
Abstract
Alkaline water electrolysis is a promising low-cost strategy for clean and sustainable hydrogen production but is largely limited by the sluggish anodic oxygen evolution reaction and the challenges in maintaining adequate separation between H2 and O2. Here, we reveal an anodic-cathodic sequential oxygen evolution process via electrochemical oxidation and subsequent reduction of Ni hydroxides, enabling much lower overpotentials than conventional anodic oxygen evolution. By using (isotope-labeled) differential electrochemical mass spectrometry and in situ Raman spectroscopy combined with density functional theory calculations, we evidence that the sequential oxygen evolution originates from the electrochemical oxidation of Ni hydroxides to NiOO– active species while undergoing a different, reductive step of NiOO– for the final release of O2 due to weakened Ni–O covalency. Based on this sequential process, we propose and demonstrate a hybrid water electrolysis and energy storage device, which enables time-decoupled hydrogen and oxygen evolution and electrochemical energy storage in the Ni hydroxides.
Similar content being viewed by others

Introduction
Water electrolysis powered by renewable solar/wind energies has been a promising strategy for clean and sustainable hydrogen (H2) energy production and utilization1,2,3. In this process, water is oxidized to O2 at the anode (oxygen evolution reaction, OER), releasing protons and electrons for H2 production at the cathode (hydrogen evolution reaction, HER). However, the sluggish multi-step kinetics of the anodic OER results in a high overpotential, greatly limiting the efficiency of water electrolysis4,5,6. Among different types of water electrolysis technologies, alkaline water electrolysis (AWE) represents a relatively low-cost choice due to the use of non-noble metal OER electrocatalysts such as Ni-based (oxy)hydroxides (e.g., Ni-Fe)7,8, which nevertheless exhibit an overpotential generally more than 200 mV9,10,11. An in-depth understanding of the underlying principle governing the OER kinetics is crucial to improve the OER activities significantly.
Conventionally, OER electrocatalysis occurs through the metal cation oxidation driven by increasing overpotentials12,13, e.g., the oxidation of Ni hydroxides produces high valent Ni oxyhydroxides (NiOOH) on which the OER initiates14,15,16. The increased holes in the high valent metal cations are considered to enhance the metal–oxygen covalency to promote O–O coupling, which was generally considered to be the rate-determining step (RDS) of OER6,14,17. On the other hand, increasing the metal–oxygen covalency may also drive holes into the O-2p orbital (a so-called oxygen redox reaction), which creates an electrophilic nature that facilitates nucleophilic attack of water and hence activates the OER electrocatalysis. The increased holes in the O-2p orbital may also promote the direct evolution of the lattice oxygen (lattice oxygen mechanism, LOM)5,18,19,20. Understanding the complex roles of metal/oxygen redox chemistry in the OER electrocatalysis is a nontrivial task. For instance, the high valent Ni oxyhydroxides exhibited rather low OER activity. Instead, when doping a second transition metal such as Fe or Co, the Ni oxidation was largely inhibited, whereas the OER activity was remarkably enhanced7,21,22,23,24. This contradiction poses essential questions about whether the metal or the oxygen redox chemistry dominantly controls the O–O coupling and, if the O–O coupling step is the RDS for the OER on Ni-based oxyhydroxides.
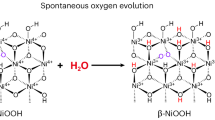
Besides the sluggish anodic OER electrocatalysis associated with the complex metal/oxygen redox chemistry, the AWE technology also suffers from considerable challenges in adequately separating H2 and O2 during electrolysis, since crossover of H2 into the anodic chamber via the alkaline electrolyte would be possible and hazardous. To ensure absolute separation of H2 and O2, efforts have been made to explore decoupled water splitting with temporal and/or spatial separation of HER and OER. For instance, a (thermo)chemically driven OER has also been reported25,26,27. In this context, H2 evolved at the cathode, and meanwhile, the Ni(OH)2 anode was electrochemically oxidized to NiOOH, which then spontaneously released O2 through chemical reduction by water (4NiOOH + 2H2O → 4Ni(OH)2 + O2). Using the two-step electrochemical-chemical cycle, decoupled water splitting was achieved with a high efficiency. Nevertheless, to reach a considerable reaction rate of the chemical step, the reaction temperature has to be elevated (~95 ˚C or even higher at 120 ˚C)25,26, which consumes additional thermal energy and makes the whole system relatively more complex.
In this study, we discover an anodic-cathodic sequential OER (ACS-OER) process through the electrochemical Ni(OH)2/NiOOH redox reaction, which exhibits fast kinetics and operates at much lower overpotentials compared to conventional anodic OER electrocatalysis. We further explore the molecular mechanism of the ACS-OER process by using in situ (isotope-labeled) differential electrochemical mass spectrometry (DEMS) and electrochemical Raman spectroscopy combined with density functional theory (DFT) calculations. Our results show that the ACS-OER process consists of the first formation of NiOO– active species through electrooxidation of Ni(OH)2 as in the conventional anodic OER14, while undergoing a different, reductive final step for the release of O2 from the NiOO– species caused by a weakened Ni-O covalency. The active sites and the influence of a second transition metal (Fe) on the ACS-OER process were further studied. Since the ACS-OER intrinsically decouples from HER, we further propose and demonstrate a hybrid energy device, which enables decoupled HER (during charging) and OER (during discharging) at a low cell voltage and room temperature and, as a natural result, the energy storage at the Ni (oxy)hydroxide electrodes during the cyclic electrolysis.
Results and discussion
Structures and electrochemical measurement of the Ni-based hydroxides
Two Ni hydroxides with different lateral sizes were synthesized to investigate the structural factors controlling the (sequential) OER mechanism. One was synthesized by impregnating nickel nitrate onto Vulcan XC carbon support and then decomposing at 200 ˚C in air, denoted as Ni(OH)2/C. The as-prepared catalyst exhibits an α-Ni(OH)2 phase (pdf#00-038-0715) based on X-ray diffraction (XRD) analysis (Fig. 1a for samples sprayed on carbon paper electrodes, and Supplementary Fig. 1a for powder samples) according to the characteristic (003) and (006) diffraction peaks16,28. A nanosheet morphology was revealed by high-angle annular dark-field scanning transmission electron microscopy (HADDF-STEM) (Supplementary Figs. 1b, c). This catalyst was then aged by a further solvothermal treatment in water at 120 ˚C for 6 h (denoted as Ni(OH)2/C–120), leading to significantly larger lateral sizes ranging from 50 to 500 nm (Supplementary Fig. 1d). Additionally, both XRD analysis (Fig. 1a and Supplementary Fig. 1a) and HAADF-STEM images (Supplementary Fig. 1e) indicate the transformation of the α-Ni(OH)2 phase to β-Ni(OH)2 (pdf#00-014-0117)29,30 after the solvothermal treatment, with the majority of the interlayer distance decreased from 0.67 to 0.48 nm.

a XRD patterns of the Ni(OH)2/C and Ni(OH)2/C–120 catalysts deposited on a carbon paper before and after OER tests. The carbon paper contains a microporous layer with polytetrafluoroethylene (PTFE) as the binder, resulting in strong diffraction peaks of C (002) at ca. 26˚ (marked by #) and PTFE (100) at 18˚ (marked by *). The bottom lines show the reference patterns of α-Ni(OH)2 (blue, pdf#00-038-0715), β-Ni(OH)2 (red, pdf#00-014-0117), graphite (gray) and PTFE (black). b Cyclic voltammograms of the OER recorded in 0.1 M KOH at a scan rate of 10 mV s-1. Inset shows structural models of α- and β-Ni(OH)2, where Ni and O atoms are represented by gray and red balls, respectively, and H atoms are not shown here. c–d HAADF-STEM images of the OER-tested Ni(OH)2/C (c) and the OER-tested Ni(OH)2/C–120 catalyst (d). Insets are the high-resolution images showing the laminar structure as well as the (001) interplane distance. Source data are provided as a Source Data file.
Cyclic voltammograms (CVs) were conducted in Fe-free KOH solution to avoid the impact of Fe substitution (Fig. 1b). The Ni(OH)2/C catalyst shows much higher Ni(OH)2/NiOOH redox reaction peaks and OER activity compared to the Ni(OH)2/C–120 catalyst. Despite having different initial structures, both Ni(OH)2/C and Ni(OH)2/C–120 catalyst electrodes show a similar β-Ni(OH)2-like phase after the OER test, as shown by their XRD patterns in Fig. 1a. This suggests an electrochemical aging of the α-Ni(OH)2 toward β-Ni(OH)2 for the Ni(OH)2/C catalyst7,31. Note that both the (001) peaks of the OER-tested Ni(OH)2/C and OER-tested Ni(OH)2/C–120 catalyst are shifted to a lower angle and broadened compared to the standard β-Ni(OH)2 phase. This can be attributed to intercalated hydrated anions such as hydrated K+ and H2O after OER cycling, which has also been observed in Ni(OH)2-based catalysts previously32,33. Regardless of the detailed intercalation structure, the main difference between the OER-tested Ni(OH)2/C and Ni(OH)2/C–120 catalysts is the smaller lateral size (~10 nm) of the former, as shown by the HAADF-STEM images in Figs. 1c, d. The larger Ni(OH)2/NiOOH redox peaks and the higher OER activities of the Ni(OH)2/C catalyst suggest that the active sites for both two reactions are located at the edges of the nanosheets due to the presence of catalytically more active µ1/µ2-(hydro)oxyl motifs7. In addition, the smaller lateral size of the Ni(OH)2 nanosheets may also facilitate higher electron conduction on the carbon support, which favors a faster Ni(OH)2/NiOOH redox kinetics16. Compared to the two Ni(OH)2 catalysts, incorporation of Fe by adding 1 mM Fe3+ into the electrolyte results in spontaneous Fe adsorption preferentially at the edges of the Ni(OH)2 nanosheets (denoted as Fe–Ni(OH)2/C) and leads to significantly enhanced OER activity (Fig. 1b)23. Nevertheless, the Ni(OH)2/NiOOH redox capability is significantly inhibited due to electronic interaction between Fe and Ni cations21,23,34,35. Therefore, it appears that one cannot establish a direct relation between the roles of high valent metal cations and the OER activity.
The ACS-OER and the Role of Metal Redox Reaction
To differentiate between the Ni(OH)2/NiOOH redox activities and the OER activities, we performed online DEMS measurement using a commercial dual thin-electrolyte layer cell (Hiden, Type B DEMS cell36) as schemed in Supplementary Fig. 2. This measurement has been shown an efficient way to deconvolute the faradic charges used to generate the molecular O2 from those stored on the catalyst surface such as the high valent metal cations16,30. Fig. 2a shows the CVs of the Ni(OH)2/C catalyst scanned to three different upper potential limits (UPL) and the online DEMS signal of O2 evolved. When the CV was scanned to a UPL of 1.6 V (panel I), a pronounced peak attributed to the oxidation of Ni(OH)2 to NiOOH occurs at about 1.42 V37,38,39, while the DEMS signal of O2 evolution does not appear until 1.56 V. Reversing the electrode potential to 1.5 V, the DEMS signal of evolved O2 gradually decreases and disappeared. Unexpectedly, when further lowering the electrode potential to 1.38 V, O2 evolution emerges in the DEMS signal again, accompanying the reduction of NiOOH between 1.28 V and 1.38 V. We refer to the O2 evolution occurring during the electrochemical reduction process as cathodic OER to distinguish it from the conventional anodic OER at high potentials.

a CVs (red lines) of the Ni(OH)2/C catalyst in 0.1 M KOH scanned to different UPLs (I, 1.60 V; II, 1.50 V; III, 1.45 V) at a scanning rate of 1 mV s−1 and the operando DEMS signals of O2 (m/z = 32, blue lines) using a commercial double thin layer flow cell. b Comparison of CVs and operando DEMS signals of O2 on the Ni(OH)2/C, the Ni(OH)2/C–120, and the Fe–Ni(OH)2/C catalyst formed by adsorbing Fe from 0.1 M KOH containing 0.1 mM Fe(NO3)3. The inset shows schematically the formation of Fe–Ni(OH)2/C catalyst. c Faradaic current density-time curve (j-t, red lines) of the Ni(OH)2/NiOOH redox reaction on the Ni(OH)2/C catalyst and the operando DEMS signal of O2 (blue lines). The left panel shows the signals for a whole Ni(OH)2/NiOOH redox cycle, while in the right figure, the potential control was withdrawn after reaching 1.45 V, with the open circuit potential recorded during the reduction process. The potential-time curves (E-t, black line) during the processes are shown to identify the specific emerging potential of the ACS-OER and the chemical OER process. Source data are provided as a Source Data file.
The amount of O2 evolved during the NiOOH reduction is found to decrease with the decreasing extent of Ni(OH)2 oxidation. When lowering the UPL to 1.50 V (panel II), a potential where the first oxidation wave of Ni(OH)2 to NiOOH completes but before the anodic OER starts, the cathodic OER proceeds at a similar amount detected by DEMS. Further lowering the UPL to 1.45 V leads to a partial oxidation of Ni(OH)2 (panel III), and accordingly, the amount of the cathodically evolved O2 decreases. These results suggest that the cathodic OER is highly related to the reduction of the Ni oxyhydroxides. Indeed, the Ni(OH)2/C–120 catalyst with a significantly lower Ni(OH)2/NiOOH redox activity shows nearly no DEMS signal of the cathodic OER (Fig. 2b). Furthermore, once Fe3+ ions are incorporated into the Ni(OH)2/C catalyst, the Ni(OH)2/NiOOH redox peaks gradually decreased in intensity during potential cycling, and accordingly the cathodic OER decreased as revealed by DEMS (Fig. 2b and Supplementary Fig. 3). Nevertheless, the oxygen-bridged Fe–O–Ni motifs constitute new active sites, which exhibit much higher anodic OER activities as expected32,40,41.
The cathodic OER is relevant but shows significantly enhanced kinetics compared to recently reported chemical O2 evolution on the NiOOH surface25,39. In the chemical O2 evolution process, the NiOOH is reduced to Ni(OH)2 by water via a spontaneous and exergonic chemical reaction and produces O2 (4NiOOH + 2H2O → 4Ni(OH)2 + O2), where the evolved O2 has been identified to originate from water by using O-isotope-labeled DEMS analysis26. As shown in Fig. 2c, when scanning the electrode potential from 1.0 to 1.45 V (for the oxidation of Ni(OH)2 to NiOOH) and then withdrawing the electrode potential control, the monitored open circuit potential (OCP) gradually decreased. Once the OCP decreased to 1.38 V (the onset potential of NiOOH reduction), the chemical O2 evolution is observed by DEMS but proceeds much slower compared to the cathodic OER process. Although the apparent effect of applying voltage is to increase the reaction rate, the essence behind the increased reaction rate could be a different reaction mechanism. For instance, a different self-circulating electrochemical–thermal OER mechanism was proposed to account for an increased reaction kinetics of thermo-chemical O2 evolution at elevated temperatures (e.g., 120 ˚C) compared to the room temperature chemical O2 evolution26.
In situ electrochemical Raman spectroscopy was further implemented to probe the Ni(OH)2/NiOOH redox chemistry and the OER mechanism using a thin-layered electrochemical cell in conjunction with a confocal Raman spectrometer, as illustrated in Supplementary Fig. 4a. Figure 3a shows the contour plot of the potential-dependent Raman spectra of the Ni(OH)2/C catalyst recorded at different constant potentials (the original spectra are presented in Supplementary Fig. 4b). No obvious signal can be found below 1.38 V due to the small Raman scattering cross-section of the pristine nano-sized Ni(OH)242. When the oxidation of Ni(OH)2 starts at 1.38 V, two Raman bands appear at 474 and 555 cm−1, designated as the eg bending (marked as δ (NiIII–O)) and A1g stretching (marked as ν (NiIII–O)) vibrational modes of Ni–O in the NiOOH active phase, respectively7,14,43. The intensity of these two Ni–O vibrational bands increases significantly at higher potentials. In addition, a broad Raman band is observed between 850–1200 cm−1 at potentials above 1.42 V, which has been designated to the superoxo (O–O) vibration of negatively charged NiOO– species formed by deprotonation of NiOOH and subsequent O–O coupling14. This charged NiOO– species was considered to be the crucial precursor for the last O2 release step during the conventional anodic OER, thus being called active oxygen14,44. Importantly, the NiOO– forms at a relatively low potential (1.42 V) on the Ni(OH)2/C surface, soon after the onset of Ni oxidation (1.38 V) but ahead of the anodic OER (1.56 V), suggesting that OER on this NiOOH surface is largely limited by the O2 release instead of the O–O coupling step14,16,45. Upon decreasing the electrode potential from 1.50 to 1.25 V, the Raman signal of NiOO– attenuates gradually and coincides well with the cathodic OER process detected by DEMS. In contrast, on the Ni(OH)2/C–120 catalyst, the Raman band of NiIII–O does not appear until around 1.45 V, and a very weak signal of the active NiOO– species can not be detected until 1.52 V (Fig. 3b and Supplementary Fig. 4c). Once Fe3+ was incorporated into Ni(OH)2, the intensity of NiOO– active species also gradually decreased when holding at the constant potential of 1.5 V (Supplementary Fig. 5). The significantly reduction of NiOO– active species is consistent with the fact that no cathodically evolved O2 was detected on the Ni(OH)2/C–120 and the Fe-Ni(OH)2/C catalysts by DEMS.

Potential-dependent in situ Raman spectra collected on (a) the Ni(OH)2/C and (b) the Ni(OH)2/C–120 catalyst in 0.1 M KOH solution during potential cycling, showing the Ni–O vibration signal (left panels) of the NiOOH active phase between 400–600 cm-1 and NiOO– active species (right panels) between 900–1050 cm-1. Source data are provided as a Source Data file.
The strong correlation between the formation of NiOO– and the amount of the cathodically evolved O2 indicates that the active species NiOO– through the oxidation of Ni(OH)2 could be also the key precursor for the revealed cathodically evolved O2. We therefore define this OER process as an anodic-cathodic sequential OER (ACS-OER) process. This also implies that both the anodic OER and the revealed ACS-OER exhibit the same RDS, namely the decomposition of NiOO– to release O246, but proceed in different subsequent ways to overcome the RDS. In contrast, the formation of OO– through O–O coupling could become the RDS of the anodic OER on the Fe-doped Ni(OH)2 surface, which explains the absence of NiOO– species yet much higher anodic OER activity (Supplementary Fig. 5, Fig. 1b).
O18 Isotope Labeling on the Contribution of LOM in the ACS-OER
The footprint of the active oxygen species was further investigated using 18O-isotope-labeled DEMS and in situ Raman spectroscopy, which allows to discriminate whether or not a LOM is present for both the conventional anodic and the ACS-OER. Figure 4a shows schematically the labeling procedures, explaining how the catalysts were labeled by 18O first and then tested in 16O electrolyte (details are described in the experimental section of SI). The O2 product of different isotopes was continuously analyzed by DEMS, where the signal at m/z = 34 is relevant to the participation of the 18O labeled lattice oxygen or bridge site O (μ2-O) in the Ni(OH)230,32. The pronounced signal of 18O16O in Fig. 4b indicates a substantial level of surface LOM for both the anodic and the ACS OER on the Ni(OH)2/C catalyst. Conversely, a significantly lower level of LOM is identified on both the Ni(OH)2/C–120 (Fig. 4c) and a homemade carbon-supported Ni–Fe hydroxide (Ni–Fe/C) catalyst (Fig. 4d), an analogy of the Fe–Ni(OH)2/C catalyst but more convenient for the isotope labeling experiment. The participation of lattice oxygen in the ACS-OER is always in the same ratio as in the conventional anodic OER. This suggests that the ACS-OER shares the same oxygen footprint with the anodic OER, specifically the common NiOO– precursor, but with a different O2 release step. Moreover, we found a positive correlation between the ACS-OER activity and the extent of LOM on Ni-based hydroxides, as compared in Fig. 4e, suggesting that the higher the extent of LOM, the higher the ACS-OER activity. The steady-state in situ Raman spectra at 1.5 V of 18O-labeled Ni hydroxides in 16O-rich 0.1 M KOH in Figs. 4f, g also indicates a faster exchange between lattice 18O and adsorbed 16O from the electrolyte on the Ni(OH)2/C catalyst than the Ni(OH)2/C-120 catalyst, evidencing the much larger extent of LOM and hence faster O–O coupling for the formation of NiOO– on the former32,47,48.

a Scheme of the 18O isotopic labeling procedure for the DEMS and in situ Raman spectroscopy measurements. b–d Time-dependent faradaic current density and the operando DEMS signals of O2 with different mass-to-charge at 18O-labeled (b) Ni(18OH)2/C, (c) Ni(18OH)2/C–120 catalyst and (d) 18O–Ni–Fe/C catalyst in 0.1 M K16OH solution during OER cycle at the scanning rate of 1 mV s–1. e A positive correlation between the lattice oxygen participation (defined as the ratio of 18O in all oxygen evolved) and the ACS-OER activity (defined as the ratio of O2 evolved via ACS-OER over those via the anodic OER) of different Ni-based hydroxides. The error bars represent the standard deviations of three independent measurements. f–g Quasi-steady-state in situ Raman spectra of 18O-labeled Ni(18OH)2/C (f) and Ni(18OH)2/C–120 (g) catalysts in 0.1 M 16O-KOH solution under 1.5 V for different periods. Source data are provided as a Source Data file.
DFT Calculations on the ACS-OER Reaction Pathways
To investigate the dynamics of NiOO– formation and O2 release in both the anodic and the ACS OER processes, we performed DFT calculations on the catalytically active edge plane of the Ni(OH)2 surface. On the edge plane, there are two types of undercoordinated O atoms: one is the top OH (μ1-OH), and the other is the bridge site OH (μ2-OH). Upon Ni oxidation at elevated electrode potentials, both μ1-OH and μ2-OH deprotonated, resulting in μ1-O and μ2-O, respectively. Ab initio molecular dynamics (AIMD) simulations performed using the explicit solvent model (see details in the Methods) indicate that μ1-O rapidly coupled with the nearby μ2-O to form the active oxygen (OO–) species, which would eventually release O2 from the surface (Fig. 5a). Crystal orbital Hamilton populations (COHP) were further analyzed to evaluate the Ni–O bonding strength in the optimized NiOO structures at different applied potentials combining the projected density of states (PDOS) of NiOO (Fig. 5b, Supplementary Figs. 6, 7). It can be found that there is a strong hybridization between the Ni-3d band and the O-2p band around the Fermi level. The bonding strength of Ni–O was quantified by the integrated COHP (ICOHP) at different applied potentials (Fig. 5c). A smaller ICOHP suggests a weakening of the Ni–O covalency and, consequently a weakened Ni–O bond49,50. Notably, the Ni–O bond reaches its maximum strength at approximately 1.35 V, the potential at which the NiOO– species formed. Either increasing or decreasing the potential can weaken the Ni–O bonding and thus facilitate the release of O2, which nicely accounts for the conventional anodic OER and the ACS-OER pathway, respectively. Bader charge analysis of the coupled OO at the edge sites of NiOOH further suggests that a partial oxidation of the coupled OO species occurs when decreasing the electrode potential, showing an electron transfer from the OO species to the nearby Ni (as schemed in the inset of Fig. 5c). This implies that both the reduction of the Ni cation and the oxidation of oxygen anions contribute to the ACS-OER pathway.

a AIMD simulations of the O–O coupling and O2 release on the edge site of NiOOH surface using the explicit solvent model. The gray, red, and pink color balls represent Ni, O, and H. The yellow color balls represent the O participated in O–O coupling and O2 release. b Crystal orbital Hamilton populations (COHP) and Projected density of states (PDOS) of the Ni-O bond of NiOO active species formed on the NiOOH edge surface at 1.34 VRHE. c Comparison of integrated -COHP up to Fermi level of Ni-OO bond under different potentials as well as bader charge analysis of the coupled O–O species. d Schematic diagram of the ACS-OER pathway (red cycle) in comparison with conventional anodic OER pathway (blue cycle) at Ni(OH)2 edge sites. Source data are provided as a Source Data file.
In Fig. 5d, we consolidate the conventional anodic and the ACS OER pathways at the Ni(OH)2 edges. Starting from the oxidation of Ni(OH)2 to NiOOH (step (i)), the further deprotonation of NiOOH (step (ii)) leads to the fast O–O coupling (step (iii)) between the μ1-O and the μ2-O and the formation of NiOO–. Due to a specific pKa of NiOOH7,14, not all the NiOOH edges can deprotonate and induce a fast O–O coupling. For those un-deprotonated NiOOH at the edges, O–O coupling can be only achieved between μ2-O and adsorbed OH– driven by further elevating the electrode potential, similar to the Mars-van-Krevelen mechanism as reported recently30. The further release of O2 from the NiOO– (step (iv, v)) becomes the RDS of the anodic OER process. Our work demonstrates a sequential O2 release pathway which proceeds through the same former three steps as the conventional anodic OER to form the active oxygen (OO–) but a subsequent reduction of NiOO– (step (vii)). Such a reduction weakens the Ni–O bond and promotes the O2 release. This ACS-OER operates at much lower overpotentials than the conventional anodic OER process.
A hybrid water splitting and energy storage device
Based on the ACS-OER process, we propose a hybrid energy device that allows time-decoupled HER and OER during the charging and discharging, respectively, as shown schematically in Fig. 6a. During the charging of the device, Ni(OH)2 is oxidized to NiOOH at the positive (anodic) electrode, as depicted by step (i) in Fig. 5d. It should be noted that only a portion of the NiOOH can be further deprotonated and undergo O–O coupling to form the NiOO– species (step (ii-iii) in Fig. 5d). This is first due to the fact that NiOO− active species, formed by the O–O coupling between the μ1-O and the μ2–O, can only be generated at the edge sites, while the rest (e.g., the in-plane sites) remains NiOOH. This is also supported by the much higher intensity of the Raman band assigned to NiOO− at the Ni(OH)2/C nanosheet catalyst with smaller lateral sizes compared to the Ni(OH)2/C–120 catalyst (Fig. 3). Secondly, even at the edge sites, only a portion of NiOOH can be deprotonated to NiOO– due to a specific pKa value14. Therefore, taking as an example, one out of six NiOOH units deprotonates and undergoes O–O coupling to form NiOO– during charging, the equation for the positive electrode reaction during charging is shown in Fig. 6b (corresponding to step (i-iii) in Fig. 5d).

a Scheme of the hybrid device showing time-decoupled HER and OER during charging and discharging of the Ni hydroxide, respectively. b Positive electrode reactions during charging and discharging, where the red dashed rectangles denote the portion of Ni(OH)2 undergoing the ACS-OER process. c Charge-discharge voltage profiles at 5 mA cm−2 using the Ni(OH)2/C catalyst on a carbon paper (5 mg cm−2) as the positive electrode and a homemade Pt/C catalyst on a carbon paper (1 mg cm-2) used as the negative electrode in 1 M KOH solution. The inset shows the production of H2 bubbles at the negative electrode. d The specific capacity and coulombic efficiency of the hybrid device over 100 cycles. Source data are provided as a Source Data file.
Meanwhile, hydrogen is produced at the negative electrode (Eq. (1)):
During the discharging, the NiOOH/NiOO– species at the positive (cathodic) electrode is reduced back to Ni(OH)2, with the simultaneous release of O2 (step (vii) in Fig. 5d), as shown in Fig. 6b (Discharging).
Here, the Ni oxyhydroxide positive electrode receives less electrons from the external circuit during discharging than those released during the charging step due to an internal electron transfer between the high valent Ni and the negatively charged OO– species. Meanwhile, a portion of the produced H2 or other fuels (e.g., waste pollutive organics) is oxidized at the negative (anodic) electrode, outputting the electric energy (Eq. 2).
Combining Fig. 6b and Eqs. 1–2, the whole reaction in a charging-discharging cycle can be written as an overall water-splitting reaction, as shown in Eq. 3:
Through the charging-discharging cycle, time-decoupled H2/O2 evolution can be achieved, and the whole water electrolysis process can be completed with much lower overpotentials (< 200 mV). Noteworthy, one can see that the coulomb efficiency of the charging-discharging cycle must be less than 100% since part of the charge input is used for water splitting. Moreover, the coulomb efficiency largely depends on the amount of NiOO– formed; a larger amount of NiOO– would lead to a lower coulomb efficiency.
To demonstrate the capability of the hybrid device with decoupled water splitting, galvanostatic charging-discharging of the Ni(OH)2 /C catalyst at 5 mA·cm−2 was performed in a two-compartment H-type cell, as presented in Supplementary Fig. 8a. To avoid the anodic OER process, the cutoff voltage was set at 1.5 V. As shown in the charging-discharging profile in Fig. 6c, a voltage plateaus associated with Ni(OH)2 oxidation can be observed in the charging process; meanwhile, obvious H2 bubbles are found on the negative electrode. During the discharging, there is also a voltage plateau corresponding to the NiOOH reduction, with O2 produced at the Ni (oxy)hydroxide positive electrode. The specific capacity and the coulomb efficiency for energy storage at the Ni (oxy)hydroxide positive electrode during the first cycle are calculated to be around 299 mA·h·g−1 and 85%, respectively, implying that 15 % of the total charge input is used for water splitting. This evidences the hybrid nature of this device with both water splitting and energy storage in a charging-discharging cycle. Upon 100 charging-discharging cycles (Figs. 6c,d), the specific capacity gradually drops to 186 mA·h·g−1, while the coulomb efficiency increases from 85% to 96%, suggesting decreased performance of the decoupled water splitting. This can be intrinsically tied to a decreased ACS-OER activity of the Ni(OH)2/C catalyst. Online DEMS measurement (Supplementary Fig. 9) shows that repeated ACS-OER signal can be obtained on the Ni(OH)2/C catalyst over extended cycles but gradually decreases. HAADF-STEM images (Supplementary Fig. 8b) reveal an apparent growth (fewer edge sites) and agglomeration of the Ni(OH)2 nanosheets after 100 charging-discharging cycles, which accounts for both the capacity degradation and the lowered ACS-OER activity. Therefore, the stability of the hybrid device is mainly restricted by the structural stability of the Ni(OH)2/C catalyst. Further improvement on the stability of the nano-sized Ni(OH)2/C catalysts, e.g., by enhancing the catalyst-support interaction or by controlling the charging/discharging depth51, may enable a sustainable decoupled water splitting and improved cycling performance.
Although the charge-discharge cycling life is preliminarily insufficient at present and requires further optimization, our findings demonstrate the possibility of the hybrid device that enables water splitting with time-decoupled H2 and O2 production at a low cell voltage, and, a natural result of this, the energy storage in the Ni hydroxides. This device could find possible applications in integrating short-term power grid stabilization and peak load shifting (as a battery) and long-term energy storage in hydrogen (through the decoupled water splitting) during extended charge/discharge cycles.
In conclusion, we reveal a sequential ACS-OER process on Ni (oxy)hydroxides, which proceeds through a first electrochemical oxidation of Ni(OH)2 to NiOO– active species but a second reduction of NiOO– to Ni(OH)2 for O2 release, in contrast to the conventional four-electron anodic OER process. By using in situ (isotope-labeled) DEMS and electrochemical Raman spectroscopy combined with ab initial MD simulation and DFT calculations, we evidence a fast, LOM-dominated O–O coupling during the formation of NiOO– active species, from which the O2 release step becomes the RDS of OER on the Ni (oxy)hydroxide catalyst. Unlike the conventional anodic OER process that activates the O2 release by further increasing the overpotential, the electro-reduction of Ni(III) to Ni(II) also facilitates the weakening of the Ni–O bond and therefore promotes the O2 release. Incorporation of Fe decreases the LOM level and thus the O–O coupling became the RDS, with no ACS-OER observed. Based on the ACS-OER process, we further demonstrate a hybrid device for alkaline water electrolysis coupled to the energy storage process, which enables a time-decoupled HER and OER along with electric energy storage. This work not only offers insights into the alkaline OER mechanism (i.e., the roles of metal/oxygen redox and the RDS) on the Ni-based hydroxides but also provides a hybrid energy device for energetically efficient decoupled water splitting and energy storage.
Methods
Reagents and materials
Nickel nitrate hexahydrate (Ni(NO3)2·6H2O, 99.99%) was purchased from Thermo Scientific. Ltd. Potassium hydroxide (KOH, 99.99% metals basis) and isopropyl alcohol (C3H8O, AR, ≥ 99.5%) were purchased from Shanghai Macklin Biochemical Co., Ltd. Nafion® solution (5 wt.%) was purchased from DuPont Company. Analytically ethanol was obtained from Sinopharm Chemical Reagent Co., Ltd. Conductive carbon black (Vulcan XC-72) was purchased from Shanghai Cabot Chemical Co., Ltd. The water used throughout all experiments was ultrapure water (Millipore, 18.0 MΩ).
Catalyst synthesis
The Ni(OH)2/C catalyst was synthesized by impregnation of Vulcan XC-72 carbon black support (160 mg) with an ethanol solution (600 µL ultrapure water and 200 µL ethanol) and 0.68 mL of 1.0 M Ni(NO3)3 at a nominal metal loading of 20 wt.%. The mixture was sonicated for 2 min and then magnetically stirred for 24 h. Finally, after 12 h of freeze drying, the obtained powder material was placed in a muffle furnace and heated in air at 200 ˚C for 2 h. The Ni(OH)2/C–120 catalyst was prepared by hydrothermal (hydrolysis) treatment of the Ni(OH)2/C catalyst in ultrapure water at 120 ˚C for 6 h.
The Ni–Fe layered double hydroxide (Ni–Fe/C) catalyst was prepared by a similar method as the Ni(OH)2/C catalyst, except that a mixture of 1.0 M Ni(NO3)3 (2.7 mL) and 1.0 M Fe(NO3)3 (0.9 mL) with a Ni/Fe molar ratio of 3: 1 was added together with 836 mg Vulcan XC-72 carbon black support in the impregnation solution (1 mL absolute ethanol and 400 µL ultrapure water). The Fe-Ni(OH)2/C catalyst was prepared by in situ adsorption of Fe ions on the Ni(OH)2/C catalyst from 0.1 M KOH electrolyte containing 0.1 mM Fe(NO3)3 during repeated potential cycling between 0.05 and 1.5 V.
Materials characterizations
X-ray diffraction (XRD) was performed on an X-ray powder diffractometer (RIGAKU D/max 2500/PC) with a Cu Kα radiation source (λ = 0.154 nm) at the scanning speed of 5˚/min between 10–90˚. To compare XRD patterns before and after the OER test, the catalysts were evenly sprayed (at 4.0 mg/cm2) on a 1 cm × 1 cm carbon paper electrode with a microporous layer consisting of carbon black and PTFE as the binder. The catalyst electrodes were then subjected to 10 cycles between 1.1–1.6 V, after which the potential control was withdrawn at 1.6 V. The OER-tested electrodes were rinsed by ultrapure water, dried and then used for XRD measurement. Transmission electron microscopy was performed on a spherical-aberration-corrected scanning transmission electron microscope (Thermo Fisher Scientific, Spectra 300) operated at 300 kV using high-angle annular dark field imaging at a camera length of 91 mm. To minimize the sample damage, a relatively low electron dose (5250 e-/Å2) was controlled.
Electrochemical measurements
To prepare the catalyst working electrode (WE), 1.6 mg catalyst was ultrasonically dispersed in 1.0 mL mixed solution of isopropanol (IPA, 0.2 mL), ultrapure water (0.8 mL), and 5 wt. % Nafion (4 μL) for 30 min to form a homogeneous ink. Then, 10 μL of the catalyst ink was pipetted onto a pre-polished and cleaned glassy carbon electrode (5 mm in diameter, 0.196 cm2) and then dried at 50 °C for 15 min, leading to a catalyst loading of 0.08 mg cm−2. Electrochemical measurement was performed in an H-type three-compartment cell in conjunction with a Bio-Logic VSP-300 electrochemical workstation, where the WE compartment and the counter electrode (CE) compartment are physically separated by porous ceramics to avoid potential effects of any products formed at the CE52. A Hg/HgO (1 M KOH) electrode and a graphite rod were used as the reference (RE) and the CE, respectively. The RE was calibrated by using a homemade reversible hydrogen electrode (RHE) consisting of a polished Pt disk electrode in H2-saturated 0.1 M KOH solution. The Hg/HgO (1 M KOH) electrode is calibrated to be -0.895 V vs. RHE in 0.1 M KOH solution. All potentials reported in this study were referred to RHE and corrected by 85% iR compensation.
Purification of the KOH electrolyte
To minimize the influence of residual Fe impurities that normally exist in commercial KOH reagents, 500 mL 1 M KOH electrolyte was purified by prolonged electrolysis (at a constant current of 1 mA for 48 h) using a nickel wire (0.5 mm, 99.999% metals basis, Alfa Aesar) as both the WE and CE based on the previous report53. The absence of Fe impurities was evidenced by the continuous decaying in the OER performance of Ni(OH)2/C catalyst upon cycling in the purified electrolyte22.
Differential electrochemical mass spectrometry (DEMS)
DEMS measurements were conducted with a commercial double-thin-layer electrochemical flow cell made by Teka-Peek (as schemed in Supplementary Fig. 2)36, the volume of which is about 30 μL. Nafion membrane (Nafion® 211) was used to isolate the working and counter electrode chamber to avoid the effects of products formed at the Pt CE. The electrolytes were refreshed by a peristaltic pump (the electrolyte flow rate was usually 2 mL/min in this experiment). Here, leak-free Ag/AgCl (Innovative Instruments, Inc., LF-1, 1 mm outer diameter) was used as the RE and Pt wire as the CE. The RE potential was calibrated to be -0.929 V vs. RHE in 0.1 M KOH. Before each measurement, the background signals for m/z = 32, 34, and 36 were scanned for at least 2 h to reach a steady baseline mass signal of volatile species54. Here, the CV measurements were performed at a scanning rate of 1 mV s-1, which is also simultaneously recorded by the DEMS with an extra circuit connection to the electrochemical workstation.
In situ electrochemical Raman spectroscopy
In situ Raman measurements were conducted using a three-electrode electrochemical Raman cell with a Hg/HgO electrode as the RE and a Pt wire as the CE in 0.1 M KOH electrolyte (as schemed in Supplementary Fig. 4a)55, which was then placed under a confocal Raman spectrometer (Horiba LabRAM HR800). Due to the limitation of the electrochemical Raman cell, a Pt wire, instead of graphite electrodes as suggested for noble-metal-free electrocatalyst56 was used as the CE. Nevertheless, the graphitic rod was also used as CE for comparative EC-Raman and electrochemical performance characterization under the same condition. In the potential region of this work, the same Ni(OH)2/C catalyst showed almost the same Raman characteristics and electrochemical performance regardless of the CE employed. The working electrode was prepared by drop-casting a thin catalyst film onto a glassy carbon electrode (3 mm in diameter), which was then placed against a thin sapphire optical window to ensure a thin electrolyte formed between the optical window and the working electrode. A 633 nm excitation laser at a power of ca. 1.0 mW was used as the light source and focused on the sample surface through a dry objective lens with a numerical aperture of 0.5 (Olympus, 50X, working distance = 10.6 mm). Raman frequencies were calibrated using a Si wafer and ClO4− solution spectra before each measurement.
O18 isotopic labeling procedures
For 18O isotopic labeling DEMS measurement, the Ni-based hydroxide catalysts were 18O-labeled by holding at 1.65 V for 10 min in 2.0 mL 18O-enriched 0.1 M KOH using 98% H2O18 as the solvent (isotopechina, Wuhan) and a small-volume electrochemical cell, following a previous protocol19. The 18O-labeled catalysts were then rinsed with H216O to remove residual H218O. Afterward, OER measurements were carried out using the 18O-labeled catalysts in 0.1 M KOH electrolyte using H216O as the solvent, and the gaseous products of 16O2 (m/z = 32), 16O18O (m/z = 34), and 18O2 (m/z = 36) were constantly monitored by on-line DEMS. For 18O-labeling in-situ Raman spectroscopy, the 18O isotopic labeling was performed by applying a constant potential of 1.5 V on the catalysts in 0.1 M KOH with 98% H218O as the solvent. Then, the Raman spectra were recorded at a constant potential of 1.5 V in 0.1 M KOH with the conventional H216O solvent.
Construction and measurement of the hybrid water splitting and energy storage device
A homemade carbon-supported Pt catalyst (Pt/C, 2-nm Pt nanoparticles supported on Vulcan XC carbon black, with a Pt content of 20 wt%) and the Ni(OH)2/C catalyst were evenly coated onto a 1 cm × 1 cm carbon paper by the ink drop-casting at a catalyst loading of 1 and 5 mg cm-2, respectively. The Pt/C- and the Ni(OH)2/C-coated carbon papers were then used as the negative and the positive electrode, respectively, immersed in 0.1 M purified KOH in a two-compartment H-type cell (as schemed in Supplementary Fig. 8). Nafion membrane (Nafion®115) was used to isolate positive and negative electrode chamber to avoid the effects of products formed at the Pt negative electrode. Galvanostatic charging-discharging tests were conducted at a current density of 5 mA·cm-2 and a charging- and discharging-cutoff voltage at 1.5 V and 1.2 V, respectively, to avoid the anodic OER process. The specific capacity was calculated based on the metal loading of 20 wt.%.
Density functional theory (DFT) calculations
Spin-polarized density functional theory calculations were performed by using the Vienna ab initio simulation package (VASP 5.4.4) in conjunction with VASPsol57,58,59,60. The projector augmented wave61 (PAW) potential and the generalized gradient approximation (GGA) with the Perdew-Burke-Ernzerhof62 (PBE) functional were used to describe the electron-ion interactions and the exchange-correlation energy, respectively. The van der Waals interactions were described using the DFT-D3 correction63. A Hubbard correction of U = 5.5 eV was applied to Ni atoms to describe the proper magnetic moment correctly. For ab initio molecular dynamics (AIMD) simulations, the Nose-Hoover thermostat was used for canonical sampling at 300 K, and the time step was set to 0.5 fs. The energy cutoff for the plane wave basis expansion was set to 500 eV. For the Brillouin zone sampling, only the gamma point was used for AIMD simulations, while a denser 3 × 3 × 1 grid and 5 × 5 × 1 grid were used for structure relaxation and electronic structure calculations, respectively. The COHP of the Ni–O bond was calculated using the Lobster code (freely available from www.cohp.de)64,65,66,67.
The Ni(OH)2 slab is modeled using a (4 × 2) supercell of (100) edge containing two layers. A vacuum space of ~15 Å along the z-direction is used to separate the interaction, and two layers of water molecules are placed on the edge of Ni(OH)2.
For a given structure with a specific number of electrons, we can get the electron potential Φ with respect to the standard hydrogen electrode (ΦSHE) can be calculated as Eq. 468.
where EF and Ees are the Fermi level of the system and the electrostatic energy in the implicit electrolyte region, and EFSHE – EesSHE is benchmarked to 4.6 eV for VASPsol69. The applied potential with respect to the RHE (ΦRHE) can be obtained as Eq. 5.
where the pH value is set to 13.
Data availability
The data generated and analyzed in this study have been included in the published article and its Supplementary Information files. All other relevant data supporting the findings are available from the authors upon request. Source Data are provided with this paper in the Source Data file. Source data are provided with this paper.
Change history
09 April 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-58750-1
References
Chatenet, M. et al. Water electrolysis: from textbook knowledge to the latest scientific strategies and industrial developments. Chem. Soc. Rev. 51, 4583–4762 (2022).
Yu, M., Budiyanto, E. & Tüysüz, H. Principles of water electrolysis and recent progress in Cobalt‐, Nickel‐, and Iron‐based oxides for the oxygen evolution reaction. Angew. Chem. Int. Ed. 1, e202103824 (2022).
Guo, J. et al. Direct seawater electrolysis by adjusting the local reaction environment of a catalyst. Nat. Energy 3, 264–272 (2023).
Zhao, Y. et al. Oxygen evolution/reduction reaction catalysts: from in situ monitoring and reaction mechanisms to rational design. Chem. Rev. 123, 6257–6358 (2023).
Son, Y. J.; et al. Key concepts for understanding alkaline oxygen evolution reaction at the atomic/molecular scale. Curr. Opin. Electrochem. 39, 101298, (2023).
Song, J. et al. A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 7, 2196–2214 (2020).
Dionigi, F. & Strasser, P. NiFe-based (oxy)hydroxide catalysts for oxygen evolution reaction in non-acidic electrolytes. Adv. Energy Mater. 23, 1600621 (2016).
Song, F. et al. Transition metal oxides as electrocatalysts for the oxygen evolution reaction in alkaline solutions: an application-inspired renaissance. J. Am. Chem. Soc. 25, 7748–7759 (2018).
He, Z. et al. Activating lattice oxygen in NiFe-based (oxy) hydroxide for water electrolysis. Nat. Commun. 1, 1–12 (2022).
Zhang, N. et al. Lattice oxygen activation enabled by high-valence metal sites for enhanced water oxidation. Nat. Commun. 1, 4066 (2020).
Song, F. & Hu, X. Exfoliation of layered double hydroxides for enhanced oxygen evolution catalysis. Nat. Commun. 1, 4477 (2014).
Rossmeisl, J., Qu, Z.-W., Zhu, H., Kroes, G.-J. & Nørskov, J. K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 1-2, 83–89 (2007).
Duan, Y. et al. Anodic oxidation enabled cation leaching for promoting surface reconstruction in water oxidation. Angew. Chem. Int. Ed. 13, 7418–7425 (2021).
Diaz-Morales, O., Ferrus-Suspedra, D. & Koper, M. T. M. The importance of nickel oxyhydroxide deprotonation on its activity towards electrochemical water oxidation. Chem. Sci. 4, 2639–2645 (2016).
Son, Y. J. et al. Effects of electrochemical conditioning on nickel-based oxygen evolution electrocatalysts. ACS Catal. 16, 10384–10399 (2022).
Gorlin, M. et al. Tracking catalyst redox states and reaction dynamics in Ni-Fe oxyhydroxide oxygen evolution reaction electrocatalysts: the role of catalyst support and electrolyte pH. J. Am. Chem. Soc. 5, 2070–2082 (2017).
Grimaud, A. et al. Activation of surface oxygen sites on an iridium-based model catalyst for the oxygen evolution reaction. Nat. Energy 1, 1–10 (2016).
Grimaud, A., Hong, W. T., Shao-Horn, Y. & Tarascon, J.-M. Anionic redox processes for electrochemical devices. Nat. mater. 2, 121–126 (2016).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 5, 457–465 (2017).
Huang, Z.-F. et al. Tuning of lattice oxygen reactivity and scaling relation to construct better oxygen evolution electrocatalyst. Nat. Commun. 1, 3992 (2021).
Bao, F. et al. Host, suppressor, and promoter—the roles of Ni and Fe on oxygen evolution reaction activity and stability of NiFe alloy thin films in alkaline media. ACS Catal. 16, 10537–10552 (2021).
Kuai, C. et al. Revealing the dynamics and roles of iron incorporation in nickel hydroxide water oxidation catalysts. J. Am. Chem. Soc. 44, 18519–18526 (2021).
Qian, H. et al. In situ quantification of the active sites, turnover frequency, and stability of Ni–Fe (oxy) hydroxides for the oxygen evolution reaction. ACS Catal. 22, 14280–14289 (2022).
Salmanion, M. & Najafpour, M. M. Oxygen-evolution reaction performance of nickel (hydr)oxide in alkaline media: iron and nickel impurities. J. Phys. Chem. C. 37, 18340–18349 (2023).
Dotan, H. et al. Decoupled hydrogen and oxygen evolution by a two-step electrochemical–chemical cycle for efficient overall water splitting. Nat. Energy 9, 786–795 (2019).
Deng, B. et al. A self-circulating pathway for oxygen evolution reaction. Energy Environ. Sci. 16, 5210–5219 (2023).
Ifkovits, Z. P., Evans, J. M., Meier, M. C., Papadantonakis, K. M. & Lewis, N. S. Decoupled electrochemical water-splitting systems: a review and perspective. Energy Environ. Sci. 9, 4740–4759 (2021).
Gorlin, M. et al. Oxygen evolution reaction dynamics, faradaic charge efficiency, and the active metal redox states of Ni-Fe oxide water splitting electrocatalysts. J. Am. Chem. Soc. 17, 5603–5614 (2016).
Gao, M. et al. Efficient water oxidation using nanostructured α-nickel-hydroxide as an electrocatalyst. J. Am. Chem. Soc. 19, 7077–7084 (2014).
Ferreira de Araujo, J., Dionigi, F., Merzdorf, T., Oh, H. S. & Strasser, P. Evidence of mars-van-krevelen mechanism in the electrochemical oxygen evolution on Ni-based catalysts. Angew. Chem. Int. Ed. 27, 14981–14988 (2021).
Li, S. et al. Operando tailoring of defects and strains in corrugated β‐Ni(OH)2 nanosheets for stable and high‐rate energy storage. Adv. Mater. 2, 2006147 (2021).
Dionigi, F. et al. In-situ structure and catalytic mechanism of NiFe and CoFe layered double hydroxides during oxygen evolution. Nat. Commun. 1, 1–10 (2020).
Pimsawat, A., Tangtrakarn, A., Pimsawat, N. & Daengsakul, S. Effect of substrate surface roughening on the capacitance and cycling stability of Ni(OH)2 nanoarray films. Sci. Rep. 1, 16877 (2019).
Zhou, Y. & López, N. The role of Fe species on NiOOH in oxygen evolution reactions. ACS Catal. 11, 6254–6261 (2020).
Trotochaud, L., Young, S. L., Ranney, J. K. & Boettcher, S. W. Nickel–iron oxyhydroxide oxygen-evolution electrocatalysts: the role of intentional and incidental iron incorporation. J. Am. Chem. Soc. 18, 6744–6753 (2014).
Clark, E. L., Singh, M. R., Kwon, Y. & Bell, A. T. Differential electrochemical mass spectrometer cell design for online quantification of products produced during electrochemical reduction of CO(2). Anal. Chem. 15, 8013–8020 (2015).
Smith, R. D. et al. Geometric distortions in nickel (oxy) hydroxide electrocatalysts by redox inactive iron ions. Energy Environ. Sci. 9, 2476–2485 (2018).
Garcia, A. C., Touzalin, T., Nieuwland, C., Perini, N. & Koper, M. T. Enhancement of oxygen evolution activity of nickel oxyhydroxide by electrolyte alkali cations. Angew. Chem. Int. Ed. 37, 12999–13003 (2019).
Smith, R. D. & Berlinguette, C. P. Accounting for the dynamic oxidative behavior of nickel anodes. J. Am. Chem. Soc. 5, 1561–1567 (2016).
Li, C. F. et al. Interfacial Fe-O-Ni-O-Fe bonding regulates the active Ni sites of Ni-MOFs via iron doping and decorating with FeOOH for super-efficient oxygen evolution. Angew. Chem. Int. Ed. 17, e202116934 (2022).
Bai, L., Lee, S. & Hu, X. Spectroscopic and electrokinetic evidence for a bifunctional mechanism of the oxygen evolution reaction. Angew. Chem. Int. Ed. 6, 3095–3103 (2021).
Yeo, B. S. & Bell, A. T. In situ Raman study of nickel oxide and gold-supported nickel oxide catalysts for the electrochemical evolution of oxygen. J. Phys. Chem. C. 15, 8394–8400 (2012).
Hu, C. et al. Surface‐enhanced Raman spectroscopic evidence of key intermediate species and role of NiFe dual‐catalytic center in water oxidation. Angew. Chem. Int. Ed. 36, 19927–19931 (2021).
Zhang, M., de Respinis, M. & Frei, H. Time-resolved observations of water oxidation intermediates on a cobalt oxide nanoparticle catalyst. Nat. Chem. 4, 362–367 (2014).
Yang, C., Fontaine, O., Tarascon, J. M. & Grimaud, A. Chemical recognition of active oxygen species on the surface of oxygen evolution reaction electrocatalysts. Angew. Chem. Int. Ed. 30, 8778–8782 (2017).
Du, K. et al. Interface engineering breaks both stability and activity limits of RuO2 for sustainable water oxidation. Nat. Commun. 1, 5448 (2022).
Wang, Z., Goddard, W. A. 3rd & Xiao, H. Potential-dependent transition of reaction mechanisms for oxygen evolution on layered double hydroxides. Nat. Commun. 1, 4228 (2023).
Zhang, N. & Xiong, Y. Lattice oxygen activation for enhanced electrochemical oxygen evolution. J. Phys. Chem. C. 5, 2147–2159 (2023).
Chen, Z. et al. Fe1N4–O1 site with axial Fe–O coordination for highly selective CO2 reduction over a wide potential range. Energy Environ. Sci. 6, 3430–3437 (2021).
Li, Y. et al. Synergistic effect of atomically dispersed Ni–Zn pair sites for enhanced CO2 electroreduction. Adv. Mater. 41, 2102212 (2021).
Fujita, S.; Baranton, S.; Coutanceau, C.; Jerkiewicz, G. Design, synthesis, and characterization of carbon‐supported β‐Ni(OH)2 nanosheets for miniaturized nickel–metal hydride batteries. Energy Technol. 12, 2301268, (2024).
Xia, D. et al. Degradation and regeneration of Fe–Nx active sites for the oxygen reduction reaction: the role of surface oxidation, Fe demetallation and local carbon microporosity. Chem. Sci. 34, 11576–11584 (2021).
Chung, D. Y. et al. Dynamic stability of active sites in hydr(oxy)oxides for the oxygen evolution reaction. Nat. Energy 3, 222–230 (2020).
He, F. et al. The effect of water on the quantification of volatile species by differential electrochemical mass spectrometry. Anal. Chem. 13, 5547–5555 (2021).
Wei, J. et al. Probing the oxygen reduction reaction intermediates and dynamic active site structures of molecular and pyrolyzed Fe–N–C electrocatalysts by in situ Raman spectroscopy. ACS Catal. 13, 7811–7820 (2022).
Chen, J. G., Jones, C. W., Linic, S. & Stamenkovic, V. R. Best practices in pursuit of topics in heterogeneous electrocatalysis. ACS Catal. 9, 6392–6393 (2017).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 3B, B864 (1964).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 4A, A1133 (1965).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B. 1, 558 (1993).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1, 15–50 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B. 24, 17953 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 18, 3865 (1996).
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 15, 154104 (2010).
Dronskowski, R. & Blöchl, P. E. Crystal orbital Hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 33, 8617–8624 (1993).
Maintz, S., Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. Analytic projection from plane‐wave and PAW wavefunctions and application to chemical‐bonding analysis in solids. J. comput. chem. 29, 2557–2567 (2013).
Maintz, S., Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane‐wave based DFT. J. Comput. Chem. 11, 1030 (2016).
Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A. 21, 5461–5466 (2011).
Zhao, X. & Liu, Y. Origin of selective production of hydrogen peroxide by electrochemical oxygen reduction. J. Am. Chem. Soc. 25, 9423–9428 (2021).
Mathew, K., Kolluru, V., Mula, S., Steinmann, S. N. & Hennig, R. G. Implicit self-consistent electrolyte model in plane-wave density-functional theory. J. Chem. Phys. 151, 234101 (2019).
Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFA1203400, 2021YFA1202802), National Natural Science Funds of China (22102083, 52173222, 12274254), Key Area Research and Development Program of Guangdong Province (No. 2020B0909040003), Shenzhen Science and Technology Innovation Committee (ZDSYS20230626091100001, JCYJ20230807111605012) and Overseas Research Cooperation Funding of Tsinghua SIGS (HW2021006). Y.S. acknowledges support from the National Science Foundation of China (12204263) and Guangdong Basic and Applied Basic Research Foundation (2022A1515010628). This work made use of the TEM facilities at the Institute of Materials Research, Tsinghua Shenzhen International Graduate School (Tsinghua SIGS).
Author information
Authors and Affiliations
Contributions
J.W. and L.G. conceived the study and wrote the paper. J.W. performed the electrochemical, Raman, and DEMS measurements, including the data analysis. Y.S. and F.Y. conducted the DFT calculations supervised by J.L. J.X. and Z.L designed and performed the measurements of the hybrid device for water splitting and energy storage. H.Q. and Y. H. contributed to the synthesis of catalysts. L.C. and Y.W. contributed to the TEM characterization of the catalysts. All authors contributed to the discussion and interpretation of the data and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The Authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wei, J., Shao, Y., Xu, J. et al. Sequential oxygen evolution and decoupled water splitting via electrochemical redox reaction of nickel hydroxides. Nat Commun 15, 9012 (2024). https://doi.org/10.1038/s41467-024-53310-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53310-5
This article is cited by
-
Engineering oxygen nonbonding states in high entropy hydroxides for scalable water oxidation
Nature Communications (2025)
-
Boosting and stabilizing oxygen evolution reaction through Ru single atoms anchored amorphous NiMoOx electrocatalyst
Nature Communications (2025)
-
Reserved charges in a long-lived NiOOH phase drive catalytic water oxidation
Nature Chemistry (2025)
-
Cathodic electrodeposition activation of NiFe-based metal–organic frameworks for enhanced oxygen evolution reaction
Rare Metals (2025)
-
Industrial symbiosis concept applied to green hydrogen production: a critical review based on bibliometric analysis
Discover Sustainability (2024)
