
Abstract
The intracellular bacterial pathogen Legionella pneumophila utilizes the Dot/Icm system to translocate over 330 effectors into the host cytosol. These virulence factors modify a variety of cell processes, including pathways involved in cell death and survival, to promote bacterial proliferation. Here, we show that the effector LegK3 is a eukaryotic-like Ser/Thr kinase that functions to suppress host apoptosis. Mechanistically, LegK3 directly phosphorylates multiple caspases involved in apoptosis signaling, including Caspase-3, Caspase-7, and Caspase-9. LegK3-induced phosphorylation of these caspases occurs at serine (Ser29 in Caspase-3 and Ser199 in Caspase-7) or threonine (Thr102 in Caspase-9) residues located in the prodomain or interdomain linkers. These modifications interfere with the suitability of the caspases as the substrates of initiator caspases or upstream regulators without impacting their proteolytic activity. Collectively, our study reveals a novel strategy used by L. pneumophila to maintain the integrity of infected cells for its intracellular growth.
Similar content being viewed by others


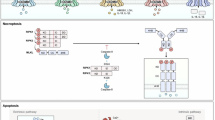
Introduction
Successful pathogens have acquired various strategies to manipulate cellular functions to evade immune clearance and promote infection1. During the complex interactions with their hosts, many intracellular bacterial pathogens utilize specialized protein translocation apparatuses, such as type III and IV secretion systems (T3SS and T4SS)2, to deliver effector proteins into the cytosol of host cells, where they modulate signaling pathways to construct environments permissive for bacterial survival and proliferation1. These effectors regulate host processes via distinct mechanisms, among which induction of post-translational modification (PTM) of critical host signaling proteins represents one of the most effective and widely used mechanisms3. Protein phosphorylation conducted by kinases is arguably the most prevalent and important PTM in cells, which controls virtually every cellular process4,5. This PTM often alters the activity, localization, or stability of the involved proteins, leading to changes in signaling transduction and subsequent physiological responses6. Given the critical roles of protein phosphorylation in cell signaling, it is not unexpected that many bacterial pathogens encode effectors with kinase activity to target important signaling hubs to promote their virulence. For example, the Salmonella SPI-1 substrate SteC is a eukaryotic-like Ser/Thr protein kinase (eSTPK) capable of phosphorylating the actin sequestering protein Hsp27 and the mitogen-activated protein (MAP) kinase MEK, whose activity causes alterations in the structure of the host actin cytoskeleton7,8. Similarly, the T3SS effectors NleH1 and NleH2 of enteropathogenic Escherichia coli are kinases that phosphorylate microvillus protein Eps8 to alter the cytoskeletal composition of attaching and effacing (AE) lesions9.
The facultative intracellular bacterium Legionella pneumophila is the causative agent of Legionnaires’ disease, a potentially fatal pneumonia10. This bacterium utilizes its Dot/Icm T4SS to inject over 330 effector proteins directly into the host to construct an intracellular niche that supports its survival and growth11. These effectors interfere with a wide range of host cell processes to promote the establishment of Legionella-containing vacuole (LCV), a membrane-enclosed compartment that avoids lysosomal degradation and supports bacterial proliferation12. To successfully hijack host functions, many L. pneumophila effectors adopt structural domains typical for proteins of eukaryotes that are involved in protein-protein interactions or in catalyzing distinct activities (e.g., kinases, ubiquitin ligases, guanine nucleotide exchange factors, deubiquitinases, phospholipases). Among these, at least six L. pneumophila effectors, named LegK1-4, LegK7, and Lem28 had been identified as potential protein kinases due to the presence of structural elements similar to kinase domains of eukaryotic proteins and such activity has been detected by biochemical reactions using self-phosphorylation as a readout13,14,15. Of these kinases, LegK1 functions to mimic the host IKK kinase to phosphorylate IκBα on Ser32 and Ser36, thereby activating NF-κB signaling16. LegK2 targets subunits of the ARP2/3 complex to suppress actin nucleation on the LCV, thus contributing to the evasion of the late endosome/lysosome trafficking pathway by the bacterium17. LegK4 induces threonine phosphorylation of HSP70 to inhibit protein synthesis in host cells18 and LegK7 is a mimicry of the host Hippo kinase that phosphorylates the scaffolding protein MOB1, thus altering host gene expression to promote infection14.
Programmed cell death (PCD) pathways, including apoptosis, necroptosis, and pyroptosis are essential in determining the outcome of bacterial infections. The death of infected cells will eliminate the replication niche of the pathogen and thus is disastrous for the pathogen. As a result, intracellular bacterial pathogens have evolved various mechanisms to manipulate host PCDs to accommodate their replication19. Earlier studies suggest that infection by L. pneumophila causes apoptosis via the activation of Caspase-320,21. Later studies reveal that L. pneumophila inhibits infected cells from undergoing apoptosis by either targeting pro-death proteins22 or by activating signaling cascades that induce expression of anti-apoptotic proteins23.
Caspases are a family of cysteine-dependent aspartate-specific proteases that function as central mediators of inflammatory responses and/or apoptosis24. Caspases are synthesized as inactive zymogens consisting of prodomain of variable size followed by large (p20) and small (p10) catalytic subunits25. Apoptotic caspases are functionally sub-categorized into initiator caspases (Caspases-8/−9/−10) and effector (or executioner) caspases (Caspases-3/−6/−7)26. Initiator caspases undergo autoactivation by dimerization-induced conformational changes to release the large and small catalytic subunits, which can cleave and activate executioner caspases26. In this study, we found that executioner Caspase-3 and Caspase-7 are not activated in cells infected by L. pneumophila, and subsequent screening identified the Dot/Icm effector LegK3 as the factor responsible for such inhibition. Further study revealed that this kinase induces phosphorylation on multiple caspases including Caspase-3, Caspase-7, and Caspase-9. Although this modification did not affect the enzymatic activity of these caspases, it impacts their suitability as substrates for upstream initiator caspases. Our results reveal a novel strategy employed by L. pneumophila to ensure the integrity of infected cells for its intracellular replication.
Results
Identification of Dot/Icm effectors involved in inhibition of host cell death
To determine the mechanism of host cell death manipulation by L. pneumophila, we infected FcγRII-expressing HeLa cells with wild-type L. pneumophila for the indicated durations and detected the active forms of Caspase-3 and Caspase-7 and found that these caspases were not activated in infected cells, which was in sharp contrast to cells treated with the cell death inducer staurosporine (STS) (Fig. S1). This observation suggests that, in line with earlier studies22,23, L. pneumophila inhibits host apoptosis.
To identify the Dot/Icm effector responsible for the suppression of caspase activation, we utilized several cluster deletion strains of L. pneumophila, each lacking distinct chromosomal regions27, for similar infection experiments. Notably, only the Δ7 mutant exhibited significantly higher levels of caspase activation than the wild-type strain (Figs. 1A–E and S2). We thus individually examined the ability of effectors carried by the deleted region of strain ∆7 by expressing their GFP fusions in HeLa cells and examined caspase activation28. After STS treatment, expression of LegK3 (Lpg2556) markedly blocked the activation of Caspase-3 and Caspase-7, leading to a marked reduction of cell death (Fig. 1F–H). Other Dot/Icm effectors carried by the missed region in strain ∆7 did not detectably inhibit cell death induced by STS (Fig. 1F–H). These results indicate that LegK3 functions as an apoptosis inhibitor during L. pneumophila infection.

A, B The Δ7 mutant has lost the ability to inhibit apoptosis. HeLa cells were infected with wild-type, dotA-, or one of the L. pneumophila chromosomal deletion mutants (Δ 2, Δ3, Δ4, Δ6, and Δ7) at an MOI of 10 for 3 h. Cells were lysed and the levels of Caspase-3/-7 in the lysates were detected by immunoblotting using antibodies specific for Caspase-3 and Caspase-7 (A). Caspase-3/-7 activity in the supernatant was determined by the substrate Ac-DEVD-pNA (B). C, E. Infection with the L. pneumophila Δ7 mutant caused significantly higher percentage of apoptosis. Cells infected with the indicated L. pneumophila strains for 3 h were determined for apoptosis by measuring the ATP levels (C) or by TUNEL staining (D, E). Representative images of L. pneumophila-infected cells that were TUNEL-positive or -negative (D). For quantitation, at least 100 infected cells were counted for each sample (E). Bar, 5 μm. F–H. LegK3 suppresses host apoptosis. HeLa cells were transfected with plasmids expressing effectors coded in the deleted region of the Δ7 mutant. The activation of Caspase-3/-7 was assessed either by immunoblotting using anti-Caspase-3/-7 antibodies (F) or by measuring the cleavage of the substrate Ac-DEVD-pNA (G). Cell viability of the transfected cells was evaluated by determining the ATP levels (H). Data in A and F are representative from three independent experiments. For B, C, E, G, and H, data shown are mean ± SD from three replicates. Unpaired two-tailed Student’s t tests were performed. Source data are provided as a Source Data file.
The S/T kinase motif of LegK3 is required for its ability to inhibit apoptosis
LegK3 was previously established as a protein kinase owing to the presence of an eSTPK domain13,29,30, but its cellular target remains elusive (Fig. 2A). To investigate whether LegK3’s kinase activity is required to block apoptosis, we constructed a kinase-dead mutant of LegK3 via mutation of D187 in its catalytic loop (HRD187IKPEN) to alanine (referred to as LegK3D/A hereafter). Although expressed at levels similar to the wild-type allele in both HeLa and HEK293 cells, LegK3D/A has lost the ability to inhibit STS- or etoposide (also known as VP-16)-triggered production of mature Caspase-3/−7 (Figs. 2B, C and S3A–C). In line with this, the production of cleaved poly-ADP-ribose polymerase (PARP), one of the cellular targets of activated caspases and a hallmark of apoptosis31, was remarkably reduced in cells expressing wild-type LegK3 but not LegK3D/A (Figs. 2B and S3B). Similarly, LegK3 also suppressed the maturation of Caspase-3/−7 induced by overexpression of pro-death members of the Bcl-2 family, such as BAX or BimEL (Fig. S3E, F). In line with LegK3-mediated Caspase-3/−7 inactivation, the ratio of STS-, VP-16-, or BAX/BimEL-induced apoptotic cell death significantly decreased in samples transfected to express wild-type but not the kinase-dead LegK3 mutant when cell death was determined by measuring the ATP levels, flow cytometry after staining with propidium iodide (PI) and DEVD-GreenNucTM or by TUNEL staining (Figs. 2D, E, S3D, S3G, and S4A–C).

A Predicted domain structure of LegK3 and alignment of its catalytic loop with several known bacterial S/T kinases. The conserved residues are highlighted in red. B, C HeLa cells expressing GFP-LegK3 or GFP-LegK3D/A were stimulated with STS for 4 h, the cell lysates were subjected to immunoblotting with anti-Caspase-3, anti-Caspase-7, and anti-PARP antibodies (B) or measuring Caspase-3/-7 activity using the substrate Ac-DEVD-pNA (C). D, E Flow cytometry analysis of apoptotic cell death of cells expressing LegK3 or LegK3D/A that had been treated with STS. HeLa cells were transfected to produce BFP, BFP-LegK3, or BFP-LegK3D/A for 24 h, cells were either left untreated or treated with 1 μM STS for 4 h. After staining the cells with PI and DEVD-GreenNucTM, samples were analyzed by a flow cytometry under dark. Representative images and quantification of apoptotic rates were shown in (D) and (E), respectively. F–I LegK3 inhibits Caspase-3/-7 activation in L. pneumophila-infected cells. FcγRII-expressing HeLa cells were infected with the indicated L. pneumophila strains for 3 h. Infected cells were either left untreated (F, G) or incubated with 1 μM of STS for 4 h (H–I). The amounts of mature Caspase-3/-7 were assessed by immunoblotting (F, H) or by the cleavage of the substrate Ac-DEVD-pNA (G, I). Data in B, F, and H are representative from three independent experiments. For C, E, G, and I, data shown are mean ± SD from three replicates. Unpaired two-tailed Student’s t tests were performed. Source data are provided as a Source Data file.
Next, we investigated the role of LegK3 in apoptotic status of cells infected by L. pneumophila. To this end, we first measured the gene expression of intracellular L. pneumophila by quantitative real-time PCR (qRT-PCR). Remarkably, transcription of legK3 was detected in bacteria retrieved from wild-type and dotA- L. pneumophila-infected cells (Fig. S4D). In addition, we employed the TEM (β-lactamase) reporter system to visualize Dot/Icm-dependent translocation of LegK328. At 3 h post-infection, approximately 50% of the cells infected with the wild-type L. pneumophila strain expressing TEM-LegK3 exhibited blue fluorescence, which was generated via the cleavage of CCF4/AM by TEM (Fig. S4E, F). By contrast, only green fluorescence was observed in cells similarly infected with the dotA- mutant strain producing TEM-LegK3. These analyses establish that LegK3 is delivered into host cells during L. pneumophila infection in a Dot/Icm-dependent fashion (Fig. S4E, F). We then infected FcγRII-expressing HeLa cells with each of the following L. pneumophila strains: wild-type, dotA-, Lp02ΔlegK3, Lp02ΔlegK3 (pLegK3), and Lp02ΔlegK3 (pLegK3D/A). The level of active p17 of Caspase-3/−7 was higher in cells infected with strain Lp02ΔlegK3 than those infected with the wild-type strain (Figs. 2F, G and S5). Expression of LegK3, but not the LegK3D/A mutant from a plasmid fully restored the capacity of ΔlegK3 strain to suppress Caspase-3/−7 activation (Figs. 2F, G and S5). In addition, cells infected by strain Lp02ΔlegK3 (pLegK3) exhibited resistance to STS-induced Caspase-3/−7 activation (Fig. 2H, I). Consistent with the defect in Caspase-3/−7 activation, the apoptotic rates were significantly higher in cells infected with the Lp02ΔlegK3 mutant than those infected with the wild-type strain (Fig. S4G–K). Similarly, the elevation in apoptosis exhibited by cells infected with the Lp02ΔlegK3 mutant can be completely complemented to levels of wild-type infected samples by expressing LegK3 but not the LegK3D/A mutant (Fig. S4G–K). Thus, the kinase activity of LegK3 is required for its ability to inhibit apoptosis.
LegK3 directly phosphorylates Caspase-3
The requirement of kinase activity for LegK3 to inhibit Caspase-3 activation suggested that LegK3 targets key components of the apoptotic pathway by phosphorylation. When co-expressed in mammalian cells, GFP-LegK3 extensively colocalized with HA-Caspase-3 (Fig. S6). In addition, HA-Caspase-3 co-transfected with GFP-LegK3 in HEK293T cells consistently migrated slower in SDS-PAGE gels. Such retardation did not occur in samples that were co-expressed with LegK3D/A (Fig. 3A). This observation suggests Caspase-3 as a substrate of LegK3. Indeed, probing HA-Caspase-3 isolated from cells co-expressing LegK3 with anti-Phospho-(Ser) antibodies detected phosphorylation signals (Fig. 3A). We determined the LegK3-induced phosphorylation site on Caspase-3 using LC-MS/MS and found that Ser29 is the sole modified residue (Figs. 3B and S7). This site lies between the prodomain and the large subunit of Caspase-3. To confirm phosphorylation on Ser29 by LegK3, we created and co-expressed the Caspase-3S29A mutant with GFP-LegK3 and probed phosphorylation by immunoblotting with phospho-specific antibodies. LegK3-induced phosphorylation no longer occurred in this mutant when co-expressed with LegK3 (Fig. 3A).

A HEK293T cells were transfected to express GFP-LegK3 and HA-Caspase-3, samples transfected with the indicated mutants were also established. After immunoprecipitation with anti-HA agarose, the beads-bound proteins were detected by immunoblotting using anti-HA or anti-Phospho-(Ser) antibodies. B HA-Caspase-3 obtained in (A) by elution with 3xHA peptide was analyzed by mass spectrometry. Extracted ion chromatograms of the Ser29-phosphorylated peptide (IIHGSESMDpSGISLDNSYK) and a control peptide (SGTDVDAANLR) were shown. C Phosphorylation of Caspase-3 by LegK3 in E. coli. Flag-tagged LegK3 or LegK3D/A and His6-tagged Caspase-3* were co-expressed in E. coli. His6-Caspase-3* was obtained by affinity purification. Phosphorylation of Caspase-3* was detected by phospho-protein staining, immunoblotting with anti-Phospho-(Ser) antibodies or native PAGE. Expression of LegK3 was probed by immunoblotting with the Flag antibody. Caspase-3* represents for Caspase-3C163A, a catalytic inactive mutant of Caspase-3 that can be purified as full-length protein from the E. coli expression system. D, E LegK3 targets Caspase-3 for phosphorylation during L. pneumophila infection. HEK293 cells transfected to express the FcγRII receptor and Flag-Caspase-3 were either left uninfected or challenged with the indicated L. pneumophila strains for 3 h. After immunoprecipitation with the Flag antibody, the beads-associated proteins were analyzed by immunoblotting using Flag or Phospho-(Ser)-specific antibodies, respectively (D). Post-exponential bacteria were lysed and probed for the production of LegK3 by immunoblotting using Flag antibody (E). The housekeeping protein ICDH (isocitrate dehydrogenase) was probed as a loading control (E). F Flag-Caspase-3 immuniprecipitated from wild-type and dotA- L. pneumophila infected samples (D) were eluted by 3xFlag peptide and further detected by mass spectrometry. Extracted ion chromatograms of the Ser29-phosphorylated peptide (IIHGSESMDpSGISLDNSYK) and a control peptide (KQIPCIVSMLTK) were shown. Data in A, C–E are representative from three independent experiments. Source data are provided as a Source Data file.
Next, we attempted to use biochemical reactions with recombinant proteins to prove that LegK3 directly phosphorylates Caspase-3. However, we were unable to obtain recombinant LegK3 of high purity owing to extensive degradation in E. coli expression systems. We thus co-expressed Caspase-3C/A in which the catalytic cysteine (C163) was mutated into alanine with LegK3 or LegK3D/A in E. coli, which revealed that His6-Caspase-3C/A co-expressed with LegK3 exhibited a marked molecular weight shift in native PAGE gels and such shift did not occur for samples co-expressed with the kinase-defective mutant (Fig. 3C). Similarly, probing by a phosphoprotein staining agent or immunoblotting with phospho-specific antibodies detected robust phosphorylation signals associated with His6-Caspase-3C/A co-expressed with wild-type but not LegK3D/A (Fig. 3C). Furthermore, Caspase-3S29A can no longer be modified by LegK3 in these assays (Fig. 3C).
We next investigated LegK3-induced phosphorylation of Caspase-3 in cells infected with L. pneumophila. To this end, we transfected HEK293 cells to express the FcγRII receptor and 4xFlag-Caspase-3 and infected these cells with relevant L. pneumophila strains. Flag-Caspase-3 isolated by immunoprecipitation was analyzed for phosphorylation by immunoblotting. Flag-Caspase-3 purified from cells infected with the wild-type L. pneumophila strain displayed phosphorylation signals, but not from cells infected with the dotA- or the Lp02ΔlegK3 mutant (Fig. 3D). Introduction of a plasmid expressing LegK3 into strain Lp02ΔlegK3 allowed the mutant to cause Caspase-3 phosphorylation and similarly expressed LegK3D/A failed to induce the modification (Fig. 3D, E). Finally, mass spectrometric analysis indicated that Ser29 was phosphorylated in Caspase-3 isolated from cells infected with wild-type L. pneumophila (Fig. 3F). No phosphorylation was detected in Caspase-3S29A isolated from cells infected with wild-type L. pneumophila (Fig. 3D). Taken together, these results demonstrate that LegK3 directly induces Caspase-3 phosphorylation on Ser29 during L. pneumophila infection.
Phosphorylation of Caspase-3 at Ser29 by LegK3 blocks its cleavage by initiator caspases
To determine the impact of Ser29 phosphorylation on the function of Caspase-3, we purified the active p17/12 complex of Caspase-3 from the E. coli strain harboring a plasmid carrying Caspase-3s29-C under different conditions. Co-expression of this complex with LegK3 resulted in phosphorylation of p17 as indicated by its retarded migration in acrylamide gels electrophoresis and positive signals in phosphoprotein staining (Fig. S8A). Phosphorylated p17/p12 (P-p17/p12) complex purified by an ion exchange column was used to cleave several established Caspase-3 substrates (Fig. S8B). P-p17/p12 cleaved GSDMD, GSDME, and PARP indistinguishably to its native unmodified counterpart (Fig. S8C–I). Thus, LegK3-catalyzed phosphorylation of Caspase-3 does not affect its enzymatic activity.
Activation of Caspase-3 involves two steps: initiator caspases (−8/−9/−10) cleave procaspase-3 at the interdomain linker (IETD175-S176) to generate an immature p20 subunit with an exposed active Cys163 and a p12 subunit; the p20 subunit subsequently underdoes self-processing at the prodomain linker (ESMD28-S29) to remove the N-terminal fragment, yielding a mature p17 large subunit that forms a complex with the p12 small subunit capable of executing proteolytic activity32. To determine whether LegK3-induced Ser29 phosphorylation of Caspase-3 affects its processing by upstream caspases, we purified phosphorylated Caspase-3C/A co-expressed with wild-type LegK3 from E. coli (Fig. S9) and examined its cleavage by Caspase-8 and Caspase-9. Both initiator caspases cleaved P-Caspase-3C/A at rates significantly lower than those of native Caspase-3C/A (Fig. 4A–D). Similar results were observed when 4xFlag-Caspase-3C/A co-expressed with GFP-LegK3 in HEK293T cells was used as the substrate for recombinant active Caspase-9 (Fig. 4E). Moreover, phosphomimetic mutants of Caspase-3 (S29D, S29Q, and S29E) exhibited markedly lower sensitivity to Caspase-8/-9 than the native form (Figs. 4F–I and S10A–D). Taken together, these results indicate that LegK3-mediated phosphorylation of Caspase-3 at Ser29 made it less cleavable by upstream caspases without detectably impacting its enzymatic activity.

A–D. His6-Caspase-3*, phosphorylated His6-Caspase-3* or His6-Caspase-3*S29A was incubated with His6-Caspase-9140-C (A, B) or His6-Caspase−8 (C, D) for the indicated durations. Cleavage of Caspase-3* was visualized by Coomassie brilliant blue staining (CBB) after SDS-PAGE. Quantitation was determined by measuring the density of the bands corresponding to uncleaved Caspase-3* (B, D). Caspase-3* represents for Caspase-3C163A. Caspase-9140-C expressed in E. coli produces the active form of Caspase-9. E 4xFlag-Caspase-3* purified from HEK293T cells co-transfected with GFP-LegK3 or GFP-LegK3D/A was incubated with His6-Caspase-9140-C. Cleavage of Caspase-3 was detected by immunoblotting. Caspase-3* represents for Caspase-3C163A. F–I Phosphomimetic mutants of Caspase-3* purified from E. coli were reacted with recombinant active Caspase-9140-C and the cleavage was evaluated by CBB staining. Quantitation was determined by measuring the density of the bands corresponding to uncleaved Caspase-3* (G and I). Caspase-3* represents for Caspase-3C163A. Data in A, C, E, F, and H are representative from three independent experiments. For B, D, G, and I, data shown are mean ± SD from three replicates. Unpaired two-tailed Student’s t tests were performed. Source data are provided as a Source Data file.
LegK3 targets multiple caspases involved in the apoptosis pathway by phosphorylation
To study whether LegK3 targets other caspases involved in apoptosis, we first co-expressed cleavage-deficient mutants of Caspase-7/-8-9 with LegK3 in E. coli. Detection by phosphoprotein staining or immunoblotting with phospho-specific antibodies revealed that Caspase-7/-9 but not Caspase-8 can be modified by this bacterial kinase (Fig. 5A). Mass spectrometric analysis identified Ser199 within the interdomain linker (IQAD198-S199) of Caspase-7 and Thr102 within the prodomain linker (KLSKPT102) of Caspase-9 as the sole phosphorylation site, respectively (Figs. 5B, C and S11A, B). Indeed, mutants of Caspase-7 and Caspase-9 with a substitution in Ser199 or Thr102 can no longer be modified by LegK3 (Fig. 5D, E). When co-expressed in mammalian cells, both HA-Caspase-7 and HA-Caspase-9 extensively colocalized with GFP-LegK3 (Fig. S12), and phosphorylation of these caspases by LegK3 was similarly observed in a kinase activity-dependent manner (Fig. 5F, G). By contrast, LegK3-induced phosphorylation was not detected for Caspase-7S199A and Caspase-9T102A when co-expressed with LegK3 in HEK293T cells (Fig. 5F, G).

A His6-Caspase-7*, His6-Caspase-8* or His6-Caspase−9* was purified from E. coli strains co-expressing Flag-LegK3 or Flag-LegK3D/A. After separation by SDS-PAGE, phosphorylation of these caspases was either assessed by Pro-Q diamond phospho-protein staining or analyzed by immunoblotting by phospho-(Ser/Thr)-specific antibodies. Caspase-7*, Caspase-8* and Caspase-9* represent for Caspase-7C186A, Caspase-8C360A, and Caspase-9C287A, respectively. B Extracted ion chromatograms of the Ser199-phosphorylated peptide (GTELDDGIQADpSGPINDTDANPR) and a control peptide (SSFVPSLFSK) from Caspase-7 used in (A). C Extracted ion chromatograms of the Thr102-phosphorylated peptide (LSKPpTLENLTPVVLRPEIR) and a control peptide (TGSNIDCEKLR) from Caspase-9 used in (A). D, E Caspase-7*S199A and Caspase-9*T102A purified from E. coli co-expressing wild-type LegK3 or LegK3D/A were probed by Pro-Q diamond phospho-protein staining or by immunoblotting using anti-Phospho-(Ser/Thr) antibodies. Caspase-7* and Caspase-9* represent for Caspase-7C186A and Caspase−9C287A, respectively. F, G HA-Caspase-7S199A or HA-Caspase−9T102A was co-expressed with GFP-LegK3 or GFP-LegK3D/A in HEK293T cells by transfection. After immunoprecipitation, phosphorylation of Caspase-7/−9 was evaluated by immunoblotting with anti-Phospho-(Ser/Thr) antibodies. Data in A, D, E, F, and G are representative from three independent experiments. Source data are provided as a Source Data file.
We next investigated whether modification of Caspase-7/-9 by LegK3 occurs under infection conditions. To this end, HEK293 cells transfected to produce FcγRII and HA-Caspase-7/-9 were challenged with relevant L. pneumophila strains and phosphorylation was determined in proteins isolated by immunoprecipitation. Phosphorylation of Caspase-7/-9 occurred in samples isolated from cells infected with the wild-type but not the dotA- or the ΔlegK3 mutant L. pneumophila (Fig. S13A, B). Importantly, complementation of the ΔlegK3 strain with wild-type LegK3 but not LegK3D/A by a plasmid led to robust Caspase-7/-9 phosphorylation (Fig. S13A, B). Additionally, mass spectrometric analysis indicated that, similar to samples obtained by co-expression in E. coli, Ser199 and Thr102 of Caspase-7 and Caspase-9 were phosphorylated, respectively (Fig. S13C, D). Taken together, these results demonstrate that, in addition to Caspase-3, Caspase-7 and Caspase-9 are also the physiological targets of LegK3.
The Asp198-Ser199 located within the interdomain linker of Caspase-7 is the cleavage site of upstream initiator Caspases, we thus hypothesized that Ser199 phosphorylation may affect its recognition by Caspase-8 and Caspase-9. To test this, we purified active cysteine-deficient phosphomimetic mutants of Caspase-7 (C186 AS199D, C186AS199E, and C186AS199Q) and determined their cleavage by Caspase-8/-9. The cleavage efficiency of all Caspase-7 phosphomimetic mutants was markedly lower than the native protein (Fig. 6A–H). Caspase-9 is activated by the apoptosis protease-activating factor 1 (Apaf-1), which oligomerizes in response to cytochrome c release and forms a large complex33,34. To determine whether phosphorylation at Thr102 affects Caspase-9 processing, a constitutively active mutant of Apaf-1 (Apaf-11-541)35 was purified from mammalian cells and incubated with either His6-Caspase-9 or its phosphomimetic mutant His6-Caspase-9T102E. Whereas native Caspase-9 was readily cleaved by Apaf-1, as evidenced by the production of mature p35 fragment, little cleavage occurred in reactions containing His6-Caspase-9T102E (Fig. 6I). Corresponding to this observation, expression of wild-type LegK3 but not LegK3D/A in HeLa cells suppressed the maturation of Caspase-9 induced by overexpression of BimEL (Fig. S14). Taken together, these results indicate that phosphorylation of Caspase-7/-9 by LegK3 inhibits their activation by upstream regulators.

A–H Proteins of phosphomimetic mutants of Caspase-7* were incubated with recombinant active Caspase-9 (A–D) or Caspase-8 (E–H) and the reactions were terminated at indicated time points by the addition of 5xSDS sample buffer. Cleavage of Caspase-7* as well as its phosphomimetic mutants were separated by SDS-PAGE and detected by CBB staining. Quantitation was determined by measuring the density of the bands corresponding to uncleaved Caspase-7* (B, D, F, H). Caspase-7* represents for Caspase-7C186A. I Recombinant Caspase-9 or its phosphomimetic mutant (T102E) was incubated with Flag-Apaf-11-541 obtained from HEK293T cells after transfection. Cleaved Caspase-9 was analyzed by immunoblotting. Data in A, C, E, G, and I are representative from three independent experiments. For B, D, F, and H, data shown are mean ± SD from three replicates. Unpaired two-tailed Student’s t tests were performed. Source data are provided as a Source Data file.
LegK3 blocks pyroptosis initiated by the Caspase-3-GSDME axis
Caspase-3 has long been considered the hallmark of activated apoptosis36. In addition, recent studies have showed that Caspase-3 also cleaves GSDME to activate pyroptosis37. Given the inhibitory effect of LegK3 on Caspase-3 activation, we investigated whether LegK3 is able to block pyroptosis induced by GSDME activation. To test this, we treated HeLa cells that had been transfected to express GFP, GFP-LegK3 or GFP-LegK3D/A with STS or VP-16. Cleavage of GSDME occurred at significantly lower rates in cells expressing LegK3, which is accompanied with lower rates of pyroptosis (Fig. 7A–F). Consistent with this observation, infection of HeLa cells with wild-type L. pneumophila led to very limited GSDME cleavage and pyroptosis, which is in contrast to cells infected with the Lp02ΔlegK3 mutant (Fig. 7G–I). The observed inhibition can be restored by expressing LegK3 but not its enzymatically inactive mutant in strain Lp02ΔlegK3 (Fig. 7G–I).

A–F HeLa cells transfected with GFP, GFP-LegK3 or GFP-LegK3D/A were either left untreated or treated with STS (A–C) or VP-16 (D-F) for 4 h or 24 h. The levels of mature p17 of Caspase-3 and cleaved GSDME were detected by immunoblotting using antibodies specific for Caspase-3 or GSDME, respectively (A, D). Quantitation of GSDME-N: Actin ratio was performed by ImageJ and the results were shown in panels B and E. Cell lysis caused by pyroptosis was determined by measuring the release of LDH (C, F). G–I HeLa cells expressing the FcγRII receptor were infected with opsonized L. pneumophila strains at an MOI of 10 for 3 h. Cleaved Caspase-3 and GSDME were determined by immunoblotting with indicated antibodies (G). Quantitation of GSDME-N: Actin ratio was performed by ImageJ (H). LDH release was measured to evaluate the levels of pyroptosis (I). Data shown in A, D, and G are representative from three independent experiments. Data shown in B, C, E, F, H, and I are mean ± SD from three replicates. Unpaired two-tailed Student’s t tests were performed. Source data are provided as a Source Data file.
LegK3 is required for optimal bacterial growth in VP-16-treated host cells
To investigate the role of LegK3 in intracellular bacterial replication, we infected FcγRII-expressing HeLa cells with the wild-type strain and the Lp02ΔlegK3 mutant and evaluated their growth. The growth of the mutant was indistinguishable from that of the wild-type strain (Fig. S15A), indicating that LegK3 is dispensable for intracellular proliferation in this cell line under standard experimental conditions.
Because of its role in suppressing host cell death, we determined the role of LegK3 in bacterial intracellular growth under apoptosis/pyropotosis-inducing conditions. To this end, we treated infected cells with the cell death induction agent VP-16 and evaluated bacterial growth, which revealed that LegK3 is required for optimal replication (Fig. S15A). VP-16 did not detectably affect L. pneumophila growth in bacteriological medium (Fig. S15B). Thus, inhibition of host cell death is important for intracellular growth of L. pneumophila and LegK3 contributes importantly to such inhibition.
Discussion
The balance between host cell death and survival is critical for pathogenic organisms to establish infection38. Consequently, during intimate interactions with their hosts, intracellular bacterial pathogens, such as Shigella, Salmonella, Coxiella, and Legionella, have developed a variety of intricate strategies to manipulate PCD pathways to promote infection19,39. Such manipulation can be accomplished by transcriptional regulation or by directly targeting critical regulatory hubs involved in cell death signaling by secreted virulence factors22,40. L. pneumophila infection had been previously suggested to trigger apoptosis via activation of Caspase-3 in a manner that is dependent upon its Dot/Icm secretion system20,21. These observations are counterintuitive to the concept that cell integrity is essential for productive intracellular replication. Later studies demonstrated that cells harboring actively replicating L. pneumophila are resistant to cell death stimuli41. Consistent with this notion, induction of apoptosis by pharmacological inhibition of the host cell pro-survival protein BCL-XL or activation of Caspase-3 abrogates Legionella replication41,42. One rational explanation for these observations is that host apoptotic signaling is triggered in response to virulent L. pneumophila infection, but the bacteria are capable of suppressing or reversing this process, possibly via the actions of Dot/Icm substrates.
Indeed, L. pneumophila infection robustly elevates the transcription of a large number of anti-apoptotic genes through activation of the NF-κB pathway in a Dot/Icm-dependent manner23, which might be attributed to the functions of effectors LnaB and LegK116,43. Whereas the biochemical mechanism of LnaB remains to be investigated, LegK1 was proven to be a host IKK mimic capable of directly phosphorylating IκBα and other IκB family inhibitors, including p100 in the noncanonical NF-κB pathway16. In addition, effector SidF suppresses apoptosis of L. pneumophila-infected cells at least partially via directly interacting with and neutralizing the functions of BNIP3 and Bcl-rambo, two noncanonical pro-death members of the Bcl2 protein family22. Moreover, effector SdhA appears to be required for protecting host cells from death, as cells infected with the sdhA-null mutant strain exhibit elevated nuclear degradation, mitochondrial disruption, membrane permeability, and caspase activation44. Although its mechanism of action remains elusive, SdhA appears to suppress host innate immune defenses that attack the integrity of LCV membranes45,46. Our finding that LegK3 functions to inhibit host apoptosis by phosphorylating several caspases has expanded the mechanisms used by L. pneumophila to maintain the integrity of infected cells (Fig. S16).
Caspase cascades are central for host cells to execute PCD, including apoptosis, pyroptosis, and necroptosis25. Among these, Caspase−3/−7/−8/−9 play pivotal roles in controlling apoptosis25. In addition, Caspase-3 was also depicted as a regulatory knot for pyroptosis upon stimulation by chemotherapy drugs via the cleavage of GSDME37. Therefore, direct targeting of caspases represents a highly efficient strategy to manipulate PCD, which was best exemplified by OspC-like arginine ADP-riboxanase47, which utilizes NAD to modify arginine residues in Caspase-4/-11 by adenosine diphosphate riboxanation (ADP-riboxanation), thereby blocking their autoprocessing activity as well as recognition and cleavage of GSDMD47. OspC3 of Chromobacterium violaceum attacks Caspase-3/-7/-8/-9 by a similar mechanism to dysregulate apoptosis, necroptosis, and pyroptosis38,48. In addition, the E. coli effector NleF and the Yersinia pestis effector YopM can inactivate caspases via direct interactions, thus inhibiting PCDs49,50. Interestingly, phosphorylation by LegK3 does not affect the proteolytic activity of these caspases (Fig. S16). Instead, the modification thwarts their cleavage by initiator caspases or upstream regulators (Fig. S16). To the best of our knowledge, LegK3 is the first bacterial virulence factor known to hijack caspases via phosphorylation. In mammalian cells, the activities of several caspases, including Caspase-2, Caspase-3, Caspase-6, Caspase-7, Caspase-8, and Caspase-9, are regulated by phosphorylation catalyzed by various kinases51,52,53,54,55,56. For instance, p38-MAPK can directly phosphorylate Caspase-3 at Ser150, thereby inhibiting its activity and further hindering apoptosis55; PAK2 targets Caspase-7 by phosphorylation at multiple sites, in which Ser30 phosphorylation allosterically obstructs Caspase-7 activation by Caspase-9, whereas Ser239 phosphorylation renders active Caspase-7 incapable of binding substrates, thus blocking subsequent events in apoptosis56. Similarly, Caspase-9 was shown to be phosphorylated at Thr125 by multiple kinases, including ERK1/2, CDK1-cyclin B and DYRK1A, depending upon the stimulus and cell cycle57,58,59. Phosphorylation on Thr125 prevents Caspase-9 from being processed by Apaf-1, thus inhibiting apoptosis57. The fact that the catalytic motif of LegK3 is structurally similar to those found in eukaryotic proteins suggests that gene coding for this effector may be acquired by the pathogen via horizontal gene transfer60.
Protein phosphorylation is reversed by phosphatases which downregulate the signaling in response to environmental cues61. Manipulation of host targets by L. pneumophila effectors in many cases is specifically regulated by effectors with opposite biochemical activities, which impose sophisticated spatial-temporal regulation of host signaling. For example, Rab1 activity is tightly controlled by reversible AMPylation executed by SidM/DrrA62 and SidD63,64, reversible phosphorylcholination carried out by AnkX and Lem365,66, reversible phosphoribosyl-linked serine ubiquitination conducted by SidEs67,68,69 and DupA/DupB70,71. Hence, it is possible that L. pneumophila also encodes effectors to counteract the function of LegK3, thus imposing temporal regulation of PCD to promote host cell death in the final phase of L. pneumophila infection72. Dot/Icm effectors with phosphatase activity have been identified, including WipA73, WipB74, and Ceg475. It will be of interest to investigate whether any of these enzymes or other effectors with phosphatase activity counteract the activity of LegK3 to fine-tune PCD in cells infected by L. pneumophila to ensure maximal bacterial replication.
Although we have demonstrated that several caspases, including Caspase-3, Caspase-7, and Caspase-9, are bona fide substrates of LegK3, this bacterial S/T kinase might target diverse host proteins besides caspases to facilitate bacterial infection. A further unbiased proteome-wide phosphoproteomics study will potentially uncover additional phosphorylation sites and phosphorylated proteins targeted by LegK3 during L. pneumophila infection.
Methods
Bacterial strains, plasmids, cell lines, and culture methods
The bacterial strains used in this study are listed in Table S1. L. pneumophila strains were grown at 37 °C either on buffered charcoal yeast extract (CYE) agar plates or in AYE liquid media. L. pneumophila ΔlegK3 was constructed by allelic exchange using pSR47S as described previously76, and complementation of the mutant strain was done by electronic transformation of pZL50777 carrying LegK3. E. coli strains were cultured on LB agar plates or in broth supplemented with appropriate antibiotics (100 µg/ml ampicillin, 50 µg/ml kanamycin). E. coli strains DH5α and BL21(DE3) were used to maintain plasmids and express proteins, respectively. For expression of proteins in E. coli, genes were inserted into pET28a or pETDuet. For expression of proteins in mammalian cells, PCR products were ligated into pEGFP, pCDNA3.1-HA, or pCMV-4xFlag77. Site-directed mutants were constructed using the QuikChange II site-directed mutagenesis kit according to the recommended protocols. All plasmids and primers used in this study are presented in Tables S2 and S3.
HeLa, HEK293, HEK293T, and Raw264.7 cells were obtained from the American Type Culture Collection (ATCC) and were grown at 37 °C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS).
Protein expression and purification
Twenty milliliters of overnight cultures of E. coli BL21 (DE3) strains harboring relevant plasmids were transferred into 1 liter of fresh LB broth supplemented with appropriate antibiotics. The cultures were grown at 37 °C to an OD600nm of 0.6–0.8. Protein expression was induced for 12 h at 18 °C with constant shaking (200 rpm) in the presence of 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Then, bacteria were harvested by centrifugation and lysed by a homogenizer (JN-mini, JNBIO, Guangzhou, China). The cell debris was pelleted by centrifugation at 14,000×g for 1 h at 4 °C. The resulting supernatants were subsequently passed through the Ni-NTA beads (QIAGEN) for three times to retain His6-tagged recombinant proteins. After extensive washing of the beads with a buffer containing 20 mM imidazole, the proteins on the beads were eluted with 250 mM imidazole. When necessary, phosphorylated recombinant proteins were further separated by ion exchange chromatography. All purified proteins were dialyzed twice against a buffer containing 300 mM NaCl, 20 mM Tris-HCl (pH 7.5), and 10% glycerol at 4 °C.
Mammalian cell transfection, immunoprecipitation, western-blot, and immunostaining
Mammalian cells seeded on 6-well plates were grown to a confluence of 80%. Transfection of cells was performed using Lipofectamine 3000 transfection reagent (Invitrogen). At 24 h post-transfection, cells were either left untreated or treated with 1 μM of STS or 40 μM of VP-16 for 4 h and 24 h. Cells were collected by centrifugation (100×g for 10 min) at 4 °C and lysed with RIPA cell lysis reagent containing 1x protease inhibitors (Sigma) at 4 °C for 10 min. Cell debris and unbroken cells were removed by centrifugation (14,000×g) for 10 min at 4 °C. Immunoprecipitation was performed by incubating the cell lysates with anti-HA (Sigma) or anti-Flag (Sigma) agarose for 2 h at 4 °C on an end-to-end rotator. The agarose-bound proteins were either eluted by adding 1xSDS sample buffer followed by boiling at 100 °C for 5 min or by Flag/HA peptides (Sigma). For western-blot analysis, protein samples were resolved by SDS-PAGE and subsequently transferred to nitrocellulose membranes (Pall Life Sciences). After a blocking step using 5% w/v nonfat dry milk, the membranes were sequentially incubated with appropriate primary and secondary antibodies for 1 h at room temperature. The blot images were captured under the Odyssey® CLx Infrared Imaging System (LI-COR Biosciences). The antibodies used in the study are described in Table S4.
For immunostaining, HEK293 cells transfected with the indicated plasmids were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X−100 in PBS. After a blocking step using 4% goat serum (Sigma) in PBS for 30 min, the cells were immunostained with the anti-HA antibodies (ABclonal, cat# AE008, 1:1000) and Texas Red-conjugated secondary antibodies (Thermo Fisher Scientific, cat# T-6390, 1:500). The nuclei were stained by Hoechst 33342 (Beyotime, cat# C1022). Immunofluorescent signals were visualized via an Olympus IX-83 fluorescence microscope.
L. pneumophila infection
Wild-type, dotA-, ΔlegK3, ΔlegK3 (pLegK3), and ΔlegK3 (pLegK3D/A) L. pneumophila strains were grown in AYE liquid medium at 37 °C to the post-exponential phase as judged by an OD600nm of 3.3–3.8. The bacteria were opsonized with an anti-Legionella antibody (1: 5000 dilution) at 37 °C for 30 min before infection. HEK293 or HeLa cells transfected with FcγRII78 and relevant plasmids were then infected with opsonized L. pneumophila for 3 h at an MOI of 10. Infected cells were collected and lysed using the RIPA lysis buffer. The cell lysates were immunoprecipitated with anti-HA or anti-Flag beads. Proteins were further detected by western-blot analysis using the indicated antibodies, as listed in Table S4.
TEM translocation assay
Raw264.7 cells were seeded on 24-well plates at a density of 1 × 104/well. L. pneumophila strains expressing relevant TEM fusion proteins were used to infect the cells at an MOI of 20. At 3 h post-infection, the infection samples were loaded with the 6×CCF4/AM reaction mixture (K1095; Thermo Fisher) and reacted in the dark at room temperature for 1 h. Then, translocation of the TEM fusion proteins by L. pneumophila was visualized by a fluorescence microscope (IX-83; Olympus) equipped with a β-lactamase FL-Cube (U-N41031; Chroma Technology Corp., Bellows Falls, VT). The wild-type L. pneumophila strain producing TEM-RalF or TEM-Fab179 was used as the positive and negative controls, respectively.
Quantitative real-time PCR
Raw264.7 cells seeded on six-well plates (1×106/well) were infected with wild-type, dotA- or ΔlegK3 L. pneumophila strains for 3 h at an MOI of 10. Infected cells were washed with PBS for five times to remove extracellular bacteria. Then, cells were collected and RNA was extracted using TRIzol (TIANGEN). After removing DNA with RNase-free DNaseI (TIANGEN), mRNA was reverse-transcribed into cDNA using a cDNA synthesis kit (Applied Biological Materials) according to the manufacturer’s instructions. Detection of the legK3 transcription levels was performed with the BlasTaq™ 2×qPCR MasterMix (Applied Biological Materials) and the QuantStudio™ Real-Time PCR system (Thermo Fisher). The 16S rRNA gene was used as an internal control. Relative gene expression was calculated by the 2-ΔΔCT method. The primer sequences used for qRT-PCR analysis were listed in Table S3.
Detection of phosphorylated proteins
Recombinant proteins purified from E. coli strains co-expressing wild-type LegK3 or D187A mutant LegK3 were separated by either SDS-PAGE or native PAGE. SDS-PAGE gels were subjected to western-blot analysis using antibodies recognizing phosphorylated serine/threonine or by staining the gel with Pro-Q™ Diamond Phosphoprotein Gel Stain (ThermoFisher Scientific). For native PAGE analysis, protein samples were mixed with 5x native gel loading buffer (pH 8.8) and loaded onto 10% native gels. After running the gels in the Tris-glycine buffer (pH 8.8), the protein bands were observed by Coomassie brilliant blue staining (CBB).
Proteins retrieved from transfected cells or infected cells were dissolved by SDS-PAGE and subsequently analyzed by western-blot using phospho serine/threonine-specific antibodies.
Identification of phosphorylation sites by mass spectrometry
To identify phosphorylation sites, Caspase-3, Caspase-7 or Caspase-9 purified from E. coli or HEK293 cells co-expressing LegK3, or cells infected with L. pneumophila strains, were separated by SDS-PAGE. The excised protein bands of interest were further digested with trypsin and analyzed by LC-MS/MS as described below. LC-MS experiments were performed on a hybrid linear ion trap-Orbitrap mass spectrometer (ThermoFisher Scientific). A 40-min gradient was employed that ranged from 10% to 40% solvent B (100% ACN, 0.1% FA). Raw MS files were processed by Mascot for peptide assignments. The following settings were used for database search: 20 ppm for precursor ion mass tolerance, 0.8 Da for fragment ion mass tolerance, cysteine carbamidomethylation as a fixed modification, and methionine oxidation, serine/threonine/tyrosine phosphorylation as variable modifications.
Measurement of Caspase-3/-7 activity, ATP cell viability, and LDH release
HeLa or HEK293 cells plated in 24- or 6-well plates were stimulated by distinct treatments. To measure LDH levels, the culture media were collected and LDH release was detected by the LDH Cytotoxicity Assay Kit (Beyotime). For measurement of Caspase-3/-7 activity and ATP cell viability, cells were lysed in RIPA buffer. Then, the Caspase-3/-7 assay kit (Beyotime) and Enhanced ATP Assay Kit (Beyotime) were used to quantify the Caspase-3/-7 activity and ATP cell viability in the cell lysates, respectively.
Detection of cell apoptosis by TUNEL staining
HeLa cells were transfected with plasmids to express GFP, GFP-LegK3, or GFP-LegK3D/A for 24 h. These cells were either left untreated or treated with 1 μM STS for 4 h prior to fixation by 4% paraformaldehyde. The fixed cells were further stained for DNA fragmentation using the Colorimetric TUNEL Apoptosis Assay Kit (Beyotime) following the manufacturer’s instructions.
HeLa cells transfected with FcγRII were either uninfected or infected with wild-type, dotA-, ΔlegK3, ΔlegK3 (pLegK3), and ΔlegK3 (pLegK3D/A) L. pneumophila strains for 3 h at an MOI of 10. The cells were fixed by 4% paraformaldehyde in PBS for 30 min followed by permeabilization with 0.2% Triton for 5 min. Immunostaining was subsequently performed using anti-L. pneumophila antibodies and a Colorimetric TUNEL Apoptosis Assay Kit (Beyotime). Immunofluorescence was observed under an Olympus IX-83 fluorescence microscope.
Detection of cell apoptosis by flow cytometry
HeLa cells were transfected with BFP, BFP-LegK3, or BFP-LegK3D/A. At 24 h post-transfection, cells were either left untreated or treated with 1 μM STS for 4 h. Then, the cells were resuspended in PBS and incubated with propidium iodide (PI) (Beyotime) and GreenNuc™ Caspase-3 Substrate (Beyotime) for 10 min in the dark. Flow cytometry was performed on FACSCelesta (BD Biosciences) using 488 nm and 530 nm lasers.
FcγRII-producing HeLa cells were either uninfected or infected with relevant L. pneumophila strains for 3 h (MOI = 10). Infected cells were washed and resuspended in PBS for three times. After incubation of the cells with PI and GreenNuc™ Caspase-3 Substrate, apoptosis was determined via flow cytometry on FACSCelesta.
Biochemical cleavage assays
To determine the impact of LegK3-catalyzed phosphorylation on the enzymatic activity of Caspase-3, 3 μg of recombinant GSDMD or GSDME was reacted with 0.1 μg of p17/p12 or P-p17/p12 complex of Caspase-3 at 37 °C for 1 h. For cleavage of PARP, cell lysates of HEK293 cells were incubated with p17/p12 or P-p17/p12 complex of Caspase-3 at 37 °C for 20 min. Reactions were stopped by the addition of 5xSDS sample buffer and boiling for 5 min. Cleavage of the substrates was visualized by CBB staining (GSDMD and GSDME) or western-blot analysis using anti-PARP antibodies after separation by SDS-PAGE. For determination of the cleavage dynamics, aliquots of the reaction mixtures were withdrawn at the indicated time points and added with 5xSDS sample buffer. After separation by SDS-PAGE and staining by CBB, cleavage of the substrates was calculated by measuring the band intensities using ImageJ.
To evaluate the influence of Caspase-3/-7 phosphorylation on their hydrolysis by initiator caspases, cleavage reaction mixtures containing recombinant active Caspase-9 or Caspase-8 and Caspase-3C/A/Caspase-7C/A, phosphorylated Caspase-3C/A, or Caspase-3C/A/Caspase-7C/A phosphomimetics were incubated at 37 °C. At the indicated durations, aliquots of the samples were stopped by the addition of 5xSDS sample buffer and boiling for 5 min. Proteins dissolved by SDS-PAGE were observed by CBB staining. Protein intensities were measured by ImageJ.
To investigate the effect of LegK3-mediated phosphorylation on Apaf-1-induced Caspase-9 cleavage, HEK293T cells were transfected with Flag-Apaf-11-541 for 24 h. Flag-Apaf-11-541 eluted by 3xFlag peptides were reacted with recombinant wild-type Caspase-9 or Caspase-9T102E in a buffer contain 20 mM Hepes (pH 7.5), 10 mM KCl, 1.5 mM MgCl, 1 mM EDTA, 1 mM DTT and 0.1 mM PMSF for 1 h at 30 °C. The reaction mixtures were terminated by 5xSDS sample buffer. Cleaved Caspase-9 was detected by western-blot analysis using anti-Caspase-9 antibodies.
L. pneumophila intracellular replication assay
FcγRII-expressing HeLa cells were infected with anti-Legionella antibody-opsonized wild-type, ΔlegK3, ΔlegK3 (pLegK3), or ΔlegK3 (pLegK3D/A) bacteria for 2 h at an MOI of 0.05. Extracellular bacteria were removed by washing the plates three times with prewarmed PBS. The infection samples were added with fresh culture media supplemented with or without 40 μM VP-16. At 2, 24, 48, and 72 h post-infection, cells were lysed with 0.2% saponin for 30 min. Serial dilutions of the lysates were spotted onto CYE agar plates and grown at 37 °C for 4-5 days before counting the colony-forming units.
Quantification and statistical analysis
The band intensities shown in the cleavage assays were quantified by the ImageJ software. To determine the percentage of apoptotic cells by TUNEL staining, 100 transfected or L. pneumophila-positive cells were calculated. The data are presented as the mean ± SD as indicated in the figure legends. Statistical analyses were performed using unpaired two-tailed Student’s t tests, and p < 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD054358. The link to access the data: https://www.iprox.cn//page/project.html?id=IPX0009364000. The data that support the conclusions of this study are included in this published article along with its Supplementary Information files. Source data are provided with this paper.
References
Alto, N. M. & Orth, K. Subversion of cell signaling by pathogens. Cold Spring Harb. Perspect. Biol. 4, a006114 (2012).
Hayes, C. S., Aoki, S. K. & Low, D. A. Bacterial contact-dependent delivery systems. Annu. Rev. Genet. 44, 71–90 (2010).
Yang, Y. et al. Legionella pneumophila-mediated host posttranslational modifications. J. Mol. Cell Biol. 15, mjad032 (2023).
Ochoa, D. et al. The functional landscape of the human phosphoproteome. Nat. Biotechnol. 38, 365–373 (2020).
Safaei, J., Manuch, J., Gupta, A., Stacho, L. & Pelech, S. Prediction of 492 human protein kinase substrate specificities. Proteome Sci. 9, S6 (2011).
Lee, P. C. et al. The Legionella kinase LegK7 exploits the Hippo pathway scaffold protein MOB1A for allostery and substrate phosphorylation. Proc. Natl Acad. Sci. USA 117, 14433–14443 (2020).
Odendall, C. et al. The Salmonella kinase SteC targets the MAP kinase MEK to regulate the host actin cytoskeleton. Cell Host Microbe 12, 657–668 (2012).
Imami, K. et al. Global impact of Salmonella pathogenicity island 2-secreted effectors on the host phosphoproteome. Mol. Cell. Proteom. 12, 1632–1643 (2013).
Pollock, G. L. et al. Targeting of microvillus protein Eps8 by the NleH effector kinases from enteropathogenic E. coli. Proc. Natl Acad. Sci. USA 119, e2204332119 (2022).
Newton, H. J., Ang, D. K., van Driel, I. R. & Hartland, E. L. Molecular pathogenesis of infections caused by Legionella pneumophila. Clin. Microbiol. Rev. 23, 274–298 (2010).
Qiu, J. & Luo, Z. Q. Legionella and Coxiella effectors: strength in diversity and activity. Nat. Rev. Microbiol. 15, 591–605 (2017).
Isberg, R. R., O’Connor, T. J. & Heidtman, M. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7, 13–24 (2009).
Hervet, E. et al. Protein kinase LegK2 is a type IV secretion system effector involved in endoplasmic reticulum recruitment and intracellular replication of Legionella pneumophila. Infect. Immun. 79, 1936–1950 (2011).
Lee, P. C. & Machner, M. P. The effector kinase LegK7 hijacks the host hippo pathway to promote infection. Cell Host Microbe 24, 429 (2018).
Sreelatha, A. et al. effector kinase is activated by host inositol hexakisphosphate. J. Biol. Chem. 295, 6214–6224 (2020).
Ge, J. N. et al. A type IV effector activates the NF-κB pathway by phosphorylating the IκB family of inhibitors. Proc. Natl Acad. Sci. USA 106, 13725–13730 (2009).
Michard, C. et al. The legionella kinase LegK2 targets the ARP2/3 complex to inhibit actin nucleation on phagosomes and allow bacterial evasion of the late endocytic pathway. mBio 6, e00354–00315 (2015).
Moss, S. M. et al. A Legionella pneumophila kinase phosphorylates the Hsp70 chaperone family to inhibit eukaryotic protein synthesis. Cell Host Microbe 25, 454–462.e456 (2019).
Wanford, J. J., Hachani, A. & Odendall, C. Reprogramming of cell death pathways by bacterial effectors as a widespread virulence strategy. Infect. Immun. 90, e0061421 (2022).
Gao, L. Y. & Abu Kwaik, Y. Activation of caspase 3 during Legionella pneumophila-induced apoptosis. Infect. Immun. 67, 4886–4894 (1999).
Molmeret, M. et al. Activation of caspase-3 by the Dot/Icm virulence system is essential for arrested biogenesis of the Legionella-containing phagosome. Cell. Microbiol. 6, 33–48 (2004).
Banga, S. et al. Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc. Natl Acad. Sci. USA 104, 5121–5126 (2007).
Losick, V. P. & Isberg, R. R. NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 203, 2177–2189 (2006).
Wilson, C. H. & Kumar, S. Caspases in metabolic disease and their therapeutic potential. Cell Death Differ. 25, 1010–1024 (2018).
Van Opdenbosch, N. & Lamkanfi, M. Caspases in cell death, inflammation, and disease. Immunity 50, 1352–1364 (2019).
Ghorbani N., Yaghubi R., Davoodi J. & Pahlavan, S. How does caspases regulation play role in cell decisions? apoptosis and beyond. Mol. Cell. Biochem. 479, 1599–1613 (2023).
O’Connor, T. J., Adepoju, Y., Boyd, D. & Isberg, R. R. Minimization of the Legionella pneumophila genome reveals chromosomal regions involved in host range expansion. Proc. Natl Acad. Sci. USA 108, 14733–14740 (2011).
Zhu, W. et al. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS ONE 6, e17638 (2011).
de Felipe, K. S. et al. Evidence for acquisition of Legionella type IV secretion substrates via interdomain horizontal gene transfer. J. Bacteriol. 187, 7716–7726 (2005).
Linsky, M. & Segal, G. A horizontally acquired Legionella genomic island encoding a LuxR type regulator and effector proteins displays variation in gene content and regulation. Mol. Microbiol. 116, 766–782 (2021).
Rosen, A. & Casciola-Rosen, L. Macromolecular substrates for the ICE-like proteases during apoptosis. J. Cell. Biochem. 64, 50–54 (1997).
Ponder, K. G. & Boise, L. H. The prodomain of caspase-3 regulates its own removal and caspase activation. Cell Death Discov. 5, 56 (2019).
Acehan, D. et al. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol. Cell 9, 423–432 (2002).
Zou, H., Henzel, W. J., Liu, X., Lutschg, A. & Wang, X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90, 405–413 (1997).
Srinivasula, S. M., Ahmad, M., Fernandes-Alnemri, T. & Alnemri, E. S. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol. Cell 1, 949–957 (1998).
Porter, A. G. & Janicke, R. U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 6, 99–104 (1999).
Wang, Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017).
Peng, T. et al. Pathogen hijacks programmed cell death signaling by arginine ADPR-deacylization of caspases. Mol. Cell 82, 1806–1820 e1808 (2022).
Friedrich, A., Pechstein, J., Berens, C. & Luhrmann, A. Modulation of host cell apoptotic pathways by intracellular pathogens. Curr. Opin. Microbiol. 35, 88–99 (2017).
Luo, J., Hu, J., Zhang, Y., Hu, Q. & Li, S. Hijacking of death receptor signaling by bacterial pathogen effectors. Apoptosis 20, 216–223 (2015).
Abu-Zant, A. et al. Incomplete activation of macrophage apoptosis during intracellular replication of Legionella pneumophila. Infect. Immun. 73, 5339–5349 (2005).
Speir, M. et al. Eliminating Legionella by inhibiting BCL-XL to induce macrophage apoptosis. Nat. Microbiol. 1, 15034 (2016).
Losick, V. P., Haenssler, E., Moy, M. Y. & Isberg, R. R. LnaB: a Legionella pneumophila activator of NF-kappaB. Cell. Microbiol. 12, 1083–1097 (2010).
Laguna, R. K., Creasey, E. A., Li, Z., Valtz, N. & Isberg, R. R. A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl Acad. Sci. USA 103, 18745–18750 (2006).
Ge, J., Gong, Y. N., Xu, Y. & Shao, F. Preventing bacterial DNA release and absent in melanoma 2 inflammasome activation by a Legionella effector functioning in membrane trafficking. Proc. Natl Acad. Sci. USA 109, 6193–6198 (2012).
Creasey, E. A. & Isberg, R. R. The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc. Natl Acad. Sci. USA 109, 3481–3486 (2012).
Li, Z. et al. Shigella evades pyroptosis by arginine ADP-riboxanation of caspase-11. Nature 599, 290–295 (2021).
Liu, Y. et al. Calmodulin binding activates chromobacterium CopC effector to ADP-riboxanate host apoptotic caspases. mBio. 13, e0069022 (2022).
LaRock, C. N. & Cookson, B. T. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe 12, 799–805 (2012).
Blasche, S. et al. The E. coli effector protein NleF is a caspase inhibitor. PLoS ONE 8, e58937 (2013).
Cardone, M. H. et al. Regulation of cell death protease caspase−9 by phosphorylation. Science 282, 1318–1321 (1998).
Andersen, J. L. et al. Restraint of apoptosis during mitosis through interdomain phosphorylation of caspase-2. EMBO J. 28, 3216–3227 (2009).
Li, X. et al. Phosphorylation of caspase-7 by p21-activated protein kinase (PAK) 2 inhibits chemotherapeutic drug-induced apoptosis of breast cancer cell lines. J. Biol. Chem. 286, 22291–22299 (2011).
Suzuki, A. et al. Regulation of caspase-6 and FLIP by the AMPK family member ARK5. Oncogene 23, 7067–7075 (2004).
Alvarado-Kristensson, M. et al. p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J. Exp. Med. 199, 449–458 (2004).
Eron, S. J., Raghupathi, K. & Hardy, J. A. Dual site phosphorylation of caspase-7 by PAK2 blocks apoptotic activity by two distinct mechanisms. Structure 25, 27–39 (2017).
Allan, L. A. et al. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat. Cell Biol. 5, 647–U645 (2003).
Allan, L. A. & Clarke, P. R. Phosphorylation of caspase-9 by CDK1/cyclin B1 protects mitotic cells against apoptosis. Mol. Cell 26, 301–310 (2007).
Seifert, A. & Clarke, P. R. p38α- and DYRK1A-dependent phosphorylation of caspase-9 at an inhibitory site in response to hyperosmotic stress. Cell Signal. 21, 1626–1633 (2009).
Juhas, M. Horizontal gene transfer in human pathogens. Crit. Rev. Microbiol. 41, 101–108 (2015).
Day, E. K., Sosale, N. G. & Lazzara, M. J. Cell signaling regulation by protein phosphorylation: a multivariate, heterogeneous, and context-dependent process. Curr. Opin. Biotechnol. 40, 185–192 (2016).
Muller, M. P. et al. The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science 329, 946–949 (2010).
Neunuebel, M. R. et al. De-AMPylation of the small GTPase Rab1 by the pathogen Legionella pneumophila. Science 333, 453–456 (2011).
Tan, Y. & Luo, Z. Q. Legionella pneumophila SidD is a deAMPylase that modifies Rab1. Nature 475, 506–509 (2011).
Mukherjee, S. et al. Modulation of Rab GTPase function by a protein phosphocholine transferase. Nature 477, 103–106 (2011).
Tan, Y., Arnold, R. J. & Luo, Z. Q. Legionella pneumophila regulates the small GTPase Rab1 activity by reversible phosphorylcholination. Proc. Natl Acad. Sci. USA 108, 21212–21217 (2011).
Bhogaraju, S. et al. Phosphoribosylation of ubiquitin promotes serine ubiquitination and impairs conventional ubiquitination. Cell 167, 1636–1649.e1613 (2016).
Qiu, J. et al. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 533, 120–124 (2016).
Kotewicz, K. M. et al. A single legionella effector catalyzes a multistep ubiquitination pathway to rearrange tubular endoplasmic reticulum for replication. Cell Host Microbe 21, 169–181 (2017).
Wan, M. et al. Deubiquitination of phosphoribosyl-ubiquitin conjugates by phosphodiesterase-domain-containing Legionella effectors. Proc. Natl Acad. Sci. USA 116, 23518–23526 (2019).
Shin, D. et al. Regulation of phosphoribosyl-linked serine ubiquitination by deubiquitinases DupA and DupB. Mol. Cell 77, 164–179 e166 (2020).
Speir, M., Vince, J. E. & Naderer, T. Programmed cell death in Legionella infection. Future Microbiol. 9, 107–118 (2014).
Pinotsis, N. & Waksman, G. Structure of the WipA protein reveals a novel tyrosine protein phosphatase effector from Legionella pneumophila. J. Biol. Chem. 292, 9240–9251 (2017).
Prevost, M. S., Pinotsis, N., Dumoux, M., Hayward, R. D. & Waksman, G. The Legionella effector WipB is a translocated Ser/Thr phosphatase that targets the host lysosomal nutrient sensing machinery. Sci. Rep. 7, 9450 (2017).
Quaile, A. T. et al. The Legionella pneumophila effector Ceg4 is a phosphotyrosine phosphatase that attenuates activation of eukaryotic MAPK pathways. J. Biol. Chem. 293, 3307–3320 (2018).
Liu, Y. & Luo, Z. Q. The Legionella pneumophila effector SidJ is required for efficient recruitment of endoplasmic reticulum proteins to the bacterial phagosome. Infect. Immun. 75, 592–603 (2007).
Xu, L. et al. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. 6, e1000822 (2010).
Arasaki, K. & Roy, C. R. Legionella pneumophila promotes functional interactions between plasma membrane syntaxins and Sec22b. Traffic 11, 587–600 (2010).
de Felipe, K. S. et al. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. 4, e1000117 (2008).
Acknowledgements
We thank Dr. Ralph Isberg (Tufts School of Medicine, Boston, MA, USA) for L. pneumophila cluster deletion strains. This work was supported by the National Natural Science Foundation of China (grants 32170182 and 82372268 to J.Z.Q, 22174003 and 21974002 to X.Y.L) and the National Key Research and Development Program of China (2022YFA1304500 to X.Y.L); the Fundamental Research Funds for the Central Universities to J.Z.Q.
Author information
Authors and Affiliations
Contributions
J.Z.Q., X.Y.L., and Z.Q.L. conceived the study and provided critical revisions to the manuscript. J.Z.Q. and J.L.G. were responsible for the experimental design, data analysis, and composition of the manuscript. J.L.G., Y.W., X.Y.L., and Q.L. performed biochemical, cell biological, and infection assays. J.L.G. and Y.W. performed mass spectrometry analyses. H.Q.Y., H.T.L., K.L.M., and X.M.D. contributed to the experimental design and data analysis.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yuxin Mao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ge, J., Wang, Y., Li, X. et al. Phosphorylation of caspases by a bacterial kinase inhibits host programmed cell death. Nat Commun 15, 8464 (2024). https://doi.org/10.1038/s41467-024-52817-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52817-1
This article is cited by
-
Chlorella-derived extracellular vesicle-based nanogels suppress cGAS-STING for treatment of radiation-induced lung injury
Nature Communications (2026)
-
Post-translational weapons of microbial warfare: how bacterial effectors hijack host cell death and xenophagy
Cell Communication and Signaling (2025)
-
Noncoding RNA-encoded peptides in cancer: biological functions, posttranslational modifications and therapeutic potential
Journal of Hematology & Oncology (2025)
-
Stress granules and cell death: crosstalk and potential therapeutic strategies in infectious diseases
Cell Death & Disease (2025)
-
Engineering bi-directional chemically-modulated synthetic condensates for cellular control
Nature Communications (2025)
