
Abstract
Key innovations, traits that provide species access to novel niches, are thought to be a major generator of biodiversity. One commonly cited example of key innovation is pharyngognathy, a set of modifications to the pharyngeal jaws found in some highly species-rich fish clades such as cichlids and wrasses. Here, using comparative phylogenomics and phylogenetic comparative methods, we investigate the genomic basis of pharyngognathy and the impact of this innovation on diversification. Whole genomes resolve the relationships of fish clades with this innovation and their close relatives, but high levels of topological discordance suggest the innovation may have evolved fewer times than previously thought. Closer examination of the topology of noncoding elements accelerated in clades with the pharyngognathy innovation reveals hidden patterns of shared ancestry across putatively independent transitions to pharyngognathy. When our updated phylogenomic relationships are used alongside large-scale phylogenetic and ecological datasets, we find no evidence pharyngognathy consistently modifies the macroevolutionary landscape of trophic ecology nor does it increase diversification. Our results highlight the necessity of incorporating genomic information in studies of key innovation.
Similar content being viewed by others
Introduction
Understanding how novel traits influence both ecological and species diversity is a critical component of modern biodiversity studies. Perhaps the most influential framework linking these factors is the key innovation hypothesis1. This idea combines work from the Modern Synthesis, particularly Simpson2,3, who posited that phenotypic novelties will often change the relationship between the clades possessing the innovation and their environment, enabling entry into novel adaptive zones. The occupancy of novel adaptive zones is thought to provide ecological benefits that can lead to lineage diversification, which may involve both increased speciation rates as well as decreased extinction risk1,2,3,4,5,6,7.
Definitions of key innovation vary throughout the literature1,8, complicating explicit tests of their ecological and evolutionary consequences9,10. The advent of molecular phylogenetics and phylogenetic comparative methods11,12,13,14 has increased interest in testing the key innovation hypothesis across many different clades, particularly in the context of diversification and rates of morphological evolution15,16,17,18,19. However, this modern emphasis on the final consequences of innovation neglects key components of the hypothesis, particularly testing for the presence of ecological evolution associated with entry into novel adaptive zones2,3, and the possibility that innovations may affect other aspects of diversification without impacting speciation rates8,20,21,22. Comprehensive tests of innovations spanning the scope of their known transitions using modern phylogenetic comparative methods are needed to understand the consequences of key innovations on ecological and evolutionary dynamics, especially given the mixed evidence for several proposed innovations23,24.
Integrating genomic information alongside macroevolutionary analyses has the potential to increase our understanding of the patterns and processes of key innovation25,26,27,28,29. Genomes can be used to interrogate the evolutionary relationships of innovative clades using thousands of markers, which can improve on traditional inferences based on a small number of genes16,17,24,29. Genomic data can also improve our ability to detect processes such as incomplete lineage sorting and hybridization that can confound comparative analyses, even for highly resolved trees30,31. In some cases, whole genomes can also illuminate the genetic basis of key innovation, providing a powerful window into the processes that undergird exceptional adaptations26,32,33.
One classic example of a putative key innovation is pharyngognathy, which involves three substantial osteological and myological modifications to the pharyngeal jaws at the back of the throat in spiny-rayed fishes, potentially decoupling the oral jaws from the pharyngeal jaws for prey capture and processing respectively, providing access to novel trophic niches7,34,35. Pharyngognathy was first posited by Liem7 as the underlying reason for the spectacular radiation of cichlids (Cichlidae) in African lakes, but it has also been proposed to play a crucial role in the evolution of two other species-rich and diverse marine families, the wrasses (Labridae) and damselfishes (Pomacentridae)36. However, several additional marine clades, some with relatively few species, also possess the innovation34.
Historically, pharyngognathy was viewed as a synapomorphy for a majority of pharyngognathous families, uniting them in the clade Labroidei37. This was viewed as a major hindrance to testing the impacts of pharyngognathy as an innovation as it was believed to represent a singular unique evolution38. Current molecular phylogenetic evidence suggests pharyngognathy could have evolved as many as six times34, but the phylogenetic position of these clades has yet to be robustly resolved in a multispecies coalescent framework39. This lack of phylogenetic resolution continues to hinder the comparative study of pharyngognathy, particularly in cases where clades with the innovation are compared to putative sister groups29, and has meant that most studies investigating the relationship between ecology and pharyngognathy have been limited to single clades16,35.
In this work, we use comparative phylogenomics and phylogenetic comparative methods alongside a large ecological dataset to investigate the evolutionary history and genomic basis of pharyngognathy while assessing its impact as a key innovation on both diversification and trophic evolution in acanthomorph fishes. We show that pharyngognathy may have a shared genomic origin and that its macroevolutionary dynamics fail to support critical predictions for a key innovation.
Results
Phylogenomics of key innovation
We assembled the genomes from two species in previously unsequenced pharyngognath clades, the pile perch (Embiotocidae: Phanerodon vacca) assembled with PacBio Sequel data using Flye v2.840 and the false scorpionfish (Centrogenyidae: Centrogenys vaigiensis) assembled from Illumina NovaSeq and Oxford Nanopore data using MaSuRCA v4.0.3, supplemented by additional assemblies built from Illumina NovaSeq and assembled using MaSuRCA v4.0.341 (Supplementary Data 1). We then combined our data with publicly available assemblies for the remaining four pharyngognath lineages and an additional outgroup (n = 32). We identified orthologous nuclear loci (n = 122,690) in LAST v124342, then tested for recombination breakpoints using MDL43, which split our nuclear loci into 237,364 loci (Supplementary Information, Table S1). After estimating recombination breakpoints, our locus length reduces to 150 base pairs on average, which is consistent with expectations for a 32-taxon tree with short internodes, due to the inverse relationship between number of taxa and coalescence gene length44.
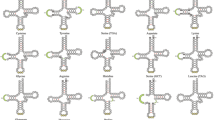
We produced topologies for our split loci in IQ-TREE 245, then generated a species tree (Fig. 1a, Supplementary Information, Fig. S1) using multispecies coalescent methods, which resolved the evolutionary relationships of every pharyngognath clade. We find strong support for the monotypic pharyngognath family Centrogenyidae as sister to Labridae, which reduces the number of potential transitions to the innovation by one. Notably, this relationship is not resolvable in a coalescent framework if fewer than 10,000 loci are used39. Within the clade Ovalentaria, which contains the remaining four pharyngognathous clades, no pharyngognathous clades are sister to each other, though two, Embiotocidae and Pomacentridae, occur in close proximity (Fig. 1a).

a ASTRAL topology for 237,364 aligned loci resolving relationships between all known pharyngognathous fish clades and their close relatives. For all parts of this figure, pharyngognathous taxa are denoted in red, while non-pharyngognaths are in blue. Pies represent branch support from black for support values of 1, black and gray for support values greater than 0.9 but less than 1, and solid gray for support values less than 0.9. Images next to the tips of pharyngognathous taxa are microCT reconstructions of their lower pharyngeal jaws. b Our topology from (a) with branch lengths in coalescent units estimated from locus trees in ASTRAL and colored by hemiplasy risk factor. As ASTRAL does not estimate terminal branch lengths, they have been truncated. c Our topology from (a) with branch lengths in coalescent units estimated from site concordance factors (sCF) calculated from IQ-Tree converted into coalescent units with HeiST. As ASTRAL does not estimate terminal branch lengths, they have been truncated. Many pharyngognath lineages are connected by branches with risk factors near 1.
When we examine our tree in coalescent units, which incorporate gene tree discordance (Fig. 1b), we find extremely short branch lengths at the base of many pharyngognath lineages, which substantially raises the risk of hemiplasy, a pattern that occurs when discordance among gene trees can cause a homologous trait to appear to have evolved independently30. We measured hemiplasy risk factor (hrf)46 across our phylogeny, finding relatively high values throughout the tree (hrf = 0.75-0.93) (Fig. 1b), with extremely high values in cichlids, surfperch, and damselfishes. To ensure results were robust to gene tree estimation error, we also calculated hrf using site-based concordance factors, which utilize concordance between individual sites rather than between trees. We find similarly high levels of hemiplasy (hrf = 0.76−0.92) (Fig. 1c). While our study contains more loci than other fish phylogenies using the multispecies coalescent, we also find high levels of hemiplasy across previously published phylogenomic datasets as well, including in areas of the phylogeny that contain pharyngognath lineages39,47,48,49 (Supplementary Information, Fig. S2−S8). These results raise the possibility that pharyngognathy did not evolve independently in all clades possessing the innovation, but within a smaller number of common ancestors.
Comparative genomics of key innovation
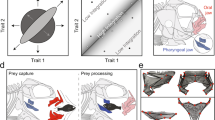
The comparative genomic signature of pharyngognathy supports the hemiplasy hypothesis. We examined coding regions (n = 121,979) for signatures of positive selection using maximum-likelihood and Bayesian methods, then examined noncoding regions (n = 679,285) for accelerated evolution using PhyloAcc v2.225,50, finding a subset of loci with acceleration in pharyngognaths (n = 315). Current methods for identifying accelerated elements assume a common species tree for all sequences50, but this assumption may not hold true under complex evolutionary scenarios51. Therefore, we generated topologies for loci that exhibited strong acceleration in at least one pharyngognath clade. In loci with convergent acceleration — two or more accelerated pharyngognath clades — we find numerous cases where pharyngognaths are clustered together (Fig. 2). Examination of stochastic character maps using a Bayesian model revealed significantly fewer transitions between pharyngognaths and non-pharyngognaths in convergently accelerated loci, supporting a complex and likely hemiplasious origin of the innovation.

a Reduced species tree topology from Fig. 1 for the16 taxa used in accelerated rate analyses. b The posterior distribution from a Bayesian mixed-model analysis of stochastic character transitions in convergent loci (accelerated in two or more pharyngognaths) versus non-convergent (one accelerated pharyngognath), with the 95% confidence interval is denoted by dashed lines. Source data are provided as a Source Data file. c Topologies for 6 (out of 20) loci with accelerated rates in two or more pharyngognathous lineages, showing sister relationships in pharyngognath clades that do not match the species tree in (a).
Eighteen of the 20 loci identified as accelerated in two or more pharyngognath lineages are found in close proximity to coding regions. We utilized a deep-learning approach to assign gene ontology terms to all coding regions within the Oreocheomis niloticus (Cichlidae) genome, then used a Bayesian model to examine differential GO enrichment in our loci (Supplementary Information, Figure S9). We found associations with processes critical for bone development, including keratan sulfate biosynthesis52 and ephrin signaling53, though we stress that the multi-trait nature of pharyngognathy precludes us from associating a particular locus with a specific component trait of the innovation.
Our hemiplasy results highlight that heavy gene tree discordance can occur on deep evolutionary timescales, particularly in rapidly radiating clades such as acanthomorph fishes during the Cretaceous39. Discordance can occur for a variety of reasons, such as hybridization and incomplete lineage sorting, causing gene topologies to vary relative to the species tree. In our case, gene tree discordance due to rapid radiation long ago in ovalentarian fishes can leave a complex pattern that persists to the present. While methods aiming to detect accelerated loci assume a fixed topology, our results highlight that examining unconstrained topologies of accelerated loci may yield valuable insights into elucidating the evolutionary history of traits of interest, especially in the context of non-treelike evolution.
Ecological and evolutionary consequences of key innovation
If pharyngognathy is behaving as a Simpsonian key innovation, we would expect that it would facilitate entry to novel trophic adaptive zones for pharyngognathous fishes. To provide an evolutionary framework for investigating the macroevolutionary dynamics of pharyngognathy, we generated a time-calibrated megaphylogeny of 9323 acanthomorph species from twenty-five nuclear and sixteen mitochondrial genes and seventy fossil calibrations, with topological constraints from previous phylogenomic studies of smaller clades (see Dryad: https://doi.org/10.5061/dryad.cz8w9gjcx). We generated a dataset for 3490 species obtained from quantitative diet studies in the literature and calculated fractional trophic levels, then used the R package BBMV v2.154 to estimate univariate macroevolutionary landscapes, a phenotypic surface in which species evolve up local peaks, for trophic level.
We find that pharyngognaths exhibit a higher peak for low trophic level diets (e.g., herbivory and detritivory), suggesting that pharyngognathy could represent movement into a new adaptive zone (Fig. 3a, b). To ensure that results were not biased by a particular pharyngognath clade, we then repeated our analyses by leaving each clade out in a manner analogous to leave-one-out cross validation (Fig. 3c, Supplementary Information, Table S2−S3). Removing cichlids from the analysis removed the low trophic level peak, but removing just the exceptionally fast-speciating Lake Malawi and Lake Victoria radiations55 did not alter the peak, suggesting that it is influenced by diet evolution across the entire family (Fig. 3c). We also tested if pharyngognathy led to an early burst in trophic evolution within each pharyngognath clade, but this model has low support compared to alternatives (Supplementary Information, Table S4). The early burst model fit poorest, with all families having Brownian motion as the best fitting model model, except for cichlids, in which case the best fit model was the Ornstein-Uhlenbeck model.

a Distribution of estimated shape parameters defining the macroevolutionary landscape of trophic level in pharyngognaths (red) and related non-pharyngognathous lineages (blue) parameterized using BBMV (n = 3490 species). Overlaid boxplots display the median and interquartile range of the distribution. b Macroevolutionary landscape of trophic level in pharyngognaths (red) and related non-pharyngognathous lineages (blue) parameterized using BBMV. Caricatures underneath the x-axis represent typical prey fed upon at the corresponding trophic levels. c Landscapes of pharyngognaths and nonpharyngognaths, excluding one pharyngognath clade at a time. d Relative model weights of character-independent diversification models (CID-4 and CID-2), hidden-state speciation and extinction models (HiSSE), and binary-state speciation and extinction model (BiSSE) (n = 9323 species). Source data are provided as a Source Data file.
When we examine the effects of pharyngognathy on the macroevolutionary landscape of trophic level, only cichlids exhibit increased peak height for low trophic level ecologies, whereas our marine pharyngognaths exhibit decreased occupation of both low and high trophic level peaks. This suggests that the primary driver of change in the macroevolutionary landscape may be related more to the occupation of freshwater habitats by cichlids, rather than an effect of pharyngognathy. In marine pharyngognaths, the reduced peak heights for extreme trophic levels suggest that the innovation is acting more as a constraint than facilitating access to novel adaptive zones22.
We fail to find an impact of pharyngognathy on lineage diversification, further undermining the proposal that this trait represents a key innovation. We used HiSSE v2.1.913 to test for state-dependent diversification in relation to pharyngognathy. HiSSE explicitly models hidden states that account for rate variation and provides a framework to test a variety of character-independent diversification models to compare to state-dependent diversification models. We find strong support for character-independent diversification, e.g. no effect of pharyngognathy, when using HiSSE relative to state-dependent diversification models for both the full tree (delta AICc = 508) and within Ovalentaria (delta AICc = 144) (Fig. 3d, Supplementary Information, Fig. S10, Supplementary Data 2−3). While we find significant rate differences in pharyngognaths relative to non-pharyngognaths using FiSSE56 (p = 0.015 and p = 0.017 for full tree and Ovalentaria respectively), this pattern disappears when removing the cichlid adaptive radiations of Lake Malawi and Lake Victoria (p = 0.753 and p = 0.269 for full tree and Ovalentaria respectively) (Supplementary Information, Table S8).
Discussion
Reevaluating the key innovation hypothesis
After Liem7 proposed pharyngognathy as a key innovation in cichlids, it was thought to constitute a synapomorphy uniting cichlids, damselfishes, labrids, and surfperches in a clade called Labroidei37. Given that cichlids as well as both damselfishes and labrids constitute some of the most species-rich fish families, it is not surprising that this shared character was thought to be responsible for the evolutionary success of the Labroidei, though its purported single origin made it difficult to fully assess the evolutionary impact of pharyngognathy38. After molecular phylogenetic studies suggested the independent origin of pharyngognathy34,57, tests of pharyngognathy as a key innovation have not included all transitions to pharyngognathy or have been limited to putative sister group comparisons29. Our integrative approach across all pharyngognaths calls into question the status of pharyngognathy as a textbook example of the key innovation hypothesis.
When pharyngognathy was originally proposed as a key innovation by Liem7, the motivation involved an explanation for the incredible ecological diversity present within the lakes of the East African radiation. However, despite the exceptional diversity of some cichlid lake species flocks, there are many lineages that do not speciate58. For example, Astatoreochromis alluaudi in Lake Victoria has not radiated while other closely related haplochromine cichlids undergo rapid diversification59. Additionally, more recent phylogenetic work has consistently shown a lag time between the evolution of pharyngognathy in cichlids and elevated diversification rates55,60. While crown age estimates for cichlids are highly variable, the youngest estimates for the crown age are around 65 million years ago around the KPG, and the oldest cichlid lake species flocks are less than 20 million years old, a substantial gap in time61,62,63. Recent genomic studies have uncovered hybridization and incomplete lineage sorting involving ancient ecologically relevant haplotypes as a much more likely driver of the exceptional species and ecological diversity seen within these cichlid radiations55,64.
At the time pharyngognathy was proposed as an explanation for the increased species richness in marine groups such as wrasses and damselfishes, information on clade ages was not readily available, but when clade age is taken into account, we see that species-rich pharyngognath families are noticeably older than related families that also harbor considerable fish species richness, indicating the exceptional species richness of these groups may not be coupled with increased diversification65. Previous studies have suggested that pharyngognathy may functionally limit access to high-trophic level niches22, and comparison of pharyngognaths with putative sister lineages found mixed evidence of pharyngognathy elevating rates of morphological or diet breadth evolution, though this result was likely hindered by incorrect sister clade identification29. Our evidence further suggests pharyngognathy may not be behaving as the key innovation Liem7 originally posited it to be.
Despite being touted as an example of a key innovation for nearly half a century, our results suggest pharyngognathy does not meet the standard for a key innovation in either the Simpsonian2,3 or Heard and Hauser1 framework, as it is not promoting either novel ecological occupancy or diversification rates. However, pharyngognathy is clearly a functional novelty. For example, pharyngognathy may have effects beyond food processing: it may promote evolvability via functional decoupling and sound production in courtship behavior35,66. Additionally, since pharyngognathy involves three constituent traits, it is possible that individual traits could influence diversification. Lineages with one or two modifications of the pharyngeal jaws are not uncommon34,65,67, and some contain notable ecological diversity and species richness. We suggest future studies take an approach of understanding the evolutionary dynamics of each constituent trait, though this will require scoring the presence and absence of each trait across the diversity of fishes.
We stress the necessity of incorporating both lineages with and without the innovation of interest, contextualizing the ecological and evolutionary consequences of a trait and its proposed benefits. Studies of pharyngognathy have traditionally been limited to single clades and lack the sampling of non-pharyngognaths in their comparative analyses and accept pharyngognathy as a key innovation a priori16,35,68. When studies have sampled clades with and without the key innovation, they have found results contrary to expectations, such as pharyngognathy not promoting functional decoupling69. While our study does not invalidate the findings of previous single-clade studies, which provide valuable information on the evolutionary and ecological dynamics of the taxa investigated, studies that lack non-pharyngognathous lineages in their comparative analysis do not directly test the assumptions of the key innovation hypothesis. We expect that key innovation studies employing broad taxonomic sampling with other clades and traits may find similar patterns, calling into question many previously proposed key innovations.
Our results highlight the pivotal necessity of utilizing genomic information in the study of key innovation. Perhaps the most widely-recognized benefit of utilizing genomes is the increased number of phylogenetic markers, which can substantially improve the chance of recovering an accurate topology over traditional methods31. Phylogenomic datasets also offer the ability to calculate branch lengths in terms of coalescent units, which can identify cases where trait histories might be more fluid than traditional concatenated ultrametric trees might suggest. We suggest that it will likely be necessary to utilize trees with branch lengths in coalescent units more frequently in phylogenetic comparative analyses, lest we discover that the “irrational exuberance” associated with resolved species trees has similar consequences for macroevolutionary trait analyses31. A classic ultrametric tree may not necessarily preserve the information on gene tree discordance, which can cause traditional phylogenetic comparative methods such as stochastic character mapping to overestimate the number of evolutionary transitions34,70. Similarly, many phylogenetic comparative methods are sensitive to the number of independent evolutionary events, which can have impacts on phylogenetic pseudoreplication71. Whole genome datasets may also be used to interrogate the topologies of trait-associated genomic regions, allowing for a deeper understanding of the molecular basis of convergent evolution51,72. Our results suggest that a whole-genome perspective now represents a vital component of key innovation research.
While the notion of a positive relationship between functional novelty and evolutionary success is inherently appealing, our results suggest that even supposedly classic examples of key innovation may not possess the signatures of accelerated lineage or ecological evolution previously thought to exemplify the process. Indeed, several other classic key innovations, such as floral nectar spurs23 and toepads24 exhibit more complex histories that do not conform to these simple models, and many older examples have yet to be re-examined with modern phylogenetic comparative methods. We suggest that it is likely necessary to move beyond the framework used by Heard and Hauser1, which emphasizes lineage, ecological, or morphological diversification as the end result of innovation, in favor of a more Simpsonian2,3 focus on understanding the ecological consequences of innovation, as well as the many alternative outcomes to invasion of a new adaptive zone that do not involve increased diversification6,8,15,22.
Methods
Animal care and use compliance
All specimens used in this study were processed using the following approved institutional protocols: Monash University AEC 11725. Animals were shipped via commercial fish suppliers using standard methods for small fishes (air travel, insulated foam boxes, plastic bags with water and added oxygen). Additionally, the targeted collection of Centrogenys vaigiensis and Congrogadus subducens specimens was performed under NT Fisheries permit S17/3418 and processed using the Charles Darwin University Animal Ethics Committee permit A12009 from the macro-tidal habitat in Darwin Harbour, Northern Australia (−12.41528, 130.81741) using dip net and spotlight at spring low tide during the night (0.6 m AHD, 1/8/2019). Ambassis elongata was collected in nearby water bodies (−16.08047, 136.31725) using a similar approach. All specimens were euthanized, tissued, and preserved as voucher specimens.
Phylogenomic topology of pharyngognaths and close relatives
Genome sequencing and assembly of Phanerodon vacca and Centrogenys vaigiensis
We obtained species representatives from the two pharyngognath families without published genome assemblies: Phanerodon vacca (Embiotocidae) and Centrogenys vaigiensis (Centrogenyidae). A live P. vacca specimen was obtained from a licensed California scientific collector (Matsu Collections, Inc.)73. Fish were euthanized with an overdose of clove oil by MDM, then fin clips, gill arches, and muscle tissue samples were removed and preserved. Extracted tissue was then sent to commercial sequencing providers (Phase Genomics, Seattle, WA) for PacBio Sequel and Hi-C sequencing. A total of 60 Gb 5 kb+ reads were produced across two Sequel flow cells, then assembled and error-corrected using Flye v2.8840 (v2.8) with default options. The PacBio assembly was used for downstream phylogenetic and comparative genomic analyses. This genome was then scaffolded with 13 GiB of Hi-C paired reads. We used the BBTools package v17.1274 for preprocessing, including identifying adapters with BBMerge, removing adapters and reads of average quality 10 or less with BBDuk, then removing PCR duplicates with Dedupe. We then used Chromap75 to align Hi-C reads to the PacBio assembly using the default Hi-C presets, creating a SAM file which was then sorted and converted to BAM using samtools. Finally, we used YaHS76 with default options to scaffold the PacBio assembly. The final assembly contained 24 linkage groups comprising 0.61 Gb, with an additional 0.012 Gb in 223 scaffolds.
Centrogenys vaigiensis was collected in tropical reef habitat (sandy areas interspersed/adjacent to rocky reef) in the macro-tidal Darwin Harbour, northern Australia, using dip net and spotlight at spring low tide during the night (0.6 m AHD, 1/8/2019). Fish were euthanized with an overdose of clove oil by MDM (Monash University AEC 11725), then tissue samples were then sent to Deakin Genomics (Geelong, VIC) for DNA extraction and library prep using PCR-free protocols. Libraries were then sequenced on S4 flowcells using Illumina Novaseq, with long-read sequencing performed using an ONT MinION flow cell. The Illumina short reads were then assembled using MaSuRCAv4.0.341 after first utilizing BBMerge from the BBTools package v17.1274 to identify insert size distributions. Nanopore reads of sizes 10 kb+ were utilized in MaSuRCA using its Flye40 module.
Draft genomes of additional species
We also sequenced representatives from eight related clades within Ovalentaria: Enneapterygius nanus (Tripterygiidae), Assessor macneilli (Plesiopidae), Opistognathus darwiniensis (Opistognathidae), Gerres filamentosus (Gerreidae), Parapercis cylindrica (Pinguipedidae), Glossolepis incisus (Melanotaeniidae), Ambassis elongata (Ambassidae), and Congrogadus subducens (Congrogadidae). Congrogadus subducens was collected simultaneously with Centrogenys, and Ambassis elongata was sampled from nearby water bodies using a similar approach. Glossolepis incisus captive-bred in Australia (Ausyfish, Inc) were obtained from the aquarium trade (Aquarium Industries, Inc), and the remainder of the samples were obtained from fish collected in Australian waters from the marine aquarium trade (Cairns Marine, Inc). DNA was extracted from preserved tissues at Deakin Genomics using PCR-free protocols, then sequenced on S4 flowcells using Illumina NovaSeq. Samples were assembled using MaSuRCAv4.0.341 after first utilizing BBMerge from the BBTools v17.1274 package to identify insert size distributions. The assembly for Glossolepis incisus did not successfully complete via MaSuRCA and we utilized Platanus 1.2.477 instead.
Additional published genomes
We added 22 additional genomes (Supplementary Data 1) from public repositories. These genomes were designed to represent all major clades within Ovalentaria and Perciformes sensu lato with hypothesized affinities to any pharyngognath group. Because our project relied heavily on the accurate assembly of small orthologous genomic regions, rather than long stretches of contiguous sequence, we re-assembled publicly available genomes where the primary assembly was produced with first-generation PacBio reads or any generation of Nanopore reads (Supplementary Data 1). MaSuRCA v4.0.341 after first utilizing BBMerge from the BBTools v17.1274 package to identify insert size distributions, using any present long reads in subsequent assembly steps after the production of super-reads from Illumina sequencing.
Genome QC
Since our project revolved primarily around phylogenomics and comparative genomics, our QC metrics focused on the completeness of individual genes, particularly orthologs common to actinopterygian fishes. As such, we used BUSCO v5.1.378 with the actinopterygii_odb10 dataset to examine completeness of both the Sequel-only assembly and the Sequel+Hi-C assembly. Our surfperch genome exhibited only 0.8% missing orthologues, the smallest missingness of any currently available teleost genome at time of manuscript submission. We attribute this to the extremely high coverage of long-read sequencing and its relatively compact genome (0.6 Gb). The remaining genomes exhibited varying levels of completeness.
Genome alignment
We assembled genomes of pharyngognaths and close relatives (Supplementary Data 1). We aligned each genome to the Thunnus albacares reference (Supplementary Data 1) using a LAST v1243 pipeline42. The Thunnus reference was first prepared using ‘last-db’ and a NEAR model to optimize for short-and-strong matches suitable for gene/locus tree creation using the command ‘lastdb -R01 -P48 -uNEAR’. We then utilized the ‘last-train’ learning algorithm using the command ‘last-train --revsym -D1e9 -R01’ on each individual sample to optimize alignment parameters, followed by ‘lastal’ to align each genome to the reference using the commands ‘lastal -R01 -D1e9 -m100’ and ‘last-split -fMAF + ’. To minimize the influence of paralogs, we also performed reverse-mapping using the command ‘last-split -r -m1e-6’, where each identified aligned sequence from the reference was then mapped to the focal genome, discarding any sequences without strong evidence of a unique match. We then used ‘maf-join’ to identify common blocks where every genome aligned to the reference. The alignments produced by maf-join are iterative, necessitating realignment in order to properly distribute any indels in a given alignment. As such, we utilized MAFFT v7.31079 to re-align each block using the L-INS-i model.
Recombination breakpoint analysis
Most commonly-utilized tree-building methods implicitly assume no intralocus recombination, which is generally appropriate for mitochondrial sequences but problematic for nuclear loci80. We used MDL43 to test for the presence of discordant topologies and split loci by recombination breakpoints, discarding any locus where one or more taxa consisted entirely of gaps, producing a total of 237,364 loci. These locus alignments were then passed to IQ-TREE 245, utilizing a GTR + R4 model to generate a tree for each locus, excluding 130 loci which did not contain enough informative sites. We then utilized ASTRAL v5.15.481 on our 237,364 locus topologies to produce a species tree (Fig. 1a, Supplementary Information, Table S1).
To ensure that the resulting shorter alignments retained power to distinguish between individual locus trees versus the species tree, we used IQ-TREE 2 to perform a series of topology tests, including the SH test82 and the AU topology test83. For each locus, we compared the topology inferred for an individual locus as well as the ASTRAL species topology, testing whether the species topology would be rejected in favor of the locus tree at a p < 0.05 level after performing a p-value adjustment using a false discovery rate correction84 for multiple comparisons using the p.adjust() function in R using method “fdr”. For the SH test, most alignments (n = 223,880) rejected the species topology in preference for the locus topology, and the AU test showed a similar pattern (n = 232,675). For the 170,582 alignments derived from MDL splitting alignments with recombination breakpoints, an even higher proportion of alignments rejected the species topology in favor of individual locus topologies for both the SH (n = 163,146) and the AU (n = 167,886) models, suggesting that alignments split into recombination breakpoints retained their discordant signal (see Dryad: https://doi.org/10.5061/dryad.cz8w9gjcx).
Locus tree-based hemiplasy analysis
We next examined whether concordance between individual gene trees and the species tree was low enough to suggest that the pharyngognathy innovation may not have independently evolved in each lineage possessing the trait. We calculated the hemiplasy risk factor using R package pepo v0.2 by inputting the ASTRAL tree with branch lengths in coalescent units46 (available at https://github.com/guerreror/pepo). Hemiplasy risk factors range from 0 to 1, with values representing no risk of hemiplasy and complete risk of hemiplasy respectively. Overall, we find high hemiplasy risk factors across the phylogeny (hrf = 0.75-0.93). Hemiplasy risk factors are quite high in Ovalentaria, where a majority of pharyngognathous lineages occur (Cichlidae, Pomacentridae, and Embiotocidae), suggesting that pharyngognathy may not have independently evolved within each clade, but originated due to ILS near the base of Ovalentaria. While our study contains more loci than other fish phylogenies using the multispecies coalescent, we also find high levels of hemiplasy across previously published phylogenomic datasets as well, including in areas of the phylogeny that contain pharyngognath lineages39,47,48,49 (Figs. S2–S7, Supplementary Materials). We caution against directly linking branch lengths in coalescent units explicitly to divergence times for several reasons. As our sampling consists of a single individual per species, we were unable to estimate terminal branch lengths using ASTRAL. Additionally, since ASTRAL estimates branch lengths in coalescent units, its branch lengths are a function of generation time and population size, and the taxa in our study are highly diverse and divergent in both population sizes and generation times85,86,87,88.
Site-based hemiplasy analysis
To ensure that our hemiplasy results were robust to both gene tree estimation error and our recombination breakpoint analysis, we concatenated all loci and calculated site-based concordance factor (sCF), the proportion of decisive alignment sites supporting a given branch in the tree, in IQ-TREE 2, using our ASTRAL topology89. We then used HeIST v0.3.190 to convert sCF into coalescent units (Fig. 1c) and estimated hemiplasy risk factor using pepo as above. We find similarly high levels of hemiplasy throughout the tree, consistent with our ASTRAL results (hrf = 0.76–0.92). As an additional approach, we identified loci of at least 300 base pairs in length in our 32 taxa, then randomly removed loci within 100 kb or less of each other using a custom R script. We then aligned and performed tree inference for the pruned locus set (n = 2574), in an identical manner to above, without testing for recombination breakpoints. We then used ASTRAL to generate a species tree (Supplementary Information, Fig. S7) and performed the same hemiplasy analysis as above. This tree exhibits reduced branch support for some taxa but shows the same pattern of short internodes as our primary results.
Analyzing the effect of hemiplasy on transitions to pharyngognathy
To further understand how hemiplasy may have influenced pharyngognath evolution, we utilized HeIST90. HeIST uses a coalescent tree with observed character states and simulates a set of gene trees from the coalescent tree and counts the number of transitions across each gene tree to provide an estimate of how likely the character of origin has a homoplastic evolutionary history. We used HeIST on a reduced dataset of 14 species (Thunnus, Istiophorus, Sander, Symphodus, Parapercis, Ambassis, Opistognathus, Amphiprion, Oreochromis, Phanerodon, Assessor, Xiphophorus, Hirundichthys, Oryzias), considering Symphodus and Centrogenys to represent the same transition, then performed approximately 10 billion simulations in order to obtain 100 trees with compatible character histories for each of 3 different transition rates (s = 0.05, s = 0.005, and s = 0.0005), for a total of 300 trees. Most of our simulations were used in the attempt to identify compatible transition histories for s = 0.0005, and it was not computationally feasible to estimate histories for slower transition rates. Attempts to estimate compatible histories for larger datasets than 14 species were also unsuccessful.
If pharyngognathy is entirely homoplastic, we would expect to recover an estimated five transitions in our HeIST analyses, one for each lineage of pharyngognaths (assuming Centrogenys and Symphodus to represent only one transition). However, if pharyngognathy is entirely the result of hemiplasy, we would expect simulations to suggest a single transition. It is also possible that pharyngognathy is the result of both hemiplasy and homoplasy, in which case, we would expect simulations to suggest more than one evolutionary transition (i.e. hemiplasy) but fewer than five (i.e. homoplasy). Our results, for all estimated transition rates, supported a hemiplasious history for pharyngognathy (Supplementary Information, Fig. S11), generally with two or fewer origins, with the caveat that HeIST was designed predominantly for analyses of younger clades than we utilize here.
Additional outgroups
To supplement our primary topology, we included additional species from other genome assemblies, transcriptomes, and phylogenomic studies (Supplementary Data 1). Assemblies were aligned to Thunnus in the same manner as described in the methods. We then used ASTRAL v5.15.481 to identify a species tree, utilizing our previous 32-taxon tree as a constraint (Fig. 1a). We then repeated our hemiplasy risk factor analysis as described in our methods section (Supplementary Information, Fig. S2). As we recover some polytomies in this topology, we set the hemiplasy risk factor value for zero-length branches to be 1. The computational limitations of our recombination analysis and the number of aligned loci precluded us from including more clades.
Additional analysis of Ovalentaria
Our primary analysis lacked several ovalentarian families, including engineer gobies (Pholidichthyidae), leaffishes (Polycentridae) and grammas (Grammatidae). We used MDL43 in the same manner as in the methods section to determine recombination breakpoints from a previous alignment of Ovalentaria48, found a total of 402 loci, used IQ-TREE 245 to generate individual locus trees, then produced a species tree using ASTRAL alongside a topological constraint from our main analysis described in the methods section. We then repeated our hemiplasy risk factor analysis as described in our methods section (Supplementary Information, Fig. S3) and set the hemiplasy risk factor value for zero-length branches to be 1.
Additional hemiplasy analyses
To further test if patterns of hemiplasy risk factors are consistent between our results and other studies, we calculated hemiplasy risk factors across three previously published phylogenomic datasets. We analyzed the ASTRAL-generated phylogeny of Hughes et al.49 which was constructed from 1105 orthologous exon sequences of 303 ray-finned fishes. The second study we analyzed was that of Alfaro et al.47 which generated a topology of 120 species from 1100 ultraconserved elements (UCE) loci. The third study was from Ghezelayagh et al.39 which generated a topology of 1084 species from 987 UCE loci. For all trees, we reduced the phylogeny to the MRCA of all pharyngognathous lineages and reduced the topology to a single representative taxa per family. We then repeated our hemiplasy risk factor analysis as described in our methods section (Supplementary Information, Figs. S4–S6) and set the hemiplasy risk factor value for zero-length branches if present to be 1.
Comparative genomics of pharyngognathy
Taxon sampling
For our analysis of the comparative genomic basis of pharygognathy, we opted for a more restrictive set of genomes, focusing on representatives of the five major transitions to pharygognathy: cichlids, surfperch, damselfishes, flyingfishes, and wrasses+false scorpionfish, as well as closely related outgroups to each transition. This resulted in a reduction to 16 species (Amphiprion, Assessor, Ambassis, Centrogenys, Glossolepis, Hirundichthys, Istiophorus, Latolabrax, Oreochromis, Oplegnathus, Parapercis, Phanerodon, Planiliza, Sebastes, Symphodus, and Thunnus). The remaining taxa were joined using ‘maf-join’ as described previously.
Genome contiguity and alignment lengths
When choosing taxa, we selected the highest-quality genome from each group, as well as a set of high-quality assemblies from close relatives, initially favoring assemblies with resolved chromosomes. However, in cases where multiple representatives were available, we discovered that assembly contiguity was not generally a good predictor of alignment length. We illustrate this with a comparison of several possible atheriniform and cyprinidontiform outgroups for Hirundichthys. For our primary analysis, we selected our draft assembly of Glossolepis incisus in our set of 16 taxa. We then compared these alignment blocks to a set of taxa where Oryzias celebensis was substituted for Glossolepis, as well as a set where Xiphophorus maculatus was substituted for Glossolepis, and a final comparison where Nothobranchius furzeri was substituted for Glossolepis. We then compared the lengths from the Thunnus reference associated with each alignment block (Supplementary Information, Table S5). The Glossolepis draft genome produced longer alignment block lengths than any of the others, despite several of the above assemblies being produced to chromosomal or near-chromosomal resolution. Similar issues were found with fish lineages near blenniforms and damselfishes, with our Assessor draft assembly outperforming the chromosomally-resolved assembly of the clingfish Gouania willdenowi. This suggests that while greater assembly quality is certainly a desirable characteristic in a comparative genomic analysis, it will often be overshadowed by the biological properties of the genomes themselves.
Limitations of the comparative genomic analyses
While we can use our set of taxa to examine the evolutionary dynamics of pharyngognathy, elucidating which genomic regions are associated with each component of the adaptation is a far more complex task. This is because several other fish clades have at least one of the adaptations. Anabas climbing perches have a fused 5th ceratobranchial, Toxotes archerfishes have a muscular sling, and many other clades have not been comprehensively investigated for the presence or absence of each character state34,65,91. We have avoided this issue by focusing the analysis only on species known to not have any of the adaptations, but this precludes us from identifying regions associated only with a particular trait.
Coding sequence analysis
We extracted exonic sequences from our combined multiple genome alignment files (MAF) as individual multi-FASTA alignments, pruning bases from each end to retain a proper reading frame based on the available CDS data for the T. albacares assembly using a custom R script. We then removed all gaps and re-aligned using default parameters in MACSE 0.9b1, a codon-aware aligner92. Sites identified with frameshifts were converted to ambiguous nucleotides. We then utilized PAML93 to screen for exons with some evidence of divergent dN/dS ratios between pharyngognaths and non-pharyngognaths. We identified terminal branches on the species tree associated with pharyngognathy and marked them as foreground branches, then run two PAML models on each exon: one where all branches were constrained to a common rate, and another, where pharyngognath (foreground) branches exhibited a different rate than non-pharyngognath branches. We then performed a likelihood ratio test at a p < 0.05 level after performing a false discovery rate correction using the p.adjust() function in R with method = “fdr”, retaining 5 exons with some evidence of positive selection.
Because the codon alignment is treated as fixed in a PAML run, it is possible for alignment ambiguity to be interpreted as positive selection94. We therefore utilized Bali-Phy95 for the remaining steps in our pipeline. We utilized the ‘branch_site’ model in Bali-Phy, fixing the species tree topology with the same identified foreground branches while allowing other parameters to vary. We then ran 10,000 Bali-Phy iterations for each of the 5 exons, with none reaching a posterior probability of positive selection higher than 0.9.
Noncoding sequence analysis
We used MafFilter v1.3.196 to extract individual loci from our 16-taxon MAF, excluding all coding sequences as well as sequences smaller than 50 bp, producing individual multi-FASTA alignments. We then used MAFFT with L-INS-i to realign each multi-FASTA, and MDL43 to split loci by recombination breakpoints in a manner identical to above, producing a total of 679,285 individual alignments of noncoding sequence (Supplementary Information, Table S6). We then used PHAST97 to extract fourfold degenerate sites from our exonic sequence, and estimated branch lengths from these, using a topology derived from our original ASTRAL topology pruned to 16 species. We then used our fourfold degenerate site tree and all locus trees with PhyloAcc v2.250 to identify loci with accelerated rates of evolution. We split the 679,285 loci into 100 runs of approximately 6800 loci each, then identified loci with at least moderate support for accelerated evolution in pharyngognaths (log Bayes factor (BF1) > 2), greater relative acceleration in pharyngognaths (log Bayes factor (BF2) > 2), and where no non-pharyngognaths exhibited a posterior probability of acceleration greater than 0.1, and at least one pharyngognath exhibited a posterior probability of acceleration of at least 0.9 (n = 315). We then identified how many loci within this set had at least two accelerated pharyngognaths with posterior probability 0.9 or greater for each (n = 20).
To verify that splitting loci by recombination breakpoints using MDL was not substantially altering our ability to infer acceleration, we repeated the PhyloAcc analysis using our loci prior to splitting by breakpoints and applied our logBF1 > 2 and logBF2 > 2 threshold. In both the split and unsplit analyses, ~2% of loci meet the Bayes factor threshold, prior to assessing posterior probabilities of acceleration in individual lineages.
Topological analysis of accelerated loci
If pharyngognathy had a hemiplasious origin in some pharyngognaths, we would expect the topologies of loci associated with the trait to exhibit a signal of shared ancestry. We used IQ-TREE 245 with a GTR + R4 model to produce individual topologies for each accelerated locus. Examination of some of these loci reveals many cases where multiple pharyngognaths exhibited sister relationships not found in the species tree (Fig. 2a). Therefore, we used the ‘make.simmap’ function from the R package phytools98 to calculate 100 simulated character histories of pharyngognathy for each accelerated locus (n = 315), then calculated the median number of transitions between states across all 100 histories for each locus using the function ‘describe.simmap’. ‘make.simmap’ was unable to produce stochastic maps for 2 of the 315 loci, which were excluded from further analysis. We then used a Bayesian model in R package brms v2.1899 to examine median transitions as a function of whether the locus had two or more accelerated pharyngognaths, revealing an effect size with a 95% credible interval that did not overlap zero (Fig. 2b, Supplementary Information, Table S7).
Gene ontology of accelerated regions
We used DeepGraphGO100, a deep learning-based method, to generate gene ontology (GO) terms for molecular function of all coding regions (n = 27,253) in the O. niloticus assembly, retaining terms with a probability of over 0.5 (n = 1,648). We then examined whether our convergently-accelerated loci (n = 20) were found within 10 kb or less of a protein-coding region. If a locus was within 10 kb of two or more regions, we took the closest region. We identified 18 loci meeting this criterion, then used a Fisher’s exact test in R with ‘fisher.test’ to select which GO terms had a significant association across all coding regions against the accelerated loci. We identified 18 GO terms significant at the p < 0.05 level and placed them into a multivariate Bayesian model using STAN via R package brms99 using a Bernoulli family and a lasso prior with degrees of freedom 3 and scale 10 to examine which GO terms were enriched in the 18 convergently-accelerated loci relative to other coding regions. Of the 18 GO terms, 12 exhibited 95% credible intervals that did not overlap zero (Supplementary Data 4). We then visualized these 12 GO terms as a network using AMIGO2’s network visualization101 (Supplementary Information, Fig. S9). As an alternative to the Bayesian approach, we also performed an enrichment analysis of all GO terms assigned to the focal loci and performed a false discovery rate (FDR)-corrected Fisher’s exact test in a similar manner to above, retaining the same 12 terms as significant at the p < 0.05 level after correction.
Macroevolutionary analyses
Phylogenetic reconstruction
We generated a 35,746 bp alignment from twenty-five nuclear and sixteen mitochondrial loci for 9323 species of acanthomorphs uniting the most recent common ancestor of all pharyngognaths and several outgroup taxa using the Phylogeny Assembly with Databases pipeline (PHLAWD v3.4b)102 (see Dryad: https://doi.org/10.5061/dryad.cz8w9gjcx). Additional mitochondrial sequences were extracted from whole mitochondrial genomes using the R package AnnotationBustR v1.2103 to supplement sequences obtained using PHLAWD. We extracted legacy markers from whole genome sequences for select taxa that did not have traditional Sanger sequence data for certain loci on GenBank. We inferred a maximum likelihood topology using RAxML 8.2.8104 under the GTR + CAT approximation of rate heterogeneity partitioned by gene, and for protein-coding sequences, also partitioned by codon position with the final optimization of the tree and branch lengths estimated in IQ-TREE 245 under GTR + Γ. We found that for very large phylogenies such as this one, RAxML was noticeably faster for topological influence in the presence of constraints, whereas IQ-TREE 2 was faster when inferring branch lengths. We employed topological constraints based on recovered phylogenetic relationships from high-throughput sequencing studies of acanthomorph fishes55,105,106,107,108,109,110,111,112,113,114,115. We converted our phylogeny into a chronogram using treePL with 70 fossil and geological constraints from the literature with the optimal smoothing parameter obtained using cross-validation116 (see Dryad: https://doi.org/10.5061/dryad.cz8w9gjcx).
Trophic level data acquisition
117,118We assembled a stomach-contents based trophic level database from 2874 published sources for 3490 species of acanthomorph fishes (see Dryad: https://doi.org/10.5061/dryad.cz8w9gjcx). We limited studies to those that used volume, weight, or compound indices using volume or weight as other diet composition methods such as frequency of occurrence or numeric can be problematic when estimating trophic levels119,120. To limit possible ontogenetic biases, we limited our dataset to studies that included adults within their sampling. Dietary studies which were performed outside of a species native range were also excluded to avoid possible shifts in trophic ecology due to invading non-native habitats. We calculated trophic levels using the R package dietr v1.1.3117, which follow the routines implemented by Pauly et al.118 to calculate fractional trophic levels (see Dryad: https://doi.org/10.5061/dryad.cz8w9gjcx). In cases where species had data from multiple studies, trophic levels were calculated for each study and the mean trophic level for that species was used in downstream analyses. Standard error estimates for species that had more than one diet record were calculated. However, for species with a single diet record, we used a regression to predict a standard error value for a single diet record using the number of diet records as a predictor. This value was then applied as the standard error value for all species that only had a single diet record for mvMorph121 analyses described below. As BBMV54 internally handles error estimation as part of its model, the calculated trophic level for every record in our dataset was used as input.
Tests for early bursts in trophic level evolution
We used the R package mvMORPH v1.1.6121 to test if trophic level evolution best fit a Brownian motion, Ornstein-Uhlenbeck, or early burst model evolution. If pharyngognathy is leading to rapid bursts of trophic divergence within pharyngognathous families, we would expect trophic evolution to follow an early burst model.
Comparative diet analyses
To investigate if the macroevolutionary landscape of trophic level is different in pharyngognaths relative to non-pharyngognaths, we generated macroevolutionary landscapes for these two trophic traits using the R package BBMV v2.154. BBMV (Bounded Brownian Motion with potential) implements the Fokker-Planck-Kolomogorov (FPK) model of evolution to determine the evolutionary potential and rate of evolution of a univariate trait. BBMV allows for bounded evolution, which is useful for trophic ecology, where bounds are realistic (e.g., trophic levels for fish must be bounded at 2 as there are no autotrophic fish). As the Fokker-Planck-Kolmogorov model implemented in BBMV is a generalization of both Ornstein-Uhlenbeck (OU) and Brownian motion (BM) models, equivalent BM and OU models can be performed for model comparison by fixing certain parameters to 054. We fit macroevolutionary landscape models of trophic level in a maximum likelihood framework under BBMV, OU, or BM models to assess which fit best using the lnL_BBMV function. We found overwhelming support for the BBMV model (Supplementary Information, Table S2) and re-fit the BBMV model using Markov chain Monte Carlo estimation. Given our comparative genomic analysis highlights the difficulty of estimating the number of distinct transitions to pharyngognathy, especially in Ovalentaria, we used the BBMV function MH_MCMC_FPK_multiclades to estimate a single macroevolutionary landscape for pharyngognaths but allowed the rate of evolution to vary between labrid (including Centrogenys) pharyngognaths and ovalentarian pharyngognaths. For non-pharyngognaths we used the MH_MCMC_FPK function, which estimates a macroevolutionary landscape with a single rate of evolution. For all landscapes, we bounded the possible values of landscape following the known range of trophic levels. MCMC chains were run for 1,000,000 steps with traits discretized across 50 points. If pharyngognathy involves invasion of a new adaptive zone, we would expect that pharyngognathous clades would have different trophic level macroevolutionary landscapes compared to non-pharyngognathous clades (Supplementary Information, Table S2).
To assess the impact of each pharyngognathous clade on the macroevolutionary landscape, we then repeated our analyses by leaving each clade out in a manner analogous to leave-one-out cross validation. Overall, landscapes were congruent across analyses. However, we found a major deviation in the macroevolutionary landscape when cichlids were removed. When cichlids were removed, the large low trophic level peak seen in pharyngognathous fishes was no longer present. To assess if this was due to the removal of the Lake Malawi and Lake Victoria adaptive radiations, which display non-treelike behavior and are known to cause issues in phylogenetic comparative studies, we also generated a macroevolutionary landscape that included all pharyngognaths except the species uniting the most recent common ancestor of these two radiations.
Diversification analyses
To test for state-dependent diversification, we used the R package HiSSE (hidden state speciation and extinction), which employs a hidden Markov model that assumes a hidden un-observed state to measure rates of speciation, extinction, and transition rates among states, while providing null models to test for character-independent diversification13. We focused our analysis to the most recent common ancestor of all pharyngognath taxa and ran a set of forty-two models on both the entire tree as well as within Ovalentarians. These models varied in the number of free parameters to be estimated. Of our models, eight were equivalent to BiSSE models, lacking a hidden state, twenty-six were hidden state models where different parameters were allowed to vary, and eight were character-independent diversification models. As transition rates between states can be difficult to estimate in an SSE framework, we constrained transitions to be equal across states13. We mapped pharyngognathy to the six pharyngognathous clades, Centrogenyidae, Cichlidae, Embiotocidae, Exocoetidae/Hemiramphidae, Labridae, and Pomacentridae following Wainwright et al. 34. We accounted for missing species in our dataset by supplying HiSSE with the known sampling fraction of taxa in our phylogeny for each state, which we calculated based on the number of known described species of pharyngognathous and non-pharyngognathous species listed in the California Academy of Sciences Catalog of Fishes122. Our phylogeny contained 7218 non-pharyngognaths of 11,429 described non-pharyngognaths and 2105 pharyngognaths of 2985 described pharyngognaths representing 63.1% and 70.5% taxon sampling respectively. Within Ovalentaria, our phylogeny contained 2118 of 3542 described non-pharyngognaths and 1565 of 2310 described pharyngognaths, representing 59.8% and 67.77% taxon sampling respectively.
We also tested for significant differences in diversification rates between pharyngognaths and non-pharyngognaths using the non-parametric method FiSSE56. This method measures the DR statistic of Jetz et al. 123 for each character state and uses the difference between these as the test statistic. It then computes a null distribution to test the test statistic against by simulating character evolution on the empirical phylogeny under a one-parameter Markov model and assesses significance by comparing the proportion of simulated datasets that are more extreme than the observed test statistic. It is important to note that the DR statistic best represents speciation rates rather than net diversification124. We ran FiSSE on both the full and Ovalentaria tree with 1000 simulations using the FiSSE.binary function. To account for patterns being driven by young exceptional clades of cichlids which have been previously shown to bias inferences due to their non-treelike behavior55, we re-ran FiSSE analyses after removing the clade that contains the most recent common ancestor of the two exceptional cichlid radiations of Lake Malawi and Lake Victoria.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The genomic data generated in this study have been deposited in NCBI BioProject PRJNA1136234. The genomic assembly, GO terms, character mapping, hemiplasy risk factor values, and hemiplasy simulation data generated in this study are provided in the Supplementary Information/Source Data file. Additional phylogenetic, trophic, and genomic data required to replicate the results of this study have been deposited in the Dryad repository and are available at the following link: https://doi.org/10.5061/dryad.cz8w9gjcx. Source data are provided as a Source Data file. Source data are provided with this paper.
Code availability
Code and scripts used in this study has been deposited in the Dryad repository and are available at the following link: https://doi.org/10.5061/dryad.cz8w9gjcx.
References
Heard, S. B. & Hauser, D. L. Key evolutionary innovations and their ecological mechanisms. Historical Biol. 10, 151–173 (1995).
Simpson, G. G. Major Features of Evolution. (Columbia University Press, 1953).
Simpson, G. G. Tempo and Mode in Evolution. (Columbia University Press, 1944).
Hunter, J. P. Key innovations and the ecology of macroevolution. Trends Ecol. Evol. 13, 31–36 (1998).
Mitter, C., Farrell, B. & Wiegmann, B. The phylogenetic study of adaptive zones- has phytophagy promoted insect diversification. Am. Naturalist 132, 107–128 (1988).
Vermeij, G. Innovation and evolution at the edge: origins and fates of gastropods with a labral tooth. Biol. J. Linn. Soc. 72, 461–508 (2001).
Liem, K. F. Evolutionary strategies and morphological innovations- cichlid pharyngeal jaws. Syst. Zool. 22, 425–441 (1973).
Rabosky, D. L. Phylogenetic tests for evolutionary innovation: the problematic link between key innovations and exceptional diversification. Philos. Trans. R. Soc. B: Biol. Sci. 372, 20160417 (2017).
Stebbins, G. L. in Studies in the Philosophy of Biology 285-306 (Springer, 1974).
Cracraft, J. in Evolutionary Innovations (eds M. H. Nitecki & D. V. Nitecki) 21-44 (University of Chicago Press, 1990).
Maddison, W. P., Midford, P. E. & Otto, S. P. Estimating a binary character’s effect on speciation and extinction. Syst. Biol. 56, 701–710 (2007).
Rabosky, D. L. Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PloS one 9, e89543 (2014).
Beaulieu, J. M. & O’Meara, B. C. Detecting hidden diversification shifts in models of trait-dependent speciation and extinction. Syst. Biol. 65, 583–601 (2016).
May, M. R. & Moore, B. R. A bayesian approach for inferring the impact of a discrete character on rates of continuous-character evolution in the presence of background-rate variation. Syst. Biol. 69, 530–544 (2020).
Miller, A. H., Stroud, J. T. & Losos, J. B. The ecology and evolution of key innovations. Trends Ecol. Evol., https://doi.org/10.1016/j.tree.2022.09.005 (2022).
Alfaro, M. E., Brock, C. D., Banbury, B. L. & Wainwright, P. C. Does evolutionary innovation in pharyngeal jaws lead to rapid lineage diversification in labrid fishes? BMC Evolut. Biol. 9, 255 (2009).
Drummond, C. S., Eastwood, R. J., Miotto, S. T. & Hughes, C. E. Multiple continental radiations and correlates of diversification in Lupinus (Leguminosae): testing for key innovation with incomplete taxon sampling. Syst. Biol. 61, 443–460 (2012).
Rainford, J. L., Hofreiter, M., Nicholson, D. B. & Mayhew, P. J. Phylogenetic distribution of extant richness suggests metamorphosis is a key innovation driving diversification in insects. PloS one 9, e109085 (2014).
Hodges, S. A. & Arnold, M. L. Spurring plant diversification: are floral nectar spurs a key innovation? P R. Soc. B-Biol. Sci. 262, 343–348 (1995).
Givnish, T. J. Adaptive radiation versus ‘radiation’and ‘explosive diversification’: why conceptual distinctions are fundamental to understanding evolution. N. Phytologist 207, 297–303 (2015).
Stroud, J. T. & Losos, J. B. Ecological opportunity and adaptive radiation. Annu. Rev. Ecol., Evolution, Syst. 47, 507–532 (2016).
McGee, M. D. et al. A pharyngeal jaw evolutionary innovation facilitated extinction in Lake Victoria cichlids. Science 350, 1077–1079 (2015).
Cacho, N. I., Berry, P. E., Olson, M. E., Steinmann, V. W. & Baum, D. A. Are spurred cyathia a key innovation? Molecular systematics and trait evolution in the slipper spurges (Pedilanthus clade: Euphorbia, Euphorbiaceae). Am. J. Bot. 97, 493–510 (2010).
Miller, A. H. & Stroud, J. T. Novel Tests Of The Key Innovation Hypothesis: Adhesive Toepads In Arboreal Lizards. Syst. Biol. 71, 139–152 (2021).
Sackton, T. B. et al. Convergent regulatory evolution and loss of flight in paleognathous birds. Science 364, 74–78 (2019).
Blount, Z. D., Barrick, J. E., Davidson, C. J. & Lenski, R. E. Genomic analysis of a key innovation in an experimental Escherichia coli population. Nature 489, 513–518 (2012).
Wang, K. et al. African lungfish genome sheds light on the vertebrate water-to-land transition. Cell 184, 1362–1376 e1318 (2021).
Kim, B. M. et al. Antarctic blackfin icefish genome reveals adaptations to extreme environments. Nat. Ecol. Evol. 3, 469–478 (2019).
Larouche, O. et al. Do key innovations unlock diversification? A case-study on the morphological and ecological impact of pharyngognathy in acanthomorph fishes. Curr. Zool. 66, 575–588 (2020).
Avise, J. C. & Robinson, T. J. Hemiplasy: a new term in the lexicon of phylogenetics. Syst. Biol. 57, 503–507 (2008).
Hahn, M. W. & Nakhleh, L. Irrational exuberance for resolved species trees. Evolution 70, 7–17 (2015).
Shimeld, S. M. & Holland, P. W. Vertebrate innovations. Proc. Natl Acad. Sci. USA 97, 4449–4452 (2000).
Gould, S. J. & Vrba, E. S. Exaptation—a missing term in the science of form. Paleobiology 8, 4–15 (1982).
Wainwright, P. C. et al. The evolution of pharyngognathy: A phylogenetic and functional appraisal of the pharyngeal jaw key innovation in labroid fishes and beyond. Syst. Biol. 61, 1001–1027 (2012).
Hulsey, C. D., de, Leon, F. J. G. & Rodiles-Hernandez, R. Micro- and macroevolutionary decoupling of cichlid jaws: A test of Liem’s key innovation hypothesis. Evolution 60, 2096–2109 (2006).
Liem, K. F. & Sanderson, S. L. The pharyngeal jaw apparatus of labrid fishes: A functional morphological perspective. J. Morphol. 187, 143–158 (1986).
Liem, K. F. & Greenwood, P. H. A functional approach to the phylogeny of the pharyngognath teleosts. Am. Zool. 21, 83–101 (1981).
Lauder, G. V. Form and function: structural analysis in evolutionary morphology. Paleobiology 7, 430–442 (1981).
Ghezelayagh, A. et al. Prolonged morphological expansion of spiny-rayed fishes following the end-Cretaceous. Nat. Ecol. Evol. 6, 1211–1220 (2022).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Zimin, A. V. et al. The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677 (2013).
Frith, M. C., Hamada, M. & Horton, P. Parameters for accurate genome alignment. BMC Bioinforma. 11, 1–14 (2010).
Ane, C. Detecting phylogenetic breakpoints and discordance from genome-wide alignments for species tree reconstruction. Genome Biol. Evol. 3, 246–258 (2011).
Springer, M. S. & Gatesy, J. Delimiting Coalescence Genes (C-Genes) in Phylogenomic Data Sets. Genes 9, https://doi.org/10.3390/genes9030123 (2018).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evolution 37, 1530–1534 (2020).
Guerrero, R. F. & Hahn, M. W. Quantifying the risk of hemiplasy in phylogenetic inference. Proc. Natl Acad. Sci. USA 115, 12787–12792 (2018).
Alfaro, M. E. et al. Explosive diversification of marine fishes at the Cretaceous-Palaeogene boundary. Nat. Ecol. Evol. 2, 688–696 (2018).
Eytan, R. I. et al. Are 100 enough? Inferring acanthomorph teleost phylogeny using Anchored Hybrid Enrichment. BMC Evolut. Biol. 15, 113 (2015).
Hughes, L. C. et al. Comprehensive phylogeny of ray-finned fishes (Actinopterygii) based on transcriptomic and genomic data. Proc. Natl Acad. Sci. USA 115, 6249–6254 (2018).
Hu, Z., Sackton, T. B., Edwards, S. V. & Liu, J. S. Bayesian detection of convergent rate changes of conserved noncoding elements on phylogenetic trees. Mol. Biol. Evolution 36, 1086–1100 (2019).
Smith, S. D., Pennell, M. W., Dunn, C. W. & Edwards, S. V. Phylogenetics is the new genetics (for most of biodiversity). Trends Ecol. Evol. 35, 415–425 (2020).
Funderburgh, J. L. MINI REVIEW Keratan sulfate: structure, biosynthesis, and function. Glycobiology 10, 951–958 (2000).
Matsuo, K. & Otaki, N. Bone cell interactions through Eph/ephrin: bone modeling, remodeling and associated diseases. Cell Adhes. Migr. 6, 148–156 (2012).
Boucher, F. C. BBMV: an R package for the estimation of macroevolutionary landscapes. Ecography 42, 558–564 (2019).
McGee, M. D. et al. The ecological and genomic basis of explosive adaptive radiation. Nature 586, 75–79 (2020).
Rabosky, D. L. & Goldberg, E. E. FiSSE: A simple nonparametric test for the effects of a binary character on lineage diversification rates. Evolution 71, 1432–1442 (2017).
Mabuchi, K., Miya, M., Azuma, Y. & Nishida, M. Independent evolution of the specialized pharyngeal jaw apparatus in cichlid and labrid fishes. BMC Evolutionary Biol. 7, 10.1186/1471-2148-7-10 (2007).
Wagner, C. E., Harmon, L. J. & Seehausen, O. Ecological opportunity and sexual selection together predict adaptive radiation. Nature 487, 366–369 (2012).
Muller, C., Moser, F. N., Frei, D. & Seehausen, O. Constraints to speciation despite divergence in an old haplochromine cichlid lineage. Evolution, https://doi.org/10.1093/evolut/qpad001 (2023).
Seehausen, O. African cichlid fish: a model system in adaptive radiation research. Proc. R. Soc. B: Biol. Sci. 273, 1987–1998 (2006).
Friedman, M. et al. Molecular and fossil evidence place the origin of cichlid fishes long after Gondwanan rifting. Proc. Biol. Sci. / R. Soc. 280, 20131733 (2013).
Irisarri, I. et al. Phylogenomics uncovers early hybridization and adaptive loci shaping the radiation of Lake Tanganyika cichlid fishes. Nat. Commun. 9, 3159 (2018).
Matschiner, M., Bohne, A., Ronco, F. & Salzburger, W. The genomic timeline of cichlid fish diversification across continents. Nat. Commun. 11, 5895 (2020).
Ronco, F. et al. Drivers and dynamics of a massive adaptive radiation in cichlid fishes. Nature 589, 76–81 (2021).
Stiassny, M. L. & Jensen, J. S. Labroid interrelationships revisited: morphological complexity, key innovations, and the study of comparative diversity. Bull. Mus. Comp. Zool. 151, 269–319 (1987).
Amorim, M. C. P., Simoes, J. M., Fonseca, P. J. & Turner, G. F. Species differences in courtship acoustic signals among five Lake Malawi cichlid species (Pseudotropheus spp.). J. Fish. Biol. 72, 1355–1368 (2008).
Springer, V. G., Johnson, G. D., Orrell, T. M. & Darrow, K. Study of the dorsal gill-arch musculature of teleostome fishes, with special reference to the Actinopterygii. Vol. 1 (Biological Society of Washington, Smithsonian Institution, 2004).
Ronco, F. & Salzburger, W. Tracing evolutionary decoupling of oral and pharyngeal jaws in cichlid fishes. Evol. Lett. 5, 625–635 (2021).
Roberts-Hugghis, A. S., Burress, E. D., Lam, B. & Wainwright, P. C. The cichlid pharyngeal jaw novelty enhances evolutionary integration in the feeding apparatus. Evolution, https://doi.org/10.1093/evolut/qpad109 (2023).
Bollback, J. P. SIMMAP: stochastic character mapping of discrete traits on phylogenies. BMC Bioinforma. 7, 88 (2006).
Maddison, W. P. & FitzJohn, R. G. The Unsolved Challenge to Phylogenetic Correlation Tests for Categorical Characters. Syst. Biol. 64, 127–136 (2015).
Fukushima, K. & Pollock, D. D. Detecting macroevolutionary genotype–phenotype associations using error-corrected rates of protein convergence. Nat. Ecol. Evol. 7, 155–170 (2023).
Eastment, R. V., Wong, B. B. M. & McGee, M. D. Convergent genomic signatures associated with vertebrate viviparity. BMC Biol. 22, 34 (2024).
Bushnell, B. BBTools software package (2014).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nat. Commun. 12, 6566 (2021).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, https://doi.org/10.1093/bioinformatics/btac808 (2023).
Kajitani, R. et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res 24, 1384–1395 (2014).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evolution 38, 4647–4654 (2021).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evolution 30, 772–780 (2013).
Neches, R. Y., McGee, M. D. & Kyrpides, N. C. Recombination should not be an afterthought. Nat. Rev. Microbiol 18, 606 (2020).
Zhang, C., Rabiee, M., Sayyari, E. & Mirarab, S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinforma. 19, 153 (2018).
Shimodaira, H. & Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evolution 16, 1114 (1999).
Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51, 492–508 (2002).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc.: Ser. B (Methodol.) 57, 289–300 (1995).
Allen, G. R. & Cross, N. J. Rainbowfishes of Australia and Papua New Guinea. (TFH Publications 1982).
Lou, R. N. et al. Full mitochondrial genome sequences reveal new insights about post-glacial expansion and regional phylogeographic structure in the Atlantic silverside (Menidia menidia). Marine Biology 165, https://doi.org/10.1007/s00227-018-3380-5 (2018).
Berois, N., García, G. & de Sá, R. O. Annual Fishes: Life History Strategy, Diversity, and Evolution. (CRC Press, 2015).
Kolora, S. R. R. et al. Origins and evolution of extreme life span in Pacific Ocean rockfishes. Science 374, 842–847 (2021).
Mo, Y. K., Lanfear, R., Hahn, M. W. & Minh, B. Q. Updated site concordance factors minimize effects of homoplasy and taxon sampling. Bioinformatics 39, btac741 (2023).
Hibbins, M. S., Gibson, M. J. & Hahn, M. W. Determining the probability of hemiplasy in the presence of incomplete lineage sorting and introgression. Elife 9, https://doi.org/10.7554/eLife.63753 (2020).
Liem, K. F. The comparative osteology and phylogeny of the Anabantoidae (Teleostei, Pisces). Ill. Biol. Monogr. 30, 1–149 (1963).
Ranwez, V., Douzery, E. J., Cambon, C., Chantret, N. & Delsuc, F. MACSE v2: toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evolution 35, 2582–2584 (2018).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evolution 24, 1586–1591 (2007).
Redelings, B. Erasing errors due to alignment ambiguity when estimating positive selection. Mol. Biol. Evolution 31, 1979–1993 (2014).
Redelings, B. D. Bali-Phy version 3: Model-based co-estimation of alignment and phylogeny. Bioinformatics, https://doi.org/10.1093/bioinformatics/btab129 (2021).
Dutheil, J. Y., Gaillard, S. & Stukenbrock, E. H. MafFilter: a highly flexible and extensible multiple genome alignment files processor. BMC Genomics 15, 1–10 (2014).
Hubisz, M. J., Pollard, K. S. & Siepel, A. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief. Bioinform 12, 41–51 (2011).
Revell, L. J. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evolution 3, 217–223 (2012).
Bürkner, P.- C. brms: An R package for Bayesian multilevel models using Stan. J. Stat. Softw. 80, 1–28 (2017).
You, R., Yao, S., Mamitsuka, H. & Zhu, S. DeepGraphGO: graph neural network for large-scale, multispecies protein function prediction. Bioinformatics 37, i262–i271 (2021).
Carbon, S. et al. AmiGO: online access to ontology and annotation data. Bioinformatics 25, 288–289 (2009).
Smith, S. A., Beaulieu, J. M. & Donoghue, M. J. Mega-phylogeny approach for comparative biology: an alternative to supertree and supermatrix approaches. BMC Evolut. Biol. 9, 37 (2009).
Borstein, S. R. & O’Meara, B. C. AnnotationBustR: an R package to extract subsequences from GenBank annotations. PeerJ. 6, e5179 (2018).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Jones, J. C., Fan, S., Franchini, P., Schartl, M. & Meyer, A. The evolutionary history of Xiphophorus fish and their sexually selected sword: a genome-wide approach using restriction site-associated DNA sequencing. Mol. Ecol. 22, 2986–3001 (2013).
Longo, G. & Bernardi, G. The evolutionary history of the embiotocid surfperch radiation based on genome-wide RAD sequence data. Mol. Phylogenet. Evol. 88, 55–63 (2015).
Takahashi, T. & Moreno, E. A RAD-based phylogenetics for Orestias fishes from Lake Titicaca. Mol. Phylogenet. Evol. 93, 307–317 (2015).
Harrington, R. C. et al. Phylogenomic analysis of carangimorph fishes reveals flatfish asymmetry arose in a blink of the evolutionary eye. BMC Evolut. Biol. 16, 224 (2016).
McGee, M. D. et al. Replicated divergence in cichlid radiations mirrors a major vertebrate innovation. Proc. R. Soc. B: Biol. Sci. 283, 20151413 (2016).
Rican, O., Pialek, L., Dragova, K. & Novak, J. Diversity and evolution of the Middle American cichlid fishes (Teleostei: Cichlidae) with revised classification. Vertebr. Zool. 66, 1–102 (2016).
Burress, E. D. et al. Phylogenomics of pike cichlids (Cichlidae: Crenicichla): the rapid evolution and trophic diversification of an incipient species flock. J. Evol. Biol. https://doi.org/10.1111/jeb.13196 (2017).
Ilves, K. L., Torti, D. & Lopez-Fernandez, H. Exon-based phylogenomics strengthens the phylogeny of Neotropical cichlids and identifies remaining conflicting clades (Cichliformes: Cichlidae: Cichlinae). Mol. Phylogenet. Evol. 118, 232–243 (2017).
Song, S., Zhao, J. & Li, C. Species delimitation and phylogenetic reconstruction of the sinipercids (Perciformes: Sinipercidae) based on target enrichment of thousands of nuclear coding sequences. Mol. Phylogenet. Evol. 111, 44–55 (2017).
Malinsky, M. et al. Whole genome sequences of Malawi cichlids reveal multiple radiations interconnected by gene flow. Nat. Ecol. Evolution 2, 1940–1955 (2018).
Rodgers, R., Roach, J., Reid, N., Whitehead, A. & Duvernell, D. D. Phylogenomic analysis of Fundulidae (Teleostei: Cyprinodotiformes) using RNA-sequencing Data. Mol. Phylogenet. Evol. 121, 150–157 (2018).
Smith, S. A. & O’Meara, B. C. treePL: divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 28, 2689–2690 (2012).
Borstein, S. R. dietr: an R package for calculating fractional trophic levels from quantitative and qualitative diet data. Hydrobiologia 847, 4285–4294 (2020).
Pauly, D. et al. TrophLab Manual. ICLARM, Manila, Philippines (2000).
Froese, R. & Pauly, D. FishBase 2000: Concepts Designs and Data Sources. (ICLARM, 2000).
Stergiou, K. I. & Karpouzi, V. S. Feeding habits and trophic levels of Mediterranean fish. Rev. Fish. Biol. Fish. 11, 217–254 (2002).
Clavel, J., Escarguel, G. & Merceron, G. mvmorph: an r package for fitting multivariate evolutionary models to morphometric data. Methods Ecol. Evolution 6, 1311–1319 (2015).
Fricke, R., Eschmeyer, W. N. & van der Laan, R. Eschmeyer’s Catalog of Fishes, <http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain> (2024).
Jetz, W., Thomas, G., Joy, J., Hartmann, K. & Mooers, A. The global diversity of birds in space and time. Nature 491, 444 (2012).
Title, P. O. & Rabosky, D. L. Tip rates, phylogenies and diversification: What are we estimating, and how good are the estimates? Methods Ecol. Evolution 10, 821–834 (2019).
Acknowledgements
We would like to thank R. Nagesan for help with microCT scanning. We thank members of the O’Meara lab and Lopez-Fernandez lab for helpful discussions about the manuscript. We thank C. Austin, Y.P. Lee, and S. Loke of Deakin University for Illumina and Nanopore sequencing assistance, and Phase Genomics for PacBio sequencing. This work was supported by the MASSIVE HPC facility (www.massive.org.au) and Monash Aquacore. This research was funded by National Science Foundation grant DEB-1701913 to S.R.B and B.C.O., National Science Foundation grant DBI-2010931 to S.R.B, and Australian Research Council grant DE180101558 to M.D.M.
Author information
Authors and Affiliations
Contributions
S.R.B. and M.D.M. conceived the study. S.R.B. and M.D.M. assembled genomes. M.D.M. produced phylogenomic topology. S.R.B. and M.D.M. performed comparative genomic analyses. S.R.B. produced large-scale phylogenetic and diet datasets. S.R.B. and M.D.M. performed comparative diet analyses. S.R.B. and B.C.O. performed diversification analyses. M.P.H. performed field collections. S.R.B. and M.D.M. wrote the manuscript with assistance from B.C.O. and M.P.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Daren Card and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Borstein, S.R., Hammer, M.P., O’Meara, B.C. et al. The macroevolutionary dynamics of pharyngognathy in fishes fail to support the key innovation hypothesis. Nat Commun 15, 10325 (2024). https://doi.org/10.1038/s41467-024-53141-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53141-4
