
Abstract
Optimizing metal catalyst structures to achieve desired states is vital for efficient surface reactions, yet remains challenging due to the lack of well-defined precursor materials and weak metal-support interaction. Palladium-based catalysts, when not properly tailored for complete methane oxidation exhibit insufficient performance. Herein, we fabricate Pd oxide nano-clusters supported on SSZ-13 using atomic ions with strong metal-support interaction (SMSI). Steam treatment of Pd/SSZ-13 transforms Pd particles into ions and induces SMSI. Subsequently, CO reduction and O2 oxidation yield mildly sintered Pd oxide nano-clusters firmly anchored on extra-framework Alpenta sites of SSZ-13, facilitating superior activity. The robustness from SMSI prevents irreversible deactivation, and water-resistance by complete dehydration suppresses reversible degradation in wet conditions. This catalyst exhibits high performance in bench-scale reactions using monolith catalysts, ensuring applicability for industrial methane abatement. The results demonstrate that sequential treatment to Pd/SSZ-13 offers a promising approach for tailoring metal structures to enable high-performance methane oxidation.
Similar content being viewed by others

Introduction
Designing the optimum and robust structure of supported metal is the ultimate goal to achieve high performance in heterogeneous catalysis1,2,3. Since Jones et al. reported the atom trapping method to prepare the single-atom catalysts in 20164, it has been used as a precursor to design the catalysts with a desired structure5,6,7. A prerequisite for effective structural control by post-treating the atomically dispersed metal on the support is a strong metal-support interaction (SMSI)8,9,10. Such SMSI suppresses excessive sintering and enables precise structural control of supported metals. Since Tauster et al. first reported a phenomenon termed SMSI in 197811, it has received tremendous attention due to its unique advantages in controlling the structure12,13. Typically, the formation of SMSI is facilitated between metal and reducible oxide support by high-temperature treatment under reducing atmospheres or ultrahigh vacuum14,15. In addition, steam treatment with water vapor at high temperatures (>700 °C) has been demonstrated to induce the redispersion of metal species and stabilize the dispersed atoms on the support, forming SMSI16,17. Particularly, Pd particles could be favorably transformed into atomic Pd ions on the SSZ-13 support through steam treatment18,19.
The pressing crisis of global warming and air pollution poses an imminent threat. Given that the shipping industry facilitates approximately 90% of world trade, its influence on atmospheric environment is directly tied to maritime transport. Compared to heavy fuel oil, liquefied natural gas (LNG) significantly reduces NOx, SOx, and particulate matter emissions, while also achieving a more than 20% reduction in CO2 emissions20. However, a notable challenge for LNG-fueled ships is unburned methane (CH4) slip, which has a global warming potential 28 times greater than CO2 over a 100-year period21,22. Thus, complete CH4 oxidation catalysts are becoming increasingly important, and Pd-based catalysts have been extensively studied as promising candidates23,24. However, they often exhibit inadequate activity at low temperatures and experience severe deactivation due to unrefined and labile Pd phases25,26. Particularly, Pd catalysts are prone to reversible poisoning and irreversible Pd deformation in the presence of moisture primarily due to competitive adsorption and insufficient metal-support interaction, respectively, posing a significant obstacle to practical applications27,28. This challenge is critical given the urgent need to comply with impending regulations on CH4 emission from LNG-fueled ships.
In this work, we designed Pd oxide nano-clusters on SSZ-13 support as an optimum catalyst for complete CH4 oxidation. SSZ-13 possesses small pores, strong acid strength, controllable water-resistance, and hydrothermal stability, making it a suitable support for tailoring the Pd structure and applying it in exhaust aftertreatments29,30. Atomic Pd ions were prepared through steam treatment of Pd/SSZ-13. Subsequent CO reduction and O2 oxidation transformed Pd ions into Pd oxide nano-clusters, forming SMSI with the external surface of SSZ-13. The Pd oxide nano-cluster catalyst was employed for complete CH4 oxidation with superior activity at low temperatures. Moreover, its robustness and water resistance, derived from SMSI and complete dehydration, endowed high durability against water-induced deactivation. It overcame the limitations of Pd oxide particles, which are readily deteriorated by moisture, or atomic Pd ions, which are less active. Furthermore, a monolith catalyst was prepared and applied in a bench-scale reaction to ensure feasibility for practical applications.
Results
Pd oxide nano-clusters strongly interacted with SSZ-13 support
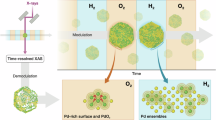
The procedure for designing the Pd oxide nano-clusters on SSZ-13 support is illustrated in Fig. 1a. Initially, the Pd/SSZ-13 catalyst (as-made), produced using the ion-exchange method, was subjected to steam treatment (ST) at 750 °C to uniformly disperse the Pd species and induce the SMSI between Pd and SSZ-13. Subsequently, the Pd ions underwent mild sintering to form nano-clusters on the external surface of SSZ-13 through CO reduction (ST-CO). Finally, the Pd oxide nano-clusters on SSZ-13 support were prepared after O2 oxidation (ST-CO-O2), anchored on extra-framework Alpenta sites of SSZ-13. Thermally treated Pd/SSZ-13 (T) without steam and its CO-O2 treated sample (T-CO-O2) were also prepared to reveal the role of steam treatment in tailoring the Pd structure into the Pd oxide nano-clusters.

a Synthetic procedure of Pd oxide nano-clusters on SSZ-13 support. Pd morphology characterizations of as-made, ST, and their CO-O2 treated catalysts through (b) cryogenic H2-TPR spectra, (c) 27Al NMR spectra, (d) k3-weighted Fourier transformed-EXAFS R-space results (Pd K edge), and (e) DRIFTS results obtained upon CO adsorption.
Even after steam or thermal treatment at 750 °C for 24 h, the Brunauer-Emmett-Teller (BET) surface area remained similar to that of the as-made catalyst (Table 1). Additionally, CO reduction and O2 oxidation did not affect the surface area of the Pd/SSZ-13 catalysts. The X-ray diffraction (XRD) patterns in Supplementary Fig. 1 confirm that the SSZ-13 structure did not change after various heat treatments. The actual Pd content was consistently close to 1 wt% for all Pd/SSZ-13 catalysts, as confirmed by inductively coupled plasma-optical emission spectroscopy (ICP-OES) analysis (Table 1). However, the dispersion and particle size of Pd changed significantly depending on the treatment conditions. While the as-made Pd/SSZ-13 showed 35.4% dispersion (3.1 nm), both ST and T samples exhibited 99.7% and 99.3% dispersion, respectively, with Pd sizes smaller than 1.0 nm. This indicates that Pd species were dispersed on the SSZ-13 support during steam and thermal treatments, consistent with widely known phenomena18,31,32. When CO-O2 treatment was employed, the Pd particle size increased, with the as-made-CO-O2 sample showing 22.0% dispersion (5.1 nm). Pd in the Pd/SSZ-13 catalyst is known to undergo sintering when exposed to a CO reducing atmosphere at high temperatures33. The formation of Pd carbonyl induces the escape of Pd from the zeolite pores, resulting in the sintering of Pd on the external surface of SSZ-13. Surely, Pd particle size increased upon CO-O2 treatment for both ST and T samples. However, the final Pd morphologies were distinctly different for each sample, despite their similar Pd dispersion before CO-O2 treatment. ST-CO-O2 showed 85.7% Pd dispersion with a 1.3 nm Pd size, whereas T-CO-O2 showed 37.1% Pd dispersion with a 3.0 nm size. Notably, for the ST-CO-O2, the extreme sintering of Pd was suppressed.
In the cryogenic H2-temperature programmed reduction (TPR) results (Fig. 1b), the as-made Pd/SSZ-13 displayed a strong H2 consumption peak at -9 °C with a small negative peak at 55 °C. The strong H2 consumption peak at lower temperatures is attributed to the reduction of Pd oxide to Pd and then Pd hydride, whereas the negative peak is ascribed to the release of H2 during the decomposition of Pd hydride34. This demonstrates that Pd oxide particles primarily exist in the as-made sample. However, steam treatment led to significant changes in the H2-TPR results. While both peaks at -9 and 55 °C were hardly identified, a broad H2 consumption peak centered at 97 °C was noticeably observed. The broad H2 consumption peak at higher temperatures is attributed to the reduction of Pd ions in the zeolite18,35. Thus, the Pd oxide particles were finely dispersed to ionic Pd through steam treatment. The T sample showed a similar reduction peak of ionic Pd as in the ST sample, but the reduction temperature was much lower at 55 °C (Supplementary Fig. 2a). The more stabilized Pd species can be reduced at the higher temperature36,37, thus, it can be inferred that steam treatment induced strong interaction as well as Pd dispersion.
After CO-O2 treatment, the H2-TPR spectra varied significantly depending on the sample. As-made-CO-O2 and T-CO-O2 showed strong H2 consumption peaks at -19 and -7 °C, respectively, derived from large Pd oxide particles. However, a much smaller peak appeared at a higher temperature of 21 °C for the ST-CO-O2 sample, suggesting the presence of smaller Pd oxide nano-clusters that were strongly interacted with SSZ-13, distinct from Pd oxide particles. The Pd ions in the ST sample were stabilized more strongly than those in the T sample, therefore, they were mildly sintered during CO-O2 treatment. The SMSI in the ST and ST-CO-O2 was further investigated by quantifying the H2-TPR peak (Supplementary Fig. 3 and Supplementary Table 1). The content of reduced Pd in the sample was calculated using the net amount of H2 molecules consumed during H2-TPR. The ST and ST-CO-O2 exhibited a reduced Pd content of 0.531 and 0.256 wt% among the loaded 1 wt% Pd, which is significantly lower than the other samples because SMSI stabilized the Pd species, thereby lowering their tendency to be reduced during H2-TPR.
The location of Pd species was investigated by comparing the surface concentration of Pd, calculated from the X-ray photoelectron spectroscopy (XPS) results (Table 1). The as-made sample exhibited 0.39 at% of surface Pd, but the surface concentration decreased to 0.20 and 0.21 at% after steam and thermal treatments, respectively, due to the dispersion of Pd species in the SSZ-13 support. After CO-O2 treatment, the surface Pd ratio considerably increased to 0.73-0.77 at%, suggesting an exposure of Pd species on the external surface of SSZ-13. Additionally, the changes in CO adsorption properties after toluene exposure were explored (Supplementary Fig. 4). Toluene has strong adsorption affinity for Pd surfaces, which blocks the CO adsorption on Pd surfaces38,39. Given that toluene molecules have a kinetic diameter of 0.58 nm, exceeding the pore size of SSZ-13 (0.38 nm), they cannot enter SSZ-13 pores. Therefore, CO molecules cannot adsorb on the Pd species on the external surface of SSZ-13 after toluene exposure, whereas CO can still adsorb on Pd species within the SSZ-13 pores. The as-made sample exhibited CO peaks with 8.1 μmol∙gcat-1 of chemisorbed CO after toluene exposure. In contrast, CO-O2 treated samples displayed no identifiable CO peaks and nearly zero μmol∙gcat−1 of chemisorbed CO, confirming the exposure of Pd species on the external surface after CO-O2 treatment. Thus, steam treatment dispersed and effectively stabilized the ionic Pd species, and the subsequent CO-O2 treatment created small-sized Pd oxide nano-clusters on the external surface of SSZ-13, forming the SMSI.
In the solid-state 27Al magic angle spinning nuclear magnetic resonance (MAS NMR) spectra (Fig. 1c), as-made exhibited a strong resonance from the tetra-coordinated framework Al (Altetra) at a chemical shift of 58 ppm. However, a significant decline in the signal intensity of Altetra was observed for ST and ST-CO-O2, indicating framework dealumination during the steam treatment40. This dealumination increased the amount of extra-framework Al, including penta-coordinated Al (Alpenta) and octa-coordinated Al (Alocta) at 32 and 0 ppm, respectively. Although thermal treatment induced some dealumination (Supplementary Fig. 2b), steam treatment resulted in more extensive dealumination. The Brønsted acid sites with bridging silanol groups (Si-OH-Al) in zeolites, based on Alpenta, are known for their anchoring capability for Pd species32,41. Consequently, Pd oxide nano-clusters were firmly anchored on Alpenta sites, abundantly formed by steam treatment, leading to the formation of the SMSI.
The geometric structures were examined by extended X-ray absorption fine structure (EXAFS) analyses (Fig. 1d). Supplementary Fig. 5 shows the EXAFS fitting results, with the best fit values provided in Supplementary Table 2. For the as-made Pd/SSZ-13, the coordination number was 4.0 for the Pd-O interaction, and 1.8 (short) and 3.8 (long) for the Pd-O-Pd interaction. However, the Pd-O-Pd interaction disappeared after steam treatment, leaving only a coordination number of 4.2 for the Pd-O interaction, confirming Pd ion structures. When the as-made sample was treated with CO-O2, the coordination number was 3.0 for the Pd-O interaction, 1.5 for the Pd-Pd interaction, and 2.1 (short) and 4.3 (long) for the Pd-O-Pd interaction. Thus, Pd in the as-made sample were sintered, forming the Pd-Pd interaction. However, for the ST-CO-O2 catalyst, the coordination number was 3.3 for the Pd-O interaction, and 0.5 (short) and 1.1 (long) for the Pd-O-Pd interaction, without a noticeable peak from the Pd-Pd interaction. This indicates that the ST-CO-O2 consisted of Pd oxide nano-clusters, distinct from the large particles. The T sample showed typical results from Pd ions, similar to the ST sample (Supplementary Fig. 2c, d). However, T-CO-O2 exhibited that the coordination number was 2.9 for the Pd-O interaction, 1.1 for the Pd-Pd interaction, and 1.5 (short) and 3.2 (long) for the Pd-O-Pd interaction, derived from the large particles like those in the as-made-CO-O2 sample.
The structural changes of Pd upon steam and CO-O2 treatments were further investigated using diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) as depicted in Fig. 1e. CO probe molecules preferentially adsorb in bridge and hollow modes on larger Pd particles due to their extended surface area, while adopting a linear mode of adsorption on smaller Pd species. For the as-made Pd/SSZ-13, linear CO peaks at 2147 and 2119 cm−1 from Pd2+-(CO), and CO peaks at 2098 and 1942 cm−1 adsorbed on Pd oxide particles in linear and bridge modes, respectively, were observed40. After steam treatment, the bridge peak was eliminated, while other linear CO peaks notably appeared. The peaks at 2183 and 2160 cm−1 are assigned to Pd2+-(CO)2 and [Pd(OH)]+-CO, respectively, and the peaks at 2142 and 2122 cm−1 are attributed to Pd2+(OH)-CO42. For the T sample, there were no CO peaks adsorbed on Pd oxide particles at 2098 and 1942 cm−1, similar to the ST sample. However, it exhibited different linear CO peaks at 2147 and 2119 cm−1 (Supplementary Fig. 2e). For steam treatment, the dispersed atomic Pd ions exhibited a morphology associated with hydroxyl groups, which was clearly distinct from that observed in the thermal treatment. Hydroxyl groups can adsorb onto the metal atoms and then contribute to the dispersion17, leading to strong Pd stabilization on the SSZ-13 support.
In the as-made-CO-O2 and T-CO-O2, linear peaks decreased, while a bridge peak increased, derived from CO adsorption on largely sintered Pd oxide particles. However, ST-CO-O2 showed markedly different results, with very large linear peaks and a tiny bridge peak, which can be attributed to mildly sintered Pd oxide nano-clusters. Additionally, these peaks were shifted to higher wave numbers because CO molecules more weakly adsorbed on the strongly stabilized nano-clusters than on large particles43. The surface hydroxyl groups, formed during steam treatment, were removed during the sequential CO reduction at 700 °C and O2 oxidation at 500 °C, which was confirmed by the temperature programmed desorption (TPD) results of ST and ST-CO-O2 (Supplementary Fig. 6). While the ST exhibited a large H2O peak from the desorption of surface hydroxyl groups, the ST-CO-O2 showed no H2O peak. Therefore, Pd ions in ST were effectively stabilized by hydroxyl groups, leading to their mild sintering to form Pd oxide nano-clusters, which strongly interacted with Alpenta sites on the external surface of SSZ-13 support, through sequential ST-CO-O2 treatment.
The high-resolution transmission electron microscopy (HRTEM) images (Fig. 2a–d and Supplementary Fig. 7) clearly illustrate the distinct Pd structure on the SSZ-13 following various treatments. The as-made, as-made-CO-O2, and T-CO-O2 samples contained numerous Pd oxide particles with approximate diameters of 2.8, 5.1, and 3.0 nm, respectively (Table 1 and Supplementary Fig. 8). The average Pd sizes closely align with the values estimated from pulsed CO chemisorption except for as-made sample. The Pd particles within the SSZ-13 pores are prone to Al decoration, which blocks the surface sites29. In the as-made sample, some Pd species were present within the pores (Supplementary Fig. 4). Thus, the average Pd size from pulsed CO chemisorption (3.1 nm) was larger than size from TEM images (2.8 nm) due to the hindrance of CO adsorption. The ST and T samples did not reveal any identifiable Pd species in the HRTEM images. However, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) mapping images (Fig. 2e and Supplementary Figs. 9a, b and 10) demonstrate that the atomic Pd ions were evenly dispersed on the SSZ-13 support. Remarkably, the ST-CO-O2 sample exhibited small Pd oxide nano-clusters and a uniform size distribution with an average size of 1.3 nm (Fig. 2d, f and Supplementary Figs. 7d, 8c, and 9c, d).

HRTEM images of (a) as-made, (b) as-made-CO-O2, (c) ST, and (d) ST-CO-O2 catalysts supported on SSZ-13. HAADF-STEM images of (e) ST and (f) ST-CO-O2. White circles indicate atomic Pd ions.
The oxidation states of Pd/SSZ-13 catalysts were investigated through XPS and X-ray absorption near-edge structure (XANES) analyses. The XPS peaks were deconvoluted into peaks representing Pd4+, Pd2+, and Pd0, and each atomic fraction was estimated from the area ratios (Fig. 3a–d). The as-made sample entirely consisted of Pd2+, however the ST sample contained 15.6% Pd4+ and 84.4% Pd2+. As the Pd species were dispersed and stabilized during steam treatment, some extent of over-oxidation to Pd4+ occurred. When the CO-O2 treatment was conducted, ST-CO-O2 consisted almost entirely of Pd2+, whereas as-made-CO-O2 possessed 76.3% Pd2+ and 23.7% Pd0. In Supplementary Fig. 11a, b, the T sample contained a lower Pd4+ fraction (6.2%) than the ST sample, due to the absence of strong stabilization by hydroxyl groups. Also, sintered Pd oxide particles of the T-CO-O2 sample showed 89.8% Pd2+ and 10.2% Pd0. After H2-TPR up to 300 °C, the ST and ST-CO-O2 samples exhibited notably lower Pd0 ratios of 38.9% and 28.5%, respectively, compared to other samples where Pd species predominantly existed in a metallic state (Supplementary Fig. 12). The content of reduced Pd estimated from XPS closely matched the H2-TPR results (Supplementary Table 1). This impeded Pd reduction in ST and ST-CO-O2 confirms the SMSI induced by steam treatment. In the XANES spectra (Fig. 3e, f and Supplementary Fig. 11c, d), all catalysts had high white-line intensities, similar to PdO, but there were slight differences in the order of ST > T > as-made, ST-CO-O2 > T-CO-O2 > as-made-CO-O2, in agreement with XPS results. Therefore, ST-CO-O2 had certainly different geometric and electronic structures from other Pd/SSZ-13 catalysts. Steam treatment induced Pd dispersion and SMSI, and the following CO-O2 treatment enabled the preparation of Pd oxide nano-clusters, anchored on the external surface Alpenta sites of SSZ-13 support, with a Pd2+ valence state.

Pd 3d XPS spectra of (a) as-made, (b) as-made-CO-O2, (c) ST, and (d) ST-CO-O2. In the XPS spectra gray dots are raw data and gray lines are fitting results. Pd K edge XANES results of as-made, as-made-CO-O2, ST, and ST-CO-O2 catalysts: (e) whole spectra and (f) enlarged spectra.
Complete CH4 oxidation using Pd oxide nano-cluster catalysts
Due to concerns regarding global warming, there is an urgent need for the development of catalysts to purify exhaust CH4 from LNG-fueled ships. However, many catalysts have shown insufficient activity and, particularly, have suffered from severe deactivation by moisture25,26. Figure 4a,b illustrates the light-off activities for complete CH4 oxidation at a gas hourly space velocity (GHSV) of 200,000 mL gcat−1 h−1. Under dry conditions (Fig. 4a), the as-made catalyst achieved a 50% CH4 conversion (T50) at 309 °C, outperforming the Pd/γ-Al2O3 reference catalyst (T50 = 335 °C). Following steam treatment, the ST catalyst exhibited significantly lower activity, however, its activity was greatly enhanced after CO-O2 treatment, achieving a T50 of 281 °C for ST-CO-O2. Conversely, the CO-O2 treatment adversely affected the as-made catalyst, resulting in decreased activity with a T50 of 325 °C for as-made-CO-O2. Generally, moisture in the feed gas interferes with catalytic complete CH4 oxidation, leading to light-off curves being shifted to higher temperatures (Fig. 4b, c). Pd/γ-Al2O3 was mostly degraded with a ∆T50 of 70 °C, and both as-made and as-made-CO-O2 catalysts also exhibited significantly lower activity with a ∆T50 of approximately 60 °C. Notably, however, ST-CO-O2 exhibited superior activity and the least degradation under wet conditions with a ∆T50 of 31 °C and a T50 of 312 °C, close to the dry condition activity of the as-made catalyst. The T sample exhibited similar activity to the ST catalyst, whereas T-CO-O2 performed much worse than ST-CO-O2 with T50 values of 302 °C (dry) and 360 °C (wet), and a ∆T50 of 58 °C (Supplementary Fig. 13). Further investigation into the effect of Pd oxidation state was conducted by measuring the activity of the ST-CO catalyst without the final O2 treatment (Supplementary Fig. 14). Under wet conditions, ST-CO showed intermediate activity between ST and ST-CO-O2, with a T50 of 398 °C. It had 83.5% dispersion (1.4 nm), similar to that of ST-CO-O2 but consisted of 27.3% Pd2+ and 72.7% Pd0. Therefore, the catalytic performance for complete CH4 oxidation was also dependent on the Pd oxidation state, and the superior activity of ST-CO-O2 could be attributed to both its small nano-clusters and the Pd2+ valence state.

Light-off curves of CH4 conversion of as-made, as-made-CO-O2, ST, ST-CO-O2, and Pd/γ-Al2O3 catalysts under (a) dry and (b) wet conditions. c The temperatures at which CH4 conversion reached 50% (T50) for dry and wet reactions. Durability tests at 350 °C for (d) 100 h long-term under wet conditions and (e) 5-cycles humid switch on/off (2 h each) mode for a total of 20 h using as-made, as-made-CO-O2, ST-CO-O2, and Pd/γ-Al2O3 catalysts. f Light-off and (g) isotherm at 350 °C reactions using ST-CO-O2 under various moisture contents. h Heating-cooling and (i) repeated reactions up to 850 °C (inset: up to 450 °C) using as-made, as-made-CO-O2, and ST-CO-O2 catalysts under wet conditions. The reactions were performed using 60 mg of each catalyst, diluted with 540 mg of quartz sand. Total feed flow rate was 200 mL min−1, maintaining a GHSV of 200,000 mL gcat−1 h−1. The feed gas is composed of 5000 ppm CH4, 15% O2, 5% CO2, and 5% H2O (for wet condition reaction) in a N2 balance.
To elucidate the relationship between the location of Pd species and catalytic performance, the ST-H2-O2 sample was prepared using H2 reduction instead of CO reduction because Pd species tend to sinter within the pores under H2 atmosphere33. Indeed, the CO adsorption property of ST-H2-O2 was barely affected after toluene exposure, suggesting that most Pd species were located within the pores of SSZ-13 (Supplementary Fig. 15). The ST-H2-O2 had Pd oxide nano-clusters similar to those of ST-CO-O2 (Supplementary Fig. 16 and Supplementary Table 3). However, when both catalysts were employed to complete CH4 oxidation under wet conditions (Supplementary Fig. 17), ST-H2-O2 exhibited significantly lower activity (T50 = 338 °C) compared to ST-CO-O2 (T50 = 312 °C). The surface active sites of Pd within the SSZ-13 pores were blocked by Al decoration, resulting in lower CH4 oxidation performance29. This Al decoration was also evident in the CO adsorption results (Supplementary Fig. 15). Despite having similar sizes of Pd oxide nano-clusters, ST-H2-O2 showed a smaller amounts of adsorbed CO molecules (59.8 μmol gcat−1) compared to ST-CO-O2 (80.5 μmol gcat−1), attributable to surface site blockage by Al decoration. Thus, the Pd oxide nano-clusters on the external surface of SSZ-13, achieved through the ST-CO-O2 sequential treatment, represent the optimum and most active structure for complete CH4 oxidation.
Intrinsic activities of Pd/SSZ-13 catalysts were investigated (Supplementary Fig. 18 and Supplementary Table 4). The activation energies and turnover frequency (TOF) values were calculated when the CH4 conversion was below 20% to avoid mass transfer limitations. In this region, the reaction rates were nearly identical at both space velocities of 200,000 and 400,000 mL gcat−1 h−1, achieved by changing the total flow rate from 200 to 400 mL·min−1 while keeping a fixed catalyst amount of 60 mg (Supplementary Fig. 19). Under dry conditions at 250 °C, the ST-CO-O2 catalyst exhibited a reaction rate of 229.4 μmolCH4·gPd−1·s−1 and a TOF value of 28.5 × 10−3 s−1. However, other Pd/SSZ-13 showed no more than 68.0 μmolCH4·gPd−1·s−1 of reaction rate and 19.5·10−3 s−1 of TOF. When the activation energies were estimated, the values were found to be 132.4, 144.1, 130.8, and 123.0 kJ mol−1 for as-made, as-made-CO-O2, T-CO-O2, and ST-CO-O2, respectively. Therefore, the ST-CO-O2 is the intrinsically most active for complete CH4 oxidation. This excellent activity became even more pronounced under wet conditions. The ST-CO-O2 achieved a reaction rate of 240.1 μmolCH4 gPd−1 s−1 and a TOF value of 29.8·10−3 s−1, whereas other catalysts showed no more than 19.7 μmolCH4 gPd−1 s−1 of reaction rate and 5.6 × 10−3 s−1 of TOF at 285 °C. Additionally, the activation energy of ST-CO-O2 slightly increased to 129.1 kJ mol−1 under wet conditions, however, other catalysts showed a significant increase in activation energy, with values ranging 144.2 to 172.8 kJ·mol−1.
To study the mechanism, in situ DRIFTS experiments were conducted using ST-CO-O2 and as-made-CO-O2 catalysts (Supplementary Fig. 20). The IR peaks were assigned according to previous in situ DRIFTS studies on Pd catalysts for complete CH4 oxidation44,45. As the temperature increased, the CH4 peaks at 1304 and 3014 cm−1 gradually disappeared, while CO2 peaks at 2270–2390 cm−1 emerged. For the ST-CO-O2, the reaction began at a lower temperature of 225 °C, with small peaks at 3200–3800 cm−1 from surface hydroxyl groups, suggesting effective CH4 oxidation with resistance to water poisoning. Conversely, the as-made-CO-O2 showed significant peaks from accumulated hydroxyl groups, inhibiting complete CH4 oxidation with a higher onset temperature of 325 °C. Furthermore, for ST-CO-O2, CH3 intermediate peaks at 1457 and 2930 cm−1 appeared at 225 °C, followed by a formate peak (1340 cm−1) at 250 °C. As the temperature reached 300 °C, formate species transitioned to carbonate species at 1380, 1430, and 1480 cm−1. The CH3 and formate peaks diminished beyond 350 and 400 °C, respectively, due to their instability. The as-made-CO-O2 exhibited a similar trend, but intermediates appeared at higher temperatures. Based on these findings, a model for the complete CH4 oxidation over the ST-CO-O2 was developed (Supplementary Fig. 21). The Pd oxide nano-clusters are highly active, particularly under wet conditions, efficiently oxidizing CH4 through CH3, formate, and carbonate intermediates with less water poisoning.
Durability under wet conditions is one of the most demanding yet challenging tasks in complete CH4 oxidation catalysts. We evaluated the long-term durability of as-made, as-made-CO-O2, and ST-CO-O2 compared to the Pd/γ-Al2O3 catalyst at 350 °C for 100 h (Fig. 4d). While as-made, as-made-CO-O2, and Pd/γ-Al2O3 catalysts experienced severe deactivation, notably, ST-CO-O2 maintained about 85% of CH4 conversion under wet conditions. Catalytic durability was further investigated through alternate dry/wet cycles of CH4 oxidation at 350 °C (Fig. 4e). For the ST-CO-O2 catalyst, the CH4 conversion decreased to about 85% under wet conditions but fully recovered to about 95% under dry conditions during the five cycles of humid switch on/off mode. This implies that there is only reversible water inhibition but no irreversible deactivation for the ST-CO-O2 catalyst. On the other hand, as-made, as-made-CO-O2, and Pd/γ-Al2O3 suffered severely from not only reversible water inhibition but also irreversible deactivation under wet conditions. The CH4 conversion decreased rapidly when moisture was introduced to the feed gas, and the catalytic performance continued to deteriorate under wet conditions which was not fully restored in dry conditions. As the moisture content increased from 3% to 10%, the light-off curves of ST-CO-O2 gradually shifted to higher temperatures, and the CH4 conversion at 350 °C decreased (Fig. 4f) because the water inhibition became more severe. However, the CH4 conversion of ST-CO-O2 was fairly stable under each moisture content, and it completely recovered upon returning to dry conditions without any irreversible deactivation during both isotherm and iterative light-off tests (Fig. 4g and Supplementary Fig. 22).
Figure 4h shows the light-off curves of as-made, as-made-CO-O2, and ST-CO-O2 as the temperature was ramped from 200 to 850 °C, then cooled back to 200 °C. The CH4 conversion of the as-made and as-made-CO-O2 samples transiently decreased at temperatures between 600 and 750 °C during the downward cooling because the active Pd oxide decomposes to the thermodynamically stable Pd metal, which is much less active21. Moreover, a permanent activity loss was confirmed even after the transient degradation disappeared. However, ST-CO-O2 exhibited considerably stable activity over the entire range of temperatures without any deactivation. The light-off curve during the cooling process was shifted to lower temperatures (by approximately 26 °C) compared to that of the heating process. Such hysteresis is typical for exothermic CH4 oxidation and has been attributed to the surface temperature exceeding the gas inlet temperature due to the heat released at the catalyst surface46,47. Furthermore, wet condition reaction was conducted iteratively; the reaction temperature was elevated up to 850 °C and subsequently cooled back to 200 °C (Fig. 4i). This cycle was repeated five times. The CH4 conversion remained relatively consistent for the ST-CO-O2 catalyst, whereas the activity of the as-made-CO-O2 diminished with repeated reactions, even only up to 450 °C. Additionally, the catalytic performance of ST-CO-O2 remained stable even after hydrothermal aging at 750 °C for 24 h in 10% H2O/air (Supplementary Fig. 23). Thus, Pd oxide nano-clusters, anchored on external surface Alpenta sites of SSZ-13, exhibited high durability under harsh conditions during complete CH4 oxidation.
Robust and water-resistant Pd oxide nano-clusters for practical use
In complete CH4 oxidation, catalysts are susceptible to deactivation by moisture through two main phenomena: (i) fast and reversible inhibition by water poisoning and (ii) slow and irreversible loss of active sites27,28. Remarkably, the Pd oxide nano-clusters did not experience irreversible deactivation at all, whereas as-made and as-made-CO-O2 underwent irreversible deterioration as shown in Fig. 4. The conversion of CH4 in wet conditions is known to induce the deformation of Pd structures, leading to the irreversible loss of active sites27,28. Therefore, we investigated the structural changes in Pd species after long-term CH4 oxidation under wet conditions. The SSZ-13 structure did not change for all catalysts, as described in the XRD (Supplementary Fig. 24a, b). However, large peaks at 33.9° and 42.1° appeared for the as-made and as-made-CO-O2 samples after the long-term reaction due to the formation of large-sized Pd oxide particles. In the DRIFTS spectra (Supplementary Fig. 24c), the adsorbed CO peaks on the as-made and as-made-CO-O2 significantly decreased, and in particular, the decrease in the linear peak was noticeable after the long-term reaction due to the sintering of Pd particles. For ST-CO-O2, however, there was no meaningful difference, implying no significant change in the Pd structure. Steam treatment provided strong anchoring sites of extra-framework Alpenta, leading to the formation of the robust Pd oxide nano-clusters with SMSI.
The as-made and as-made-CO-O2 exhibited considerable changes in the EXAFS (Supplementary Fig. 24d and Supplementary Table 5). The coordination numbers were 3.2 and 3.4 (short), and 6.5 and 6.9 (long) for the Pd-O-Pd interaction, indicating much larger Pd oxide particles than before the long-term reaction. However, for ST-CO-O2, EXAFS results were almost unchanged. In the TEM images (Supplementary Figs. 24e–g and 25), large Pd oxide particles ranging from 20 to 40 nm were primarily observed for the as-made and as-made-CO-O2, whereas Pd oxide nano-clusters were evenly dispersed on the SSZ-13 support (with 84.9% Pd dispersion) without any sintered particles for the ST-CO-O2 catalyst. Moreover, the Pd oxide nano-clusters displayed remarkable robustness, preserving their structure without any Pd deformation after iterative reactions up to 850 °C and hydrothermal aging at 750 °C, as confirmed in Supplementary Fig. 26. After long-term reaction, all catalysts exhibited the oxidation state of Pd2+, and there was no change in ST-CO-O2 compared to before the long-term reaction (Supplementary Fig. 27). Consequently, the irreversible deformation of the Pd structure was effectively prevented for ST-CO-O2 because the Pd oxide nano-clusters strongly interacted with Alpenta sites on the external surface of SSZ-13 support.
Water poisoning obstructs the access of CH4 molecules and inhibits the initiation of the reaction, resulting in reversible degradation. To directly compare the degree of water poisoning, H2O-TPD was performed (Fig. 5a). The samples were exposed to water vapor (3% H2O/He) for 30 min, and the desorbed water was monitored while being heated to 400 °C under a He stream. The desorbed water peaks of ST and ST-CO-O2 were smaller than those of as-made and as-made-CO-O2, indicating the improved resistance to water poisoning by steam treatment. Steam treatment can increase the water-resistance of Pd species by dehydrating Pd-hydrate ions48. Additionally, steam treatment led to extensive dealumination (Fig. 1c), which was evidently associated with an increase in water-resistance of SSZ-1348. This water-resistance of SSZ-13 can further enhance the water-resistance of Pd species, particularly when there is a SMSI49,50. The water-resistance of Pd/SSZ-13 catalysts was assessed using CO-DRIFTS in the presence of water vapor (Fig. 5b, c). In the as-made and as-made-CO-O2, CO adsorption peaks were significantly reduced, indicating surface Pd sites blocking by water poisoning. In contrast, the ST-CO-O2 retained substantial CO adsorption peaks, suggesting enhanced water resistance.

Comparison of water poisoning on as-made, as-made-CO-O2, ST-CO-O2, and ST-CO-N2-O2 catalysts through (a) H2O-TPD, (b) DRIFTS results obtained upon CO adsorption, (c) DRIFTS results in the presence of water vapor, and (d) UV-vis spectra. Catalytic performance of ST-CO-N2-O2 for complete CH4 oxidation. e Light-off curves of CH4 conversion under dry, 5% H2O, and 10% H2O conditions. f Durability tests at 350 °C for 5-cycles humid switch on/off (2 h each) mode for a total of 20 h (inset: 100 h long-term tests under wet conditions). The reactions were performed using 60 mg of each catalyst, diluted with 540 mg of quartz sand. Total feed flow rate was 200 mL·min−1, maintaining a GHSV of 200,000 mL gcat−1 h−1. The feed gas is composed of 5000 ppm CH4, 15% O2, 5% CO2, and 5% or 10% H2O (for wet condition reaction) in a N2 balance.
However, the Pd oxide nano-clusters still experienced water poisoning, leading to reversible degradation. As shown in Fig. 4, although the Pd oxide nano-cluster catalyst did not suffer from irreversible deactivation, its performance was reversibly degraded due to competitive adsorption between reactants and water molecules. Ultraviolet-visible (UV-vis) spectra revealed that the Pd species in the ST-CO-O2 were not fully dehydrated, as indicated by a peak at 410 nm−1, attributed to a Pd-hydrate complex (Fig. 5d). To achieve further dehydration, N2 treatment at 300 °C for 1 h was additionally conducted (ST-CO-N2-O2) during the preparation of the Pd oxide nano-clusters. The Pd oxide nano-clusters of ST-CO-N2-O2 exhibited structures nearly identical to those of ST-CO-O2, as confirmed by DRIFTS, EXAFS, HAADF-STEM, and XPS analyses (Fig. 5b, Supplementary Fig. 28, and Supplementary Table 6). However, in the UV-vis spectra, the ST-CO-N2-O2 sample showed only a peak at 450 nm−1, derived from Pd2+, with no observed Pd-hydrate peak. Additionally, no significant desorbed water peak was observed (Fig. 5a), and CO adsorption was not hindered by water vapor (Fig. 5c, d), indicating that complete dehydration significantly enhanced the water resistance of the Pd oxide nano-clusters.
When the ST-CO-N2-O2 catalyst was employed to complete CH4 oxidation (Fig. 5e), water-induced degradation of the Pd oxide nano-clusters was entirely mitigated under wet conditions, even with either 5% or 10% H2O. The Pd oxide nano-clusters, particularly in the ST-CO-N2-O2 catalyst, demonstrated a distinct advantage in activity under wet conditions, compared to high-performance Pd-based catalysts in the literature (Supplementary Table 7). Moreover, the ST-CO-N2-O2 consistently achieved approximately 95% CH4 conversion at 350 °C during five cycles of alternating dry/wet tests and 100 h long-term reactions under wet conditions (Fig. 5f). Consequently, the water-resistance of the Pd oxide nano-clusters, endowed by steam treatment, SMSI, and complete dehydration, effectively suppressed the interference of water molecules with reactants adsorption. This indicates that the robust and water-resistant Pd oxide-nano clusters did not experience either irreversible deactivation or reversible degradation during complete CH4 oxidation under wet conditions.
The powder catalysts pose challenges for practical applications in exhaust CH4 removal, especially under high space velocity conditions due to issues such as pressure drop, mass transfer limitations, catalyst loss, and handling complexities51. From the standpoint of catalyst commercialization, monolith catalysts offer significantly greater suitability. Therefore, we prepared monolith catalysts of as-made, as-made-CO-O2, ST-CO-O2, ST-CO-N2-O2, and Pd/γ-Al2O3. Scanning electron microscopy (SEM) images of catalyst slurries reveal that the ball-milled particles exhibited uniform particle size distributions with an average diameter of 0.45–0.48 μm (Fig. 6a–e). The catalyst slurries were coated onto the cordierite honeycomb substrates with 1-inch diameter, 1-inch length, and 400 channels per square inch (cpsi) (Supplementary Fig. 29). The SEM images in Fig. 6f–j demonstrate that the catalysts were efficiently coated onto the walls of the channels of the honeycomb substrate, exhibiting a uniform catalyst layer without visible cracks or pores. The thickness of the coating layer was approximately 5–10 μm.

SEM images and particle size distribution analysis results of ball-milled catalyst slurries of (a) as-made, (b) as-made-CO-O2, (c) ST-CO-O2, (d) ST-CO-N2-O2, and (e) Pd/γ-Al2O3. SEM images of cross-sectional microstructures of monolith catalysts of (f) as-made, (g) as-made-CO-O2, (h) ST-CO-O2, (i) ST-CO-N2-O2, and (j) Pd/γ-Al2O3. k Light-off activity and (l) 100 h long-term durability tests at 350 °C using as-made, as-made-CO-O2, ST-CO-O2, ST-CO-N2-O2, and Pd/γ-Al2O3 monolith catalysts under wet conditions. m Durability test with NO and SO2 at 350 °C using ST-CO-O2 and ST-CO-N2-O2 monolith catalysts under wet conditions. The reactions were performed using 1-inch diameter, 1-inch length, and 400 cpsi of each catalyst and 4.3 L min−1 of total feed flow at the same GHSV of 20,000 h−1. The feed gas is composed of 5000 ppm CH4, 15% O2, 5% CO2, 5% H2O, 200 ppm NO (when applicable), and 10 ppm SO2 (when applicable) in a N2 balance.
The prepared monolith catalysts were placed in a laboratory bench reactor, and complete CH4 oxidation was performed under wet conditions. The loading amount of catalyst for the monolith was 100 g · L−1, and the corresponding GHSV was 20,000 h−1 from powder reaction conditions, resulting in a total flow rate of 4.3 L·min−1. The composition of the feed gas was the same as that for the powder catalyst test. Figure 6k shows that all monolith catalysts exhibited quite similar activities compared to those of the powder samples. The Pd oxide nano-cluster, particularly ST-CO-N2-O2, is the most active, with a T50 of 282 °C under both dry and wet conditions, demonstrating high resistance to water-induced degradation. Additionally, Pd oxide nano-cluster monolith catalysts were highly durable against irreversible deactivation during long-term reactions under wet conditions (Fig. 6l). CH4 conversions were stably maintained at 350 °C for 100 h without any activity loss, whereas the other catalysts experienced severe deactivation.
The exhaust gas of LNG-fueled ships contains tiny amounts of NO and SO2, which can deteriorate the catalysts52,53. Therefore, we introduced 200 ppm NO and 10 ppm SO2 into the feed gas to verify the feasibility of the Pd oxide nano-cluster catalysts for practical applications (Fig. 6m). The ST-CO-O2 and ST-CO-N2-O2 monolith catalysts stably maintained their high performance despite the addition of 200 ppm NO. When 10 ppm SO2 was additionally introduced into the feed gas, the ST-CO-O2 catalyst experienced reversible degradation, with CH4 conversion dropping to approximately 45% at 350 °C. The zeolite-supported Pd catalysts are generally susceptible to sulfur poisoning27,53. However, the ST-CO-N2-O2 monolith catalyst exhibited high tolerance against sulfur-induced degradation, maintaining high performance with approximately 95% CH4 conversion. The water-resistant nature of the Pd oxide nano-clusters likely repels polar molecules such as H2O and SO2, facilitating the preferential adsorption of non-polar reactants (CH4 and O2). Therefore, the robust and water-resistant Pd oxide nano-clusters on the SSZ-13 support overcame reversible degradation and irreversible deactivation issues in complete CH4 oxidation under real exhaust gas conditions from LNG-fueled ships, ensuring their feasibility for practical applications.
In conclusion, steam treatment transformed Pd particles into atomic Pd ions and induced SMSI with SSZ-13. Leveraging the beneficial effects of atomic Pd ions and SMSI, Pd nano-clusters, distinct from Pd particles, were fabricated through mild sintering via CO reduction. Notably, without steam treatment or its substitution with thermal treatment, the process did not yield the Pd nano-cluster structure. The O2 oxidation modulated valence states to Pd2+, resulting in Pd oxide nano-clusters strongly anchored on Alpenta sites of SSZ-13 external surface. When various Pd/SSZ-13 catalysts were employed for complete CH4 oxidation, the Pd oxide nano-clusters exhibited superior activity, indicating that they represent the optimum and most active structures. The robustness of Pd oxide nano-clusters, derived from SMSI, prevented the irreversible loss of active sites by Pd structure deformation. Also, the water-resistance, further enhanced by complete dehydration, suppressed reversible degradation caused by water and sulfur poisoning. Therefore, the Pd oxide nano-cluster catalyst achieved high durability in complete CH4 oxidation under harsh conditions. However, the Pd oxide particles displayed severe deactivation caused by both reversible water poisoning and irreversible Pd deformation. The excellent catalytic characteristics of Pd oxide nano-clusters were also observed in bench-scale tests using monolith catalysts. Their robustness and water resistance enabled high-performance CH4 oxidation without any deactivation, even in the presence of moisture and sulfur. The Pd oxide nano-cluster catalyst holds significant potential for practical exhaust CH4 abatement in LNG-fueled ship applications, while providing the sequential treatment strategies for designing the desired structures of metal catalysts.
Methods
Catalysts preparation
Pd/SSZ-13 powder catalysts were synthesized using the ion-exchange method. Commercial NH4-SSZ-13 zeolite powder (CCG-CHA-1) with a Si to Al ratio of 15, confirmed by inductively coupled plasma-optical emission spectroscopy (ICP-OES), was obtained from China Catalyst Holding. 990 mg of SSZ-13 powder was dispersed in 100 mL of deionized water, and then 44.2 mg of Pd(II) nitrate solution from SNS Corp., with a concentration of 22.63%, was introduced into the SSZ-13 solution, resulting in a loading amount of 1 wt% Pd. The solution was stirred at 80 °C for 15 h, and then filtered and washed several times with deionized water until the pH of the solution reached 7.0. The obtained powder was dried at 120 °C for 4 h and then subjected to calcination in static air at 500 °C (2 °C·min−1) for 4 h, resulting in as-made sample. The Pd/SSZ-13 powder catalysts underwent steam treatment in a quartz glass cell by flowing gas containing 10% H2O and 15% O2 in a N2 balance at 750 °C for 24 h, resulting in a sample denoted as ST. When the Pd/SSZ-13 catalysts were thermally treated without steam, the resulting powder was denoted as T sample. Subsequently, as-made, ST and T samples were reduced in a quartz glass cell by flowing 1% CO/N2 at 700 °C for 5 min and then further oxidized by flowing 10% O2/N2 at 500 °C for 30 min, producing as-made-CO-O2, ST-CO-O2 and T-CO-O2 samples, respectively. The ST-H2-O2 sample was prepared by flowing 1% H2/N2 instead of 1% CO/N2 at 700 °C for 5 min during the reduction step. The N2 treatment was additionally conducted by flowing N2 at 300 °C for 1 h between CO reduction and O2 oxidation, resulting in ST-CO-N2-O2 sample. The reference Pd/γ-Al2O3 catalyst was synthesized via the impregnation method. Initially, 990 mg of commercial γ-Al2O3 powder (MI307, Solvay) was dispersed in 100 mL of deionized water in a round-bottom evaporation flask. Then, a solution containing 44.2 mg of Pd(II) nitrate was put into the γ-Al2O3 solution, resulting in an impregnation of 1 wt% Pd. Subsequently, the evaporation flask was placed in a rotary evaporator (Hei-VAP, Heidolph), and the solution was completely evaporated at 80 °C under vacuum with continuous stirring. The obtained powder was then subjected to drying at 120 °C for 4 h, followed by calcination in static air at 500 °C (2 °C·min−1) for 4 h.
The prepared powder catalysts were applied onto a cordierite substrate (Corning) with a honeycomb shape, which has 1-inch diameter, 1-inch length, and 400 channels per square inch (cpsi). To obtain the catalyst slurry with uniform particle size, 30 g of either Pd/SSZ-13 or Pd/γ-Al2O3 powder was dispersed in 49.5 mL of deionized water and subjected to milling with zirconia balls for 12 h. Commercial boehmite binder (Disperal P2, Sasol) 0.93 g was then introduced to this slurry. The resulting catalyst slurry was then transferred into a container installed in a custom-made coating machine. The honeycomb substrate was positioned on a rack beneath the container, and a low vacuum was applied beneath the honeycomb substrate, facilitating the wash-coating process. During this process, the Pd/SSZ-13 or Pd/γ-Al2O3 slurry was evenly distributed onto the inner walls of the honeycomb substrate channels. Subsequently, the wash-coated sample underwent drying at 120 °C for 4 h and calcination at 500 °C (2 °C·min−1) for 4 h. This process yielded Pd/SSZ-13 and Pd/γ-Al2O3 monolith catalysts, with a loading of approximately 100 g·L−1 in each sample.
Characterizations
The Brunauer-Emmett-Teller (BET) surface area of the Pd/SSZ-13 catalyst was estimated using volumetric adsorption equipment (BELSORP MINI X, MicrotracBEL). X-ray diffraction (XRD) analysis was conducted using a high-resolution powder X-ray diffractometer (SmartLab, Rigaku) with Cu Kα radiation to explore the crystalline structure of the Pd/SSZ-13 catalysts. ICP-OES analysis was performed using an iCAP7400DUO (Thermo Scientific) instrument to identify the content of Pd in the catalysts. The dispersion of Pd in the Pd/SSZ-13 catalyst was determined by a pulsed CO chemisorption method using a BELCAT-II (MicrotracBEL). Initially, 30 mg of catalyst powder was placed in a quartz cell and pretreated with a 50 mL·min−1 flow of He at 100 °C for 1 h. After cooling to 30 °C, CO pulses in a He stream were then injected every minute until CO chemisorption onto the Pd reached saturation. Cryogenic H2-temperature programmed reduction (TPR) was carried out using a BELCAT-II (MicrotracBEL) equipped with a thermal conductivity detector (TCD). The powder sample (60 mg) was placed in a U-shaped quartz cell and pretreated in the following sequence: (i) 50 mL·min−1 of 5% O2/He (150 °C, 30 min), (ii) 50 mL min−1 of Ar (−120 °C, 10 min), and (iii) 50 mL·min−1 of 5% H2/Ar (−120 °C, 5 min). Subsequently, the sample was heated to 300 °C with a ramping rate of 10 °C·min−1 under a 50 mL·min−1 flow of 5% H2/Ar. For toluene exposure on the catalysts, initially, 30 mg of catalyst powder underwent treatment with a 50 mL·min−1 flow of He at 100 °C for 1 h. The catalyst was then subjected to 30 mL·min−1 flow of 1000 ppm toluene/He at 50 °C for 30 min, followed by evacuation with a 50 mL·min−1 flow of He for 30 min. Toluene gas was generated by bubbling He through a container containing pure toluene in an ice water bath. After toluene exposure, the catalyst was immediately employed for diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and pulsed CO chemisorption analyses.
Solid-state 27Al magic angle spinning nuclear magnetic resonance (MAS NMR) experiments were conducted at room temperature using a Bruker AVANCE II+ 400 MHz NMR spectrometer (at KBSI Seoul Western Center). A 4 mm MAS probe at a spinning rate of 12 kHz was employed to collect the data. X-ray absorption spectroscopy (XAS) of the Pd K edge was performed at the 10 C Wide XAFS beamline of the Pohang Light Source (PLS). The Pd K edge spectra were acquired in fluorescence mode using a passivated implanted planar silicon (PIPS) detector (Canberra). Concurrently, reference Pd foil measurements were conducted to calibrate each sample. The XAS data were processed and fitted to obtain X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) results utilizing the ATHENA and ARTEMIS software programs. The fits of the Pd K edge were executed on the first and second coordination shells with a radial range (R-range) of 1.0–3.5 Å using Fourier-transformed k3-weighted χ(k) functions within a k-range of 3.0-13.9 Å−1. Coordination numbers were determined by maintaining a fixed amplitude reduction factor (S02) value, acquired from fitting the reference Pd foil. The R-factor, representing the relative error between the fitted values and experimental data, was assessed. A value of R-factor less than 0.05 is typically indicative of a good fit. Although this value does not guarantee absolute accuracy, it can serve as a skeptical threshold for the fitting process.
DRIFTS was conducted utilizing a praying mantis DRIFTS accessory (Harrick Scientific) installed on a Nicolet iS-50 (Thermo Scientific). The 15 mg of catalyst powder was put into a sample cup and then placed inside the DRIFTS room. Prior to analysis, the sample underwent pretreatment by passing a 100 mL·min−1 stream of He at 100 °C for 1 h to remove any water and impurities. Following pretreatment, the sample was cooled to room temperature under a flow of He. A 1% CO gas, diluted with He, was introduced to the sample at room temperature for 10 min to serve as a probe molecule. To study the effect of water vapor, a gas mixture containing 1% CO and 2% H2O in a He balance was used. The water vapor was generated by passing He through a container of water. DRIFTS spectra were then collected under a He purge to eliminate any interfering gaseous CO peaks. The existence of hydroxyl groups on the surface of the ST and ST-CO-O2 catalysts was explored using temperature programmed desorption (TPD) analysis. The samples were placed in a U-shaped quartz cell immediately after preparation and heated to 400 °C with a ramping rate of 5 °C·min−1 under a 60 mL·min−1 of He flow. The desorbed H2O (m/z = 18) was monitored by mass spectrometry.
High-resolution transmission electron microscopy (HRTEM) images of the Pd/SSZ-13 catalysts were collected using a Tecnai G2 F30 S-Twin (FEI) operating at an accelerating voltage of 300 kV. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) mapping images were obtained using a Spectra Ultra (Thermo Fisher) with an accelerating voltage of 300 kV. The obtained HRTEM and HAADF-STEM images were analyzed using DigitalMicrograph Software (GMS 3, GATAN). For X-ray photoelectron spectroscopy (XPS) analysis of the electronic properties of Pd in the Pd/SSZ-13 catalysts, a K-Alpha+ XPS System (Thermo Scientific) was utilized. A monochromated Al Kα X-ray source (energy of 1486.8 eV) with a power of 72 W (6 mA × 12 kV) was employed. The binding energies were corrected using the maximum intensity of the reference C 1 s signal at 284.6 eV.
Water adsorption on the catalyst was analyzed by H2O-TPD using a BELCAT-B instrument (BEL) equipped with a TCD. A 60 mg of powder sample underwent pretreatment by passing 60 mL·min−1 of He at 100 °C for 30 min, followed by cooling to room temperature. Subsequently, 60 mL·min−1 of 3% water vapor in He was introduced to the sample using a syringe pump at room temperature for 30 min. After purging the sample with 60 mL·min−1 of He flow for 60 min, the sample was heated to 400 °C with a ramping rate of 5 °C·min−1 under 60 mL·min−1 of He flow. Ultraviolet-visible diffuse reflectance spectroscopy (UV-vis) was conducted using a Cary 5000 (Agilent) for powder samples at room temperature. Scanning electron microscopy (SEM) images of the slurries and monolith catalysts of Pd/SSZ-13 and Pd/γ-Al2O3 were acquired using a JSM-6610LV (JEOL) operating at an accelerating voltage of 30 kV. The particle size of ball-milled catalyst slurries was determined using a LA-350 laser scattering particle size distribution analyzer (Horiba).
Catalytic reactions
The complete CH4 oxidation was performed using powder catalysts in a quartz glass fixed-bed flow reactor at atmospheric pressure. A total flow rate of 200 mL·min−1 of feed gas was introduced to 60 mg of catalyst to achieve a gas hourly space velocity (GHSV) of 200,000 mL·gcat−1 · h−1. To avoid heat transfer limitations, 60 mg of catalyst was diluted with 540 mg of inert quartz sand (99%, Sigma-Aldrich). To exclude influence on catalytic performance from impurities, the quartz sand was calcined at 875 °C for 5 h prior to dilution. The diluted mixture, consisting of 10 wt% catalyst, underwent pretreatment at 500 °C for 1 h under 15% O2/N2 atmosphere, after which the reactor was cooled down to 200 °C. Following pretreatment, the catalyst was exposed to the feed gas mixture at 200 °C for 5 h to stabilize before evaluating the catalytic activity. The feed gas composition consisted of 5000 ppm CH4, 15% O2, 5% CO2, and 5% H2O in a N2 balance, with moisture introduced by vaporizing the supplied water using a syringe pump. The reaction conditions were determined based on the exhaust gas composition from LNG-fueled four-stroke marine engines and from the literature in Supplementary Table 7. The reactor was heated to the target reaction temperature with a ramping rate of 2 °C·min−1. The product gases were analyzed in real-time using an online Fourier transform-infrared spectrometer (FT-IR, Antaris IGS gas analyzer, Thermo Fisher Scientific) equipped with a gas cell (MERCURY, 17 cm path length, 170 mL volume). Additionally, the outlet gases were analyzed using an online gas chromatography (GC) instrument (Younglin 6500GC system) equipped with a Porapak N column (13052-U, Supelco, 10 ft × 1/8 in × 2.1 mm) and a Mol Sieve 13X column (13047-U, Supelco, 3 ft × 1/8 in × 2.1 mm) with a TCD and a flame ionization detector (FID).
The mechanism of complete CH4 oxidation under wet conditions was studied through in situ DRIFTS experiments. The 15 mg of catalyst was heated to 500 °C for 1 h in a 100 mL·min−1 flow of 15% O2/He to remove the surface impurities, then cooled to 200 °C. After acquiring the background spectrum, a total flow rate of 100 mL·min−1 of feed gas was introduced at 200 °C. The feed gas contained 5000 ppm CH4, 15% O2, and 2% H2O balanced with He. The water vapor was generated by passing He through a container of water. The temperature was then increased from 200 to 450 °C at a rate of 5 °C·min−1 under the feed gas flow, pausing every 25 °C for 30 min to record the DRIFTS spectra.
Moreover, the performance of bench-scale complete CH4 oxidation was evaluated using monolith catalysts in an in-house laboratory bench reactor at atmospheric pressure. A total feed gas flow rate of 4.3 L·min−1 was introduced to 1-inch diameter and 1-inch length monolith catalyst, resulting in a GHSV of 20,000 h−1. The monolith catalyst underwent pretreatment at 500 °C for 1 h under 15% O2/N2 atmosphere, after which the reactor was cooled down to 200 °C. Following pretreatment, the catalyst was exposed to the feed gas mixture at 200 °C for 5 h to stabilize before evaluating the catalytic activity. The feed gas composition mirrored the conditions of the powder catalytic reactions, with an additional introduction of 200 ppm NO and 10 ppm SO2 to assess feasibility for practical application. The reactor was heated to the target temperature with a ramping rate of 2 °C·min−1. The product gases were analyzed in real-time using an online CH4/CO/CO2 gas analyzer (VA-5000, Horiba) and a total hydrocarbon gas analyzer (FIA-510, Horiba). The CH4 conversion was calculated using the following equation: ([CH4]inlet – [CH4]outlet) / [CH4]inlet × 100%, where [CH4]inlet and [CH4]outlet represent the CH4 concentration at the inlet and outlet, respectively.
Data availability
The data that support the findings of this study are available within the paper and its Supplementary Information files. All other relevant data supporting the findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Rong, H., Ji, S., Zhang, J., Wang, D. & Li, Y. Synthetic strategies of supported atomic clusters for heterogeneous catalysis. Nat. Commun. 11, 5884 (2020).
Mitchell, S., Qin, R., Zheng, N. & Pérez-Ramírez, J. Nanoscale engineering of catalytic materials for sustainable technologies. Nat. Nanotechnol. 16, 129–139 (2021).
Mitchell, S. & Pérez-Ramírez, J. Atomically precise control in the design of low-nuclearity supported metal catalysts. Nat. Rev. Mater. 6, 969–985 (2021).
Jones, J. et al. Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science 353, 150–154 (2016).
Xu, L. et al. Formation of active sites on transition metals through reaction-driven migration of surface atoms. Science 380, 70–76 (2023).
Jiang, D. et al. Dynamic and reversible transformations of subnanometre-sized palladium on ceria for efficient methane removal. Nat. Catal. 6, 618–627 (2023).
Zhang, Z. et al. Memory-dictated dynamics of single-atom Pt on CeO2 for CO oxidation. Nat. Commun. 14, 2664 (2023).
Pereira-Hernández, X. I. et al. Tuning Pt-CeO2 interactions by high-temperature vapor-phase synthesis for improved reducibility of lattice oxygen. Nat. Commun. 10, 1358 (2019).
Hu, S. & Li, W.-X. Sabatier principle of metal-support interaction for design of ultrastable metal nanocatalysts. Science 374, 1360–1365 (2021).
Xiong, H. et al. Engineering catalyst supports to stabilize PdOx two-dimensional rafts for water-tolerant methane oxidation. Nat. Catal. 4, 830–839 (2021).
Tauster, S. J., Fung, S. C. & Garten, R. L. Strong metal-support interactions. Group 8 noble metals supported on titanium dioxide. J. Am. Chem. Soc. 100, 170–175 (1978).
Wang, L., Wang, L., Meng, X. & Xiao, F.-S. New strategies for the preparation of sinter-resistant metal-nanoparticle-based catalysts. Adv. Mater. 31, 1901905 (2019).
Luo, Z., Zhao, G., Pan, H. & Sun, W. Strong metal-support interaction in heterogeneous catalysts. Adv. Energy Mater. 12, 2201395 (2022).
Zhang, S. et al. Dynamical observation and detailed description of catalysts under strong metal-support interaction. Nano Lett. 16, 4528–4534 (2016).
Frey, H., Beck, A., Huang, X., van Bokhoven, J. A. & Willinger, M. G. Dynamic interplay between metal nanoparticles and oxide support under redox conditions. Science 376, 982–987 (2022).
Jeong, H., Bae, J., Han, J. W. & Lee, H. Promoting effects of hydrothermal treatment on the activity and durability of Pd/CeO2 catalysts for CO oxidation. ACS Catal. 7, 7097–7105 (2017).
Jeong, H. et al. Fully dispersed Rh ensemble catalyst to enhance low-temperature activity. J. Am. Chem. Soc. 140, 9558–9565 (2018).
Ryou, Y. et al. Activation of Pd/SSZ-13 catalyst by hydrothermal aging treatment in passive NO adsorption performance at low temperature for cold start application. Appl. Catal. B 212, 140–149 (2017).
Li, D. et al. Deactivation of Pd/SSZ-13 passive NOx adsorber from the perspectives of phosphorus poisoning and hydrothermal aging. Chem. Eng. J. 446, 136779 (2022).
Yu, Y.-S., Zhang, X., Liu, J.-W., Lee, Y. & Li, X.-S. Natural gas hydrate resources and hydrate technologies: a review and analysis of the associated energy and global warming challenges. Energy Environ. Sci. 14, 5611–5668 (2021).
Cargnello, M. et al. Exceptional activity for methane combustion over modular Pd@CeO2 subunits on functionalized Al2O3. Science 337, 713–717 (2012).
Li, T. et al. Denary oxide nanoparticles as highly stable catalysts for methane combustion. Nat. Catal. 4, 62–70 (2021).
Divins, N. J. et al. Investigation of the evolution of Pd-Pt supported on ceria for dry and wet methane oxidation. Nat. Commun. 13, 5080 (2022).
Oh, J., Boucly, A., van Bokhoven, J. A., Artiglia, L. & Cargnello, M. Palladium catalysts for methane oxidation: old materials, new challenges. Acc. Chem. Res. 57, 23–36 (2024).
Goodman, E. D. et al. Monolayer support control and precise colloidal nanocrystals demonstrate metal-support interactions in heterogeneous catalysts. Adv. Mater. 33, 2104533 (2021).
Lott, P., Casapu, M., Grunwaldt, J.-D. & Deutschmann, O. A review on exhaust gas after-treatment of lean-burn natural gas engines - from fundamentals to application. Appl. Catal. B 340, 123241 (2024).
Friberg, I., Sadokhina, N. & Olsson, L. The effect of Si/Al ratio of zeolite supported Pd for complete CH4 oxidation in the presence of water vapor and SO2. Appl. Catal. B 250, 117–131 (2019).
Mortensen, R. L. et al. Understanding the reversible and irreversible deactivation of methane oxidation catalysts. Appl. Catal. B 344, 123646 (2024).
Cui, Y. et al. Onset of high methane combustion rates over supported palladium catalysts: from isolated Pd cations to PdO nanoparticles. JACS Au 1, 396–408 (2021).
Sinha Majumdar, S. et al. Impact of Mg on Pd-based methane oxidation catalysts for lean-burn natural gas emissions control. Appl. Catal. B 341, 123253 (2024).
Khivantsev, K. et al. Palladium/ferrierite versus palladium/SSZ-13 passive NOx adsorbers: adsorbate-controlled location of atomically dispersed palladium(II) in ferrierite determines high activity and stability. Angew. Chem. Int. Ed. 61, e202107554 (2022).
Wang, Y. et al. The study of Pd-SSZ-13 as low-temperature passive NOx adsorber materials: high dispersal of Pd in small-pore CHA zeolites by thermal treatment. Appl. Catal. B 324, 122254 (2023).
Ryou, Y. et al. Effect of reduction treatments (H2 vs. CO) on the NO adsorption ability and the physicochemical properties of Pd/SSZ-13 passive NOx adsorber for cold start application. Appl. Catal. A 569, 28–34 (2019).
Chen, Z. et al. Investigation of crystal size effect on the NOx storage performance of Pd/SSZ-13 passive NOx adsorbers. Appl. Catal. B 291, 120026 (2021).
Kim, Y. et al. Uniform synthesis of palladium species confined in a small-pore zeolite via full ion-exchange investigated by cryogenic electron microscopy. J. Mater. Chem. A 9, 19796–19806 (2021).
Jeong, H. et al. Highly durable metal ensemble catalysts with full dispersion for automotive applications beyond single-atom catalysts. Nat. Catal. 3, 368–375 (2020).
Ryu, S.-H. et al. Improved resistance to water poisoning of Pd/CeO2 monolithic catalysts by heat treatment for ozone decomposition. Chem. Eng. J. 485, 149487 (2024).
Zhao, S., Hu, F. & Li, J. Hierarchical core-shell Al2O3@Pd-CoAlO microspheres for low-temperature toluene combustion. ACS Catal. 6, 3433–3441 (2016).
Bi, F., Zhang, X., Chen, J., Yang, Y. & Wang, Y. Excellent catalytic activity and water resistance of UiO-66-supported highly dispersed Pd nanoparticles for toluene catalytic oxidation. Appl. Catal. B 269, 118767 (2020).
Zhao, H., Chen, X., Bhat, A., Li, Y. & Schwank, J. W. Insight into hydrothermal aging effect on deactivation of Pd/SSZ-13 as low-temperature NO adsorption catalyst: effect of dealumination and Pd mobility. Appl. Catal. B 286, 119874 (2021).
Wang, Z. et al. Brønsted acid sites based on penta-coordinated aluminum species. Nat. Commun. 7, 13820 (2016).
Chen, D. et al. Unravelling phosphorus-induced deactivation of Pd-SSZ-13 for passive NOx adsorption and CO oxidation. ACS Catal. 11, 13891–13901 (2021).
Tang, H. et al. Strong metal-support interactions between gold nanoparticles and nonoxides. J. Am. Chem. Soc. 138, 56–59 (2016).
Duan, Q. et al. Atomically dispersed palladium-based catalysts obtained via constructing a spatial structure with high performance for lean methane combustion. J. Mater. Chem. A 8, 7395–7404 (2020).
Tian, J. et al. Enhancing methane combustion activity by modulating the local environment of Pd single atoms in Pd1/CeO2 catalysts. ACS Catal. 14, 183–191 (2024).
Amin, A., Abedi, A., Hayes, R., Votsmeier, M. & Epling, W. Methane oxidation hysteresis over Pt/Al2O3. Appl. Catal. A 478, 91–97 (2014).
Xiong, J. et al. Pd-promoted Co2NiO4 with lattice CoONi and interfacial PdO activation for highly efficient methane oxidation. Appl. Catal. B 292, 120201 (2021).
Lee, J. et al. Improving NOx storage and CO oxidation abilities of Pd/SSZ-13 by increasing its hydrophobicity. Appl. Catal. B 277, 119190 (2020).
Losch, P. et al. Modular Pd/zeolite composites demonstrating the key role of support hydrophobic/hydrophilic character in methane catalytic combustion. ACS Catal. 9, 4742–4753 (2019).
Li, L. et al. Integration of Pd nanoparticles with engineered pore walls in MOFs for enhanced catalysis. Chem 7, 686–698 (2021).
Ercolino, G., Karimi, S., Stelmachowski, P. & Specchia, S. Catalytic combustion of residual methane on alumina monoliths and open cell foams coated with Pd/Co3O4. Chem. Eng. J. 326, 339–349 (2017).
Sadokhina, N., Smedler, G., Nylén, U., Olofsson, M. & Olsson, L. The influence of gas composition on Pd-based catalyst activity in methane oxidation - inhibition and promotion by NO. Appl. Catal. B 200, 351–360 (2017).
Zhang, Y. et al. Sulfur poisoning and regeneration of Rh-ZSM-5 catalysts for total oxidation of methane. Appl. Catal. B 277, 119176 (2020).
Acknowledgements
This research was financially supported by the Fundamental Research Program (PNK9750) of the Korea Institute of Materials Science. The experiments at PLS were supported in part by MSIP and POSTECH. The authors thank Prof. Ja Hun Kwak from Ulsan National Institute of Science and Technology for H2O-TPD analysis. Additionally, we appreciate the KAIST Analysis center for Research Advancement (KARA) for characterization equipment.
Author information
Authors and Affiliations
Contributions
S.-H.R. and H.J. conceived and designed the study. S.-H.R. carried out the catalyst preparation and catalytic reaction tests. S.-H.R. and H.J. performed characterizations. S.K. and H.L. participated in part of characterizations. J.-H.C. and H.J. supervised the project. S.-H.R. and H.J. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Eleni Kyriakidou, Daiqi Ye and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ryu, SH., Kim, S., Lee, H. et al. Robust palladium oxide nano-cluster catalysts using atomic ions and strong interactions for high-performance methane oxidation. Nat Commun 15, 8348 (2024). https://doi.org/10.1038/s41467-024-52698-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52698-4
