
Abstract
Vinca alkaloids, a class of tubulin-binding agent, are widely used in treating cancer, yet the emerging resistance compromises their efficacy. Hepatoma up-regulated protein (HURP), a microtubule-associated protein displaying heightened expression across various cancer types, reduces cancer cells’ sensitivity to vinca-alkaloid drugs upon overexpression. However, the molecular basis behind this drug resistance remains unknown. Here we discover a tubulin-binding domain within HURP, and establish its role in regulating microtubule growth. Cryo-EM analysis reveals interactions between HURP’s tubulin-binding domain and the vinca domain on β-tubulin -- the site targeted by vinca alkaloid drugs. Importantly, HURP competes directly with vinorelbine, a vinca alkaloid-based chemotherapeutic agent, countering microtubule growth defects caused by vinorelbine both in vitro and in vivo. Our findings elucidate a mechanism driving drug resistance in HURP-overexpressing cancer cells and emphasize HURP tubulin-binding domain’s role in mitotic spindle assembly. This underscores its potential as a therapeutic target to improve cancer treatment.
Similar content being viewed by others

Introduction
Cell division requires the precise segregation of sister chromatids, a process dependent on the assembly of a microtubule-based bipolar spindle. The mitotic spindle comprises three distinct populations of microtubules, polar polymers formed by α/β-tubulin: kinetochore microtubules (K-fibers), astral microtubules, and interpolar microtubules1. While cellular and proteomics studies have identified numerous proteins (e.g., spindle assembly factors (SAFs)2) that facilitate microtubule formation and are required for mitotic spindle assembly, mechanistic insight of how these proteins conduct their functions to build the spindle machinery remain elusive. Furthermore, although anti-mitotic drugs targeting mitotic components have been used in cancer treatment, their efficiency is limited by toxic side effects and drug resistance3. Therefore, comprehending the function and structure of proteins involved in mitotic spindle assembly holds not only profound implications for our understanding of cell division but also potential for the development of more effective anti-cancer agents.
SAFs exert control over various aspects of microtubule behavior during spindle assembly, including nucleation, stabilization, and bundling2. These activities are modulated by the Ran-nuclear transport receptor pathway2,4. According to current models, Ran•GTP interacts with nuclear transport receptors, releasing SAFs like NuMA5,6,7 and Targeting Protein for Xklp2 (TPX2)8,9, thereby facilitating mitotic spindle organization. One such SAF is hepatoma up-regulated protein (HURP), initially identified in hepatocellular carcinoma (HCC)10. HURP serves as a microtubule-associated protein (MAP) and modulates microtubule dynamics by altering catastrophe and rescue frequency11,12,13. Furthermore, HURP localizes predominantly to K-fibers and promotes the microtubules stabilization, facilitating chromosome congression during mitosis12,14. Notably, HURP’s interaction with microtubules can be modulated by Importin-β, a nuclear transport receptor, placing it within the SAF family12,15.
In addition to hepatoma, many human cancer types, such as non-small cell lung cancer (NSCLC)16 and oral squamous cell carcinoma (OSCC)17, exhibit elevated levels of HURP mRNA transcripts (Supplementary Table 1). Knockdown of HURP expression has been shown to suppress cancer cell proliferation16,18,19, implying its involvement in carcinogenesis. Furthermore, there is a significant correlation between the half-maximal inhibitory concentration (IC50) values of vinorelbine, a vinca alkaloid drug that is widely used chemotherapeutic20, and the expression level of HURP in cancer cell lines17. This suggests that HURP also confers resistance to the anti-cancer agent vinorelbine, enabling cancer cells to proliferate. Notably, vinorelbine binds to the vinca domain on β-tubulin and induces self-association of tubulin dimers into aggregates, thereby impeding microtubule assembly21,22. However, the precise mechanisms underlying how HURP reduces the sensitivity of cancer cells to vinorelbine remain unknown.
Here, we unveil a tubulin-binding domain (TBD) in HURP that suppresses metaphase spindle size in Xenopus egg extracts and operates independently of the Ran-importin pathway. The tubulin binding activity allows HURP to control microtubule catastrophe and rescue frequency. Moreover, the cryo-EM structure of the HURP-TBD with α/β-tubulin dimer reveals that the HURP-TBD, composed of three extended α-helices, binds to the vinca domain on the β-tubulin. We further show that HURP directly competes with vinorelbine binding on tubulin and rectifies the microtubule assembly deficiencies induced by vinorelbine. Our findings elucidate a molecular mechanism through which HURP regulates microtubule dynamics, providing resistance to clinically relevant agents. Significantly, this study opens avenues for the development of chemical inhibitors targeting HURP for cancer treatment.
Results
HURP functions as a CLASP-like protein
HURP is a MAP, and the N-terminal 280 residues have been shown to mediate microtubule binding (Fig. 1a)13. HURP, therefore, binds and bundles microtubules, facilitating K-fiber stabilization12,13,14. Recently, a total internal reflection fluorescence (TIRF) microscopy-based assay showed that HURP regulates the dynamic behavior of single microtubules by reducing catastrophe frequency and increasing rescue11, contributing to microtubule stability. To further examine whether the microtubule-binding activity of HURP accounts for the alternation of microtubule catastrophe and rescue rates, we conducted the TIRF-based dynamic microtubule assays in the presence of different concentrations of recombinant human GFP-tagged HURP full-length (hereafter GFP-HURP FL) or microtubule-binding domain (a.a. 1-280; hereafter GFP-HURP (1-280)) (Supplementary Videos 1-3). While GFP-HURP FL and GFP-HURP (1-280) displayed no effect on microtubule polymerization, the depolymerization rate of microtubules in the presence of GFP-HURP FL or GFP-HURP (1-280) was reduced ~ 2-fold when compared with control (500 nM protein concentration; Fig. 1b-g). These results indicate that the binding of HURP to microtubules modulate the microtubule depolymerization rate. Notably, compared to GFP-HURP (1-280), GFP-HURP FL further reduced the frequency of microtubule catastrophe and increased the rescue rate in a concentration-dependent manner (Fig. 1h-j and Supplementary Fig. 1a-e; also see Supplementary Table 2). Hence, we observed longer dynamic microtubules after 20-minute incubation with GFP-HURP FL (Fig. 1j). These findings indicate that the HURP microtubule-binding domain regulates the rate of microtubule depolymerization, whereas other regions of HURP modulate microtubule catastrophe and rescue. Thus, overall, HURP generally stabilizes microtubule dynamics.

a Domain structures of HURP. The microtubule-binding domain is colored black. Residue numbers indicate domain boundaries of HURP. b Schematic representation of a kymograph illustrating microtubule growth events from a microtubule seed. Microtubule seed is colored by red and dynamic microtubule is shown by blue. Microtubule growth events, including polymerization, depolymerization, catastrophe and rescue, are indicated. Plus and minus ends of microtubules are indicated. c–e Representative kymographs show microtubules grown from X-rhodamine-labeled GMPCPP-stabilized seeds with HiLyte FluorTM647 tubulin in the (c) absence and presence of 500 nM (d) GFP-HURP (1-280) and (e) GFP-HURP FL. Vertical scale bar = 90 sec, and horizontal scale bar = 10 μm. f–j Quantification of microtubule dynamicity, including (f) polyermization rate, (g) depolymerization rate, (h) catastrophe frequency, (i) rescue frequency and (j) microtubule length, as a function of the indicated GFP-HURP (1-280) and GFP-HURP FL concentrations. Data are mean ± SD (Standard deviation) from three independent experiments (n = 25 (Control), 17 (GFP-HURP (1-280) and 26 (GFP-HURP FL) microtubules). Two-tailed Student’s t test; Statistical differences: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n.s., not significant. k A summary of recruitment of tubulin to the microtubule in the presence of HURP. X-rhodamine-labeled microtubules were immobilized on the glass surface. GFP-HURP (indicated as HURP; GFP is shown as a black dot) and HiLyte FluorTM 647 labeled tubulin (indicated as α and β; HiLyte FluorTM647 is shown as an open dot) were imaged using TIRF microscopy. l, m GMPCPP-stabilized microtubules (X-rhodamine-labeled) were incubated with HiLyte FluorTM647 tubulin in the (l) absence and (m) presence of GFP-HURP FL. Scale bars, 10 μm. n HiLyte FluorTM 647 intensity for each condition (arbitrary units (arb. units)) was analyzed and plotted. Data are mean ± SD from three independent experiments (n = 99 microtubules for each condition). Two-tailed Student’s t test; Statistical differences: ****, P < 0.0001.
Furthermore, GFP-HURP FL, but not GFP-HURP (1-280), was enriched in polymerized GDP tubulin of dynamic microtubules, apart from at microtubule tips where EB1-GFP is bound (Supplementary Fig. 1f–j). This observation aligns with the behavior in vivo of HURP reported previously11. These findings imply that full-length HURP exhibits distinct affinities for tubulin in different nucleotide states. Additionally, the region outside HURP’s microtubule-binding site is involved in how HURP distinguishes between the different nucleotide states of tubulin. Notably, EB1-GFP elicited an increased frequency of microtubule catastrophe, causing microtubules to be more dynamic (Supplementary Fig. 1k, r)23,24,25. Given that HURP reduces the frequency of microtubule catastrophe and enhances rescue events in the presence of EB1 (Supplementary Fig. 1l–s), HURP and EB1 exert antagonistic roles in microtubule dynamics. The dynamic instability parameters in the presence of GFP-HURP (1-280) and GFP HURP FL were quantitated and are shown in Supplementary Table 2.
Many microtubule-associated proteins have been shown to control microtubule stability by independently regulating microtubule growth/depolymerization rates, catastrophe frequency, and rescue rate. Among them, the CLASP family of proteins contain microtubule-binding and tubulin-binding tumor overexpressed gene (TOG) domains that regulate microtubule dynamics26,27,28,29. Specifically, Cls1p, a Saccharomyces pombe homolog of CLASP that binds tubulin dimers, does not affect microtubule growth but instead induces microtubule rescue and suppresses microtubule catastrophe30. Based on the results revealed by the TIRF-based dynamic microtubule assays (Fig. 1b–j), we hypothesize that the HURP protein may bear tubulin-binding domains and thereby functions as the yeast CLASP protein. We performed multiple sequence alignments by ClustalW using protein sequences of HURP and TOG domains that have been structurally determined. The phylogenetic tree revealed that the protein sequence of HURP is closely related to the TOG2 domain in CLASP1 and CLASP2 (Supplementary Fig. 2a). Moreover, the pairwise protein alignment showed a similarity between HURP residues 250–600 and the CLASP TOG2 domain, despite the limited secondary structure agreement (Supplementary Fig. 2b). These results suggest that HURP contains TOG-like domains and may bind α/β-tubulin dimers.
Next, we applied TIRF microscopy to experimentally examine whether soluble α/β-tubulin dimers can be recruited to microtubules in the presence of HURP protein (Fig. 1k). The fluorescence signal of HiLyte FluorTM 647-labeled tubulin was essentially not detectable on the microtubules immobilized on coverslips in the absence of GFP-HURP FL (Fig. 1l). However, in the presence of GFP-HURP FL, the fluorescence signal of HiLyte FluorTM 647-labeled tubulin was evenly decorated along the microtubule filaments (~4.3-fold difference in HiLyte FluorTM 647 intensity in the absence and presence of GFP-HURP FL; Fig. 1m, n). These results suggest that GFP-HURP FL binds and recruits tubulin to the microtubules.
HURP bears tubulin-binding domains
We next performed pull-down assays to validate the interaction between the HURP tubulin-binding region and α/β-tubulin dimer. His-tagged GFP-HURP FL, GFP-HURP (1-280), and GFP-HURP tubulin binding region (a.a. 200-620; hereafter GFP-HURP (200-620)) were incubated with 1 μM bovine tubulin. Nocodazole was included in the reaction to suppress tubulin assembly into microtubules. The mixture containing His-tagged HURP proteins was subjected to nickel bead pulldown, and tubulin in the bound and supernatant fractions were analyzed by Coomassie blue staining. The GFP-HURP (1-280) could hardly pull down tubulin, whereas the majority of α/β-tubulin dimer could be pulled down by GFP-HURP FL and GFP-HURP (200-620) and found in the bound fractions (Fig. 2a, b). These results corroborate that HURP (200-620) contains tubulin-binding domains interacting with soluble α/β-tubulin dimers. This interaction was further validated by size exclusion chromatography (SEC), as we observed co-migration of GFP-HURP (200-620) and α/β-tubulin dimer in SEC (Fig. 2c–f), thereby suggesting that they form a stable complex in solution.

a Nickel pull-down assays of tubulin in the presence of his-tagged GFP-HURP FL, GFP-HURP (1-280) or GFP-HURP (200-620). Unbound (S) and bound (B) samples were analyzed by SDS-PAGE and stained with Coomassie blue. b Tubulin pulled down by HURP in a was analyzed. The bar graph shows the fraction of tubulin bound. Data are mean ± SD from three independent experiments. Two-tailed Student’s t test; Statistical differences: **, P < 0.01; n.s., not significant. c, SEC (Superdex 200) elution profiles (arb. units) for the GFP-HURP (200-620)•tubulin complex (red line), GFP-HURP (200-620) (green line), and tubulin (blue line). The void volume (Vo) is indicated. d–f The peak fractions of the (d) GFP-HURP (200-620)•tubulin complex, (e) GFP-HURP (200-620), (f), and tubulin were analyzed by SDS-PAGE and stained with Coomassie blue. g, h The BLI analysis displayed the binding kinetics of (g) GFP-HURP FL or (h) GFP-HURP (200-620) (the ligand) to tubulin (the analyte) over time. Various concentrations of tubulin were assessed. Binding affinities are indicated. i SDS-PAGE analysis of microtubule cosedimentation assays in the presence of GFP-HURP FL, GFP-HURP (1-280) or GFP-HURP (200-620). Supernatant and pellet fractions are indicated as S and P, respectively. j, HURP proteins co-pelleting with the microtubules in i were analyzed. The bar graph shows the fraction of HURP proteins bound. Data are mean ± SD from three independent experiments. Two-tailed Student’s t test; Statistical differences: *, P < 0.05; n.s., not significant. k, l Representative kymographs showing microtubules grown from X-rhodamine-labeled GMPCPP-stabilized seeds with HiLyte FluorTM647 tubulin in the (k) absence or (l) presence of 500 nM GFP-HURP (200-620). Vertical scale bar = 90 sec, and horizontal scale bar = 10 μm. m–p Quantification of microtubule dynamicity, including (m) polyermization rate, (n) depolymerization rate, (o) catastrophe frequency and (p) microtubule length, as a function of the indicated GFP-HURP (200-620) concentrations. Data are mean ± SD from three independent experiments (n = 30 (m–o) and n = 24 (p) microtubules). Two-tailed Student’s t test; Statistical differences: **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n.s., not significant.
We next applied biolayer interferometry (BLI) binding method to determine the binding affinities between the HURP and α/β-tubulin dimer. His-tagged GFP-labeled HURP FL, or HURP (200-620) was immobilized on Ni-NTA biosensor tips and its tubulin binding was analyzed at different concentrations of tubulin (Fig. 2g, h; 150 mM NaCl). The binding data were fit best using a 2∶1 heterogeneous ligand binding model, suggesting that two species of HURP may present and bind with different binding constants. The binding affinity (Kd1) value for full-length HURP and tubulin is stronger than that between GFP-HURP (200-620) and tubulin (Fig. 2g, h). Thus, we cannot rule out that weaker binding sites outside HURP (200-620) also contribute to tubulin binding. Notably, since the Kd2 value is 0.5 nM for GFP-HURP (200-620) binding to tubulin, we expect that the Kd2 value of GFP-HURP FL is smaller than that of GFP-HURP (200-620), i.e., approaching the detection limit of BLI and so it could not be determined. Nevertheless, our BLI data reveal a binding affinity in the sub-nanomolar range between HURP and tubulin dimer, corroborating the strong binding of HURP (200-620) to tubulin dimer and emphasizing its crucial role in facilitating tubulin interaction with HURP.
Microtubule cosedimentation assays were also conducted to examine whether the HURP (200-620) binds the microtubule. GFP-HURP FL, GFP-HURP (1-280), and GFP-HURP (200-620) were incubated with taxol-stabilized microtubules, and the mixture was then pelleted by centrifugation. GFP-HURP FL and GFP-HURP (1-280) were found mainly in the pellet fraction (>75%; Fig. 2i, j), consistent with previous studies13. However, only a small fraction of GFP-HURP (200-620) (<25%) was found in the pellet with microtubules (Fig. 2i, j), suggesting that GFP-HURP (200-620) plays a minimal role in microtubule binding. We also conducted TIRF-based dynamic microtubule assays in the presence of varying concentrations of GFP-HURP (200-620) (Fig. 2k, l). Whereas GFP-HURP (200-620) exhibited no effect on microtubule polymerization (Fig. 2m), the rate of depolymerization and frequency of catastrophe for microtubules were both increased at a protein concentration of 500 nM, resulting in shorter microtubules at higher HURP concentrations (Fig. 2n–p). These results support that GFP-HURP (200-620) sequesters tubulin dimers in solution, thereby enhancing microtubule dynamicity. Taken together, we propose that the HURP protein contains two distinct regions that facilitate microtubule binding and tubulin interaction.
The tubulin binding activity of HURP is independent of Ran-Importin-β pathway regulation
The N-terminus of HURP (a.a. 1-201) has been shown to contain a nuclear localization signal (NLS), allowing the HURP protein to interact with Importin-β directly12. The HURP microtubule-binding domain can be further divided into two motifs, and the Importin-β binding blocks the microtubule-binding activity of the motif that lies near the N-termius15,31. Thus, Importin-β at least partially prevents full length HURP and microtubule interaction12,15, suggesting that the Ran-dependent pathway modulates HURP-mediated microtubule stabilization. Here we further demonstrate that the N-terminal 60 amino acids of HURP co-eluted with Importin-β as revealed by the SEC analysis (Supplementary Fig. 2c, d), indicating that the NLS is located in the region between amino acids 1 and 60 of the HURP protein (Fig. 3a).

a Domain structure of HURP showing the microtubule-binding domain and NLS. Gray line indicates tubulin interaction region. b, c GMPCPP-stabilized microtubules were incubated with GFP-HURP FL in the (b) absence or (c) presence of Importin-β. Scale bars, 10 μm. d GFP intensity of indicated GFP-HURP proteins with and without Importin-β was analyzed and plotted (arb. units). Data are mean ± SD from three independent experiments (without Importin-β: n = 143 (GFP-HURP(1-110)) and n = 119 (GFP-HURP (1-280) and FL) microtubules and with Importin-β: n = 120 microtubules for all conditions). e SDS-PAGE analysis of GFP-HURP FL microtubule cosedimentation assays in the absence and presence of Importin-β. Supernatant and pellet fractions are indicated as S and P, respectively. f Quantification of GFP-HURP FL bound to microtubules. Data are mean ± SD from four independent experiments. Two-tailed Student’s t test; Statistical differences: **, P < 0.01; n.s., not significant. g Nickel pull-down assays of tubulin by his-tagged GFP-HURP FL with or without Importin-β. Unbound (S) and bound (B) samples were analyzed by SDS-PAGE and stained with Coomassie blue. h Fraction of tubulin bound by GFP-HURP FL in the absence and presence of Importin-β. Data are mean ± SD from five independent experiments. Two-tailed Student’s t test; Statistical differences: n.s., not significant. i–k Microtubule aster assembly in Xenopus egg extracts with (i) GFP-HURP FL, (j) GFP-HURPΔ(1-60) or (k) GFP-HURP (200-620), imaged using X-rhodamine tubulin (100 nM). Scale bars, 100 μm. l The number of microtubule asters was counted in multiple microtubule images and averaged (open circles; four extract preparations). Bars are mean ± SD. Two-tailed Student’s t test; Statistical differences: **, P < 0.01; ***, P < 0.001; n.s., not significant. m Representative fluorescence images of metaphase spindles (red: X-rhodamine-tubulin) assembled in Xenopus egg extracts with or without GFP-HURP (200-620). Scale bars, 25 μm. n Bipolar structures as in m were analyzed to determine individual spindle length (from left to right; n = 165, 154, and 162 spindles pooled from three extract preparations). Bars are mean ± SD. Two-tailed Student’s t test; Statistical differences: ****, P < 0.0001; n.s., not significant.
We next sought to recapitulate that Importin-β suppresses the microtubule-binding activity of HURP and applied TIRF microscopy to analyze the interaction of GFP-HURP FL, GFP-HURP (1-280) or GFP-HURP (a.a. 1-110; hereafter GFP-HURP (1-110)) with microtubules in the presence or absence of Importin-β. Consistent with microtubule cosedimentation results (Fig. 2i,j), both GFP signals of HURP FL and HURP (1-280) decorated along microtubules (Fig. 3b and Supplementary Fig. 2f). However, under the same acquisition conditions, GFP fluorescence signals of GFP-HURP (1-110) were not detectable along microtubules (Supplementary Fig. 2e). This result suggests that the HURP (1-110) contains the NLS and does not significantly contribute to microtubule interaction (Fig. 3a). Notably, a substantial reduction (6 to 4-fold) in the GFP signals of GFP-HURP FL and GFP-HURP (1-280) along microtubules in the presence of Importin-β was observed compared to that in the absence of Importin-β (Fig. 3b–d and Supplementary Fig. 2g). Moreover, in the microtubule cosedimentation assays, the amount of GFP-HURP FL associated with the microtubule pellet was also substantially reduced in the presence of Importin-β (~ 2 fold; Fig. 3e, f). These results indicate that Importin-β suppresses the microtubule-binding activity of HURP, consistent with previous findings12,15.
Interestingly, the amount of α/β-tubulin dimer pulled down by His-tagged GFP-HURP FL was comparable in the presence and absence of Importin-β (Fig. 3g, h), suggesting that Importin-β does not inhibit the tubulin-binding activity of HURP. Moreover, (1) GST-tagged Importin-β could pull down both GFP-HURP and tubulin (Supplementary Fig. 2h). (2) Importin-β, tubulin, and GFP-HURP FL were co-eluted in the same fractions in the SEC analysis as shown by SDS-PAGE and Western blots (Supplementary Fig. 2i–m). These results suggest that GFP-HURP FL, tubulin, and Importin-β indeed can form a tetrameric protein complex (HURP•α-tubulin•β-tubulin•Importin-β).
The HURP activities that promote microtubule assembly and sequester tubulin dimer modulate microtubule array formation
In current models, at the onset of mitosis, importins form complexes with spindle assembly factors (SAFs) (e.g., nuclear mitotic apparatus protein [NuMA]5,6,7,32 and targeting protein for Xklp2 [TPX2])8,9), and thereby block their activities2. Subsequently, RanGTP induces the release of SAFs from inhibitory complexes, promoting mitotic spindle assembly2. However, in HURP, Importin-β only suppresses microtubule-binding activity but not tubulin binding activity. This finding leads us to propose that HURP conducts two activities upon Importin-β binding and release: (1) Importin-β binds HURP and blocks its microtubule-binding activity while allowing HURP to sequester tubulin from solution (Like Stathmin33); and (2) When Importin-β is released from HURP (by Ran), HURP functions as a CLASP-like protein and stabilizes microtubule assembly (Fig. 7l).
To examine how HURP-dependent microtubule assembly promoting and tubulin sequestering activities respectively regulate microtubule array formation, two constitutively active HURP proteins (GFP-HURPΔ(1-60) and GFP-HURP (200-620)) were examined using Xenopus egg extracts. First, we performed a microtubule aster assembly assay using GFP-HURP FL, GFP-HURP (200-620), or GFP-HURPΔ(1-60). We found that the addition of 4 μM GFP-HURP FL led to substantial microtubule aster assembly compared with the buffer and taxol controls (Fig. 3i, l and Supplementary Fig. 3a, b). However, GFP-HURP (200-620) that binds tubulin but not microtubules essentially induced no microtubule aster formation even at a concentration of 15 μM (Fig. 3k, l). Consistently, GFP-HURP-FL, but not GFP-HURP (200-620), could be detected on aster microtubules (Supplementary Fig. 3d–k). Notably, a one-quarter concentration of GFP-HURPΔ(1-60), which lacks the major Importin-β binding (Supplementary Fig. 3c), is sufficient to promote a comparable aster number compared to GFP-HURP FL (1 μM versus 4 μM; Fig. 3j, l). These suggest that endogenous Importin-β in egg extracts regulates microtubule binding of HURP via the N-terminal region (residues 1-60), and its release promotes microtubule assembly and aster formation.
GFP-HURP (200-620) did not show any prominent phenotype in the aster assembly assay (Fig. 3k, l). We therefore examined its effect on metaphase spindle assembly in Xenopus egg extracts. Since GFP-HURP (200-620) cannot bind microtubules and shows an Importin-β-independent tubulin binding activity in vitro, we hypothesize that it will constitutively sequester tubulin in solution and limit spindle growth34. Unlike GFP-HURP FL, GFP-HURP (200-620) essentially was not detectable in metaphase spindles assembled in Xenopus egg extracts (Supplementary Fig. 3l–q). Importantly, addition of GFP-HURP (200-620) (15 μM) to Xenopus egg extracts reduced the length of metaphase spindles by ~15% relative to the buffer control (41.1 ± 5.3 µm to 35.7 ± 7.3 µm) (Fig. 3m, n). Therefore, we propose that the two HURP activities, microtubule assembly promoting and tubulin sequestering, are critical in the mitotic cytoplasm and control spindle assembly (Fig. 7l; See Discussion section).
Single-particle cryo-EM of the HURP•α/β-tubulin complex
Our biochemical and biophysical analyses revealed the interaction between HURP residues 200-620 and α/β-tubulin, thereby regulating microtubule dynamics. However, unlike the CLASP TOG2 domain that is an α-solenoid protein comprising six HEAT repeats35, protein secondary structural prediction revealed an equal abundance of α-helix and coiled-coil in the region of HURP residues 250-600 (Supplementary Fig. 2b). Thus, to gain structural insights into how HURP binds to α/β-tubulin, we applied cryo-EM to determine the structure of the HURP•α/β-tubulin complex.
To prepare homogeneous samples, we purified GFP-HURP (200-620) from E. coli and then biochemically assembled the GFP-HURP (200-620)•α/β-tubulin complex by mixing GFP-HURP (200-620) and bovine α/β-tubulin at an equal molar ratio (Fig. 2c–f). The peak fractions of SEC were subsequently vitrified on cryo-EM grids, and micrographs were acquired using a Titan Krios microscope (300 keV) (Supplementary Fig. 4a). Two-dimensional (2D) class averaging revealed two globular densities corresponding to α/β-tubulin dimer with an additional density placed on the top (Supplementary Fig. 4b). Next, multiple three-dimensional (3D) reconstruction rounds were performed using the best-resolved classes across each classification (Supplementary Fig. 4c). Focused refinement masking α- and β-tubulin respectively further improved the resolution of α-tubulin, and β-tubulin and HURP region (Supplementary Fig. 5). The focus-refined maps were stitched together to obtain a composite map for final atomic model building and refinement (Supplementary Fig. 4c). Ultimately, a cryo-EM structure of the GFP-HURP (200-620)•α/β-tubulin complex was determined to have an average resolution of 3.54 Å using 248,567 particles (Fig. 4a, Supplementary Fig. 6a, b and Table 1). The α and β-tubulin dimer interface in the cryo-EM volume density map could be well resolved, showing well-defined side-chain densities of most residues. Thus, α/β-tubulin could be unambiguously orientated and assigned to the two globular densities (Supplementary Fig. 6e, f).

a A cryo-EM map of the HURP (428-534)•β-tubulin•α-tubulin complex. The map is segmented into HURP (428-534) (Green), β-tubulin (light gray) and α-tubulin (dark gray) (EMD-38181). b Cartoon representation of the HURP (428-534)•β-tubulin•α-tubulin structure. HURP (428-534), β-tubulin and α-tubulin are shown in green, dark blue and light blue, respectively. The secondary structures of β-tubulin that contribute to the HURP interaction are colored differently. (PDB ID: 8X9P) c A zoomed-in view of the HURP (428-534)•β-tubulin•α-tubulin complex highlighted by the black dashed box in b is shown. The α-helix H, a loop between α-helix G and β-strand E, and the α-helix N of β-tubulin that mediate the HURP interaction are highlighted in pink, red and orange, respectively. d A schematic representation of the interaction between the HURP (428-534) and β-tubulin using the same color code as shown in a. Yellow boxes highlight two binding patches. e A close up view showing the interaction between the HURP (428-534) and β-tubulin. The HURP (428-534) and β-tubulin are shown as cartoon representations with the same color code in c. Key residues involved in the binding are indicated and shown as sticks. f SEC (Superdex 200) elution profiles (arb. units) for the GST-HURP (430-550) WT•tubulin complex (black line), GST-HURP (430-550) (light gray), and tubulin (dark gray). The void volume (Vo) is indicated. g The peak fractions of the GST-HURP (430-550) WT•tubulin complex (Top panel), GST-HURP (430-550) (Middle panel), and tubulin (Low panel) in f were analyzed by SDS-PAGE and stained with Coomassie blue. Representative gels are shown (n = 3). h–j SEC (Superdex 200) elution profiles (arb. units) for (h) GST-HURP (430-550) patch 1 mutant, (i) GST-HURP (430-550) patch 2 mutant or (j) GST-HURP (430-550) patch 1&2 mutant in the presence of tubulin (Black lines). Tubulin alone profiles are shown as light gray lines. The void volume (Vo) is indicated.
In the cryo-EM maps, we identified an extra density on top of β-tubulin and exhibited structural features of a three-α-helix bundle (Fig. 4a and Supplementary Fig. 6c, d). To determine the region in GFP-HURP (200-620) that forms the α-helix bundle and interacts with β-tubulin, GFP-HURP (200-620)•α/β-tubulin complex was subjected to limited proteolysis using elastase over different time courses (Supplementary Fig. 6g). In addition to the tubulin and GFP, a stable peptide that appeared in SDS-PAGE after 1-h elastase treatment was examined by mass spectrometry analysis (indicated by a star; Supplementary Fig. 6g). The mass spectrometry analysis identified the protease-resistant HURP segment starting from residue 398 to 588, and this stable region may bind to tubulin (Supplementary Fig. 6h). Additionally, we also carried out AlphaFold2 to predict the structure of HURP, and the region of HURP residues 428-534 (hereafter HURP (428-534)) was predicted to be a three α-helix bundle (Supplementary Fig. 6i). Ultimately, the predicted HURP (428-534) structure could be assigned into the cryo-EM density map, based on the structural features resolved in our cryo-EM maps (Fig. 4a, b and Supplementary Fig. 6d).
Two binding patches facilitate HURP and β-tubulin interaction
In the refined model of HURP (428-534)•α/β-tubulin protein complex, HURP (428-534), composed of three long α-helices, forms a 1:1 stoichiometric complex with α/β-tubulin (Fig. 4b and Supplementary Fig. 7a) (PDB ID: 8X9P). The HURP (428-534) binds to the longitudinal surface of the β-tubulin (Fig. 4b, c). We next performed interface area calculations and identified a large buried surface area between HURP (428-534) and β-tubulin (~1000 Å2, as calculated using the software Protein Interfaces, Surfaces and Assemblies (PISA)). Two binding patches are identified in this interface, contributing to the interaction between HURP (428-534) and β-tubulin (Fig. 4c–e and Supplementary Fig. 7c). In the binding patch 1, the α-helix 2 of HURP (428-534) interacts with the α-helix H and a loop between α-helix G and β-strand E of β-tubulin (Fig. 4d, e). In the binding patch 2, the α-helix 1 of HURP (428-534) and the α-helix N of β-tubulin interact with each other (Fig. 4d, e). In general, multiple polar and non-polar (e.g., F212 and Y206 of β-tubulin) residues are identified in the protein-protein interaction interface (Fig. 4d, e).
Next, we introduced point mutations in the two binding patches in HURP, and biochemically examined mutant HURP (a.a. 430-550; hereafter HURP (430-550)) and tubulin interaction using SEC analysis. Wild type GST-HURP (430-550) and α/β-tubulin exhibited an elution peak shift toward a higher value in the SEC analysis (Fig. 4f, g), confirming the interaction between HURP (430-550) and tubulin. However, mutants of GST-HURP (430-550) (Patch 1 (R468A, E480A and Q484A), Patch 2 (M512A and Q516A) and Patch 1&2) cannot bind tubulin as no peak shifts were observed in the SEC (Fig. 4h–j).
Our TIRF-based assays on microtubules demonstrate that the HURP (200-620) fragment accelerated the rate of microtubule depolymerization and increased the frequency of microtubule catastrophe, leading to shorter microtubules (Fig. 2k–p). Thus, HURP (200-620) binds and sequesters tubulin in solution, resulting in the formation of a smaller mitotic spindle in Xenopus egg extracts (Fig. 3m, n). To investigate the impact of mutations in Patch 1 and 2 on the activity of GFP-HURP (200-620) and GFP-HURP FL in regulating microtubule dynamics, we compared the effects of wild-type (WT) and mutant variants under equivalent protein concentrations (Fig. 5a and Supplementary Fig. 7d). Despite the fact that microtubule length was not supposed to change at this concentration (500 nM), the GFP-HURP (200-620) Patch 1&2 mutant exhibited a reduced depolymerization rate and catastrophe frequency of microtubules relative to GFP-HURP WT (Figs. 2p and 5b–e). Moreover, both the GFP-HURP FL Patch1 and Patch 1&2 mutants led to an increased frequency of microtubule catastrophe and a decreased frequency of rescue, as well as reduced dynamic microtubule lengths, relative to control (Fig. 5f–j). These findings validate our structural model and underscore the crucial role of residues 430-550 in HURP for mediating interactions with tubulin and regulating microtubule dynamics.

a GFP-HURP (200-620) wild type and the Patch 1&2 mutant were analyzed by SDS-PAGE and stained with Coomassie blue. b–e Quantification of microtubule dynamicity, including (b) polyermization rate, (c) depolymerization rate, (d) catastrophe frequency, and (e) microtubule length, in the presence of GFP-HURP (200-620) wild type or the Patch 1&2 mutant. Data are mean ± SD from three independent experiments (n = 25 (b–d) and n = 19 (e) microtubules). Two-tailed Student’s t test; Statistical differences: *, P < 0.05; n.s., not significant. f–j Quantification of microtubule dynamicity, including (f) polyermization rate, (g) depolymerization rate, (h) catastrophe frequency, (i) rescue frequency and (j) microtubule length, in the presence of GFP-HURP FL wild type, and the GFP-HURP FL Patch 1 or GFP-HURP FL Patch 1&2 mutants. Data are mean ± SD from three independent experiments (n = 20 microtubules for each condition). Two-tailed Student’s t test; Statistical differences: **, P < 0.01; ***, P < 0.001; n.s., not significant. k Superimposed structures of HURP (423-534) (green), DARPin (pink), and PN2-3 (purple) with β-tubulin (gray). l Overlapping regions in β-tubulin that contact HURP (428-534), PN2-3 and vinorelbine. Protein secondary structures of the contact regions in β-tubulin are shown. Interfacing residues in β-tubulin that facilitate HURP (428-534), PN2-3 and vinorelbine interaction are highlighted in green, purple and brown, respectively. m Superimposition of the HURP (428-534) (green), vinblastine (orange) and vinorelbine (brown) structures. n SDS-PAGE analysis of vinblastine-mediated tubulin association with or without HURP-TBD. Supernatant and pellet fractions are indicated as S and P, respectively. o The bar graph of tubulin in supernatant. Data are mean ± SD from four independent experiments. Two-tailed Student’s t test; Statistical differences: **, P < 0.01; ****, P < 0.0001. p GMPCPP-stabilized microtubules were incubated with GFP-HURP FL with or without vinorelbine. Scale bars, 10 μm. q GFP signal intensity for each condition (arb. units). Data are mean ± SD from three independent experiments (n = 50 microtubules). r Schematic representation of how HURP prevents vinorelbine-induced tubulin aggregation. HURP contains a microtubule-binding domain (MBD; black) and a tubulin-binding domain (TBD; green).
HURP binds the vinca domain on β-tubulin longitudinal surface
The TOG domain structures were determined from XMAP215 family members, showing a solenoid-like structure containing six HEAT repeats connected by loops26,36. Structural studies further revealed that these evolutionarily conserved intra-HEAT loops bind to the lateral surface of tubulin dimer. However, the HURP (428-534) interacts with the β-tubulin longitudinal surface (Fig. 4b). Thus, although the HURP (428-534) and TOG domains both interact with tubulin dimer, they bind to distinct binding sites on tubulin (the longitudinal and lateral surfaces, respectively), facilitated by different binding modes (α-helices and intra-HEAT loops, respectively) (Supplementary Fig. 7b).
Currently, two proteins, designed ankyrin repeat proteins (DARPins), a genetically engineered antibody mimetic protein, and PN2-3 peptide, N-terminus of centrosomal-P4.1-associated-protein (CPAP) have been structurally shown to bind the β-tubulin longitudinal surface exposed at microtubule plus ends (Fig. 5k and Supplementary Fig. 7b)37,38. Particularly, the N-terminal disordered region of PN2-3 that shows high mobility binds the vinca domain on β-tubulin, as revealed by crystal structures (Fig. 5k, l)39. However, because of the structural and biochemical properties, the PN2-3 domain binds β-tubulin in a different way from the HURP (428-534) (See Discussion Section).
HURP directly competes with vinorelbine binding on β-tubulin, promoting microtubule growth
Vinca alkaloids are a subset of anti-cancer drugs inhibiting microtubule dynamics and binding to the vinca domain at the longitudinal interface between α/β-tubulin dimers (Supplementary Fig. 7e)40. One of the binding patches (Patch 1) of HURP shares interfacing residues with the vinca domain pocket (Fig. 5l, m). Moreover, the HURP binding site overlaps with vinorelbine binding site on β-tubulin (Fig. 5l, m). Vinorelbine is one of the vinca alkaloids and carries out its anti-cancer effects by poisoning microtubule dynamics41. Interestingly, the sensitivity to vinorelbine significantly correlates with HURP expression level in oropharyngeal squamous cell carcinoma (OPSCC)17.
We next examined whether HURP directly competes with vinorelbine for β-tubulin binding, contributing to the drug resistance in tumor cells. The mechanism of action for vinorelbine is to induce tubulin self-association into aggregates, suppressing microtubule assembly22. We conducted cosedimentation assays to examine whether the HURP (428-534) (hereafter HURP-tubulin binding domain (TBD)) can reduce tubulin aggregation induced by vinorelbine. Tubulin was incubated with vinorelbine at room temperature in the absence and presence of HURP-TBD. After centrifugation, tubulin in the pellet fraction was substantially reduced in the presence of HURP-TBD, corroborating that binding of HURP-TBD and vinorelbine to tubulin is mutually exclusive (Fig. 5n, o). Notably, unlike the tubulin-binding activity, microtubule binding of HURP is not perturbed by vinorelbine as the fluorescence intensity of GFP-HURP FL and GFP-HURP (1-280) was comparable in the absence and presence of vinorelbine revealed by TIRF assays (Fig. 5p, q and Supplementary Fig. 7f).
We next sought to examine whether HURP can recover the microtubule dynamics impaired by vinorelbine treatment, conferring survival advantage to cancer cells. In the presence of vinorelbine, microtubule depolymerization rate, catastrophe frequency, and rescue rate did not change (Fig. 6a, b, g, h, i, j), whereas the microtubule polymerization rate was reduced compared to the control (Fig. 6a, b, f). Ultimately, vinorelbine treatment led to a shorter length of dynamic microtubules (20 minutes; Fig. 6k). Interestingly, while HURP did not recover the microtubule polymerization rate altered by vinorelbine, the presence of HURP decreased depolymerization rate and catastrophe frequency as well as enhanced rescue rate of microtubules (Fig. 6c, d, e, h, i). Thus, HURP elevates the ratio of catastrophe frequency and rescue rate, resulting in that nearly every single catastrophe event could be rescued in the 1μM of HURP (250 nM of vinorelbine) (Fig. 6j). HURP reduces microtubule dynamics in the presence of vinorelbine, promoting net microtubule assembly (Fig. 6l, m).

a A representative kymograph show a microtubule grown from X-rhodamine-labeled GMPCPP-stabilized seeds with HiLyte FluorTM647 tubulin without addition of vinorelbine and GFP-HURP FL. b–e Under the same experimental setting as in (a), representative kymographs show microtubules grown in the presence of 250 nM vinorelbine without and with the indicated concentration of GFP-HURP FL. Plus and minus ends of microtubules are indicated. Vertical scale bar = 90 sec, and horizontal scale bar = 10 μm. f–k Quantification of microtubule dynamicity, including (f) polymerization rate, (g) depolymerization rate, (h) catastrophe frequency, (i) rescue frequency, (j) ratio of rescue and catastrophe frequency, and (k) microtubule length, as a function of vinorelbine with the indicated GFP-HURP FL concentrations. Control experiment (no addition of vinorelbine and GFP-HURP FL protein) is indicated. Data are mean ± SD from three independent experiments (n = 26 (control, vinorelbine only, 250 nM) and n = 20 (500 nM and 1 µM) microtubules). Two-tailed Student’s t test; Statistical differences: *, P < 0.05; **, P < 0.001; ****, P < 0.0001; n.s., not significant. l A representative image shows microtubules grown from X-rhodamine-labeled GMPCPP-stabilized seeds (red) with HiLyte FluorTM 647 tubulin (blue) without addition of vinorelbine and GFP-HURP FL (left panel). Under the same experimental setting, representative images show microtubules grown in the presence of vinorelbine without and with GFP-HURP FL. Incubation time is 40 minutes. Scale bars, 10 μm. m Quantification of length of dynamic microtubule (blue), as a function of vinorelbine with and without GFP-HURP FL in the indicated time points. Data are mean ± SD from three independent experiments (n = 59 microtubules for each condition). Two-tailed Student’s t test; Statistical differences: ****, P < 0.0001; n.s., not significant.
Moreover, the analytic ultracentrifugation with sedimentation velocity (SV-AUC) was performed to characterize the size distributions of HURP. SV-AUC revealed that GFP-HURP (1-280), GFP-HURP (200-620) and GFP-HURP FL primarily behave as a monomer in solution, with molecular masses consistent with calculated values (Supplementary Fig. 7g-i). Hence, the monomeric HURP directly competes with vinorelbine for β-tubulin binding, countering microtubule assembly defects caused by vinorelbine (Fig. 5r). The dynamic instability parameters in the presence of vinorelbine and GFP-HURP FL were quantitated and are shown in Supplementary Table 3.
TBD in HURP modulates K-fiber assembly and accounts for drug resistance to vinorelbine
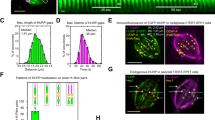
To understand the cellular relevance of HURP’s TBD, CRISPR/Cas9 genome editing was employed to remove a section of exon12 (encompassing residues 463-504) in a hepatoma cell line, Hep3B (Supplementary Fig. 8a–c). The exon 12 encodes the α-helix 2 in TBD, mediating tubulin interaction within binding patch 1 (Fig. 4d, e). The editing process resulted in a deletion of 41-residue, approximately 5 kDa, from the HURP protein (HURP-tubulin binding segment (TBS) deletion mutants; hereafter HURP-TBS deletion mutants; Supplementary Fig. 8d). Notably, while the SDS-PAGE analysis did not distinguish the size difference between the deletion mutant and wild type (Supplementary Fig. 8e; In SDS-PAGE, the observed molecular weight of the wild-type protein is >110 kDa, despite the theoretical molecular weight being ~95 kDa.), it indicated that the amount and stability of HURP protein were comparable in both wild type and mutant cell lines. Subsequently, we isolated two independent colonies exhibiting these deletions, indicating homozygous mutations (Supplementary Fig. 8a–c). These mutant cells showed an approximate 7% increase in the G2/M phase, suggesting that HURP’s TBD is crucial for mitosis progression compared to the wild-type (Fig. 7a). Moreover, cell growth curve experiments demonstrated that these mutants exhibited increased sensitivity to vinorelbine compared to the wild-type cells (Fig. 7b–d), corroborating that the HURP’s TBD accounts for the drug resistance to vinorelbine in the cellular context.

Cell cycle phases (G1, S, G2/M) represented as blue, purple, and green bars. Data are mean ± SD from five independent experiments. One-tailed Student’s t-test; Statistical differences: *p < 0.05 for the G2/M phase in the wild-type and mutant cell lines. b–d, Growth curves for (b) wild-type and (c, d) two homozygous mutant Hep3B cell lines in the absence and presence of 0.005 μM vinorelbine. Data are mean ± SD from five independent experiments. The cell number differences at the day 10 between untreated and treated with vinorelbine (0.005 μM) are indicated. e, f Representative immunofluorescence staining images of (e) wild-type and (f) HURPΔTBS-1 mutant cell lines using α-tubulin (green, left panels) and CENP-A (red, middle panels) antibodies. The merged fluorescent images are shown in the right panels. Scale bar: 5 μm. g, h Quantification plots (arb. units) of (g) total microtubule and (h) K-fiber (α-tubulin fluorescence intensities) in wild-type and mutant cells at metaphase. Data are mean ± SD from three independent experiments (n = 21 (WT) and n = 19 (mutant) cells). Two-tailed Student’s t-test; Statistical differences: *p < 0.05; ***, P < 0.001; ****, P < 0.0001. i, j Representative images of (i) wild-type and (j) HURPΔTBS-1 mutant cells after cold treatment and immunofluorescently stained using antibodies against α-tubulin (green, left panels) and CENP-A (red, middle panels). The merged fluorescence images are shown in the panels at right. Scale bar: 5 μm. k The quantification plot (arb. units) of cold stable K-fiber (α-tubulin fluorescence intensities) in wild-type and mutant cells at metaphase. Data are mean ± SD from three independent experiments (n = 24 cells). Two-tailed Student’s t-test; Statistical differences: ***, P < 0.001; ****, P < 0.0001. l A schematic model: Importin-β suppresses HURP’s microtubule-binding activity during mitosis, preventing spindle assembly. Ran-GTP releases HURP, promoting tubulin recruitment and microtubule assembly. m, n In the dividing cell, (m) vinorelbine decreases the microtubule polymerization rate, blocking mitotic spindle assembly. However, (n) elevated HURP counters vinorelbine’s effects by stabilizing K-fibers.
The immune-fluorescent studies revealed a reduction in HURP signal along the mitotic spindle in mutant cells compared to their wild-type counterparts (Supplementary Fig. 8f, g). Our investigations, along with previous studies (Figs. 2i and 3d), have indicated that HURP (1-280) predominantly mediates microtubule binding, with residues 464-504 in HURP playing a minimal role in this interaction. This led us to speculate that the decrease in the number of K-fiber might be responsible for the weakened HURP staining on the spindle. In the immune-fluorescent analysis, we observed an overall weaker intensity of microtubules in the region surrounding the kinetochore (Fig. 7e–g and Supplementary Fig. 8h). Considering that K-fibers are bundled microtubules, we further conducted an assessment of microtubule intensity that surpassed the average intensity of spindle microtubules (Supplementary Fig. 8i). The microtubules exhibiting significantly higher fluorescent intensity, considered as K-fibers, were notably diminished in the mutant cell lines (Fig. 7e, f, h). Moreover, since K-fibers display better resistance to cold treatment, we conducted cold treatment experiments in wild-type and mutant cell lines and analyzed microtubule signal intensity post-treatment. Consistently, the fluorescence intensity of the leftover microtubules was reduced in the mutant cell lines compared to wild type (Fig. 7i–k). Taken together, the results corroborate that TBD in HURP is critical for K-fiber assembly, thereby accounting for drug resistance to vinorelbine (Fig. 7m, n).
Discussion
The HURP gene was originally found to be overexpressed in HCC10, and the knockdown of HURP expression suppresses cancer cell growth16,18,19. Notably, HURP knockout mice typically develop with no distinguishable phenotypes compared to wild-types42. These results suggest that HURP may play a much more critical role in cancer cell growth compared to regular cells. Moreover, the HCC with high invasiveness further displayed a ~ 10-fold higher HURP expression than those with low invasiveness43. HURP mRNA expression analysis also revealed a five- to hundred-fold higher expression of HURP transcripts in other cancer cells (e.g., head and neck squamous cell carcinoma17, urothelial carcinoma44, and breast cancer45) (Supplementary Table 1). Since the concentration of HURP with a molecular weight of 120 kDa in Xenopus egg extracts is ~ 370 nM46, the HURP cellular concentration in cancer cells could be in the sub-micromolar range. Hence, the HURP amount in cancer cells should be sufficient to regulate microtubule assembly from highly abundant tubulin.
In this study, we identify a TBD in HURP that regulates microtubule dynamics, particularly in response to the microtubule catastrophe and rescue frequency. Moreover, our structural study shows that HURP’s TBD binds to the vinca domain on β-tubulin, allowing HURP to directly compete with vinorelbine and providing drug resistance for cancer cell. Hence, our study here allows us to develop chemical inhibitors for HURP. Conventional chemotherapy’s effectiveness is hindered by the development of drug resistance in cancer cells. To overcome this limitation, researchers have been exploring more potent treatment strategies, such as combination chemotherapy. This approach involves using multiple drugs with distinct mechanisms, reducing the chances of cancer cells becoming resistant. Vinorelbine has combined with other chemotherapeutic agents (e.g., Carboplatin47 and Cisplatin48 that block DNA synthesis) to enhance treatment efficiency. Thus, on top of the development of HURP inhibitors, our findings presented here further pave the way to create an alternative pathway for combined therapy with vinorelbine. This could significantly boost the effectiveness of cancer treatment, especially for cancer cells that exhibit high levels of HURP proteins.
The microtubule is a dynamic filament composed of α-/β-tubulin heterodimers. Individual microtubules grow by the addition of tubulin dimer to the growing tips. The frequency of the switch between microtubule growth and shortening phases is partly controlled by the availability of α-/β-tubulin dimer in the solution. Many proteins have been shown to interact with α-/β-tubulin heterodimers, thereby modulating microtubule dynamics. Among them, some interact with the lateral contact surface of α-/β-tubulin heterodimers (e.g., stathmin49 and TOG domain proteins27,50), whereas others bind to the longitudinal surfaces (e.g., CPAP). Stathmin can interact with tubulin heterodimers and prevent tubulin from being incorporated into microtubules, which suppresses microtubule formation49,51,52. TOG domain proteins (e.g., XMAP215 homologs27,50) contain multiple TOG-like domains that bind to free tubulin. Combined with their microtubule-binding activity, they have been proposed to function as microtubule polymerases. Moreover, CLASP also contains TOG-like domains and has a homolog in Saccharomyces pombe, i.e., Cls1p. This homolog has been demonstrated to interact with free tubulin, thereby stabilizing microtubules by recruiting tubulin dimers to the microtubule lattice30.
Besides the artificial proteins (e.g., DARPin37 and αRep53), a centrosomal CPAP protein in mammals uses a PN2-3 domain that contains three discrete binding motifs to bind the longitudinal surfaces of β-tubulin39. Notably, Eribulin, an anti-cancer drug that binds to a site on β-tubulin that is distinct from the vinca domain54,55, impairs the binding of PN2-3 domain for tubulin38. These results suggest the PN2-3 domain binds near the vinca domain of β-tubulin and is different from the HURP-tubulin binding site. Moreover, at present, no evidence suggests CPAP that carries the PN2-3 domain can provide drug resistance for cancer cells because (1) Low cellular concentration: CPAP has never been found to overexpress in cancer cells. Thus, cellular concentration of CPAP most likely is comparable to other centrosomal proteins that are in the nanomolar range (e.g., ~50 nM of Cep192 and pericentrin in the early embryos of C. elegans56). (2) Different β-tubulin interacting mode: the binding affinity between individual motifs of PN2-3 and tubulin is in the range of micromolar38,57, suggesting that the nanomolar affinity of PN2-3 for the longitudinal surface of β-tubulin comes from the cooperative effect of these three motifs38. Under physiological conditions, the binding affinity between the individual motifs of PN2-3 domain and β-tubulin (micromolar range) is unlikely sufficient to compete with anti-cancer drugs (e.g., Erbulin and vinorelbine). (3) Cellular localization: CPAP is located at the centrosome, limiting its function in microtubules at centrioles, not in spindle microtubules. In current models, the PN2-3 and microtubule-binding domains allow CPAP to cap the microtubule plus end, facilitating persistent slow growth of the centriolar microtubule38,57,58,59,60.
The binding modes between tubulin dimer and the TOG domain or HURP-TBD differ. However, CLASPs do not affect microtubule growth, but instead induce microtubule rescue and suppress catastrophe30,61,62. Therefore, we propose that HURP and yeast CLASP (Cls1p) share molecular mechanisms by which microtubule catastrophe and rescue are regulated, which are facilitated by the HURP TBD. HURP captures α-/β-tubulin heterodimers and binds to the microtubule lattice. Consequently, when microtubule plus-ends shrink to the sites where HURP-tubulin complexes are located, microtubules stop depolymerizing and re-grow (i.e., a rescue event). Notably, HURP is not enriched at the GTP cap of microtubules11, so it does not behave like CPAP, which blocks longitudinal tubulin-tubulin interactions and suppresses microtubule growth38. In terms of the suppression of microtubule catastrophe, HURP may promote local rescue events close to the GTP cap, thereby reducing the disassembly phase. In general, HURP elevates the local tubulin dimer concentration along the microtubule lattice and stabilizes microtubules.
The SAFs influence diverse microtubule behaviors like nucleation, stabilization, and bundling and generally promote spindle assembly2. RanGTP operates by regulating the interaction between SAF and nuclear transport receptor, thereby unleashing the actions of SAFs and promoting the assembly of spindle necessary for cell division2. Consequently, importins are expected to suppress SAFs’ actions, thereby modulating the assembly of the mitotic spindle. However, our study here reveals that Importin-β specifically suppresses the activity of HURP in binding to microtubules but not in its interaction with tubulin. This discovery led us to propose a hypothesis: at the onset of mitosis, when Importin-β binds to HURP, HURP primarily binds to tubulin dimers. This binding potentially reduces the concentration of free tubulin dimers in the cytosol, effectively hindering microtubule assembly, functioning akin to stathmin49,51,52. Next, once RanGTP triggers the release of Importin-β from HURP, the liberated HURP can then engage with tubulin dimers on the microtubule lattice. This action facilitates the assembly of microtubules, thereby aiding in the formation of the spindle required for cell division. The ability of HURP to bind tubulin independently of Importin-β likely regulates the availability of free tubulin in the cytosol. This regulatory function potentially synchronizes the timing of mitotic spindle assembly, as depicted in Fig. 7l.
Methods
Protein expression and purification
HURP-FL wild type and mutant variants were cloned into pACEBac1 (MultiBacTM, GENEVA BIOTECH), consisting of His and GFP tags at the N-terminus and a TEV cleavage site between the two tags. Recombinant His-GFP-HURP-FL was expressed in Hi5 cells. Cells were harvested at 1000xg for 30 min and resuspended in lysis buffer containing 50 mM K-phosphate, 300 mM KCl, and 1 mM β-mercaptoethanol, pH 7.4 supplemented with EDTA-free protease inhibitor. After sonication, lysed cells were centrifuged at 40,000 g for 30 minutes. The supernatant was applied to Ni-NTA beads, followed by equilibrating with 50 mM K-phosphate, 300 mM KCl, and 1 mM β-ME, pH 7.4. Non-specific interactions were eluted with 25 mM Imidazole. Protein samples were eluted with 250 mM Imidazole. Fractions were further purified onto the HiLoad Superdex 200 (16/600 pg) column (GE Healthcare). Protein quality from the size exclusion column was analyzed and confirmed by SDS-PAGE. The protein concentration was measured using extinction coefficient by a Nano spectrophotometer and was stored in 50 mM K-phosphate, 300 mM KCl, and 1 mM β-ME, 10% sucrose pH 7.4. at −80 °C. Company names and catalog numbers of all reagents are provided in Supplementary Data 1.
The GFP-tagged HURP constructs and EB1 were cloned into pET-Duet-1 (Novagen) containing an N-terminal His tag with a TEV cleavage site and GFP at the C-terminus. The GFP-HURP (1-110), GFP-HURP (1-280), GFP-HURP (200-620) and GFP-EB1 constructs were expressed in BL21 (DE3) Rosetta (Novagen). Bacterial cells were induced for 16 hrs with 0.5 mM IPTG at 18 °C. The cells were pelleted and then suspended in lysis buffer (50 mM K-phosphate, 150 mM KCl, and 3 mM β-ME, pH 7.4). The cells were lysed by passing through a French press, and the lysate obtained was centrifuged at 40,000xg for 30 min. The supernatant was mixed with Ni-NTA beads, calibrated with 50 mM K-phosphate, 150 mM NaCl, and 3 mM β-ME, pH 7.4. Non-specific interactions were eliminated with 25 mM Imidazole. Protein samples were eluted with 250 mM Imidazole. The protein was further loaded onto ionic exchange columns (GE Healthcare), and bound proteins were eluted in 500 mM NaCl. Further, the recombinant protein was loaded onto HiLoad Superdex 200(16/600 pg) (GE Healthcare) to obtain the pure form. The protein quality was confirmed by SDS-PAGE stained with coomassie blue.
His tag was removed by incubating HURP proteins and TEV protease in 20 mM HEPES, pH7.4, 100 mM NaCl, and 3 mM DTT during overnight dialysis. Plasmids used in this study are shown in Supplementary Table 4.
Microtubule binding by Total Internal Reflection (TIRF) microscopy
Taxol-stabilized X-rhodamine-biotinylated microtubules were polymerized in the presence of guanylyl-(α, β)-methylene diphosphate (GMPCPP) (Jena Bioscience) at 37 °C. Flow chambers were assembled by applying two strips of double-sided tape on the PEGlyated glass slide and sealing it with a biotinylated coverslip. The chamber was first coated with neutravidin (0.2 mg/ml) (Thermo Fischer). With subsequent incubation of 5 min, the chamber was coated with κ-casein (0.2 mg/ml) to prevent non-specific interactions. After a brief incubation, biotin-labeled microtubules were immobilized on the glass surface, followed by a subsequent wash to remove the excess microtubules. To examine the microtubule-binding, a final reaction mixture of 1XBRB80 (80 mM PIPES, pH6.8, 1 mM MgCl2, 1 mM EGTA), GFP-HURP constructs, and κ-casein (0.2 mg/ml) was added into the flow chamber. TIRF images were obtained using an Eclipse microscope Ti-E TIRF module (Nikon) hosting a 100× 1.45 oil immersion objective lens and equipped with an iXON Ultra 897 EMCCD camera (Andor). GFP was excited using a 488-nm laser (Andor) and X-rhodamine with a 561-nm laser (Andor). To examine whether Importin-β regulates the microtubule-binding activity of HURP. Microtubule binding of 100 nM of GFP-HURP constructs was visualized in the presence and absence of 500 nM of Importin-β. The GFP intensities were quantified using ImageJ with and without Importin-β.
To visualize whether HURP can recruit tubulin on the microtubule lattice, the reaction comprising of 1XBRB80, κ-casein (0.2 mg/ml), 100 nM GFP-HURP FL in the presence and absence of 1 µM HiLyte FluorTM 647 tubulin was added into the chamber. HiLyte FluorTM 647 tubulin (Cytoskeletal, Inc.) was excited using 640-nm (Andor) and quantified the fluorescence in the presence and absence of GFP-HURP FL using ImageJ.
To assess the effect of vinorelbine on GMPCCPP microtubules. The microtubule binding of GFP-HURP (1-280) & GFP-HURP-FL at 100 nM was performed in the presence and absence of vinorelbine. The intensity at 488 nm was quantified.
In vitro microtubule dynamics analysis
In vitro microtubule dynamics assays of GFP-HURP (1-280), GFP-HURP FL, GFP-HURP (200-620) and EB1 (with or without a GFP tag) were performed by immobilizing biotin-labeled X-rhodamine. The dynamic ends were reconstituted by mixing HiLyte FluorTM 647 and recycled tubulin in the presence of 1 mM GTP. The assay mix comprised of 1XBRB80 supplemented with 80 mM KCl, 13 µM bovine tubulin, κ-casein (1 mg/ml), Oxygen scavenger (OS) mix (catalase (35 µg/ml), glucose (4.5 µg/ml), glucose oxidase (200 µg/ml) and 3 mM β-mercaptoethanol, 5% sucrose and GFP-tagged HURP proteins at indicated concentrations. A fully sealed reaction in a chamber was mounted on a pre-warmed microscope objective to 37 °C by a temperature collar. Microtubule seeds and the dynamic ends were excited at 561 and 640 nm, respectively. The movies were captured within 2 sec/frame for 20 min. All the quantifications were performed using MetaMorph online.
To examine the microtubule dynamics in the presence of the drug, 250 nM of vinorelbine was added with increasing concentrations of GFP-HURP FL (250, 500 nM and 1 µM). To determine the effect of HURP on microtubule length, vinorelbine (250 nM) was incubated with GFP-HURP FL (1 µM), and snapshots were taken at different time intervals.
Ni-NTA pull down analysis of tubulin dimer
To examine the HURP interaction with tubulin dimer, 5 µM His tagged GFP-HURP constructs were mixed with 1 µM of bovine tubulin in 50mM K-phosphate, 50 mM KCl, 3 mM β-mercaptoethanol, and incubated with Ni-NTA beads for 30 min on shaking condition at 4 °C. The dilution and wash buffers used in the assay are supplemented with 30 µM nocodazole to prevent any higher-order tubulin assembly. After subsequent incubation, the unbound fractions were collected upon centrifugation at 1000 x g. The beads were washed with 25 mM imidazole to remove non-specific binding. The bound protein was eluted using 250 mM imidazole. Both unbound and bound fractions were analyzed in the SDS-PAGE on a coomassie stained gels. The band intensity of tubulin being pulled down by GFP-HURP constructs was quantified in Analytik Jena Vision Works software. To test whether Importin-β regulates the activity of HURP, 5 µM of Importin-β was included in the reaction.
Microtubule co-pelleting assay
5 µM of Taxol-stabilized microtubules were incubated in the presence and absence of 5 µM GFP-HURP proteins for 15 min at room temperature in a buffer comprising of 0.5x BRB80, 40 µM Taxol, 0.25 mg/ml κ-casein and 2 mM DTT. After sedimentation using TLA120.1 rotor at 213,400 x g for 15 min at 30 °C, the supernatant and pellet proteins were examined by SDS-PAGE. The band intensities of HURP bound and unbound fractions were quantified using Analytik Jena Vision Works software. Microtubule co-pelleting assay of GFP-HURP FL was performed in the presence of 5 µM Importin-β.
Size exclusion chromatography analysis of HURP and tubulin interaction
15 µM of GFP-HURP (200-620) and tubulin were allowed to form a complex for 30 min on ice. The reaction was loaded onto the SuperdexTM 200 (10/300GL) (GE Healthcare) column pre-calibrated in 20 mM HEPES, 50 mM NaCl, 3mMDTT, pH 7.4. The peak fractions were analyzed on SDS-PAGE.
To validate the interaction of GST-HURP (430-550) and mutants with tubulin, 15 µM proteins were mixed with 15 µM tubulin and incubated for 30 min on ice in 20 mM HEPES, 50 mM NaCl, 3 mM DTT, pH 7.4. The peak fractions were analyzed by SDS-PAGE.
Equal molar ratio of GFP-HURP FL, tubulin dimer, and Importin-β proteins were biochemically reconstituted on ice for 30 minutes. The mixture was then subjected to the pre-equilibrated column with 50 mM K-phosphate, 50 mM KCl, 1 mM β-mercaptaethanol pH 7.4. The peak fractions were analyzed on the SDS-PAGE. In addition, western blots were performed for the tubulin dimer using primary antibody α-tubulin (1:2000, GTX628802, GeneTex) for the complex.
Vinorelbine-induced tubulin aggregation assay
2 µM bovine tubulin was incubated with 20 µM vinorelbine (Sigma-Aldrich) in the absence and presence of HURP (430-550) with indicated concentrations at room temperature in 1XBRB80 supplemented with 100 µM GDP (Sigma-Aldrich). After 45 minutes, the reaction was centrifugated at 213,400 x g in TLA 120.1. The supernatant and pellet were analyzed using SDS-PAGE. Tubulin in the supernatant and pellet fractions was determined.
Limited proteolysis
The GFP-HURP (200-620) •tubulin complex was subjected to limited proteolysis with elastase (1 mg/ml) at different time courses (10, 20, 30, and 60 minutes). The high-intensity band observed on the SDS-PAGE was examined using mass spectrometry.
GST-pull down analysis
GST-Importin-β (5 µM) mixed with tubulin (1 µM) and GFP-HURP FL(3 µM) was incubated with glutathione-coupled sepharose beads (GE Healthcare) for 30 min in wash buffer (20 mM HEPES, pH7.4, 50 mM NaCl and 3 mM DTT). Upon centrifugation, the unbound fractions were collected. After pre-washing the beads with wash buffer (20 mM HEPES, 50 mM NaCl, 3 mM DTT, pH 7.4), the protein was eluted in additional 50 mM reduced glutathione and visualized on SDS-PAGE.
GST- Importin-β interaction was validated with His-GFP-HURP FL and His-GFP-HURP (Δ1-60). Each protein was mixed with Importin-β in wash buffer with 500 mM NaCl and incubated with GST beads. Upon incubation for 30 min at 4 °C, the unbound fractions were collected by centrifuging at 1000 x g. The beads were washed with 1 M NaCl, and the proteins were eluted in 50 mM reduced glutathione.
Analytical ultracentrifuge
Sedimentation velocity-analytical ultracentrifugation (SV-AUC) experiments was conducted using an XL-A analytical ultracentrifuge (Beckman Coulter, USA) hosted with an AnTi60 rotor (Beckman Coulter, USA). GFP-HURP FL, GFP-HURP (1-280), &GFP-HURP (200-620) was diluted to 1.5, 1, 1.3 mg/ml in 50mM K-phosphate, 300 mM NaCl, 1 mM β-mercaptoethanol, pH 7.4. The data collection was performed at 201,240 x g and 280 nm under 20 °C. The partial specific volume, buffer density, and viscosity were calculated by SEDNTERP. Data was analyzed with the standard c(s) method in SEDFIT and the continuous c(s) distribution was plotted.
Biolayer interferometry
The binding kinetics of His-GFP-HURP FL or His-GFP-HURP (200–620) with tubulin were measured using BLI (Gator® Plus). 200 nM of His-tagged GFP-HURP FL or GFP-HURP (200–620) was loaded on the Ni-NTA probe (Gator) and incubated with tubulin in 20 mM HEPES pH 7.4, 150 mM NaCl, and 3 mM DTT. The experiment was carried out at different tubulin concentrations (HURP (200-620): 0.94 nM-30 nM and HURP FL: 62.5 nM-2 μM). The association and dissociation were performed for 180 seconds at 30 °C. The dissociation constant (Kd) was calculated using a 2:1 fitting model of association and dissociation function with the global fit and Rmax linked.
Cryo-electron microscopy sample preparation
To prepare cryo-EM grids, the GFP-HURP (200-620)•α/β-tubulin complex was diluted to ~ 0.4 mg/mL. Four microliters of the sample were loaded onto a glow-discharged holey carbon (Quantifoil R1.2/1.3) grid (Quantifoil Micro Tools GmbH). After blotting (3 sec with blot force 0), grids were plunge-vitrification into precooled liquid ethane using an FEI Vitrobot system (Thermo Fisher Scientific). The temperature in the chamber was set to 4 °C and 100% humidity.
Cryo-electron microscopy data collection
Cryo-EM grids were first checked on a 200 keV Talos Arctica transmission electron microscope equipped with a Falcon III detector (Thermo Fisher Scientific) operated in linear mode. The images were recorded at a nominal magnification of 120,000x, corresponding to a pixel size of 0.86 Å/pixel, with a defocus setting of −2.5 μm. Suitable cryo-EM grids were recovered for further data collection on a 300 keV Titan Krios transmission electron microscope (Thermo Fisher Scientific) hosting a K3 detector (with GIF Bio-Quantum Energy Filters, Gatan) operating in super-resolution mode and (gun lens 4, spot size 4, C2 aperture 50 μm) using EPU-2.7.0 software (Thermo Fisher Scientific). The raw movie stacks were recorded at a magnification of 105,000×, corresponding to a pixel size of 0.83 Å/pixel (super-resolution 0.415 Å/pixel). The defocus range was set to −1.4 to −2.2 μm and the slit width of energy filters was set to 15 eV. Fifty frames of non-gain-normalized tiff stacks were recorded with a dose rate of ~26.5 e-/Å2 per second and the total exposure time was set to 2.0 s, resulting in an accumulated dose of ~53 e-/Å2 ( ~ 1.06 e-/Å2 per frame). The parameters for cryo-EM data acquisition are summarized in Table 1.
Single-particle image processing and 3D reconstruction
The super-resolution image stacks were motion-correction and dose-weighting using MotionCor263, with a 7 × 5 patch and two-fold binning (resulting in a pixel size of 0.83 Å). The motion-corrected micrographs were then imported into cryoSPARC64 for further single-particle reconstruction. The contrast transfer function (CTF) was determined from the motion-corrected images using the “Patch CTF estimation” function in cryoSPARC. Selected particles were extracted with a box size of 256 pixels for further 2D classification. Poor 2D class averages were removed after multiple rounds of 2D classification, and the remaining particles were used for ab initio map generation, followed by 3D heterogeneous refinement (separated into four classes). Poor 3D classes were removed via several rounds of 3D heterogeneous refinement, and particles in good 3D classes were merged before conducting an additional 2D classification to remove any remaining bad particles. The good particles were further refined by 3D non-uniform refinement without imposed symmetry. Focused refinement of each subunit is performed with a mask to further improve the resolution and density maps. Finally, the results of the focused refinement are combined to generate a composite map for further analysis. Map sharpening and resolution estimation were automatically performed via 3D non-uniform refinement and focus refinement in cryoSPARC64. The overall resolution was estimated using the Fourier Shell Correlation (FSC) = 0.143 criterion and the local resolution was also calculated in cryoSPARC64. The 3D density maps were visualized in ChimeraX65. The procedures for single-particle image processing and details of cryo-EM reconstruction are summarized in Supplementary Fig. 3. Statistical information for cryo-EM reconstructions are summarized in Table 1.
Atomic model building and refinement
The coordinate model was constructed based on the α/β-tubulin dimer (PDB ID: 1JFF) and the HURP model predicted by AlphaFold266, and refined using the Phenix software suite67 and COOT68. The statistics of model validations refer to the final output files from the real space refinement in PHENIX67. All statistics of data collection and model refinement are presented in Table 1).
Microtubule aster assembly in Xenopus egg extracts
Cell-free extracts from Xenopus laevis unfertilized eggs were prepared. For each extract preparation, female frogs were primed using pregnant mare serum gonadtropin (hor-272; Prospec) and induced to ovulate using human chorionic gonadotropin (CG-10; Sigma). Eggs were collected ∼16 h after the gonadotropin injection, washed with MMR (5 mM Na-Hepes, pH 7.85, 0.1 mM EDTA, 100 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 2 mM CaCl2), treated with 2% (w/v) cysteine solution, and then rinsed with buffers in the following order: XB (10 mM K-Hepes, 100 mM KCl, 1 mM MgCl2, 0.1 mM CaCl2, and 50 mM sucrose, pH 7.7), CSF-XB (XB plus 1 mM MgCl2, and 5 mM EGTA), and CSF-XB + PI (CSF-XB plus 10 μg/ml of leupeptin, pepstatin A, and chymostatin). The rinsed eggs were supplemented with 10 μg/ml cytochalasin D, packed in centrifuge tubes (344057; Beckman) using a tabletop centrifuge (5702 R; Eppendorf), and then crushed using a SW-55 rotor at 12,130 x g for 15 min at 16oC (Optima XE-90; Beckman). After centrifugation, a cytoplasmic fraction was obtained using a 16-gauge needle and supplemented with Energy mix (150 mM creatine phosphate, 25 mM ATP, 25 mM MgCl2, and 2.5 mM EGTA). Protease inhibitors (leupeptin, pepstatin A, and chymostatin) and cytochalasin D (each at 10 μg/ml) were added to the fraction. The entire procedure was performed in a temperature-controlled room at 19 ± 1oC. The prepared extracts were stored in 1.5-ml test tubes on ice until required and used within 6 h. For microtubule aster assembly, each reaction was prepared using 20 μl of extracts supplemented with X-rhodamine-labeled tubulin (10 μg/ml). HURP constructs were then added to reactions at different concentrations and incubated at 18oC. Negative and positive control samples were prepared with CSF-XB only and with CSF-XB plus 20 μM Taxol and 0.5% DMSO, respectively. At 10–40 min after the addition of HURP, 4 μl of each extract reaction was squashed between a clean slide and a coverslip (Matsunami), sealed using VaLaP (Desai et al., 1999), and then immediately imaged using a confocal microscope (Ti; Nikon) equipped with a 20× objective lens (0.45 NA, S Plan Fluor; Nikon), a spinning-disk confocal unit (CSU-X1, Yokogawa), a 561-nm excitation laser (OBIS, Coherent) and a sCMOS camera (Neo, Andor). Image acquisition was performed by raster-scanning ∼2200 × 2700-μm areas of each sample using image acquisition software (NIS-Elements, ver. 4.2; Nikon). The number of microtubule asters was counted using an object detection algorithm in the software, after calibrating the detection parameters using positive control samples.
Spindle assembly in Xenopus egg extracts and imaging
For each reaction, freshly prepared, cytostatic factor (CSF)–arrested extract was placed in test tubes and cycled into interphase by the addition of Ca2+ with demembranated sperm (at 1000 nuclei/µL). Subsequently, the extract was cycled back into metaphase by dilution with an equal volume of fresh CSF extract along with HURP in CSF-XB and X-rhodamine tubulin, incubated for 60 min, and then sealed in an imaging chamber (volume: 5 µL). Image acquisition was performed using the same microscopy setup as in the aster assay. To collect data, microtubules were visualized in the X-rhodamine channel and bipolar spindles were randomly sampled by scanning the microscope stage. The assay was performed at 19 ± 1 °C.
Hepatoma Hep3B cell culture
Hep3B cells (ATCC® HB-8064) were grown in MEM (Gibco™, 11095080) supplemented with 15% fetal bovine serum, 1% penicillin streptomycin (Gibco™, 15140122), 1X GlutaMAX (Gibco™, 35050061), 1 mM sodium pyruvate (Gibco™, 11360070) at 37 °C in 5% CO2. We used TrypLE (Gibco™, 12604021) for routine cell passaging.
CRISPR/Cas9 targeting strategy
sgRNA targeting human HURP sequences (Supplementary Table 5) were designed using the website https://chopchop.cbu.uib.no/. The sgRNA sequences were cloned into the PX330 vector (Addgene, plasmid #42230). Cells were dissociated into single cells using TrypLE for 6 min. The cells were then electroporated using a cell line nucleofector kit V (Lonza, VCA-1003) according to the manufacturer’s guidelines. In brief, 2 million cells were resuspended in 100 μL nucleofector mix, to which was added 10 μg CRISPR targeting plasmid. Single cells were re-plated on 6 well-plates with culture medium. After about three weeks, we picked single colonies in a 96 well-plate. When cells were 80–90% confluent, we expanded cells and then added lysis buffer to isolate genomic DNA for genotyping. PCR products were confirmed by DNA sequencing.
Growth curves
Hep3B cells were seeded in a 24-well plate with a concentration of 2×104 cells per well. Cells were treated with or without 0.005 μM vinorelbine. Cell numbers were determined by counting trypan blue-negative cells using a hemocytometer under microscopy. Cells were counted every 48 h after seeding, and growth curves were generated based on five independent experiments.
Cell-cycle analysis
Cells were dissociated with TrypLE and washed once with 1X PBS then fixed with 70% ethanol overnight. Cells were then centrifuged and washed three times with 1X PBS and then re-suspended in 200 μl of Propidium Iodide (Invitrogen, P3566) and analyzed using Muse Cell Analyzer software (Merck, Millipore).
Western blot
Cells were cultured until reaching 80% confluence, followed by scratch-off using a cell scraper. Cell lysates were obtained by shaking the cells in RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.1% SDS, 1% TritonX-100, 0.5% sodium deoxycholate) containing a protease inhibitor cocktail (Roche). The cell extracts were then cleared of debris via centrifugation at 16,000 g for 20 minutes. Western blot analysis was performed using antibodies against HURP (1:1000, Abcam, ab84509) and GAPDH (1:2000, GeneTex, GTX100118). A horseradish peroxidase (HRP) chemiluminescence kit (GeneTex) along with an HRP-conjugated secondary antibody (1:5000, GeneTex, GXT213110) were utilized for detection. Band signals were captured using the iBright FL1500 Western Blot Imaging Systems (Invitrogen).
Immunofluorescence and image analysis
Sample preparation
Hep3B cells were seeded onto cover slips and cultured accordingly. Upon reaching 80% confluency, cells were either cold-treated on ice for 5 minutes or directly fixed using 100% ice-cold methanol for 10 minutes. Subsequently, cells were blocked with 10% bovine serum albumin (BSA) (Sigma-Aldrich) and then incubated with primary antibodies HURP (1:500, Abcam, ab70744), CENP-A (1:500, GeneTex, GTX13939), alpha-tubulin (1:1000, GeneTex, GTX112141 & GTX628802) diluted in PBS containing 3% BSA overnight at 4 °C. After thorough washing with PBS, cells were incubated with secondary antibodies conjugated with Alexa Fluor-488 and −555 (1:1000, Invitrogen) for 1 hour at room temperature. Finally, nuclei were counterstained with DAPI (1:5000, Merck) and coverslips were mount with Antifade ProLong™ Gold (Invitrogen). For imaging, K-fibers are identified by their notably high intensity among microtubules, typically surpassing 1500 arbitrary units (arb. units) after subtracting the background intensity value from each voxel.
Imaging conditions and processing
The cell images were acquired using an LSM980 confocal microscope with Airyscan2 in multiplex-4Y mode, employing a 63×/1.4 Oil objective (Carl ZEISS). The 3D reconstructed images were processed using ZenBlue (version 3.6, Carl ZEISS) and Imaris software (version 10.1.0, OXFORD Instruments), with modeling based on fluorescence signals. For quantification, the same experimental conditions were acquired under consistent confocal microscopy settings, and no post-acquisition adjustments were made in the analysis.
Statistics and reproducibility
Data are either pooled from all quantifications and presented collectively, or quantifications from a single representative experiment are shown, as indicated in the figure legends. Statistical analyses were performed using Prism software (version 10). The figure legends provide the n values and the types of error bars used for each experiment. At least three independent replicate experiments were performed. To determine statistical differences in the microtubule dynamics assay (Figs. 1b–j, 2k–p, 5a–j, 6a–m and Supplementary Fig. 1a–s), microtubule binding assay (Figs. 1l–n, 3b–d, 5p–q and Supplementary Fig. 2e–g), and aster formation assays (Fig. 3i–n), two-tailed Student’s t-tests were used. The P-value notation is as follows: * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.001, and P < 0.001; ****.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this manuscript are available from the corresponding author upon request. The source data underlying panels in Figs. 1–7 and Supplementary Figs. 1, 2, 3, 6, 7, and 8 are provided in a source data file. Protein structural coordinates and maps have been deposited in the PDB and the Electron Microscopy Data Bank (EMD) with the accession codes PDB8X9P, EMD-38178, EMD-38179, EMD-38180, and EMD-38181). The validation reports of the corresponding coordinate files are available upon request. Source data are provided with this paper.
References
Glotzer, M. The 3Ms of central spindle assembly: microtubules, motors and MAPs. Nat. Rev. Mol. Cell Biol. 10, 9–20 (2009).
Forbes, D. J., Travesa, A., Nord, M. S. & Bernis, C. Nuclear transport factors: global regulation of mitosis. Curr. Opin. Cell Biol. 35, 78–90 (2015).
Chan, K. S., Koh, C. G. & Li, H. Y. Mitosis-targeted anti-cancer therapies: where they stand. Cell Death Dis. 3, e411 (2012).
Chang, C. C. & Hsia, K. C. More than a zip code: global modulation of cellular function by nuclear localization signals. FEBS J. 288, 5569–5585 (2021).
Nachury, M. V. et al. Importin beta is a mitotic target of the small GTPase Ran in spindle assembly. Cell 104, 95–106 (2001).
Wiese, C. et al. Role of importin-beta in coupling Ran to downstream targets in microtubule assembly. Science 291, 653–656 (2001).
Chang, C. C., Huang, T. L., Shimamoto, Y., Tsai, S. Y. & Hsia, K. C. Regulation of mitotic spindle assembly factor NuMA by Importin-beta. J. Cell Biol. 216, 3453–3462 (2017).
Schatz, C. A. et al. Importin alpha-regulated nucleation of microtubules by TPX2. EMBO J. 22, 2060–2070 (2003).
Giesecke, A. & Stewart, M. Novel binding of the mitotic regulator TPX2 (target protein for Xenopus kinesin-like protein 2) to importin-alpha. J. Biol. Chem. 285, 17628–17635 (2010).
Tsou, A. P. et al. Identification of a novel cell cycle regulated gene, HURP, overexpressed in human hepatocellular carcinoma. Oncogene 22, 298–307 (2003).
Castrogiovanni, C. et al. Evidence for a HURP/EB free mixed-nucleotide zone in kinetochore-microtubules. Nat. Commun. 13, 4704 (2022).
Sillje, H. H., Nagel, S., Korner, R. & Nigg, E. A. HURP is a Ran-importin beta-regulated protein that stabilizes kinetochore microtubules in the vicinity of chromosomes. Curr. Biol. 16, 731–742 (2006).
Wong, J., Lerrigo, R., Jang, C. Y. & Fang, G. Aurora A regulates the activity of HURP by controlling the accessibility of its microtubule-binding domain. Mol. Biol. Cell 19, 2083–2091 (2008).
Wong, J. & Fang, G. HURP controls spindle dynamics to promote proper interkinetochore tension and efficient kinetochore capture. J. Cell Biol. 173, 879–891 (2006).
Tsuchiya, K. et al. Ran-GTP is non-essential to activate numa for mitotic spindle-pole focusing but dynamically polarizes hurp near chromosomes. Curr. Biol. 31, 115–127 e113 (2021).
Wang, Q., Chen, Y., Feng, H., Zhang, B. & Wang, H. Prognostic and predictive value of HURP in non‑small cell lung cancer. Oncol. Rep. 39, 1682–1692 (2018).
Al-Khafaji, A. S. K. et al. Overexpression of HURP mRNA in head and neck carcinoma and association with in vitro response to vinorelbine. Oncol. Lett. 19, 2502–2507 (2020).
Kuo, T. C., Chang, P. Y., Huang, S. F., Chou, C. K. & Chao, C. C. Knockdown of HURP inhibits the proliferation of hepacellular carcinoma cells via downregulation of gankyrin and accumulation of p53. Biochem Pharm. 83, 758–768 (2012).
Zhang, H. et al. Knockdown of DLGAP5 suppresses cell proliferation, induces G(2)/M phase arrest and apoptosis in ovarian cancer. Exp. Ther. Med. 22, 1245 (2021).
Budman, D. R. Vinorelbine (Navelbine): a third-generation vinca alkaloid. Cancer Invest 15, 475–490 (1997).
Chengyong, W. et al. The high-resolution X-ray structure of vinca-domain inhibitors of microtubules provides a rational approach for drug design. FEBS Lett. 595, 195–205 (2021).
Lobert, S., Vulevic, B. & Correia, J. J. Interaction of vinca alkaloids with tubulin: a comparison of vinblastine, vincristine, and vinorelbine. Biochemistry 35, 6806–6814 (1996).
Maurer, S. P., Fourniol, F. J., Bohner, G., Moores, C. A. & Surrey, T. EBs recognize a nucleotide-dependent structural cap at growing microtubule ends. Cell 149, 371–382 (2012).
Mohan, R. et al. End-binding proteins sensitize microtubules to the action of microtubule-targeting agents. Proc. Natl Acad. Sci. USA 110, 8900–8905 (2013).
Maurer, S. P. et al. EB1 accelerates two conformational transitions important for microtubule maturation and dynamics. Curr. Biol. 24, 372–384 (2014).
Al-Bassam, J., Larsen, N. A., Hyman, A. A. & Harrison, S. C. Crystal structure of a TOG domain: conserved features of XMAP215/Dis1-family TOG domains and implications for tubulin binding. Structure 15, 355–362 (2007).
Ayaz, P., Ye, X., Huddleston, P., Brautigam, C. A. & Rice, L. M. A. TOG:alphabeta-tubulin complex structure reveals conformation-based mechanisms for a microtubule polymerase. Science 337, 857–860 (2012).
Spittle, C., Charrasse, S., Larroque, C. & Cassimeris, L. The interaction of TOGp with microtubules and tubulin. J. Biol. Chem. 275, 20748–20753 (2000).
Al-Bassam, J., van Breugel, M., Harrison, S. C. & Hyman, A. Stu2p binds tubulin and undergoes an open-to-closed conformational change. J. Cell Biol. 172, 1009–1022 (2006).
Al-Bassam, J. et al. CLASP promotes microtubule rescue by recruiting tubulin dimers to the microtubule. Dev. Cell 19, 245–258 (2010).
Song, L., Craney, A. & Rape, M. Microtubule-dependent regulation of mitotic protein degradation. Mol. Cell 53, 179–192 (2014).
Gallini, S. et al. NuMA phosphorylation by Aurora-A orchestrates spindle orientation. Curr. Biol. 26, 458–469 (2016).
Howell, B., Larsson, N., Gullberg, M. & Cassimeris, L. Dissociation of the tubulin-sequestering and microtubule catastrophe-promoting activities of oncoprotein 18/stathmin. Mol. Biol. Cell 10, 105–118 (1999).
Biswas, A., Kim, K., Cojoc, G., Guck, J. & Reber, S. The Xenopus spindle is as dense as the surrounding cytoplasm. Dev. Cell 56, 967–975 e965 (2021).
Leano, J. B. & Slep, K. C. Structures of TOG1 and TOG2 from the human microtubule dynamics regulator CLASP1. PLoS One 14, e0219823 (2019).
Slep, K. C. & Vale, R. D. Structural basis of microtubule plus end tracking by XMAP215, CLIP-170, and EB1. Mol. Cell 27, 976–991 (2007).
Pecqueur, L. et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proc. Natl Acad. Sci. USA 109, 12011–12016 (2012).
Sharma, A. et al. Centriolar CPAP/SAS-4 imparts slow processive microtubule growth. Dev. Cell 37, 362–376 (2016).
Campanacci, V. et al. Structural convergence for tubulin binding of CPAP and vinca domain microtubule inhibitors. Proc. Natl Acad. Sci. USA 119, e2120098119 (2022).
Duflos, A., Kruczynski, A. & Barret, J. M. Novel aspects of natural and modified vinca alkaloids. Curr. Med Chem. Anticancer Agents 2, 55–70 (2002).
Ngan, V. K. et al. Novel actions of the antitumor drugs vinflunine and vinorelbine on microtubules. Cancer Res. 60, 5045–5051 (2000).
Tsai, C. Y. et al. Hurp deficiency in mice leads to female infertility caused by an implantation defect. J. Biol. Chem. 283, 26302–26306 (2008).
Zhao, L. et al. KIAA0008 gene is associated with invasive phenotype of human hepatocellular carcinoma-a functional analysis. J. Cancer Res Clin. Oncol. 130, 719–727 (2004).
Chiu, A. W. et al. Potential molecular marker for detecting transitional cell carcinoma. Urology 60, 181–185 (2002).
Chen, J. et al. Hepatoma upregulated protein expression is involved in the pathogenesis of human breast carcinogenesis. Oncol. Lett. 8, 2543–2548 (2014).
Casanova, C. M., Rybina, S., Yokoyama, H., Karsenti, E. & Mattaj, I. W. Hepatoma up-regulated protein is required for chromatin-induced microtubule assembly independently of TPX2. Mol. Biol. Cell 19, 4900–4908 (2008).
Alici, S. et al. Vinorelbine in combination with carboplatin followed by single-agent consolidation therapy for unresectable localized or metastatic non-small-cell lung carcinomas. Asian Pac. J. Cancer Prev. 10, 1051–1055 (2009).
Delord, J. P., Puozzo, C., Lefresne, F. & Bugat, R. Combination chemotherapy of vinorelbine and cisplatin: a phase I pharmacokinetic study in patients with metastatic solid tumors. Anticancer Res 29, 553–560 (2009).
Gigant, B. et al. The 4 A X-ray structure of a tubulin:stathmin-like domain complex. Cell 102, 809–816 (2000).
Nithianantham, S. et al. Structural basis of tubulin recruitment and assembly by microtubule polymerases with tumor overexpressed gene (TOG) domain arrays. Elife 7 https://doi.org/10.7554/eLife.38922 (2018).
Wallon, G., Rappsilber, J., Mann, M. & Serrano, L. Model for stathmin/OP18 binding to tubulin. EMBO J. 19, 213–222 (2000).
Gupta, K. K. et al. Mechanism for the catastrophe-promoting activity of the microtubule destabilizer Op18/stathmin. Proc. Natl Acad. Sci. USA 110, 20449–20454 (2013).
Campanacci, V. et al. Selection and characterization of artificial proteins targeting the tubulin alpha subunit. Structure 27, 497–506 e494 (2019).
Prota, A. E. et al. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proc. Natl Acad. Sci. USA 111, 13817–13821 (2014).
Doodhi, H. et al. Termination of protofilament elongation by eribulin induces lattice defects that promote microtubule catastrophes. Curr. Biol. 26, 1713–1721 (2016).
Saha, S. et al. Polar positioning of phase-separated liquid compartments in cells regulated by an mrna competition mechanism. Cell 166, 1572–1584 e1516 (2016).
Zheng, X. et al. Molecular basis for CPAP-tubulin interaction in controlling centriolar and ciliary length. Nat. Commun. 7, 11874 (2016).
Kohlmaier, G. et al. Overly long centrioles and defective cell division upon excess of the SAS-4-related protein CPAP. Curr. Biol. 19, 1012–1018 (2009).
Schmidt, T. I. et al. Control of centriole length by CPAP and CP110. Curr. Biol. 19, 1005–1011 (2009).
Tang, C. J., Fu, R. H., Wu, K. S., Hsu, W. B. & Tang, T. K. CPAP is a cell-cycle regulated protein that controls centriole length. Nat. Cell Biol. 11, 825–831 (2009).
Lawrence, E. J., Arpag, G., Norris, S. R. & Zanic, M. Human CLASP2 specifically regulates microtubule catastrophe and rescue. Mol. Biol. Cell 29, 1168–1177 (2018).
Moriwaki, T. & Goshima, G. Five factors can reconstitute all three phases of microtubule polymerization dynamics. J. Cell Biol. 215, 357–368 (2016).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. Biol. Crystallogr 66, 213–221 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. Biol. Crystallogr 60, 2126–2132 (2004).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. Biol. Crystallogr 66, 12–21 (2010).
Acknowledgements
The cryo-EM experiments were performed at the Academia Sinica Cryo-EM Facility (ASCEM) and Academia Sinica Grid-computing Center (ASGC). ASCEM is jointly supported by Academia Sinica Core Facility and Innovative Instrument Project Grant No. AS-CFII-111-210 and Taiwan Protein Project Grant No. AS-KPQ-109-TPP2. We acknowledge the use of the AUC and BLI systems in the Biophysics Core Facility, funded by Academia Sinica Core Facility and Innovative Instrument Project AS-CFII1-111-201. We also thank John O’Brien for manuscript editing, IMB imaging Core facility for technical assistance. This work was supported by National Taiwan University (112L895405 and 113L893105 to S.-Y.T.), Academia Sinica (AS-IVA-112-L05 to K.-C.H), JSPS KAKENHI (JP22K18362 and JP23K23854 to Y.S., JP24KJ0225 to A.T.) and the National Science and Technology Council (NSTC- 111-2628-B-002-027-MY3 to S.-Y.T., NSTC-112-2320-B-001-006-MY3 to K.-C.H).
Author information
Authors and Affiliations
Contributions
A.S., P-P.C., and Y.-T.T. performed biochemical experiments. P.-P.C. and C.-H.W. determined the cryo-EM structure. A.S. and C.-C.C. conducted TIRF-based experiments. P.-P.C., T.-H.W. and S.-Y.T. performed cell-based analysis. A.T. and Y.S. carried out Xenopus egg extract-based experiments. All authors analyzed the data, discussed the results and helped write the manuscript. K.-C.H. directed the project and prepared the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Saju, A., Chen, PP., Weng, TH. et al. HURP binding to the vinca domain of β-tubulin accounts for cancer drug resistance. Nat Commun 15, 8844 (2024). https://doi.org/10.1038/s41467-024-53139-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53139-y
