the Cu-based catalysts underwent notable morphological transformations distinct from those observed in the H-type cell, particularly after a 10 min operation at 3 V (Fig. 4e–h and Supplementary Fig. 13d–f). The Cu2O cube disintegrated into small Cu domains, resulting in interconnected and agglomerated nanostructures. The Cu2O aggregate, on the other hand, separated into individual nanoparticles, each evolving into hollow structures with defective shells containing small Cu debris. The cCu showed highly aggregated nanostructures with bumped surfaces. During this period, both the Cu2O cube and cCu were fully reduced to metallic Cu, whereas Cu2O aggregate uniquely exhibited mixed Cu and Cu2O characteristics in XRD patterns, HRTEM, and FFT images (Supplementary Fig. 14g–i). An extended operation time of 120 min at 3.5 V led to further morphological changes (Fig. 4i–l, Supplementary Fig. 13g–i and 14j–l). The Cu2O cube completely disintegrated into subnanometer-sized Cu debris with few irregular Cu nanoparticles. Similarly, the cCu experienced a significant disintegration of its aggregated Cu nanostructures. Interestingly, the Cu2O aggregate maintained its hollow and defective structure, albeit with the emergence of sharp, branch-like features on its surface. XRD patterns revealed a significant reduction in the crystal size of both the Cu2O cube and cCu to 0.77 and 0.84 nm, respectively, indicating a transition to polycrystallinity. In contrast, the size of the Cu2O aggregate remained largely unchanged (Supplementary Table 4).
Reconstruction mechanisms of Cu and Cu2O catalysts during CO2RR depending on cell configurations
Through both operando and ex-situ characterizations, it becomes evident that the Cu catalyst develops a unique intrinsic surface during CO2RR, influenced not only by its initial nanostructures but also by the cell configuration. To elucidate the Cu reconstruction process, including Cu2O reduction, Cu detachment, and Cu dissolution and redeposition, it is essential to consider their effects. The reduction of Cu2O leads to the formation of grain boundaries, which arise from the lattice mismatch between Cu2O and Cu domains (Fig. 5a and Supplementary Fig. 15)17,38. Cu also easily detaches from the original nanoparticles and forms small Cu debris (Fig. 4i and k)39. The elevated current density in the system raises the local pH, facilitating the dissolution of Cu(OH)2- ions. Under the highly reductive conditions of CO2RR, these Cu(OH)2- ions are then sequentially reduced back into metallic Cu30,31.

a Reconstruction mechanism on the Cu surface. The reduction, detachment, dissolution, and redeposition steps are described. The morphological changes during the CO2RR of the (b) Cu2O cube, c Cu2O aggregate and (d) cCu are depicted.
Due to the slower Cu2O reduction in the H-type cell, all Cu2O catalysts preserved their original nanostructures without significant disintegration, by forming grain boundaries. The crystal size in Cu2O cube and cCu was reduced because of Cu2O reduction and Cu detachment during CO2RR, yet the size of individual nanoparticles remained largely unchanged, likely due to a balance between Cu dissolution and redeposition (Fig. 5b). Uniquely, the Cu2O aggregate demonstrated a distinct resistance to Cu2O reduction during CO2RR (Fig. 5c). This resistance is attributed to the original aggregated Cu2O domains delaying the reduction of Cu+, as indicated by literature on similar nanostructures40,41. Consequently, the Cu2O aggregate’s robustness helped maintain its crystal size and oxidation states.
Meanwhile, the accelerated Cu2O reduction in the MEA cell leads to a complete reconstruction of the Cu2O cube through grain boundary formation within 10 min at 3 V. The high current density, a result of elevated CO2 concentration, facilitates Cu detachment and initiates a cycle of Cu dissolution followed by re-deposition. Therefore, Cu2O cube and cCu construct complex networks by agglomeration of individual nanoparticles (Fig. 4e, g). In contrast, a hollow Cu2O aggregate nanostructure without an agglomeration was exclusively discovered in Cu2O aggregate (Fig. 4f). A plausible mechanism is that defective outer frameworks provide a solid framework; such framework can induce faster oxygen removal from inside.
Following an extended period of operation in the MEA cell for 120 min at 3.5 V, there was a notable disintegration of nanostructures into small Cu debris, accompanied by a significant reduction in crystal size for both the Cu2O cube and cCu (Fig. 4i, k, and Supplementary Table 3). In contrast, the Cu aggregate developed unique hollow and branched Cu nanostructures characterized by numerous sharp branches on their surface (Fig. 4j). These findings indicate that the abrupt conditions of CO2RR in the MEA cell tend to favor Cu dissolution over Cu redeposition. As a result, Cu catalysts lacking a solid framework, such as the Cu2O cube, tend to break down into small and irregular nanodebris within the MEA cell, regardless of their original nanostructures.
It should be noted that the different reconstruction behaviors of Cu-based catalysts observed in H-type cell and MEA cell are multifaceted. The different cell configurations provide different reaction environments, including local pH, local CO2/H2O ratio, *CO coverage, and surface bubble formations etc., which lead to different catalyst performances in terms of current density and FE. The critical point is that the microenvironments affect the Cu reconstructions, which determine the intrinsic properties of catalysts, leading to further variations in CO2RR performances.
Interpretation of CO2RR performance in each cell based on Cu reconstruction
A detailed understanding of Cu reconstruction and oxidation states, derived from comprehensive characterization, gives insight into the interpretation of CO2RR performance trends. In the H-type cell, characterized by a notable presence of Cu+ species during CO2RR, it is crucial to consider both the surface morphologies and oxidation states to understand the performance effectively. The presence of a mixed Cu+/Cu0 surface is recognized for its role in facilitating C-C coupling, leading to the formation of C2+ chemicals40,41,42. The Cu2O cube, with its partially oxidized surface, demonstrated a lower onset potential for the production of C2+ chemicals production (Fig. 2a). The Cu2O aggregate, on the other hand, has a Cu+ dominant surface but appears to achieve the highest C2+/C1 ratio due to its grain boundary-rich surface that favors the production of C2+ chemicals (Fig. 2b).
In the MEA cell, only the Cu2O aggregate represented a trace amount of Cu+ species at 3 V. Consequently, the Cu2O aggregate exhibited the lowest onset potential of C2+ productions due to the presence of mixed Cu+/Cu0 surfaces (Fig. 2c). Beyond this voltage (≥3.25 V), as Cu+ species were reduced to metallic Cu regardless of their initial nanostructures, suggesting that CO2RR performances could be governed by the surface morphologies of Cu. A potential explanation for the relatively low C2+/C1 ratio of Cu2O aggregate in the MEA cell involves the concept of a *CO reservoir43,44. Because high *CO concentrations are required to activate C-C coupling on the catalyst surface, the MEA cell may need a particularly larger *CO reservoir compared to the H-type cell due to its higher current density condition. During the CO2RR in the MEA cell, integrated nanostructures in Cu2O cube and cCu seem to provide a larger *CO reservoir, enhancing the activation of C-C coupling sites and thereby achieving a higher C2+/C1 ratio. On the other hand, the individually dispersed Cu2O aggregate has insufficient *CO reservoir to promote the C-C coupling reaction, resulting in an inferior C2+/C1 ratio in the MEA cell, contrasting with the conditions observed in the H-type cell.
We further investigated the variations in CO2RR performance in the MEA cell over time at a static applied voltage of 3.5 V after 10 min of pre-activation at 3 V, under the same operation conditions as operando XAS analysis (Fig. 6 and Supplementary Fig. 16). It is reasonable to note that the CO2RR performance changes were originated by the Cu reconstruction, because the control experiment using Ag electrode with the identical IrO2 anode showed stable performances in this voltage and reaction time range (Supplementary Fig. 17 and Supplementary Table 5). Both Cu2O cube and cCu, which represented a gradual decrease of Cu domain size during the CO2RR, exhibited a significant decline in FEC2+ and C2+/C1 ratio only after 40 min of operation. In particular, the rapid and consistent decrease of the selectivity of C2+ chemicals was observed in the Cu2O cube over time. It is noted that the sub-nanometer Cu domains in the Cu2O cube are unfavorable to produce C2+ chemicals from CO2RR. It is noteworthy that the Cu2O aggregate, which prefers to maintain its original nanostructures, had only a minor drop in the FEC2+ and C2+/C1 ratio.

Faradaic efficiencies of H2, C1, and C2+ of the (a) Cu2O cube, b Cu2O aggregate, and (c) cCu. The gas products were analyzed every 30 min, and the liquid products were collected for 15 min in pure electrolyte after GC injection. These data were obtained from single experiments for each catalyst.
Previous studies have suggested that the diminished stability observed in MEA cells is due to factors such as flooding45,46, hydrophilization of the cathode47, and salt formations33,48. However, based on the trends observed in the variations of CO2RR performance, particularly in relation to the morphological changes of Cu catalysts, it is indicated that the substantial morphological transformations of Cu catalysts under the abrupt CO2RR conditions in the MEA cell could be a primary cause of CO2RR deactivation towards C2+ chemicals. These findings imply that MEA cells necessitate a distinct approach to catalyst design compared to H-type cells to ensure stability and efficiency in CO2RR.
Discussion
In this study, we utilized three distinct Cu-based catalysts—Cu2O cube, Cu2O aggregate, and cCu—within both H-type and MEA cell configurations to study the impact of cell environment on Cu catalyst reconstruction during CO2RR. In the H-type cell, the Cu2O aggregate exhibited the highest selectivity for C2+ chemical production. Conversely, in the MEA cell, the Cu2O cube outperformed the Cu2O aggregate, which exhibited the lowest selectivity among the catalysts tested. Through operando XAS and ex-situ characterizations, we established a correlation between CO2RR performance, and the varying Cu2O reduction rate and Cu surface reconstruction influenced by the cell configuration. The solid Cu2O aggregate, characterized by grain boundary-rich surfaces, retained its original structure and exhibited a slower Cu2O reduction rate, thereby enhancing C2+ chemical production in the H-type cell. Meanwhile, the Cu2O cube and cCu, more prone to reconstruction, formed a network of small Cu domains within the MEA cell, leading to increased selectivity for C2+ chemicals compared to the Cu2O aggregate. However, in the long-term MEA operation, both the Cu2O cube and cCu disintegrated into small Cu nanodebris, resulting in the deactivation of CO2RR over time, which underscores their lower stability. These findings emphasize the importance of considering cell configuration when designing highly efficient and stable Cu-based catalysts, highlighting the distinct requirements for MEA versus H-type cells.
Methods
Synthesis of Cu2O cube
Initially, 5.3 g of polyvinylpyrrolidone (PVP) (MW = 55,000, Sigma Aldrich), was dissolved in 45 mL of 1,5-pentanediol (96%, Sigma Aldrich). This solution was purged with N2 for 15 min and heated to 215 °C. Subsequently, a solution of 1.05 g of copper (II) acetylacetonate (99.9%, Sigma Aldrich) in 15 mL of 1,5-pentanediol was added to the PVP mixture. The combined solution was then stirred vigorously at 215 °C under a N2 atmosphere for 21 min, during which the mixture turned bright yellow. Following the reaction, the mixture was rapidly cooled in ice water, washed with acetone (99.5%, Duksan) and ethanol several times, and finally redispersed in 30 mL of ethanol for subsequent use49.
Synthesis of Cu2O aggregate
To prepare the reaction mixture, 240 mg of copper (II) sulfate (>99%, Sigma Aldrich) and 2 g of D-(+)-glucose (>99.5%, Sigma Aldrich) were dissolved in 100 mL of deionized water (Arium mini, Sartorius). While stirring at room temperature, 25 mL of a 0.04 M ammonia solution (35%, Thermo Fisher) followed by 25 mL of a 0.2 M sodium hydroxide (97%, Sigma Aldrich) solution were added dropwise and in sequence. The solution was stirred for an additional 10 min, during which it turned blue. Subsequently, 50 mL of a 0.03 M ascorbic acid solution (99%, Sigma Aldrich) was added to the mixture. After stirring for 1 h, the reaction mixture changed to a yellowish-orange color. It was then washed with deionized water and ethanol several times, followed by overnight drying under vacuum at room temperature.
Preparation of electrodes
(1) Working electrode. To activate the ionomer, 60 μL of Sustainion XA-9 5% in ethanol (Dioxide Materials) was mixed with 60 μL of 2 M potassium hydroxide (KOH, 90%, Sigma Aldrich) in ethanol (99.9%, Duksan) and pretreated for 1 h at room temperature. 20 mg of Cu catalyst was dispersed in 2 mL of ethanol with 30 min of sonication. Then, pretreated Sustainion ionomer solution was mixed with catalyst dispersion to obtain catalyst ink. Then, the catalyst ink was air-brushed with a spray gun (Gunpiece GP-2, Fuso Seiki) on a GDE (Sigracet 39BB GDL, SGL carbon). The loading amount was determined by weighing the GDE before and after spraying (0.5 mg cm−2) on an active area of 5 cm2 for MEA cell. The uniformity of Cu catalyst loading was confirmed by X-ray fluorescence spectrometer mapping (Supplementary Fig. 18). For H-type cell test, the 0.5 cm2 Cu electrode was used by cutting and masking the as-prepared electrode for MEA cell testing. (2) Counter electrode. Platinum foil (thickness 0.2 mm, WizMac) was used as the counter electrode in the H-type cell. For the MEA cell, iridium oxide (IrO2, 99.99%, Alfa-Aesar) was used as a catalyst for the oxygen evolution reaction. 30 mg of IrO2 was sonicated and dispersed in 1 mL of isopropyl alcohol with 30 mg of 5 wt% of Nafion ionomer solution (Sigma Aldrich). The IrO2 ink was sprayed on a platinized titanium mesh (thickness 0.004 inch, FuelCellStore) with a 1 mg cm−2 loading amount. The stability of the IrO2 catalyst on the anode was evaluated by monitoring the concentration of Ir ions in the anolyte over time using inductively coupled plasma optical emission spectroscopy (iCAP 7000 Series, Thermo Scientific). The Ir concentration was determined using an Ir standard solution (1 mg ml−1 in 20% HCl, Acros Organics). (3) Reference electrode. Ag/AgCl (3 M KCl, RE-5B, BASi Research Products) was used as a reference electrode in the H-type cell. The potential of the reference electrode was calibrated by measuring its potential difference relative to a known reference electrode with a constant potential that has not been used for electrochemical experiments.
Cell configurations
The CO2RR performance was evaluated in two representative cell types, a three-electrode H-type cell and a MEA cell with an active area of 0.5 cm2 and 5 cm2, respectively. The H-type cell consists of a catholyte chamber and an anolyte chamber. Each Chamber contained an inlet and outlet for feed gas and gas products. The catholyte chamber was separated from the anolyte chamber by a Selemion AMV-N AEM (130 μm, Asahi Glass), which was activated in deionized water more than 24 h. 28 mL of 0.1 M potassium bicarbonate (KHCO3, 99.95%, Sigma Aldrich) was used as catholyte, and anolyte as well. A 0.1 M KHCO3 solution was stirred for more than 24 h after the KHCO3 salt was added to deionized water, and then constantly bubbled with 20 mL min−1 CO2 for more than 2 h before the experiments. In the MEA cell, Cu cathode and IrO2 anode were assembled by direct contact in the form of a Sustainion X37-RT AEM (50 μm, dry, Dioxide Materials) fixed between the electrodes. Prior to assembly, the membrane was pretreated in 1 M KOH solution for 24 h and washed with deionized water. The MEA was mounted on a stainless steel bipolar plate and a titanium bipolar plate for the cathode and anode, respectively. CO2 gas flowed through a humidifier (36 °C) to be humidified with a relative humidity of 100%, and then fed to the cathode at 100 mL min−1. 40 mL of 0.1 M KHCO3 solution flowed to the anode using a peristaltic pump (EMP-600A, EMS-Tech) with a flow rate of 16 mL min−1. All the CO2 flow was controlled with a mass flow controller (El-flow, Bronkhorst). Custom-built H-type cell and MEA cell were adapted for operando XAS analysis. For X-ray transmission, a single-chamber batch cell without a membrane was used for H-type cell. A Kapton film was used to transmit the X-ray to and through the catalyst surface. To avoid X-ray disturbance by electrolyte, the working electrode was directly attached to the Kapton film window. Reference electrode and counter electrode were not arranged in a straight line not to block the X-ray path. Custom-built MEA cell with holes in the anode and cathode bipolar plate was used for operando XAS analysis. Holes were covered with the Kapton film to transmit the X-ray.
Electrochemical measurements
In the H-type cell, 0.1 M CO2-saturated KHCO3 solution was used as an electrolyte. A Selemion AMV-N AEM was used as a membrane to separate both cathode and anode counterparts. In the MEA cell, Sustainion X37-RT AEM was used as a membrane to fabricate MEA configuration. Electrochemical experiments were performed as chronoamperometry at fixed voltages using a VSP potentiostat (BioLogic) with a 10 A booster. In a three-electrode system H-type cell, the applied potential was converted to RHE with 85% iR compensation by the following equation:
where i and RS denote current and resistance, respectively, and these parameters were determined using electrochemical impedance spectroscopy. The pH of the CO2-bubbled 0.1 M KHCO3 was 6.82 ± 0.02, which was measured with a pH meter (SevenCompact, Mettler Toledo). The resistance of each electrode was measured before and after the reaction using potentiostatic electrochemical impedance spectroscopy at −1.1 V vs. Ag/AgCl, at which CO2RR barely occur. In the experiments involving a two-electrode system, MEA cell, various cell voltages were applied and subsequently recorded. For the H-type cell, four discrete potentials ranging from −0.6 V to −1.2 V versus RHE were sequentially applied, with each potential level being maintained for a duration of 15 min. In contrast, within the MEA cell configuration, a step-wise voltage increase was implemented, starting from 3 V to 4 V, with increments of 0.25 V at each step. To evaluate the MEA cell’s stability, an initial potential of 3 V was applied for a short span of 10 min, followed by a prolonged application of 3.5 V for 120 min. All electrochemical experiments were conducted at room temperature, maintained between 20 and 25 °C.
Product analysis
Gas chromatography (7890B GC system, Agilent Technologies) with a flame ionization detector and a thermal conductivity detector with methanizer was conducted to analyze the gas products. ShinCarbon ST 100/200 (1 m, 1 mm/D 1/16 OD silco, Restek) column was used. Ar (99.999%) was used as the carrier gas for gas chromatography. Gas samples were collected and analyzed 10 min after the specific potential, and every 15 min thereafter. The liquid products were analyzed using 1H nuclear magnetic resonance (NMR) spectroscopy (Avance III HD 400, Bruker) with H2O suppression, using D2O (99.9 atom% D, contains 0.05 wt.% 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid, sodium salt, Sigma Aldrich) as the lock solvent and internal reference. The average CO2 flow rate was measured using a flow meter (ADM 2000, Agilent Technologies) at the gas chromatography system outlet. The productivity of each product from the CO2RR was converted to FE. The FEs of the products were calculated as the ratio of each partial current of the products to the total current using the following equations:
where moleproduct is the actual number of moles per second of the product quantified by gas chromatography or NMR, F is the Faraday constant (96,485 C mol-1), and N is the number of electrons consumed to produce each product from CO2. All cathodic current densities were expressed without minus signs.
Ex-situ and operando characterizations
The physical characteristics of the catalysts and electrodes were examined using SEM (Inspect F-50, FEI) at 10 kV, TEM (F20 G2, TECNAI) at 200 kV, and XRD (D8 ADVANCE LynxEye, Bruker). TEM images were processed using Gatan Microscopy Suite Software. Lattice fringes were calculated as an average length of 10 fringes. The scan rate of the XRD data was 0.05 degree s−1 and the data were processed using Jade Software. XRD peak analysis was conducted with reference to Cu (JCPDS Card No. 03-1018) and Cu2O (JCPDS Card No. 78-2076). The 2θ values for Cu (111), (200), and (220) were assigned at 43.42°, 50.62°, and 74.76°, respectively. Similarly, the 2θ values for Cu2O (110), (111), (200), (220), and (311) were indexed at 29.56°, 36.46°, 42.34°, 61.42°, and 73.52°, respectively. The carbon peak from the substrate was observed at 54.5°. From the XRD patterns, the full width at half maximum (FWHM) of the major peaks was measured and the average sizes of the grains in the particles were given by the Scherrer equation:
where τ is the average grain size, Κ is the shape factor, which is generally 0.9, λ is the X-ray wavelength, and θ is the Bragg angle.
The same electrodes utilized in the CO2RR performance assessments were employed for operando XAS measurements. The XAS spectra were measured and the 1D KIST beamline of Pohang Accelerator Laboratory. These operando XAS experiments were conducted at room temperature in a commercial H-type cell and MEA cell with necessary modifications for spectroscopy measurements. The measurements were carried out in fluorescence mode, using a Si (111) channel-cut monochromator. Photon energy scanning in the XANES and EXAFS regions was finely tuned, with increments of 1.0 eV for incident radiation energy and 0.03 Å−1 for photoelectron wavenumbers. Data collection at the Cu K-edge was conducted simultaneously in both fluorescence and transmission modes. Calibration of the XANES and EXAFS analyses was achieved using data from Cu foil standards. Reference foil data were also used for all experimental runs to facilitate accurate energy alignment and normalization of the data. Processing and fitting of the XAS data were performed using the IFEFFIT-based software programs, Athena50 and Artemis51. The entire dataset underwent calibration, alignment, and normalization, with background subtraction executed via the IFEFFIT suite52. The χ(R) were modeled using single scattering paths calculated by FEFF653.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files. Source data are provided as a Source data file. Source data are provided with this paper.
References
Overa, S., Ko, B. H., Zhao, Y. & Jiao, F. Electrochemical approaches for CO2 conversion to chemicals: A journey toward practical applications. Acc. Chem. Res. 55, 638–648 (2022).
Stephens, I. E. L. et al. 2022 roadmap on low temperature electrochemical CO2 reduction. J. Phys. Energy 4, 042003 (2022).
Yu, J. et al. Recent progresses in electrochemical carbon dioxide reduction on copper-based catalysts toward multicarbon products. Adv. Funct. Mater. 31, 2102151 (2021).
Langie, K. M. G. et al. Toward economical application of carbon capture and utilization technology with near-zero carbon emission. Nat. Commun. 13, 7482 (2022).
Bushuyev, O. S. et al. What should we make with CO2 and how can we make it? Joule 2, 825–832 (2018).
Ruiz-López, E., Gandara-Loe, J., Baena-Moreno, F., Reina, T. R. & Odriozola, J. A. Electrocatalytic CO2 conversion to C2 products: Catalysts design, market perspectives and techno-economic aspects. Renew. Sust. Energ. Rev. 161, 112329 (2022).
Na, J. et al. General technoeconomic analysis for electrochemical coproduction coupling carbon dioxide reduction with organic oxidation. Nat. Comm. 10, 5193 (2019).
Lee, W. H. et al. The green-ol (green-alcohol) economy. Nano Energy 110, 108373 (2023).
Choi, W., Won, D. H. & Hwang, Y. J. Catalyst design strategies for stable electrochemical CO2 reduction reaction. J. Mater. Chem. A 8, 15341–15357 (2020).
Sun, B. et al. Challenges and strategies towards copper-based catalysts for enhanced electrochemical CO2 reduction to multi-carbon products. Fuel 332, 126114 (2023).
Woldu, A. R., Huang, Z., Zhao, P., Hu, L. & Astruc, D. Electrochemical CO2 reduction (CO2RR) to multi-carbon products over copper-based catalysts. Coord. Chem. Rev. 454, 214340 (2022).
Gao, W., Xu, Y., Fu, L., Chang, X. & Xu, B. Experimental evidence of distinct sites for CO2-to-CO and CO conversion on Cu in the electrochemical CO2 reduction reaction. Nat. Catal. 6, 885–894 (2023).
Lv, X., Liu, Z., Yang, C., Ji, Y. & Zheng, G. Tuning Structures and Microenvironments of Cu-Based Catalysts for Sustainable CO2 and CO Electroreduction. Acc. Mater. Res. 4, 264–274 (2023).
Tabassum, H., Yang, X., Zou, R. & Wu, G. Surface engineering of Cu catalysts for electrochemical reduction of CO2 to value-added multi-carbon products. Chem Catal 2, 1561–1593 (2022).
Xue, Y., Guo, Y., Cui, H. & Zhou, Z. Catalyst Design for Electrochemical Reduction of CO2 to Multicarbon Products. Small Methods 5, e2100736 (2021).
Verdaguer-Casadevall, A. et al. Probing the Active Surface Sites for CO Reduction on Oxide-Derived Copper Electrocatalysts. J. Am. Chem. Soc. 137, 9808–9811 (2015).
Feng, X., Jiang, K., Fan, S. & Kanan, M. W. A Direct Grain-Boundary-Activity Correlation for CO Electroreduction on Cu Nanoparticles. ACS. Cent. Sci. 2, 169–174 (2016).
Choi, C. et al. Highly active and stable stepped Cu surface for enhanced electrochemical CO2 reduction to C2H4. Nat. Catal. 3, 804–812 (2020).
Zhang, J. et al. Reconstructing two-dimensional defects in CuO nanowires for efficient CO2 electroreduction to ethylene. Chem. Commun. 57, 8276–8279 (2021).
Gu, Z. et al. Efficient Electrocatalytic CO2 Reduction to C2+ Alcohols at Defect-Site-Rich Cu Surface. Joule 5, 429–440 (2021).
Kim, S., Shin, D., Park, J., Jung, J. Y. & Song, H. Grain Boundary-Rich Copper Nanocatalysts Generated from Metal-Organic Framework Nanoparticles for CO2 -to-C2+ Electroconversion. Adv. Sci. 10, e2207187 (2023).
Li, C. W. & Kanan, M. W. CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 134, 7231–7234, (2012).
Pang, Y. et al. Efficient electrocatalytic conversion of carbon monoxide to propanol using fragmented copper. Nat. Catal. 2, 251–258 (2019).
Cheng, D. et al. The nature of active sites for carbon dioxide electroreduction over oxide-derived copper catalysts. Nat. Commun. 12, 395 (2021).
Lum, Y., Yue, B., Lobaccaro, P., Bell, A. T. & Ager, J. W. Optimizing C–C Coupling on Oxide-Derived Copper Catalysts for Electrochemical CO2 Reduction. J. Phys. Chem. C 121, 14191–14203 (2017).
Kibria, M. G. et al. A Surface Reconstruction Route to High Productivity and Selectivity in CO2 Electroreduction toward C2+ Hydrocarbons. Adv. Mater. 30, e1804867 (2018).
Jung, H. et al. Electrochemical Fragmentation of Cu2O Nanoparticles Enhancing Selective C-C Coupling from CO2 Reduction Reaction. J. Am. Chem. Soc. 141, 4624–4633 (2019).
Kim, J. et al. Branched Copper Oxide Nanoparticles Induce Highly Selective Ethylene Production by Electrochemical Carbon Dioxide Reduction. J. Am. Chem. Soc. 141, 6986–6994 (2019).
Choi, W. et al. Microenvironments of Cu catalysts in zero-gap membrane electrode assembly for efficient CO2 electrolysis to C2+ products. J. Mater. Chem. A 10, 10363–10372 (2022).
Jiang, Y. et al. Structural Reconstruction of Cu2O Superparticles toward Electrocatalytic CO2 Reduction with High C2+ Products Selectivity. Adv. Sci. 9, e2105292 (2022).
Vavra, J., Shen, T. H., Stoian, D., Tileli, V. & Buonsanti, R. Real-time Monitoring Reveals Dissolution/Redeposition Mechanism in Copper Nanocatalysts during the Initial Stages of the CO2 Reduction Reaction. Angew. Chem. Int. Ed. Engl. 60, 1347–1354 (2021).
Mom, R. V. et al. Assessment of the Degradation Mechanisms of Cu Electrodes during the CO2 Reduction Reaction. ACS Appl. Mater. Interfaces 15, 30052–30059 (2023).
Sassenburg, M., Kelly, M., Subramanian, S., Smith, W. A. & Burdyny, T. Zero-Gap Electrochemical CO2 Reduction Cells: Challenges and Operational Strategies for Prevention of Salt Precipitation. ACS Energy Lett. 8, 321–331 (2023).
Ge, L. et al. Electrochemical CO2 reduction in membrane-electrode assemblies. Chem 8, 663–692 (2022).
Liang, S., Altaf, N., Huang, L., Gao, Y. & Wang, Q. Electrolytic cell design for electrochemical CO2 reduction. J. CO2 Util. 35, 90–105 (2020).
Kim, D., Chae, Y., Lee, U., Kim, W. & Won, D. H. Exploring the recent developments in membrane electrode assembly electrolyzer for the conversion of CO2 to CO. Curr. Opin. Electrochem. 39, 101295 (2023).
Weng, L.-C., Bell, A. T. & Weber, A. Z. A systematic analysis of Cu-based membrane-electrode assemblies for CO2 reduction through multiphysics simulation. Energy Environ. Sci. 13, 3592–3606 (2020).
Wang, L. et al. Tunable intrinsic strain in two-dimensional transition metal electrocatalysts. Science 363, 870–874 (2019).
Huang, J. et al. Potential-induced nanoclustering of metallic catalysts during electrochemical CO2 reduction. Nat. Commun. 9, 1–9 (2018).
Zhang, R. et al. Highly stability Cu+ species in hollow Cu2O nanoreactors by modulating cavity size for CO2 electroreduction to C2+ products. Chem. Eng. J. 461, 142052 (2023).
Lv, X. et al. Grain refining enables mixed Cu+/Cu0 states for CO2 electroreduction to C2+ products at high current density. Appl. Catal. B: Environ. 324, 122272 (2023).
Li, H. et al. C2+ Selectivity for CO2 Electroreduction on Oxidized Cu-Based Catalysts. J. Am. Chem. Soc. 145, 14335–14344 (2023).
Weitzner, S. E. et al. Toward Engineering of Solution Microenvironments for the CO2 Reduction Reaction: Unraveling pH and Voltage Effects from a Combined Density-Functional-Continuum Theory. J. Phys. Chem. Lett. 11, 4113–4118 (2020).
Louisia, S. et al. The presence and role of the intermediary CO reservoir in heterogeneous electroreduction of CO2. Proc. Natl. Acad. Sci. 119, 1–9 (2022).
Yang, K., Kas, R., Smith, W. A. & Burdyny, T. Role of the Carbon-Based Gas Diffusion Layer on Flooding in a Gas Diffusion Electrode Cell for Electrochemical CO2 Reduction. ACS Energy Lett. 6, 33–40 (2021).
Choi, W. et al. Origin of Hydrogen Incorporated into Ethylene during Electrochemical CO2 Reduction in Membrane Electrode Assembly. ACS Energy Lett. 7, 939–945 (2022).
Kovalev, M. K., Ren, H., Zakir Muhamad, M., Ager, J. W. & Lapkin, A. A. Minor Product Polymerization Causes Failure of High-Current CO2-to-Ethylene Electrolyzers. ACS Energy Lett. 7, 599–601 (2022).
Wheeler, D. G. et al. Quantification of water transport in a CO2 electrolyzer. Energy Environ. Sci. 13, 5126–5134 (2020).
Chae, Y. et al. Tailoring electrochemical CO2 reduction via substrate-induced gas diffusion. J. Mater. Chem. A 11, 7025–7033 (2023).
Newville, M. et al. IFEFFIT: interactive XAFS analysis and FEFF fitting. EJ. Synchrotron Radiat. 8, 322–324 (2001).
Ravel, B. & Gallagher, K. Atomic structure and the magnetic properties of Zr-doped Sm2Co17. Phys. Scr. 606, T115 (2005).
Newville, M., Līviņš, P., Yacoby, Y., Rehr, J. J. & Stern, E. A. Near-edge X-ray-absorption fine structure of Pb: A comparison of theory and experiment. Phys. Rev. B 47, 14126 (1993).
Ankudinov, A. L., Ravel, B., Rehr, J. J. & Conradson, S. D. Real-space multiple-scattering calculation and interpretation of X-ray-absorption near-edge structure. Phys. Rev. B 58, 7565 (1998).
Acknowledgements
This work was supported by ‘Nano & Materials Technology Development Program’ (No. RS-2024-00406517) and ‘Carbon Upcycling Project for Platform Chemicals’ (No. 2022M3J3A10500533) through the National Research Foundation (NRF) funded by the Ministry of Science and ICT, Republic of Korea, and a KIST institutional project. XAS analysis by E.L. and W.S.D. was supported by the Laboratory Directed Research and Development Program of the Lawrence Berkeley National Laboratory under the U.S. Department of Energy Contract (No. DE-AC02-05CH11231).
Author information
Authors and Affiliations
Contributions
W.C. and Y.C. contributed equally to this work, and mainly performed the experimental work. E.L. and W. S. D. contributed to analysis of XAS experiments and data process. D.K., H.S.O., J.H.K., D.K.L., and U.L. contributed to the system development and data analysis. W.C., Y.C., E.L., and D.H.W. wrote the manuscript with contributions from all authors. D.H.W. supervised this study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yuhang Wang, Alessandro Longo, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Choi, W., Chae, Y., Liu, E. et al. Exploring the influence of cell configurations on Cu catalyst reconstruction during CO2 electroreduction. Nat Commun 15, 8345 (2024). https://doi.org/10.1038/s41467-024-52692-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52692-w
This article is cited by
-
Using pseudo-steady-state operation to redefine stability in CO2 electrolysis
Nature Chemical Engineering (2025)
-
Self-healing Cu single-atom catalyst for high-performance electrocatalytic CO2 methanation
Nature Communications (2025)
-
Accelerated dissolution of iridium anode in the presence of organic compounds
Nature Communications (2025)
-
Cu supraparticles with enhanced mass transfer and abundant C-C coupling sites achieving ampere-level CO2-to-C2+ electrosynthesis
Nature Communications (2025)
