
Abstract
Extrachromosomal circular DNA (eccDNA) is crucial in oncogene amplification, gene transcription regulation, and intratumor heterogeneity. While various analysis pipelines and experimental methods have been developed for eccDNA identification, their detection efficiencies have not been systematically assessed. To address this, we evaluate the performance of 7 analysis pipelines using seven simulated datasets, in terms of accuracy, identity, duplication rate, and computational resource consumption. We also compare the eccDNA detection efficiency of 7 experimental methods through twenty-one real sequencing datasets. Here, we show that Circle-Map and Circle_finder (bwa-mem-samblaster) outperform the other short-read pipelines. However, Circle_finder (bwa-mem-samblaster) exhibits notable redundancy in its outcomes. CReSIL is the most effective pipeline for eccDNA detection in long-read sequencing data at depths higher than 10X. Moreover, long-read sequencing-based Circle-Seq shows superior efficiency in detecting copy number-amplified eccDNA over 10 kb in length. These results offer valuable insights for researchers in choosing the suitable methods for eccDNA research.
Similar content being viewed by others
Introduction
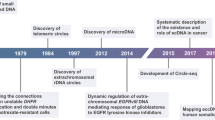
Sequencing-based studies have greatly advanced our understanding of extrachromosomal circular DNA (eccDNA), on its roles in oncogene amplification1,2,3,4, gene expression regulation5, genome rearrangements6,7, and intratumor heterogeneity4. Diverse analysis pipelines and experimental methods have been developed to detect eccDNA (Table 1). Viraj Deshpande et al. introduced the AmpliconArchitect (AA) algorithm to predict amplicon structures and eccDNA from short-read (SR) whole-genome sequencing (WGS) (WGS-SR) data8. CReSIL utilizes coverage depths and breakpoint reads to identify eccDNA from long-read (LR) WGS (WGS-LR) data9. Kumar et al. developed Circle_finder to identify eccDNA from short-read ATAC-Seq (ATAC-Seq-SR) data by analyzing split reads for eccDNA coordinates10. However, the performance of these analysis pipelines might be limited by the data generated from the corresponding experimental methods. For example, WGS and ATAC-Seq may have low eccDNA detection efficiency because vast majority of the sequencing reads were generated from linear DNA, and WGS-SR can only detect the copy number amplified eccDNA (ecDNA)4,6,11.
To enhance eccDNA detection, researchers have developed methods such as Circle-Seq7,12,13 and 3SEP14,15 for eccDNA enrichment from crude DNA. Circle-Seq utilizes rolling circle amplification (RCA) for circular DNA amplification, whereas 3SEP employs Solution A for selective circular DNA recovery. Post-enrichment, eccDNA undergoes library construction for sequencing on platforms like Illumina (Circle-Seq-SR/3SEP-SR) or Oxford Nanopore Technology (ONT) (Circle-Seq-LR/3SEP-LR). Concurrently, various analysis pipelines have been developed to process eccDNA sequencing data. Circle-Map16, ECCsplorer17, Circle_finder10, and ecc_finder (map-sr)18 are tailored for short-read data analysis. For long-read data, pipelines such as CReSIL9, NanoCircle7, eccDNA_RCA_nanopore14 and ecc_finder map-ont mode are used. Additionally, ecc_finder offers de novo assembly options: Spades in the asm-sr mode and Tidehunter in the asm-ont mode as distinct algorithms to identify eccDNA from SR and LR sequencing profiles, respectively. These eccDNA-enriched methods and tailored pipelines facilitate eccDNA identification without reliance on copy number information6.
Choosing the most suitable analysis pipeline and experimental method for eccDNA research is a complex task. Existing evaluations of these pipelines often have limited scope, focusing on single aspects like accuracy9 or computational needs18, and rely on oversimplified simulations that fall short of representing the intricacies of actual sequencing data. Additionally, detection efficiency for specific eccDNA types varies significantly between enriched (such as Circle-Seq and 3SEP) and non-enriched experimental methods (such as WGS-SR, WGS-LR, and ATAC-Seq-SR). For example, the rolling circle amplification (RCA) step is known to preferentially amplify circular DNA under 10 kb19, while the bias of Solution A enrichment remains unclear.
In this work, we conducted an in-depth evaluation of 7 analysis pipelines. The comparative analysis scopes included assessing accuracy (F1-score), identity (base pair difference between identified eccDNA and simulated eccDNA), duplication rate, and computational resource cost using seven simulated datasets designed to mirror real eccDNA characteristics. These datasets replicated the length distribution, chimeric eccDNA composition and chromosomal origins as previously identified7,9,13,20,21,22. Additionally, we compared the detection efficiencies of 7 methods on twenty-one real sequencing datasets for different eccDNA types. Our comparative analysis highlights the most effective pipelines for analyzing short-read and long-read data from eccDNA-enriched methods and underscores the variation in eccDNA detection efficiency across different experimental approaches. Our findings are intended to guide researchers in choosing the most suitable methodologies for their eccDNA studies and to foster the development of novel approaches for efficient eccDNA detection.
Results
Study design
To evaluate the performance of analysis pipelines in eccDNA identification, we developed a Python script to generate simulated eccDNA datasets. This script extrapolated length distribution, chromosomal origins, and chimeric eccDNA proportions from existing data to create a mix of simulated circular DNA (true positives) and linear DNA (true negatives). It also simulated the rolling circle amplification (RCA) process and subsequent sequencing on short-read (Illumina) and long-read (ONT) platforms (Fig. 1a). Seven simulated datasets were produced, mirroring eccDNA identified in human sperm cells7, EJM cell line9, JJN3 cell line9, Kelly cell line20, medulloblastoma21, muscle cells13 and OVCAR8 cell line22 (Supplementary Fig. 1 and Supplementary Fig. 2), each comprising 10,000 circular and 10,000 linear DNA sequences at a depth of 50X.

a Schematic overview of the benchmarking workflow used to compare the performance of bioinformatic pipelines. The cell line, healthy tissue and tumor illustration were created in BioRender. Gao, X. (2024) BioRender.com/h74t202. ‘Std’ represents standard deviation. b Performance comparison of analysis pipelines at a simulated sequencing depth of 50X (bms, bwa-mem-samblaster; mIs, microDNA.InOne.sh). Data are presented as mean values +/- SEM. c Impact of simulated sequencing depth on eccDNA identification accuracy. Data are presented as mean values +/- SEM. d Impact of simulated sequencing depth on eccDNA identification duplication rates. Centre line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range. e Impact of chimeric DNA proportion on eccDNA identification recall. Data are presented as mean values +/- SEM. The ‘n’ in the figure represents the number of datasets successfully analyzed by corresponding analysis pipeline and is used as the sample size to evaluate the performance of the respective analysis pipeline in the analysis. For panels (b, c and d) n = 7, except for ecc_finder (asm-sr) with n = 6 and ECCsplorer with n = 6 when depth ≤ 10, n = 5 for depths between 15X and 25X, n = 4 for depths between 30X and 40X, and n = 3 for depths higher than 40X. For panel (e) n = 4 for only 4 datasets contain chimeric eccDNA. Source data are provided as a Source Data file.
We evaluated 11 modes of 7 pipelines, including Circle-Map, Circle_finder (bwa-mem-samblaster and microDNA.InOne.sh), ECCsplorer, and ecc_finder (map-sr/asm-sr) for short-read data analysis, and CReSIL, eccDNA_RCA_nanopore, NanoCircle, and ecc_finder (map-ont/asm-ont) for long-read data analysis. True positive identification was defined as having over 90% sequence identity and less than 250 base pair (bp) difference with the simulated eccDNA. Performance metrics included F1-score and base pair difference between the identified eccDNA and the simulated eccDNA (see “Methods”). Additionally, we down-sampled the datasets to test pipeline robustness at low sequencing depths and generated datasets with varying chimeric DNA proportions (0–50%) to assess impact of chimeric DNA on eccDNA identification. We also introduced a duplication rate metric to address the issue of multiple detections of the same eccDNA sequence (see “Methods”) and analyzed the computational resource consumption for each pipeline.
For experimental method assessment, we selected Circle-Seq (SR/LR), 3SEP (SR/LR), WGS (SR/LR), and ATAC-Seq (SR) based on their non-targeted nature and sequencing compatibility with Illumina (SR) and ONT (LR) platforms. To minimize batch effects, eccDNA was extracted from a uniform pool of HeLa cells. Controls included a pUC-19 plasmid (2686 bp) and a mouse Egfr gene fragment (2651 bp), spiked into the cell lysate at a 1:1000 ratio to crude circular DNA. We then evaluated eccDNA detection efficiency of each method across various lengths and copy number statuses, quantifying detection efficiency as the number of eccDNA per gigabase (Gb) of sequencing data (see “Methods”).
Assessment of analysis pipelines in eccDNA identification
In our evaluation of the performance of each analysis pipeline in eccDNA identification at a simulated sequencing depth of 50X, Circle_finder (bwa-mem-samblaster) and Circle-Map outperformed the others for short-read data analysis, achieving F1-score of 0.912 and 0.908, respectively. However, Circle-Map had a lower base pair difference between the identified eccDNA and simulated eccDNA (1.354 bp difference) than Circle_finder (bwa-mem-samblaster) (4.344 bp difference). Circle_finder (microDNA.InOne.sh) performed better than Circle_finder (bwa-mem-samblaster) in terms of the base pair difference (1.383 bp difference), but its F1-score was lower (0.825) (Fig. 1b). In the long-read data category, CReSIL led with an F1-score of 0.918 and a base pair difference of 4.160 bp, outperforming eccDNA_RCA_nanopore (F1-score: 0.859, 3.592 bp difference) and NanoCircle (F1-score: 0.905, 4.214 bp difference) (Fig. 1b). Furthermore, ecc_finder asm-ont mode had the lowest F1-score (0.179) and the highest base pair difference (66.158 bp) among all pipelines for long-read data analysis. Meanwhile, ECCsplorer could identify eccDNA from dataset 2, 3 and 7 but failed in other datasets at sequencing depth 50X (Supplementary Data 1).
Impact of sequencing depth on eccDNA identification
Previous research indicates that low eccDNA coverage adversely affects the performance of analysis pipelines in eccDNA identification9. To explore this, we down-sampled our simulated datasets to various sequencing depths, assessing the performance of each pipeline in eccDNA identification. For short-read data analysis, Circle_finder (bwa-mem-samblaster), followed by Circle-Map, consistently achieved the highest F1-scores across all investigated sequencing depths (Fig. 1c). Though ECCsplorer failed in analyzing simulated dataset 5, it had the lowest base pair difference (Supplementary Fig. 3a). Circle-Map and Circle_finder (microDNA.InOne.sh) maintained stable base pair differences when sequencing depth decreased from 50X to 5X, while the base pair difference of Circle_finder (bwa-mem-samblaster) decreased from 4.344 bp at 50X to 2.890 bp at 5X (Supplementary Fig. 3a). ecc_finder (asm-sr) showed the lowest F1-score across all the simulated sequencing depths (Fig. 1c). In the realm of long-read data, CReSIL led with the highest F1-scores at depths over 10X, while eccDNA_RCA_nanopore showed superior performance below a depth of 10X (Fig. 1c). eccDNA_RCA_nanopore kept lowest base pair differences across all the simulated sequencing depths (Supplementary Fig. 3b). The base pair difference of ecc_finder (map-ont) decreased from at 9.015 bp at 50X to 5.976 bp at 5X, while ecc_finder (asm-ont) showed the lowest F1-score and highest base pair difference among all the pipelines in analyzing long-read data (Fig. 1c and Supplementary Fig. 3b).
We observed a pattern of redundancy in eccDNA identification by eccDNA_RCA_Nanopore at all simulated depths, aligning with findings from another study9. Circle_finder (bwa-mem-samblaster) also demonstrated redundancy in its results. Upon calculating the duplication rates, it was evident that both Circle_finder (bwa-mem-samblaster) and eccDNA_RCA_nanopore could identify multiple similar copies from a single eccDNA sequence (Fig. 1d). These substantial duplication rates present considerable obstacles for the experimental validation of their predictions.
Impact of chimeric DNA proportion on eccDNA identification
In addition to sequencing depth, we investigated the influence of chimeric DNA on eccDNA identification performance. We created simulated datasets with varying proportions of chimeric DNA, from 0% to 50%, maintaining a fixed sequencing depth of 20X. For short-read data analysis, the change of chimeric DNA proportion did not affect the recall for simple eccDNA identification of Circle-finder (bwa-mem-samblaster), Circle-Map, and ecc_finder (map-sr). However, the recall for simple eccDNA identification of ECCsplorer decreased from 0.414 at 0% to 0.056 at 50%. (Fig. 1e). ecc_finder (asm-sr) showed the lowest recall. The base pair differences between identified eccDNA and simulated eccDNA for these pipelines remained relatively stable except ecc_finder (asm-sr) (Supplementary Fig. 3c). Among long-read data analysis pipelines, most maintained consistent recall (with changes less than 0.1) for both simple eccDNA and chimeric eccDNA identification (Fig. 1e). The base pair differences of CReSIL, eccDNA_RCA_nanopore and ecc_finder (map-ont) showed a relatively slight increase compared to NanoCircle, of which the base pair difference increased from 2.492 at 0% to 16.899 at 50% (Supplementary Fig. 3d). Unlike the other pipelines, ecc_finder (asm-ont) showed a decreased base pair difference as the chimeric eccDNA proportion increased (Supplementary Fig. 3d).
Computational resources consumed by different analysis pipelines
In our evaluation of computational resources consumed by each pipeline, we utilized a computer cluster equipped with two Intel Xeon Scale 6248 CPUs (2.5 GHz, 320 CPU cores), 384 GB of DDR4 memory, and 2 TB AEP memory. We observed that both the time and memory consumption of most pipelines increased with mean coverage rising (Supplementary Fig. 3e and f). For identifying eccDNA from short-read data, Circle_finder (bwa-mem-samblaster) was the fastest pipeline to identify eccDNA and Circle_finder (microDNA.InOne.sh) used the least memory across all the investigated sequencing depths (Supplementary Fig. 3e). When considering the long-read data analysis pipelines, ecc_finder used the shortest time (map-ont) and the least memory (asm-ont) (Supplementary Fig. 3f). For dataset 5, ecc_finder (asm-sr) and ECCsplorer experienced memory errors on our platform (Supplementary Data 1). Besides, ECCsplorer also encountered memory errors in analyzing dataset 1 (depth over 25X), dataset 4 (depth over 40X), and dataset 6 (depth over 10X) (Supplementary Data 1).
Based on the above analysis, we concluded that Circle_finder (bwa-mem-samblaster) and Circle-Map were the most appropriate analysis pipelines for analyzing eccDNA-enriched short-read data, and CReSIL outperformed the other analysis pipelines to analyze eccDNA-enriched long-read data, due to their high detection accuracy and low base pair difference. In the following experimental methods benchmarking, we selected Circle-Map for analyzing the eccDNA-enriched short-read sequencing data because of its fewer redundant results compared to Circle_finder (bwa-mem-samblaster). High redundancy may cause the high eccDNA detection efficiency bias to the eccDNA-enriched experimental methods (Fig. 2a). Besides, we used AmpliconArchitect for analyzing WGS-SR data, CReSIL for analyzing WGS-LR data and Circle_finder (bwa-mem-samblaster) for analyzing ATAC-Seq-SR data.

a Schematic overview of the experimental methods comparison. b eccDNA detection efficiency comparison. Data are presented as mean values +/- SEM. c Circular DNA enrichment efficiency. Data are presented as mean values +/- SEM. LDD, Linear DNA Digestion; Solution A, using Solution A for circular DNA purification; RCA, Rolling Cycle Amplification. d Detection efficiency for eccDNA with different length ranges. Data are presented as mean values +/- SEM. e Correlation between eccDNA density and coding gene density. Dots represent individual experiments and the shaded area represents 95% confidence interval. For all experiments, n = 3. Statistical analyses were performed using one-way ANOVA with Tukey correction for panel (b, c and d,) and two-sided Pearson correlation was performed for panel (e). The ‘p’ represents p-value. Source data are provided as a Source Data file.
Impact of eccDNA enrichment steps on eccDNA identification
We assessed eccDNA detection efficiency by the number of eccDNA detected per gigabyte (Gb) of data. The results indicated that methods incorporating RCA steps achieved significantly higher eccDNA detection efficiencies compared to those without RCA (Fig. 2b). Notably, qPCR analyses revealed that both Solution A purification and the RCA step considerably increased the log2 ratio of circular to linear spike-in DNA (Solution A: from 2.26 to 9.60 and from 18.20 to 26.19, RCA: from 2.26 to 18.20 and from 9.60 to 26.19) (Fig. 2c). To validate these findings, we randomly selected nine simple and seven chimeric eccDNA for testing (See “Methods”), observing validation rates above 0.5 in RCA-utilizing methods (3SEP-LR: 8/16, Circle-Seq-SR: 8/9, Circle-Seq-LR: 11/16) (Supplementary Fig. 4 and Supplementary Data 2). Due to the notable efficiency of circular DNA enrichment through RCA and the use of solution A, we hypothesized that eccDNA-enriched experimental methods could effectively detect such DNA entities without the need for copy number amplification. We investigated the association between genome copy numbers and the coverage of overlapped eccDNA. Our analysis revealed a positive correlation between genome copy numbers and the coverage of overlapped eccDNA. Notably, the correlation coefficients (r values) derived from the WGS-LR (0.80) and ATAC-Seq-SR (0.41) datasets were higher than those obtained from eccDNA-enriched experimental methods (< 0.25) (Supplementary Fig. 5).
Further analysis of the eccDNA length distribution and chromatin origins revealed that Circle-Seq-LR had the highest detection efficiency for > 10 kb eccDNA and enriched methods (except for 3SEP-SR) could detect significantly more ≤ 10 kb eccDNA per Gb data than non-enriched methods (Fig. 2d). However, over 97% of the identified eccDNA from eccDNA-enriched methods were shorter than 10 kb (Circle-Seq-LR: 97%, Circle-Seq-SR: 99.8%, 3SEP-LR: 99.9%, 3SEP-SR: 99.5%) and over 90% of eccDNA detected by methods like 3SEP-SR and 3SEP-LR were shorter than 2 kb (Supplementary Fig. 6). In contrast, non-enriched methods showed a higher proportion of eccDNA lengths exceeding 10 kb (Supplementary Fig. 6). Additionally, except for 3SEP-SR and WGS-SR, a significant positive correlation was observed between eccDNA density (number of detected eccDNA per million base (Mb)) and protein-coding gene density across chromosomes in most methods, consistent with prior studies7,13 (Fig. 2e). 3SEP-SR showed a similar trend, though the correlation was not statistically significant (r = 0.39, p = 0.064), and no significant correlation was found in WGS-SR data (r = 0.12, p = 0.6). This could be due to the limited number of eccDNA identified by WGS-SR, suggesting the importance of eccDNA enrichment in experimental setups to obtain a comprehensive eccDNA profile.
Detection efficiency of ecDNA by different experimental methods
The eccDNA overlapping with copy number amplified regions was designated as ecDNA, while eccDNA outside these regions was categorized as nonecDNA11. Circle-Seq-SR, Circle-Seq-LR, and 3SEP-LR identified a higher average number of ecDNA per Gb of data (205.2, 165.8, and 203.9, respectively) compared to WGS-SR, WGS-LR, and ATAC-Seq-SR (0.01576, 0.9100, and 6.862, respectively) (Fig. 3a). However, a significantly higher proportions of ecDNA were found in the eccDNA detected by WGS-SR (100%), WGS-LR (57.68%), and ATAC-Seq-SR (36.67%) compared to Circle-Seq-SR (20.58%), Circle-Seq-LR (17.09%), and 3SEP-LR (19.26%) (Fig. 3b).

a ecDNA detection efficiency of 7 experimental methods. b Comparison of the proportion of ecDNA in the total detected eccDNA. c Comparison of the detection efficiency of ecDNA with different length ranges by 7 experimental methods. d Comparison of the detection efficiency of nonecDNA with different length ranges by 7 experimental methods. Dots represent individual experiments; For all experiments, n = 3. Statistical analyses were performed using one-way ANOVA with Tukey correction; The ‘p’ represents p-value. Data are presented as mean values +/- SEM. Source data are provided as a Source Data file.
Subsequently, we further analyzed the detection efficiencies for both ecDNA and nonecDNA across varying lengths (≤ 2 kb, 2–10 kb, > 10 kb Figs. 3c and d). 3SEP-LR demonstrated the highest efficiency in detecting both ecDNA and nonecDNA up to 2 kb in length. Circle-Seq-SR was the most efficient for detecting ecDNA between 2 kb and 10 kb. For eccDNA over 10 kb, Circle-Seq-LR outperformed all other methods in detecting both ecDNA and nonecDNA. Interestingly, for detecting ecDNA and nonecDNA over 10 kb, WGS-LR, despite not employing a circular DNA enrichment step, showed comparable efficiency with 3SEP-SR, 3SEP-LR, and Circle-Seq-SR (Figs. 3c and d).
EccDNA profiles showed heterogeneity across experimental methods
We investigated the correlation of eccDNA profiles across various technical replicates from different experimental methods. We considered eccDNA from different technical replicates to be highly-correlated (HC) when their eccDNA shared over 90% sequence identity. Our analysis indicated that, among the methods examined, eccDNA profiles detected from different experimental replicates within WGS-SR (> 50%) or WGS-LR (> 54.19%) exhibited higher correlations compared to other methods such as ATAC-Seq-SR (< 10%), 3SEP-SR (< 2%), 3SEP-LR (< 1%), Circle-Seq-SR (< 1%), and Circle-Seq-LR (< 1%) (Fig. 4a and Supplementary Data 3). Specifically, compared to WGS-SR (≤ 2), WGS-LR replicates showed more shared highly-correlated eccDNA (> 95) (Fig. 4b). Furthermore, we observed higher correlations between paired Circle-Seq-SR/LR replicates (HC eccDNA proportion > 15%, number of HC eccDNA > 4900) (e.g., Circle-Seq-SR1/Circle-Seq-LR1) compared to unpaired Circle-Seq replicates (e.g., Circle-Seq-SR1/Circle-Seq-SR2 or Circle-Seq-SR1/Circle-Seq-LR2) (Fig. 4a and b). Despite the DNA material for Circle-Seq-SR/LR pairs originating from the same debranched RCA product, the HC eccDNA proportions within these pairs were below 35% (Supplementary Data 3), indicating that the choice of sequencing platform and analysis pipelines can influence the final eccDNA profiles.

a Proportion of highly-correlated eccDNA between each pair experimental replicates in vertical replicates; b Number of highly-correlated eccDNA between each pair of experimental replicates; c Number of detected oncogenes shared across different methods. d Number of oncogenes shared by different replicates. e Proportion of read mapped to repeat elements (n = 3). For panel e, statistical analyses were performed using one-way ANOVA with Tukey correction; The ‘p’ represents p-value. Source data are provided as a Source Data file.
We speculated that though there existed high heterogeneity across different experimental methods or technical replicates, the copy number amplified eccDNA profiles (ecDNA profiles) of different methods might share common oncogenes. To explore this, we compiled a list of oncogenes from OnGene database23, and compared the detected oncogenes by different methods. Our analysis revealed that the ecDNA sequences obtained from all examined experimental methods mapped to a total of 125 oncogenes (Supplementary Fig. 7 and Supplementary Data 4). No oncogene was detected by all the examined experimental methods. 87 out of 125 oncogenes could be detected by at least two different experimental methods (Fig. 4c and Supplementary Data 4). A total of 18 oncogenes were detected by 4 experimental methods (Fig. 4c and Supplementary Data 4). For example, ZNF217, reported to promote HeLa cell viability24, was detected by Circle-Seq-SR, Circle-Seq-LR, 3SEP-LR, WGS-LR. Further, 16 of the 18 oncogenes were detected by eccDNA-enriched experimental methods. For example, TRIO25 and CULA426, reported to promote metastasis and invasion of HeLa cells, were detected by Circle-Seq-SR/LR and 3SEP-SR/LR. PVT1, a long non-coding RNA that can enhance proliferation27 and promote the cancer progress28,29 of cervical cancer cells, was also detected by Circle-Seq-SR/LR and 3SEP-SR/LR. Notably, experimental methods employing the RCA step demonstrated a higher capacity for oncogene detection (> 69, data from Circle-Seq-LR, Fig. 4d) compared to those lacking this step (< 20, data from 3SEP-SR, Fig. 4d). Furthermore, experimental replicates utilizing RCA exhibited a greater overlap in detected oncogenes (at least 42 oncogenes were detected in more than 2 replicates) compared to those without RCA (Fig. 4d and Supplementary Data 4).
Repeat elements are commonly detected in the sequencing data that are used for identifying eccDNA13,30,31. Considering that our sequencing data originated from the same HeLa cell pool, we postulated that the proportion of reads mapping to repeat elements would remain consistent across different experimental methods. However, our findings revealed notable disparities. WGS-LR exhibited the highest proportion of reads mapping to the examined repeat elements (Fig. 4e and Supplementary Data 5), including long terminal repeats (LTRs, 65.84%), short interspersed nuclear elements (SINEs, 73.70%), long interspersed nuclear elements (LINEs, 74.61%), and satellite elements (16.5%). Furthermore, WGS-LR, 3SEP-LR, and Circle-Seq-LR displayed significantly elevated proportions of reads mapping to LTRs, SINEs, and LINEs compared to their short-read counterparts (Fig. 4e and Supplementary Data 5). This suggests that sequencing results from different experimental methods inherently exhibit heterogeneity. Consequently, when comparing results across different studies, it is important to consider the experimental methods used.
Discussion
Benchmarking the available analysis pipelines and experimental protocols for detecting eccDNA is crucial for advancing eccDNA research. In this study, we have identified top performers for eccDNA detection by assessing 7 analysis pipelines using various metrics, and comparing 7 experimental methods via detection efficiency. Circle_finder (bwa-mem-samblaster) and Circle-Map stand out for their abilities to identify eccDNA from short-read data and CReSIL outperformed the others in long-read data analysis. In the realm of experimental methods, Circle-Seq-LR demonstrates the highest detection efficiency for longer eccDNA, while 3SEP-LR is more effective for shorter eccDNA. This information is vital for researchers in selecting the most suitable methodologies for their eccDNA studies.
Despite our simulated datasets closely mimicked the length distribution of real eccDNA data, they featured a comparatively smaller proportion of eccDNA longer than 10 kb. This imbalance posed challenges in precisely evaluating the performance of different analysis pipelines across various eccDNA length ranges. Additionally, while using DNA from a cell line sheds light on the eccDNA detection efficiency of diverse methods, the potential copy number bias introduced at different experimental stages remains a concern due to the absence of a known ground truth. Future research could benefit from employing a specially designed circular DNA pool with a defined copy number. Such a controlled approach would not only help in addressing potential biases but also allow for more accurate quantification of metrics like F1-score and base pair difference for each experimental method in eccDNA detection.
Split and discordant reads within short-read data, and breakpoint reads in long-read data, are primary sources for eccDNA identification. CReSIL utilizes the breakpoint read information to construct directed graphs, allowing for its effective identification of eccDNA from both the concatemeric tandem copies (CTC) reads and the non-CTC reads containing breakpoints. Conversely, eccDNA_RCA_nanopore only focuses on CTC reads and might limit its ability to identify larger eccDNA that were hard to generate CTC reads. Both eccDNA_RCA_nanopore and Circle_finder (bwa-mem-samblaster) exhibit a tendency for redundancy due to their approach of reporting results for each CTC read or split read, respectively. Circle_finder (bwa-mem-samblaster) showed the highest F1-score across all the investigated sequencing depth, reducing the redundancy results may further enhance its performance. Because the available pipelines are limited for analyzing eccDNA non-enriched data, we only compared the performance of these analysis pipelines for identifying eccDNA from simulated eccDNA-enriched datasets. Future study is needed to compare the performance of the analysis pipelines for detecting eccDNA from non-enriched data when more pipelines are available.
This benchmark study also helps to explain controversial findings in the field. For instance, the limited detection of ecDNA in normal cells4 may be due to the low sensitivity of WGS-SR in identifying eccDNA. Conversely, the effective identification of eccDNA in human germline cells may be facilitated by the use of the Circle-Seq-LR technique7. However, it is important to note from our analysis that non-enriched methods like WGS-SR hold their own unique advantages, such as providing copy number variation information essential for ecDNA classification32. Therefore, we do not suggest that non-enriched methods be replaced by enriched methods. Moreover, other non-enriched methods like WGS-LR33 and modified ATAC-Seq-SR34 can preserve nucleotide decorations in the sequencing reads, a feature could potentially lost in sequences generated from enrichment steps like RCA.
A significant challenge in eccDNA research is the inconsistency in the definitions of different eccDNA types used by various studies. We defined ecDNA as eccDNA colocalizing with genome copy number-amplified regions11, due to the putative gene amplification effect of ecDNA. Other studies may use size thresholds to define ecDNA35,36. Establishing a consensus definition is crucial for harmonizing research findings in this rapidly evolving field.
Lastly, the potential of eccDNA as a diagnostic marker for diseases like advanced chronic kidney disease37, medulloblastoma21, and colorectal cancer38 is promising. Increasing the efficiency of linear DNA digestion will be beneficial for enhancing the enrichment of circular DNA, and further efforts in this direction will be appreciated. Optimizing the RCA step, typically a lengthy process, could also enhance the feasibility of using eccDNA information for clinical diagnosis.
Methods
Generation of simulated datasets
Because the biogenesis of eccDNA has not been fully known, we considered findings or eccDNA simulating methods from previously published papers9,14,16 and created a python script to generate simulated eccDNA datasets for evaluation. The simulated datasets contained circular and linear DNA, according to the length distribution, chromosome origins and chimeric eccDNA proportion of the eccDNA from the given data. We collected the eccDNA profiles identified by different analysis pipelines (Supplementary Data 1) from human sperm cells7, EJM cell line9, JJN3 cell line9, Kelly cell line20, medulloblastoma21, muscle cells13 and OVCAR8 cell line22, and used these 7 datasets as input. We generated 7 simulated datasets, containing 10000 circular DNA (as positive sequences) and 10000 linear DNA fragments (as negative sequences). Then, we randomly shifted the positive sequence to mimic the RCA starting site and concatenated the 5000 bp of individual simulated eccDNA to mimic the RCA procedure. We used generated sequences as templates to further simulate short-read datasets using ART39 (--sr-platform ‘HS25’ --sr-mean ‘400’ --sr-std ‘125’ --sr-readlen ‘150’) and simulate long-read datasets using PBSIM240 (--ont-model ‘R94’, --ont-mean ‘3000’,--ont-std ‘2500’) with different sequencing depth (5X, 10X, 15X, 20X, 25X, 30X, 35X, 40X, 45X, 50X). We also used eccDNA identified from human sperm cells7, EJM cell line9, JJN3 cell line9 and Kelly cell line20 to simulate short-read datasets and long-read datasets with different chimeric DNA ratios (0%, 10%, 20%, 30%, 40%, 50%) at sequencing depth 20X.
Performance evaluation of each pipeline
The identification of eccDNA was done following the instructions on the website of each pipeline. We used hg38 genome as reference. For Circle-Map16, we used Circle Map Realign to identify eccDNA and used recommended filters (circle score > 50, split reads > 2, discordant reads > 2, coverage increase in the start coordinate > 0.33 and coverage increase in the end coordinate > 0.33). For Circle_finder10, we used the script circle_finder-pipeline-bwa-mem-samblaster.sh to identify eccDNA. For ECCsplorer17, we used mapping module to identify eccDNA. For ecc_finder18, all the 4 modes were used to identify eccDNA from either short-read or long-read data. The identified eccDNA with length longer than 107 bp was filtered out. For CReSIL9, we followed the instruction on its website to identify eccDNA and considered cyclic eccDNA as identified results. For NanoCircle7, we followed the instruction on its website and considered high_conf simple eccDNA and complex eccDNA as identified results. For eccDNA_RCA_nanopore14, we followed the instruction on its website to identify eccDNA. For the pipelines that did not supply FASTA format results, we used pysam41 to transform bed format into FASTA format. The FASTA files were then compared to the simulated eccDNA sequence by MUMmer342.
Cell culture
HeLa cells were bought from BeNa Culture Collection (Cat#BNCC342189; RRID: CVCL-0030). NIH3T3 (RRID: CRL-1658) was a gift from Prof. Shu Zhu lab of the University of Science and Technology of China. HeLa cells or NIH3T3 cells were cultured at 37˚C in DMEM (Thermo Fisher Scientific 11965092) containing 10% FBS (Thermo Fisher Scientific 10091148) and 1% penicillin‒streptomycin (Thermo Fisher Scientific 15140122). Upon reaching approximately 80%–100% confluence, the cells were rinsed with 1× PBS (Sangon Biotech, B540626-0500) and digested with 0.25% trypsin (Beyotime C0203-500 ml). The trypsinization process was terminated by adding DMEM + 10% FBS + 1% penicillin‒streptomycin, and the cells were collected by centrifugation at 500 × g for 5 min at RT. Cells were then washed twice by using 1X PBS and then centrifuged at 500 × g for 5 min at 4 °C to obtain the cell pellet for following experiments. Detailed company names and catalog numbers of reagents are recorded in Supplementary Data 6.
ATAC-seq library construction
For each replicate, approximately 50000 cells and a commercialized Tn5 kit (Vazyme, TD501) were used to construct the ATAC-Seq library. The reaction mix, consisting of 50,000 cells, 0.005% digitonin (Sigma‒Aldrich D141-100MG), 33 mM Tris-Ac (pH 7.8), 66 mM KAc, 10 mM MgAc, and 16% DMF, was incubated at 500 rpm for 30 mins at 37 °C using a thermal rotator. After the reaction, the cells were washed twice using wash buffer (10 mM Tris-HCl pH;7.5, 10 mM NaCl, 3 mM MgCl2, 0.005% digitonin) and resuspended in 14 µl of 10 mM Tris-HCl pH 7.5. Cells were then lysed by mixing with 2 µl lysis buffer (200 mM Tris-HCl pH 8.0, 0.4% SDS) and 0.2 µl proteinase K (20 mg/mL) at 500 rpm for 15 mins at 55 °C. The lysis reaction was terminated by adding 4 µL of 10% Tween-20 and 0.4 µL of 100 mM PMSF. The samples were incubated for 5 mins at RT, and then PCR was performed to add adapters to the DNA segment for sequencing. Detailed company names and catalog numbers of reagents are recorded in Supplementary Data 6.
Whole-genome sequencing
For preparing each replicate for WGS-SR, after washing the cells, more than 1 million cells were frozen using liquid nitrogen. Three replicates were sent to Sequanta Technologies for library construction and WGS-SR sequencing (Illumina NovaSeq 6000 platform). For preparing each replicate for WGS-LR, after washing the cells, more than 5 million cells were frozen using liquid nitrogen. Three replicates were sent to Novogene for library construction and WGS-LR sequencing (Oxford Nanopore PromethlON platform).
Isolation of crude circular DNA
Crude circular DNA was extracted from the same pool of HeLa cells following the published protocol15. In brief, more than 60 million HeLa cells were used to extract the crude circular DNA pool. For each reaction (approximately 30 million HeLa cells), cells were collected in a 50 mL tube by centrifugation at 2000xg for 10 mins at 4 °C. Resuspend the cells in 10 ml of suspension buffer (10 mM EDTA pH8.0, 150 mM NaCl, 1% glycerol, Lysis blue (1×, from QIAGEN Plasmid Plus Midi Kit), RNase A (0.55 mg/ml), and freshly supplemented with 20 µL of 2-mercaptoethanol). Add 10 mL Pyr buffer (0.5 M pyrrolidine, 20 mM EDTA, 1% SDS, adjust pH to 11.80 with 2 M Sodium Acetate pH 4.00, and freshly supplemented with 20 µL 2-mercaptoethanol) to the cell suspension. Gently mix by inverting the tube 5–10 times and incubate at room temperature for 5 mins. After lysis, 10 mL of Buffer S3 (From QIAGEN Plasmid Plus Midi Kit) was added to the mixture, and the tube was gently inverted until the solution color turned white. Then, the lysate was centrifuged at 4500xg for 10 mins. The clear lysate was transferred to a QIAilter Catridge (From QIAGEN Plasmid Plus Midi Kit) and incubated at room temperature for 10 mins. Then, the cell lysate was filtered into a 50 mL tube. The volume of the filtrated lysate was approximately 27 mL, and 9–10 mL of Buffer BB (1/3 of the lysate volume, From QIAGEN Plasmid Plus Midi Kit) was added. The lysate was mixed by inverting the tube 4-8 times. The lysate mixture was then transferred to the spin column, and vacuum was applied until all liquid passed through. We added 0.7 mL ETR buffer (From QIAGEN Plasmid Plus Midi Kit) to wash the column, and applied vacuum until all liquid passed through. Then, the wash was repeated by using 0.7 mL PE buffer (From QIAGEN Plasmid Plus Midi Kit). After washing, the tube was centrifuged at 10000xg for 2 mins to remove the liquid, and the column was transferred to a new clean 1.5 mL centrifuge tube. Crude eccDNA was then eluted by using 100 µL of 0.1x EB buffer (From QIAGEN Plasmid Plus Midi Kit). For each microgram crude eccDNA we spiked in 1 ng pUC1943 (was a gift from Joachim Messing, Addgene plasmid # 50005; RRID: Addgene_50005) and 1 ng Egfr fragment (amplified from NIH3T3 cell genome by using forward primer: AACTGCTGTCTTGGGTACGG (ordered from Sangon Biotech) and reverse primer: ATTGCAGTCGCCCAAGTGTA (ordered from Sangon Biotech)) to generate crude circular DNA mixture. Detailed company names and catalog numbers of reagents are recorded in Supplementary Data 6.
Linear DNA digestion
For each DNA digestion reaction, 3 µg crude circular DNA mixture was digested by using 0.5 µL Pac I and 1 µL ATP-dependent Plasmid Safe DNase in 1X ATP-dependent Plasmid-Safe DNase buffer. Then, 0.1 µL of 110 mg ml−1 RNase A and 2 µL of 25 mM ATP were added to the reaction in a total volume of 50 µL. The reaction mix was incubated at 37 °C for 16 hours. After digestion, 1.8X SPRIselect beads were used to purify the DNA. DNA was eluted with 66 µL of 2 mM Tris-HCl pH=7.0 to carry out Solution A purification or eluted with 66 µL of 0.1 X EB buffer (From QIAGEN Plasmid Plus Midi Kit) without further Solution A purification. Detailed company names and catalog numbers of reagents are recorded in Supplementary Data 6.
Solution A purification
The Solution A purification step followed the published study15 and was used in 3SEP-SR and 3SEP-LR only. In brief, we transferred 50 µL eluted circular DNA (in 2 mM Tris-HCl pH=7.0) to a 1.5 mL tube. Added 700 µL of Solution A (room temperature) to the tube, mixed by pipetting up and down, and incubated at room temperature for 5 mins. Took 10 µL DynabeadsTM MyOneTM Silane beads (resuspend by thoroughly vortex) to a 200 µL tube and stood it on a magnetic shelf. When beads were settled, removed the liquid and added 20 µL Solution A to resuspend the beads. Then we transferred the beads to DNA (incubated in Solution A) and pipetted up and down for 10 times. Put the mixture on a magnetic shelf, and removed the liquid when the beads were settled. Quickly spun down the beads and put it on the magnetic shelf again to remove the residual liquid. Took off the tube from magnetic shelf and resuspended the beads in 300 µL Solution A. Put the tube on the magnetic shelf and removed the liquid when the beads were settled. Quickly spun down the beads and put it on the magnetic shelf, removing the residual Solution A when beads were settled. Repeated the 300 µL Solution A wash once more. After the second Solution A wash, kept the tube on the magnetic shelf, added 700 µL 3.5 M NaCl, waited for 1 minute and then removed the liquid, and repeated once. After the second NaCl wash, kept the tube on the magnetic shelf, added 800 µL freshly prepared 80% ethanol, waited for 1 minute and then removed the liquid, and repeated once. Quickly spun down the beads and put it on the magnetic shelf again to remove the residual liquid. Took off the tube and used 30 µL 0.1X EB buffer (From QIAGEN Plasmid Plus Midi Kit) to resuspend the beads and incubated for more than 3 minutes. Put the tube back to the magnetic shelf and transferred the elute (contained purified circular DNA) when beads were settled. Detailed company names and catalog numbers of reagents are recorded in Supplementary Data 6.
Rolling Cycle Amplification (RCA) and debranching
We measured the DNA product concentration by using Qubit 4.0, and aliquoted 1 ng DNA to prepare the RCA reaction premix (2 µL 10X Phi 29 DNA Polymerase Reaction Buffer, 2 µL dNTPs (25 mM each), 1 µL Exo-resistant Random Primer, and add H2O to 17.6 µL). The samples were incubated at 95 °C for 5 mins and then ramped to 30 °C at −0.1 °C per sec. Then, added 1 µL of Phi29 DNA Polymerase, 1 µL of Pyrophosphatase (Inorganic) and 0.4 µL of recombinant Albumin (offered with Phi 29 DNA polymerase) to a 20 µL final reaction mix. The samples were incubated at 30 °C for 14 hours and inactivated at 65 °C for 10 mins. The product was diluted by adding 80 µL of H2O, and 1.8X SPRIselect beads were used to purify the product. Eluted the DNA product in 0.1X EB (From QIAGEN Plasmid Plus Midi Kit) buffer. T7 endonuclease I was employed to cleave the branched RCA product from circular DNA. Briefly, 6 µg RCA product was aliquoted into the reaction tube along with 30 µL 10X NEBuffer 2 and 15 µL T7 Endonuclease I, and H2O was added to 300 µL. The reaction mix was incubated at 37 °C for 15 mins. Used 0.4X SPRIselect to purify the reaction product. Detailed company names and catalog numbers of reagents are recorded in Supplementary Data 6.
DNA fragmentation
For Circle-Seq-SR, the debranched DNA materials were sent to Sequanta Technologies for ultrasonic fragmentation with the fragment size in 300–500 bp as reported in the published protocol12. For 3SEP-SR, the Solution A purified DNA material was sent to Sequanta Technologies for enzymatic fragmentation. To compare across different experimental methods, 1 ng DNA was used to generate the sequencing library by using Nextera XT DNA Library Preparation Kit (Illumina).
Sequencing
For ATAC-Seq-SR, 3SEP-SR, and Circle-Seq-SR, DNA library was sequenced by Sequanta Technologies on Illumina NovaSeq 6000 platform. For 3SEP-LR and Circle-Seq-LR, the long-read sequencing library was constructed by Novogene and sequenced on Oxford Nanopore PromethlON platform.
Identification of eccDNA from real datasets
We used the script circle_finder-pipeline-bwa-mem-samblaster.sh in Circle_finder10 to identify eccDNA from ATAC-seq-SR data and set a filter (length shorter than 107 bp) to select eccDNA. For WGS-SR data, we used AmpliconArchitect8 to identified eccDNA with options (cngain= 4, cnsize= 10000). For WGS-LR data, we used CReSIL identify_wgls command9 to identify eccDNA, and filtered cyclic eccDNA. For Circle-seq-SR and 3SEP-SR data, we used Circle Map Realign16 to identify eccDNA and used recommended filters (circle score > 50, split reads > 2, discordant reads > 2, coverage increase in the start coordinate > 0.33 and coverage increase in the end coordinate > 0.33, length< 107bp). For Circle-seq-LR and 3SEP-LR data, we used CReSIL identify command9 to identify eccDNA and filtered cyclic eccDNA.
Identification of ecDNA
We used Control-FREEC44 (breakPointThreshold = 0.6, window = 50000, step= 10000) to examine the copy number variation in 3 replicates of our WGS-LR data. We defined eccDNA as ecDNA if it had overlap with the CNV gain regions identified by Control-FREEC.
Oncogene overlapping analysis
The information of human oncogenes was obtained from ONGene database23. We annotated our identified ecDNA by using BEDTools intersect command45. We merged the overlap regions and calculated overlap proportion of each oncogene using the following formula.
We applied ComplexHeatmap package46 to visualize our results.
Repeat elements analysis
The genomic coordinates of repeat elements on the hg38 reference genome were obtained from UCSC genome browser47. We used pysam to calculate the proportion of reads mapped to different repeat elements, including LTR, LINE, SINE and satellite.
Circular DNA enrichment efficiency evaluation
qPCR was used to evaluate the circular DNA enrichment efficiency. qPCR primers for pUC19 (F: GCAGGTCGACTCTAGAGGAT, R: GGGCCTCTTCGCTATTACGC, ordered from Sangon Biotech), and Egfr fragment (F: AAACGGAAGATCCTGCCCTG; R: GTGTACCCTGAACACGAGGG, ordered from Sangon Biotech) were used to quantify the circular DNA and linear DNA, respectively. The ∆\({Ct}\)(original) was used to normalize the qPCR results.
While \({{Ct}\left({{{\rm{pUC}}}}19\right)}_{i}\) and \({{Ct}\left({Egfr}\right)}_{i}\) represent the cycle threshold (Ct) value of pUC19 and Ct value of Egfr fragment of the replicate i of the original DNA pool. N represents the number of replicates.
The circular DNA enrichment efficiency for each step was calculated by:
\({\Delta {Ct}\left({{{\rm{step}}}}\right)}_{j}\) was calculated by:
While \({{Ct}\left({{{\rm{pUC}}}}19\right)}_{j}\) and \({{Ct}\left({Egfr}\right)}_{j}\) represent the Ct value of pUC19 and Ct value of Egfr fragment of the replicate j after the specific circular DNA enrichment step. N represents the number of replicates.
PCR validation
We created a numerical index for each eccDNA from each sample and used the random number generating formula in EXCEL (=randbetween(start index:end index)) to select the eccDNA. For the eccDNA that we could not design primers (potentially due to repeat sequences or low sequence complexity), we added 1 to the rolled random number and redesigned the primer for the newly indexed eccDNA. DNA sequences spanning the breakpoint were obtained by using Genome Browser (https://genome.ucsc.edu/index.html). Primers targeting the eccDNA breakpoint were designed by using Primer-Blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) (Supplementary Data 2) and ordered from Sangon Biotech. The Hela cell genome was extracted by using the DNeasy® Blood & Tissue Kit (QIAGEN Cat. No. 69504). KOD FX (TOYOBO No. KFX-101) was used to perform the PCR. In brief, 20 ng DNA template (Genome DNA or Sample), 1.5 µL 10 µM forward primer, 1.5 µL 10 µM reverse primer, 4 µl 2 mM dNTPs, 10 µL 2X PCR Buffer for KOD FX, 1 µL KOD FX and nuclease-free water (Invitrogen 10977015) (to a 20 µL final volume) were combined. PCR was carried out by using the following thermal cycle: 94 °C for 2 minutes and then 30 cycles at 98 °C for 10 s, 60 °C for 30 s, 68 °C for 1 minute and 68 °C for 5 minutes. The PCR product was cut from the electrophoresis gel and sent for Sanger sequencing validation (by Sangon Biotech). We classified chimeric eccDNA as fully validated when all breakpoints were confirmed through Sanger sequencing (considered as 1 event when calculating the validation rate). In cases where only partial breakpoints could be validated, we categorized it as partially validated chimeric eccDNA (considered as 0.5 event when calculating the validation rate). Detailed company names and catalog numbers of reagents are recorded in Supplementary Data 6.
Benchmark metrics
F1-score
Where TP represents the number of true positive event, FP represents the number of false positive event, and FN represents the number of false negative event.
Base pair difference
Where LEN R and LEN Q are length of reference eccDNA and query eccDNA, LEN 1 and LEN 2 are length of alignment on reference and query eccDNA. N is the number of query eccDNA that has more than 90% identity and 90% overlap with reference eccDNA.
Duplication Rate
The duplication rate is defined by the number of identified eccDNA (TP2) that have at least a 90% overlap of simulated eccDNA divided by the number of simulated eccDNAs (TP1) that can be identified by each pipeline.
Detection efficiency of specific type of eccDNA
Detection efficiency of specific type of eccDNA (per Gb) was calculated by using the following formula:
Where: Eij is the detection efficiency of experimental method i in detecting eccDNA type j, nij is the number of eccDNA in type j detected by experimental method i, and Di is the size of the data (Gb) generated by experimental method i.
Statistics & reproducibility
For performance evaluation of bioinformatic pipelines. We used Seaborn48 to visualize statistical data. Each point showed the Mean ± SEM (Standard Error of the Mean) in the figure. For column chart, one-way ANOVA (by GraphPad Prism 9) was used to evaluate the statistical significance (degrees of freedom between methods are 6, and degrees of freedom within methods are 14). For group column chart we also used one-way ANOVA (degrees of freedom between methods are 6 and degrees of freedom within methods are 14), because we focused on the comparison within each length range. Each column showed the Mean ± SEM and data points were shown as black dot on the column. For correlation dot plot (Fig. 2e), we used two-sided Pearson correlation in scipy.stats49 to measure the linear relationship between the density of coding genes and the density of eccDNA for each chromosome, and used Seaborn to present the result.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequencing data (WGS-SR, WGS-LR, ATAC-Seq-SR, 3SEP-SR, 3SEP-LR, Circle-Seq-SR, and Circle-Seq-LR) generated in this study are openly available and have been deposited in the Genome Sequence Archive for Human (GSA-Human) database50 in National Genomics Data Center51, China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences under accession code HRA006020.The public data used in this study in Supplementary Fig. 1 to generate our simulation datasets are openly available from following study.
Dataset 1 (sperm cells): the raw sequencing data are available in the Sequence Read Archive (SRA) database under accession code PRJNA6558197.
Dataset 2 (EJM cell line) and Dataset 3 (JJN3 cell line): the raw sequencing data are available in the Sequence Read Archive (SRA) database under accession code PRJNA8068669.
Dataset 4 (Kelly cell line): the raw sequencing data are available in the European Nucleotide Archive (ENA) database under accession code PRJEB5051820.
Dataset 5 (medulloblastoma): the raw sequencing data are available in the Gene Expression Omnibus (GEO) repository under accession code GSE20517821.
Dataset 6 (muscle cells): the raw sequencing data are available in the Sequence Read Archive (SRA) database under accession code SRR631540013.
Dataset 7 (OVCAR8 cell line): the raw sequencing data are available in the Gene Expression Omnibus (GEO) repository under accession code GSE6864422.
We reanalyzed the data of EJM, JJN3, Kelly cell line, and muscle cells with corresponding pipelines in Supplementary Fig. 1. We used processed data of sperm cells, medulloblastoma and OVCAR8 cell line in their original paper. The processed template files can be found at [https://github.com/QuKunLab/eccDNABenchmarking/tree/main/ecsim/ecsim/resource/template].
All other data supporting the findings described in this paper are available in the article and its Supplementary Information files. Source data are provided with this paper.
Code availability
All original code has been deposited at Github [https://github.com/QuKunLab/eccDNABenchmarking]. We uploaded all codes and scripts used for the analyses and figure plotting in this study to a public Zenodo repository [https://zenodo.org/records/13769429]52.The simulated datasets can be generated by using the uploaded code.
References
Yi, E., Chamorro González, R., Henssen, A. G. & Verhaak, R. G. W. Extrachromosomal DNA amplifications in cancer. Nat. Rev. Genet. 23, 760–771 (2022).
Verhaak, R. G. W., Bafna, V. & Mischel, P. S. Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat. Rev. Cancer 19, 283–288 (2019).
Cox, D., Yuncken, C. & Spriggs, A. I. Minute chromatin bodies in malignant tumours of childhood. Lancet 1, 55–58 (1965).
Turner, K. M. et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125 (2017).
Paulsen, T., Shibata, Y., Kumar, P., Dillon, L. & Dutta, A. Small extrachromosomal circular DNAs, microDNA, produce short regulatory RNAs that suppress gene expression independent of canonical promoters. Nucleic Acids Res. 47, 4586–4596 (2019).
Noer, J. B., Hørsdal, O. K., Xiang, X., Luo, Y. & Regenberg, B. Extrachromosomal circular DNA in cancer: history, current knowledge, and methods. Trends Genet 38, 766–781 (2022).
Henriksen, R. A. et al. Circular DNA in the human germline and its association with recombination. Mol. Cell 82, 209–217.e207 (2022).
Deshpande, V. et al. Exploring the landscape of focal amplifications in cancer using ampliconarchitect. Nat. Commun. 10, 392 (2019).
Wanchai, V. et al. CReSIL: accurate identification of extrachromosomal circular DNA from long-read sequences. Brief Bioinform 23 https://doi.org/10.1093/bib/bbac422 (2022).
Kumar, P. et al. ATAC-seq identifies thousands of extrachromosomal circular DNA in cancer and cell lines. Sci. Adv. 6, eaba2489 (2020).
Koche, R. P. et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat. Genet. 52, 29–34 (2020).
Møller, H. D. Circle-seq: isolation and sequencing of chromosome-derived circular DNA elements in cells. Methods Mol. Biol. 2119, 165–181 (2020).
Møller, H. D. et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat. Commun. 9, 1069 (2018).
Wang, Y. et al. eccDNAs are apoptotic products with high innate immunostimulatory activity. Nature 599, 308–314 (2021).
Wang, Y., Wang, M. & Zhang, Y. Purification, full-length sequencing and genomic origin mapping of eccDNA. Nat. Protoc. 18, 683–699 (2023).
Prada-Luengo, I., Krogh, A., Maretty, L. & Regenberg, B. Sensitive detection of circular DNAs at single-nucleotide resolution using guided realignment of partially aligned reads. BMC Bioinforma. 20, 663 (2019).
Mann, L., Seibt, K. M., Weber, B. & Heitkam, T. ECCsplorer: a pipeline to detect extrachromosomal circular DNA (eccDNA) from next-generation sequencing data. BMC Bioinforma. 23, 40 (2022).
Zhang, P., Peng, H., Llauro, C., Bucher, E. & Mirouze, M. ecc_finder: a robust and accurate tool for detecting extrachromosomal circular DNA from sequencing data. Front Plant Sci. 12, 743742 (2021).
Norman, A. et al. An improved method for including upper size range plasmids in metamobilomes. PLOS ONE 9, e104405 (2014).
Tüns, A. I. et al. Detection and validation of circular DNA fragments using Nanopore sequencing. Front. Genet. 13, 867018 (2022).
Zhu, Y. et al. Whole-genome sequencing of extrachromosomal circular DNA of cerebrospinal fluid of medulloblastoma. Front. Oncol. 12, 934159 (2022).
Dillon, L. W. et al. Production of extrachromosomal microDNAs is linked to mismatch repair pathways and transcriptional activity. Cell Rep. 11, 1749–1759 (2015).
Liu, Y., Sun, J. & Zhao, M. ONGene: A literature-based database for human oncogenes. J. Genet. genomics 44, 119–121 (2017).
Thollet, A. et al. ZNF217 confers resistance to the pro-apoptotic signals of paclitaxel and aberrant expression of Aurora-A in breast cancer cells. Mol. Cancer 9, 291 (2010).
Hou, C. et al. Knockdown of Trio by CRISPR/Cas9 suppresses migration and invasion of cervical cancer cells. Oncol. Rep. 39, 795–801 (2018).
Mei, Q., Ye, L. J., Lin, H. & Chen, C. Y. CUL4A promotes the invasion of cervical cancer cells by regulating NF-κB signaling pathway. Eur. Rev. Med Pharm. Sci. 24, 10403–10409 (2020).
Wang, C. et al. C-Myc-activated long non-coding RNA PVT1 enhances the proliferation of cervical cancer cells by sponging miR-486-3p. J. Biochem 167, 565–575 (2020).
Wang, C. et al. Long non-coding RNA plasmacytoma variant translocation 1 gene promotes the development of cervical cancer via the NF-κB pathway. Mol. Med Rep. 20, 2433–2440 (2019).
Gao, Y. L. et al. Long noncoding RNA PVT1 facilitates cervical cancer progression via negative regulating of miR-424. Oncol. Res. 25, 1391–1398 (2017).
Yang, F. et al. Retrotransposons hijack alt-EJ for DNA replication and eccDNA biogenesis. Nature 620, 218–225 (2023).
Sun, H., Lu, X. & Zou, L. EccBase: a high-quality database for exploration and characterization of extrachromosomal circular DNAs in cancer. Computational Struct. Biotechnol. J. 21, 2591–2601 (2023).
Xi, R. et al. Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc. Natl Acad. Sci. 108, E1128–E1136 (2011).
Liu, Y. et al. DNA methylation-calling tools for oxford nanopore sequencing: a survey and human epigenome-wide evaluation. Genome Biol. 22, 295 (2021).
Sin, S. T. K. et al. Characteristics of fetal extrachromosomal circular DNA in maternal plasma: methylation Status and clearance. Clin. Chem. 67, 788–796 (2021).
Kim, H. et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat. Genet 52, 891–897 (2020).
Jiang, R., Yang, M., Zhang, S. & Huang, M. Advances in sequencing-based studies of microDNA and ecDNA: Databases, identification methods, and integration with single-cell analysis. Comput Struct. Biotechnol. J. 21, 3073–3080 (2023).
Lv, W. et al. Circle-Seq reveals genomic and disease-specific hallmarks in urinary cell-free extrachromosomal circular DNAs. Clin. Transl. Med. 12, e817 (2022).
Li, J. et al. Early detection of colorectal cancer based on circular DNA and common clinical detection indicators. World J. Gastrointest. Surg. 14, 833–848 (2022).
Huang, W., Li, L., Myers, J. R. & Marth, G. T. ART: a next-generation sequencing read simulator. Bioinformatics 28, 593–594 (2012).
Ono, Y., Asai, K. & Hamada, M. PBSIM2: a simulator for long-read sequencers with a novel generative model of quality scores. Bioinformatics 37, 589–595 (2020).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, 1–9 (2004).
Norrander, J., Kempe, T. & Messing, J. Construction of improved M13 vectors using oligodeoxynucleotide-directed mutagenesis. Gene 26, 101–106 (1983).
Boeva, V. et al. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 28, 423–425 (2012).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Nassar, L. R. et al. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 51, D1188–D1195 (2022).
Waskom, M. L. Seaborn: statistical data visualization. J. Open Source Softw. 6, 3021 (2021).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. methods 17, 261–272 (2020).
Chen, T. et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics, Proteom. Bioinforma. 19, 578–583 (2021).
Database resources of the national genomics data center, china national center for bioinformation in 2022. Nucleic Acids Research 50, D27–D38 (2022).
Liu, K. QuKunLab/eccDNABenchmarking: v1.0.1, https://doi.org/10.5281/zenodo.13769429 (2024).
Acknowledgements
We thank all members in the Qu laboratory for helpful discussions. We are grateful for the gift of NIH3T3 cells from Prof. Shu Zhu of the University of Science and Technology of China. This work was supported by the National Natural Science Foundation of China grants (T2125012 to K.Q., 32270978 to C.G., and 32100457 to J.F.), the National Key R&D Program of China (2020YFA0112200 and 2022YFA1303200 to K.Q.), the CAS Project for Young Scientists in Basic Research (YSBR-005 to K.Q.), Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB0940301 to K.Q.), USTC Research Funds of the Double First-Class Initiative (YD9100002032 to K.Q.) and the Fundamental Research Funds for the Central Universities (YD2070002019, WK9110000141, and WK2070000158 to K.Q.). We thank the USTC Supercomputing Center and the School of Life Science Bioinformatics Center for providing computing resources for this project. Declaration of generative AI and AI-assisted technologies in the writing process. During the preparation of this work the authors used ChatGPT 3.5 and ChatGPT 4.0 in order to improve the language and readability. After using these tools, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Author information
Authors and Affiliations
Contributions
K.Q. and J. F. conceived the project. X.G., K.L., S.L., S.Z.L., and J.F. designed the framework. X.G. and S.L. performed all the wet-lab experiments with the help of M.T, S.Z.L., Y.H., and C.G.; K.L. and S.L. performed all the bioinformatics analysis with the help of N.L. and C.J.; X.G., K.Q., K.L., C.G., and S.L. wrote the manuscript with inputs from all authors. K.Q. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
Jingwen Fang is the chief executive officer of HanGen Biotech. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gao, X., Liu, K., Luo, S. et al. Comparative analysis of methodologies for detecting extrachromosomal circular DNA. Nat Commun 15, 9208 (2024). https://doi.org/10.1038/s41467-024-53496-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53496-8
This article is cited by
-
Detection of primary cancer types via fragment size selection in circulating cell-free extrachromosomal circular DNA
Genome Medicine (2026)
-
Adaptive dynamics of extrachromosomal circular DNA in rice under nutrient stress
Nature Communications (2025)
-
Unveiling the mysteries of extrachromosomal circular DNA: from generation to clinical relevance in human cancers and health
Molecular Cancer (2024)
