
Abstract
C-Glycosides are essential for the study of biological processes and the development of carbohydrate-based drugs. Despite the tremendous hurdles, glycochemists have often fantasized about the efficient, highly stereoselective synthesis of C-glycosides with the shortest steps under mild conditions. Herein, we report a desulfurative radical protocol to synthesize C-alkyl glycosides and coumarin C-glycosides under visible-light induced conditions without the need of an extra photocatalyst, in which stable and readily available glycosyl thiols that could be readily obtained from native sugars are activated in situ by pentafluoropyridine. The benefits of this procedure include high stereoselectivity, broad substrate scope, and easy handling. Mechanistic studies indicate that the in situ produced tetrafluoropyridyl S-glycosides form key electron donor–acceptor (EDA) complexes with Hantzsch ester (for C-alkyl glycosides) or Et3N (for coumarin C-glycosides), which, upon irradiation with visible light, trigger a cascade of glycosyl radical processes to access C-glycosides smoothly.
Similar content being viewed by others
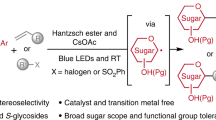
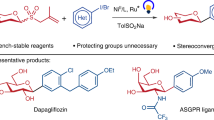
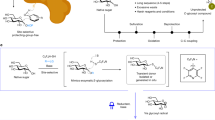
Introduction
C-Glycosides are an important class of carbohydrate scaffolds that are widely present in a variety of naturally occurring bioactive molecules and marketed drugs (Fig. 1a)1,2,3,4,5. Moreover, compared to native O-glycosides and N-glycosides, the C-glycoside analogs achieved by replacing O/N atoms in native glycosides with a carbon atom usually show better enzymatic and acidic stability; thus, this strategy is commonly applied in carbohydrate-based drug discoveries3,5,6.

a Representative examples C-glycosides as marketed drugs and bioactive compounds. b The general strategy for desulfurative glycosylation. c This work: visible light-promoted direct desulfurization of glycosyl thiols to synthesize C-glycosides. SGLT2 sodium glucose cotransporter-2, PG protective group, LG leaving group, [O] oxidation, [H] reduction, HE Hantzsch ester, EDA electron donor–acceptor.
In light of the numerous potential applications, developing practical strategies for the synthesis of C-glycosides has received continual attention1,2,3,4,5,7,8,9,10,11. Among these, the synthesis of C-alkyl glycosides via glycosyl radical strategies has mushroomed enormously in recent years12,13,14,15,16,17. Within this context, various glycosyl precursors appended with different leaving groups to assist in the generation of glycosyl radicals, which in turn facilitates cross-coupling reactions with alkene partners, have been developed over the years. In particular, recent key breakthroughs by Niu12, Koh18,19,20, Liu21, and other groups22,23,24,25 have made it possible to accomplish C-alkyl glycosylation under much milder conditions with greater functional group flexibility, providing useful tools for overcoming a variety of synthetic problems. In many instances, however, the current approaches regularly encounter issues with the utilization of unstable (such as glycosyl halides) or hard-to-access glycosyl precursors (such as glycosyl sulfones/sulfoxides and glycosyl xanthates), as well as the reliance on costly activators or initiators under harsh reaction conditions. Therefore, it remains highly desirable to develop an efficient C-alkyl glycosylation process that uses readily available yet stable glycosyl donors with a shortened synthetic pathway, and in the absence of expensive activators or initiators.
Additionally, S-glycosides are among the most frequently used glycosyl donors in carbohydrate chemistry12,26,27,28,29. The conventional desulfurative glycosylations12,30,31,32,33,34,35,36 involve multistep to prepare S-glycosides as glycosyl donors, followed by activation and coupling with glycosyl acceptors (Fig. 1b, top). In contrast, glycosyl thiols could be readily produced from native sugars in one to three steps, which makes the desulfurative glycosylation of glycosyl thiols much more straightforward. Despite being underexplored, glycosyl thiols could be activated directly to couple with acceptors, thus obviating additional C1 modification steps and would significantly improving the efficiency of glycosylation (Fig. 1b, bottom). Encouraged by recent advances in photocatalytic desulfurative coupling reactions37,38,39,40, and due to our ongoing pursuits of C-glycoside synthesis41,42 and desulflization process43. We sought to develop a desulfurative radical C-glycosylation direct from glycosyl thiols 1 (Fig. 1c). In this view, we envisioned that glycosyl radicals could be generated from glycosyl thiols 1 by in situ activation with fluoroarenes under mild photocatalytic conditions. Subsequently, it can be captured by alkenes 2 to prepare a series of C-alkyl glycosides. Mechanistically, the key to the success of such a reaction lies in the rapid generation of activated glycosyl radical precursors, while avoiding the formation of undesired S-glycosides 7 through a thiol-click reaction44. Herein, we report a visible light-mediated desulfurative radical C-glycosylation process to prepare a series of C-alkyl glycosides and coumarin C-glycosides under mild conditions, using readily available and bench-stable glycosyl thiols as glycosyl radical precursors. We found that the in situ produced tetrafluoropyridyl S-glycosides 3 from glycosyl thiols 1 and pentafluoropyridine formed electron donor–acceptor (EDA) complexes with Hantzsch ester or with Et3N in the absence of a Hantzsch ester, which under visible light irradiation generated glycosyl radicals 4. The produced glycosyl radicals could be trapped by activated alkenes and coumarins delivering C-alkyl glycosides 5 and coumarin C-glycosides 6, respectively. During preparing this manuscript, M. J. Koh, B. G. Davis, and coworkers revealed a method for the direct functionalization of native sugars, in which unprotected sugars was converted into the corresponding tetrafluoropyridyl S-glycosides with highly excessive amount of hygroscopic 2-chloro-1,3-dimethylimidazolinium chloride (DMC) (4.0 equiv.) and 2,3,5,6-tetraflupropyridine-4-thiol (5.0 equiv.) (prepared from pentafluoropyridine). The tetrafluoropyridyl S-glycosides were then converted into glycosyl radicals that were trapped by activated alkenes and disulfides to generate the corresponding C-glycosides and S-glycosides20. While employing the similar tetrafluoropyridyl S-glycoside intermediates as to Koh and Davis’s, our approach offers the following benefits: (i) easy handling and glove-box free, (ii) wider substrates scope both in α,β-unsaturated compounds and sugars (furanoses, pyranoses, and 2-amino-2-deoxypyranoses); (iii) readily performed on gram scale (up to 4.7 mmol) without loss of the yield and stereoselectivity; and (iv) much less reaction reagents were employed. Besides, we could smoothly achieve direct C–H glycosylation of coumarins in the absence of a Hantzsch ester and a photocatalyst, which is notable because conventional C–H glycosylation requires pre-installation of an extra directing group into the glycosyl acceptors to maintain high regioselectivity and efficiency.
Results and discussion
Investigation of the reaction conditions
With the consideration of directly activating glycosyl thiols to form glycosyl radicals in mind, we selected glucosyl thiol 8 and phenyl acrylate 9 as model substrates to optimization of the reaction conditions. By carefully tuning the reactivity of different fluorobenzenes and fluoropyridines, we were pleased to find out that pentafluoropyridine (A1) was the best (see Supplementary Table 1 for details). After further optimization, we found that alkyl C-glycoside 10 could be obtained in 85% yield with a more than 10:1 α:β stereoselectivity when pentafluoropyridine was employed as an activator, using Et3N as a base, Hantzsch ester as a hydrogen donor and MeCN as the solvent, and under the irradiation of a 15 W 440 nm blue LEDs without the need of a typical photocatalyst (Table 1, entry 1). For comparison, we also evaluated the influence of various solvents, and found that acetonitrile was the optimal solvent for this transformation (entries 2–4). Switching the solvent to THF slightly decreased the yield of 10 (entry 4). This is probably due to the poor solubility of Hantzsch ester in THF. Among the various organic and inorganic bases that have been investigated, Et3N was the most efficient one (entries 5–7). This reaction hardly took place in the absence of base, Hantzsch ester (entries 8, 9), or light (entry 11), or even when the reaction was exposed to air (entry 10). Therefore, it appears that the base, Hantzsch ester, and light are all integral to this transformation. Notably, this method does not require the isolation or workup of reactive glycosyl intermediate 3, and the stereochemistry of starting glycosyl thiol 8 (β:α < 7:1) does not affect the stereoselectivity of product 10 (α:β > 10:1), making it the preferred choice.
Synthesis of C-alkyl glycosides
Having identified the optimal reaction conditions, we next explored the scope of this transformation. We found that an array of bench-stable glycosyl thiols were competent substrates in our reaction, affording smoothly the corresponding C-alkyl glycosides (Fig. 2). Glycosyl thiols derived from monosaccharides, including glucose (10), galactose (12), mannose (13), glu-NAc (14), gal-NAc (15), rhamnose (16), fucose (17) and arabinose (18) participated smoothly in this reaction, and afforded the corresponding products in 32% to 80% yields with very good α-selectivity (α:β > 10:1 to α-only). Furanosyl thiols from d-ribofuranose, and d-mannofuranose were also successfully converted into C-alkyl glycosides 19 and 20 in synthetically useful yields with only one stereoisomer (α-only or β-only). Glycosyl thiols from disaccharides such as lactose, maltose, and cellobiose produced the desired products (21–23) in high yields (62% to 82%) and stereoselectivities (α:β > 10:1 to α:β > 19:1) as well. It was found that the protecting group of glycosyl substrates is not limited to the electron-withdrawing groups such as acetyl or benzoyl, as benzyl-protected glycosyl thiol produced the product 24 in even higher yield than the corresponding acetyl one (72% yield in 10 vs 82% yield in 24). Notably, this reaction could be carried out on a gram scale (with 4.7 mmol of glucosyl thiol) without significantly affecting the reaction efficiency and stereoselectivity (10, 71% yield, 1.6 g, α:β > 10:1).

a Reaction conditions: 1 (0.20 mmol), 9 (0.40 mmol), in MeCN (2.0 mL) under 440 nm illumination and argon atmosphere for 8 h. b DMSO instead of MeCN. Isolated yields were reported. HE Hantzsch ester.
The scope of the transformation was further examined by employing mannosyl thiol 25 as a glycosyl donor to investigate the scope of activated alkenes. As shown in Fig. 3, a wide variety of α,β-unsaturated compounds, including methyl acrylate, vinyl-sulfone, diethyl vinylphosphonate, acrylonitrile, acrylamides, and vinyl phenyl ketone, reacted well with mannosyl thiol 25, and the corresponding products (28–35) formed in good yields and excellent α-selectivity. Methyl 2-benzylacrylate was also successfully employed in this protocol, delivering the desired product (36) in excellent yield with exclusive α-selectivity, even though a 1.5:1 ratio of diastereoisomers was obtained in the radical hydrogen abstraction step. Notably, the method could also be leveraged to access C-linked disaccharide derivative 37 in 78% yield with exclusive α-selectivity and diastereoselectivity45,46. Moreover, activated alkenes jointed with sugars (38–41), amino acids (42, 43), and peptides (44, 45) were also successfully transformed into C-glycosylated products in good to excellent yields. Among them, the produced C-glycosyl peptides may potentially be utilized in the investigation of in vivo peptide stabilization or the development of glycopeptide drugs47.

a Reaction conditions: 25 (0.20 mmol), 26 (0.40 mmol), in MeCN (2.0 mL) under 440 nm illumination and argon atmosphere for 8 h. Isolated yields were reported. HE Hantzsch ester, dr diastereomeric ratio, Phe phenylalanine, Pro proline, Tyr tyrosine, Gly glycine.
In order to illustrate the synthetic significance of this methodology, we investigated C-glycosylation of bioactive molecules. The methacrylate ester derivatives from (±)-Menthol, Estrone, and Podophyllotoxin were subjected to the standard conditions to react with mannosyl thiol 25, producing the corresponding products (46, 47 and 48) in 72% to 83% yields, and exclusive α-selectivity. Britanin, a sesquiterpene lactone with excellent anti-infammatory, antioxidant, and antitumor activities48, was encompassed into this strategy, and afforded the C-glycosylated product 49 in 72% yield with exclusive α-selectivity as well. Additionally, the methacrylate of Diosgenin also reacted successfully with thiol 25, producing the C-glycoside 50 in satisfactory yield. It is worth mentioning that the presence of an additional free hydroxyl group (49) or double bond (50) did not thwart this transformation, which further highlighted the excellent functional group tolerance of this protocol.
C–H Glycosylation of coumarins
As one of our thrilling endeavors is establishing practical methodologies to enable the direct functionalization of natural products and on-market medicines49, in this regard, we tried to investigate the possibility of using this protocol to functionalize important natural products. Interestingly, when coumarin was explored as a glucosyl acceptor under standard reaction conditions, the product 53 from direct C–H glycosylation of coumarin was isolated in around 16% yield. Despite directing group-mediated C−H glycosylation has been extensively employed in the synthesis of C-glycosides8,50,51,52,53,54,55,56,57,58, the direct late-stage glycosylation of the bioactive natural product in our case is notable. Further optimization showed that the coumarin C-glycoside product (53) could be isolated in 65% yield when the reaction was carried out in the absence of Hantzsch ester with only an excess amount of Et3N (5.0 equiv.) (see Supplementary Table 4 for details). It is also worth noting that even though coumarin C-glycosides are uncommon in nature, their synthesis has gained increasing interest because of the significance of coumarins in both natural products and clinical drugs59,60,61,62. To assess the practicality of this direct C–H glycosylation process, different glycosyl thiols and coumarins were engaged in the reactions to synthesize coumarin C-glycosides (Fig. 4). Glycosyl thiols derived from monosaccharides (53–59), furanose (60) and disaccharides (61–63) are all eligible substrates for this transformation, produced the corresponding products in 43%-80% yields with excellent α-stereoselectivity. The glycosylation of π-extended coumarins (64–67) with interesting photophysical and biological properties63 also proceeded smoothly to afford the desired adducts. This reaction was effective for a variety of coumarins with methyl (68, 72), ester (69), alkoxy (73–75), free hydroxyl (76), and amide (77) groups at the C-6 or C-7 position of coumarin. The corresponding products were generated in 50–75% yields. Gratifyingly, halogen-substituted coumarins (70, 71, 78) were also found to be suitable for this transformation, which provided useful components for further transformations through traditional cross-coupling reactions.

a Reaction conditions: 1a (0.20 mmol), 51 (0.40 mmol), in DCM (2.0 mL) under 420 nm illumination and an argon atmosphere for 24 h. b Et3N (8.0 equiv.), 36 h. c pentafluoropyridine (3.0 equiv.), Et3N (8.0 equiv.), 36 h. d DMSO instead of DCM. Isolated yields were reported. DCM dichloromethane.
Mechanistic studies
To gain further insight into the reaction mechanism, several control experiments were conducted. First, the presence of the glycosyl radical intermediate was confirmed by trapping it with vinylcyclopropane 80, resulting in the formation of the ring-ruptured adduct 81 in 30% yield (Fig. 5a top). Furthermore, the addition of TEMPO completely inhibited the formation of C-glycosides (Fig. 5a bottom), and the TEMPO-trapped-adduct S-31 was detected by HRMS (see Supplementary Fig. 6), indicating that the reaction might follow a radical pathway. Combination of these outcomes with the control experiments mentioned in the section on optimization (Table 1, entries 8–11), it is highly possible that C-glycosylation most likely proceeds via a glycosyl radical mechanism.

a Radical trapping experiments. b Control experiments. c UV-vis absorption spectroscopy (synthesis of C-alkyl glycosides). d UV-vis absorption spectroscopy (C–H glycosylation of coumarins). condition A: pentafluoropyridine (1.5 equiv.), Et3N (3.0 equiv.), HE (2.0 equiv.), MeCN, blue LEDs (440 nm). condition B: pentafluoropyridine (1.5 equiv.), Et3N (5.0 equiv.), DCM, blue LEDs (420 nm). TEMPO 2,2,6,6-tetramethylpiperidinooxy, ND not detected, HE Hantzsch ester, DCM dichloromethane, TEA triethylamine.
Moreover, upon quenching the reaction in 0.5 h, we could isolate the pyridyl S-glucoside 82 in around 50% yield. Starting from pyridyl S-glucoside 82, under otherwise identical conditions (A or B) but without the addition of perfluoropyridine, target products 10 and 53 were obtained in 80% and 45% yields, respectively (Fig. 5b). Furthermore, in the absence of perfluoropyridine, glucosyl thiol 8 could react smoothly with phenyl acrylate to generate thiol-Michael addition product S-1 in almost quantitative yield (see Supplementary Fig. 8). It was found that in our reaction system, the rate of nucleophilic substitution of glycosyl thiol 1 with perfluoropyridine was much faster than that of the thiol-click reaction with olefin 2, thus inhibiting the formation of the undesired product 7 (see Supplementary Fig. 9). A light-on-off experiment was then conducted between glucosyl thiol 8 and phenyl acrylate 9 under standard conditions to ascertain whether a radical chain process was involved in the reaction (see Supplementary Fig. 10). No conversion occurred in the absence of light, suggesting that continuous illumination is essential and that a radical chain mechanism could be ruled out in this transformation.
These results show that continuous light triggered the in situ-formed pyridyl S-glycoside 3 to generate glycosyl radicals, which is an essential step in this transformation.
Since this process does not require an external photocatalyst, we next used UV-Vis absorption spectroscopy to identify the possible “photocatalyst” for the visible light-induced transformation. First, under standard C-alkyl glycosylation conditions but with DMSO instead of CH3CN as the solvent due to the poor solubility of Hantzsch ester in the latter (Fig. 5c), neither Et3N nor pyridyl S-glucoside 82 exhibited absorption in the visible light region (> 400 nm) (lines a and b), although a mixture of 82 and Et3N did result in a slight redshift (line d). In contrast, Hantzsch ester itself in DMSO exhibited greater absorbance (line c), which, upon treatment with Et3N, showed no noticeable changes (line f)20. Additionally, a light-yellow solution and a noticeable bathochromic shift in absorbance (line e) were observed when pyridyl S-glucoside 82 and Hantzsch ester were mixed in DMSO, which suggests that an EDA complex could be formed between the two substances. In contrast, despite a mixture of 82, Hantzsch ester and Et3N in DMSO did produce a bright yellow solution, we did not observe a significant change in the absorption spectrum (line g). Based on these results, it is reasonable that the transformation in our system relies more on the EDA complex formed between pyridyl S-glucoside 82 (as an electron acceptor) and Hantzsch ester (as an electron donor) rather than it does on the interaction between Et3N and S-glucoside 82. However, under the conditions for the preparation of coumarin C-glycosides, wherein Hantzsch ester is absent, Et3N should instead of Hantzsch ester as an electron donor to form EDA complex with pyridyl S-glucoside 82 to trigger the produce of glycosyl radicals. Indeed, a mixture of Et3N and pyridyl S-glucoside 82 in DCM showed a noticeable bathochromic shift in absorbance (Fig. 5d, line 6), and the absorbance intensities of this mixture increased as the reaction time extended (Fig. 5d, line 8).
With the above information, a plausible reaction mechanism for visible light-induced C-glycoside synthesis was rationalized as depicted in Fig. 6. For the preparation of C-alkyl glycosides 5, following path-A, in situ generated pyridyl S-glycoside 3 and Hantzsch ester forms an EDA complex I, which is capable of absorbing visible light. Upon illumination with blue LEDs, a photoinduced single-electron transfer occurs from excited state EDA complex I to give glycosyl radical 4 (stabilized by anomeric effects)14,15,64, which is accompanied by the formation of N-radical II and the departure of aryl thiolate anion III (detected by HRMS in the reaction mixture, see Supplementary Fig. 14). The glycosyl radical 4 is then trapped by alkene 2 to generate a new alkyl radical IV, which is readily hydrogenated by N-radical II65,66 (see Supplementary Figs. 15 and 16 for detail studies on the source of the proton) to deliver the desired C-alkyl glycoside 5 with high anomeric selectivity. Additionally, following path-B for the preparation of coumarin C-glycosides 6, pyridyl S-glycoside 3, and Et3N would form an EDA complex V. Upon irradiation with blue LEDs, a photoinduced single-electron transfer occurs from excited state V to give glycosyl radical 4 accompanied by the formation of Et3N+• VI. The glycosyl radical 4 is then trapped by coumarin 51 to generate the second alkyl radical VII. It is well-established that triethylamine (Et3N+•) could serve as a single-electron oxidant (Et3N: E1/2Ox = 0.78 V vs. SCE)67,68,69, therefore, the subsequent single-electron oxidation (by Et3N+• VI) and dehydrogenation (by Et3N) would generate the coumarin C-glycoside 6. Overall, in the whole process for the synthesis of coumarin C-glycosides from glycosyl thiols, Et3N plays three distinct roles: (i) as a base for the preparation of pyridyl S-glycoside intermediate 3, (ii) as an electron donor for the formation of an EDA complex with 3, and (iii) as an oxidant to facilitate the formation of the final product by converting benzyl radical VII into a cation.

EDA electron donor–acceptor.
In summary, we have developed a visible light-induced desulfurative radical protocol for the preparation of C(sp3)/C(sp2)-glycosides from readily available and bench-stable glycosyl thiols in the presence of perfluoropyridine. The reaction proceeds through the tetrafluoropyridyl S-glycoside intermediate, which forms EDA complexes with Hantzsch ester (for C(sp3)-glycosides) or Et3N (for C(sp2)-glycosides). Upon illumination with visible light, these EDA complexes could smoothly produce glycosyl radicals, which could be trapped by activated alkenes, including acrylates, vinyl-sulfones, vinylphosphonates, acrylonitrile, acrylamides, vinyl phenyl ketone, and coumarins. This protocol provides a mild way for the efficient and highly stereoselective synthesis of various C-glycosides. Moreover, the synthetic utility was showcased through the late-stage functionalization of bioactive compounds and pharmaceuticals. The significant features of the protocol include the use of bench-stable and readily available glycosyl radical precursors, easy handling, mild conditions, photocatalyst-free, and readily performed on a gram scale. Preliminary mechanistic studies provide insights into the reaction pathway and suggest that the formation of an EDA complex is critical for maintaining the efficiency of the reaction.
Methods
General procedure for visible light-promoted direct desulfurization of glycosyl thiols to access C-alkyl glycosides
To an oven-dried test tube (10 mL) equipped with a magnetic stir bar, glycosyl thiol 1 (0.2 mmol, 1.0 equiv.), olefin 26 (0.4 mmol, 2.0 equiv.) and Hantzsch ester (HE) (0.4 mmol, 2.0 equiv.) were added. The tube was capped with a rubber septum. After being evacuated and backfilled with argon three times, pentafluoropyridine (0.3 mmol, 1.5 equiv.) and Et3N (0.6 mmol, 3.0 equiv.) were dissolved in MeCN (0.10 M, 2.0 mL) and then added to the tube via a syringe. The tube was irradiated with 15 W blue LEDs (440 nm) in the photoreactor for the indicated time at room temperature. The reaction mixture was monitored by TLC. After the disappearance of the starting material, the reaction mixture was directly concentrated in vacuo, and the residue was subjected to silica gel chromatography to obtain the C-alkyl glycosides 5.
General procedure for visible light-promoted direct desulfurization of glycosyl thiols to access coumarin C-glycosides
To an oven-dried test tube (10 mL) equipped with a magnetic stir bar, glycosyl thiol 1 (0.2 mmol, 1.0 equiv.) and coumarin 51 (0.4 mmol, 2.0 equiv.) were added. The tube was capped with a rubber septum. After being evacuated and backfilled with argon three times, pentafluoropyridine (0.3 mmol, 1.5 equiv.) and Et3N (1.0 mmol, 5.0 equiv.) were dissolved in DCM (0.10 M, 2.0 mL) and then added to the tube via a syringe. The tube was irradiated with 15 W blue LEDs (420 nm) in the photoreactor for the indicated time at room temperature. The reaction mixture was monitored by TLC. After the disappearance of the starting material, the reaction mixture was directly concentrated in vacuo, and the residue was subjected to silica gel chromatography to give coumarin C-glycosides 6.
Data availability
The data supporting the findings of this study are available within this article and its Supplementary Information, which contains experimental details, mechanism studies, characterization data, copies of NMR spectra for all new compounds. All other data are available from the authors upon request.
References
Yang, Y. & Yu, B. Recent advances in the chemical synthesis of C-glycosides. Chem. Rev. 117, 12281–12356 (2017).
Kitamura, K., Ando, Y., Matsumoto, T. & Suzuki, K. Total synthesis of aryl C-glycoside natural products: strategies and tactics. Chem. Rev. 118, 1495–1598 (2018).
Liao, H., Ma, J., Yao, H. & Liu, X.-W. Recent progress of C-glycosylation methods in the total synthesis of natural products and pharmaceuticals. Org. Biomol. Chem. 16, 1791–1806 (2018).
Yang, G., Schmieg, J., Tsuji, M. & Franck, R. W. The C-glycoside analogue of the immunostimulant α-galactosylceramide (KRN7000): Synthesis and striking enhancement of activity. Angew. Chem. Int. Ed. 43, 3818–3822 (2004).
Chao, E. C. & Henry, R. R. SGLT2 inhibition-a novel strategy for diabetes treatment. Nat. Rev. Drug Discov. 9, 551–559 (2010).
Cao, X. et al. Carbohydrate-based drugs launched during 2000−2021. Acta Pharm. Sin. B 12, 3783–3821 (2022).
Wei, Y., Lin, L. Q. H., Lee, B. C. & Koh, M. J. Recent advances in first-row transition metal-catalyzed reductive coupling reactions for π-bond functionalization and C-glycosylation. Acc. Chem. Res. 56, 3292–3312 (2023).
Ghouilem, J., de Robichon, M., Le Bideau, F., Ferry, A. & Messaoudi, S. Emerging organometallic methods for the synthesis of C-branched (hetero)aryl, alkenyl, and alkyl glycosides: C−H functionalization and dual photoredox approaches. Chem. Eur. J. 27, 491–511 (2021).
Chen, A., Yang, B., Zhou, Z. & Zhu, F. Recent advances in transition-metal-catalyzed glycosyl cross-coupling reactions. Chem. Catal. 2, 3430–3470 (2022).
Das, R., Das, M., Mukherjee, D. & Kundu, T. Recent advances on the synthesis of C-glycosides from 1,2-glycals. Synthesis 56, 1070–1096 (2024).
Parida, S. P. et al. Recent advances on synthesis of C-glycosides. Carbohydr. Res. 530, 108856 (2023).
Shang, W. & Niu, D. Radical pathway glycosylation empowered by bench-stable glycosyl donors. Acc. Chem. Res. 56, 2473–2488 (2023).
Ghosh, T. & Nokami, T. Recent development of stereoselective C-glycosylation via generation of glycosyl radical. Carbohydr. Res. 522, 108677 (2022).
Xu, L.-Y., Fan, N.-L. & Hu, X.-G. Recent development in the synthesis of C-glycosides involving glycosyl radicals. Org. Biomol. Chem. 18, 5095–5109 (2020).
Chen, A. et al. Recent advances in glycosylation involving novel anomeric radical precursors. J. Carbohydr. Chem. 40, 361–400 (2022).
Jiang, Y., Zhang, Y., Lee, B. C. & Koh, M. J. Diversification of glycosyl compounds via glycosyl radicals. Angew. Chem. Int. Ed. 62, e202305138 (2023).
Wang, C., Qi, R. & Xu, Z. Glycosyl radical-based synthesis of C-glycoamino acids and C-glycopeptides. Chem. Eur. J. 29, e202203689 (2023).
Wei, Y., Wang, Q. & Koh, M. J. A photoinduced, nickel-catalyzed reaction for the stereoselective assembly of C-linked glycosides and glycopeptides. Angew. Chem. Int. Ed. 62, e202214247 (2023).
Wang, Q. et al. Visible light activation enables desulfonylative cross-coupling of glycosyl sulfones. Nat. Synth. 1, 967–974 (2022).
Jiang, Y. et al. Direct radical functionalization of native sugars. Nature 631, 319–327 (2024).
Yu, C. et al. Direct construction of C-alkyl glycosides from non-activated olefins via nickel-catalyzed C(sp3)-C(sp3) coupling reaction. Adv. Sci. 11, 2307226 (2024).
Jiao, R. et al. Visible-light-mediated synthesis of C-alkyl glycosides via glycosyl radical addition and aryl migration. Org. Lett. 25, 6099–6104 (2023).
Li, C., Zhao, Z. & Yu, S. Photoredox-catalyzed C-glycosylation of peptides with glycosyl bromides. Chin. Chem. Lett. 35, 109128 (2024).
Miura, T., Yoritate, M. & Hirai, G. Photoredox-catalyzed protecting-group-free C-glycosylation with glycosyl sulfinate via the Giese reaction. Chem. Commun. 59, 8564–8567 (2023).
Chen, A. et al. Stereoselective alkyl C-glycosylation of glycosyl esters via anomeric C–O bond homolysis: efficient access to C-glycosyl amino acids and C-glycosyl peptides. Chem. Sci. 14, 7569–7580 (2023).
Zeng, J. et al. Glycosyl sulfoxides in glycosylation reactions. Top. Curr. Chem. 376, 27–32 (2018).
Panza, M., Pistorio, S. G., Stine, K. J. & Demchenko, A. V. Automated chemical oligosaccharide synthesis: novel approach to traditional challenges. Chem. Rev. 118, 8105–8150 (2018).
Zhong, W. & Boons, G.-J. In Handbook of Chemical Glycosylation (ed. Demchenko, A. V.) (Wiley-VCH, 2008).
Pedersen, M. J. & Pedersen, C. M. Reactivity, selectivity, and synthesis of 4-C-silylated glycosyl donors and 4-deoxy analogues. Angew. Chem. Int. Ed. 60, 2689–2693 (2021).
Cai, L., Meng, L., Zeng, J. & Wan, Q. Sequential activation of thioglycosides enables one-pot glycosylation. Org. Chem. Front. 8, 3150–3165 (2021).
Deng, L.-F. et al. Palladium catalysis enables cross-coupling–like SN2-glycosylation of phenols. Science 382, 928–935 (2023).
Meng, L. et al. Glycosylation enabled by successive rhodium(II) and brønsted acid catalysis. J. Am. Chem. Soc. 141, 11775–11780 (2019).
Sun, Q. et al. N-Glycoside synthesis through combined copper- and photoredox-catalysed N-glycosylation of N-nucleophiles. Nat. Synth. 3, 623–632 (2024).
Li, G., Xiong, D.-C. & Ye, X.-S. Synthesis of α-C-glycosides by samarium diiodide mediated coupling of glycosyl pyridyl sulfones with alkenes. Synlett 16, 2410–2414 (2011).
Mazéas, D., Skrydstrup, T. & Beau, J. M. A highly stereoselective synthesis of 1,2-trans-C-glycosides via glycosyl samarium(III) compounds. Angew. Chem. Int. Ed. 34, 909–912 (1995).
Kancharla, K., Navuluri, C. & Crich, D. Dissecting the influence of oxazolidinones and cyclic carbonates in sialic acid chemistry. Angew. Chem. Int. Ed. 51, 11105–11109 (2012).
Fu, X.-P. et al. Stereoretentive post-translational protein editing. ACS Cent. Sci. 9, 405–416 (2023).
Zhou, J., Liu, Y. & Sun, Z. LADA strategy for the synthesis of unnatural amino acids and direct modifications of peptides. Sci. China Chem. 66, 1788–1794 (2023).
Panferova, L. I., Zubkov, M. O., Kokorekin, V. A., Levin, V. V. & Dilman, A. D. Using the thiyl radical for aliphatic hydrogen-atom transfer: thiolation of unactivated C−H bonds. Angew. Chem. Int. Ed. 60, 2849–2854 (2021).
Zubkov, M. O., Kosobokov, M. D., Levin, V. V. & Dilman, A. D. Photocatalyzed decarboxylative thiolation of carboxylic acids enabled by fluorinated disulfide. Org. Lett. 24, 2354–2358 (2022).
Zhang, L., Zeng, W., Xie, D., Li, J. & Ma, X. Nickel and chiral phosphoric acid cocatalysis enables synthesis of C-acyl glycosides. Org. Lett. 26, 1332–1337 (2024).
Li, J. et al. Controllable 1,3-bis-functionalization of 2-nitroglycals with high regioselectivity and stereoselectivity enabled by a H-bond catalyst. JACS Au 4, 974–984 (2024).
Ma, X., Liu, Y., Du, L., Zhou, J. & Markó, I. E. Post-functionalization of dibenzothiophene to functionalized biphenyls via a photoinduced thia-Baeyer-Villiger oxidation. Nat. Commun. 11, 914 (2020).
Hoyle, C. E. & Bowman, C. N. Thiol–ene click chemistry. Angew. Chem. Int. Ed. 49, 1540–1573 (2010).
Giese, B. & Witzel, T. Synthesis of “C-disaccharides” by radical C-C bond formation. Angew. Chem. Int. Ed. 25, 450–451 (1986).
Giese, B., Hoch, M., Lamberth, C. & Schmidt, R. R. Synthesis of methylene bridged C-disaccharides. Tetrahedron Lett. 29, 1375–1378 (1988).
Negri, L., Lattanzi, R., Tabacco, F., Scolaro, B. & Rocchi, R. Glycodermorphins: opioid peptides with potent and prolonged analgesic activity and enhanced blood–brain barrier penetration. Br. J. Pharmacol. 124, 1516–1522 (1998).
Li, K., Zhou, Y., Chen, Y., Zhou, L. & Liang, J. A novel natural product, britanin, inhibits tumor growth of pancreatic cancer by suppressing nuclear factor-κB activation. Cancer Chemother. Pharm. 85, 699–709 (2020).
Wang, A., Liu, Y., Shen, Z., Qiao, Z. & Ma, X. Regioselective synthesis of pyrazolo[1,5-a]pyridine via TEMPO-mediated [3+2] annulation–aromatization of N-aminopyridines and α,β-unsaturated compounds. Org. Lett. 24, 1454–1459 (2022).
Gou, X. et al. Ruthenium-catalyzed stereo- and site-selective ortho- and meta-C−H glycosylation and mechanistic studies. Angew. Chem. Int. Ed. 61, e202205656 (2022).
Qi, R. et al. Visible-light-promoted stereoselective C(sp3)−H glycosylation for the synthesis of C-glycoamino acids and C-glycopeptides. Angew. Chem. Int. Ed. 61, e202200822 (2022).
Wu, J. et al. Remote C−H glycosylation by ruthenium(II) catalysis: modular assembly of meta-C-Aryl glycosides. Angew. Chem. Int. Ed. 61, e202208620 (2022).
Wu, J., Kopp, A. & Ackermann, L. Synthesis of C-oligosaccharides through versatile C(sp3)−H glycosylation of glycosides. Angew. Chem. Int. Ed. 61, e202114993 (2022).
Wu, J., Wei, W., Pöhlmann, J., Purushothaman, R. & Ackermann, L. Domino meta-C−H ethyl glycosylation by ruthenium(II/III) catalysis: modular assembly of meta-C-alkyl glycosides. Angew. Chem. Int. Ed. 62, e202219319 (2023).
Wang, S. et al. C–H glycosylation of native carboxylic acids: discovery of antidiabetic SGLT-2 inhibitors. ACS Cent. Sci. 9, 1129–1139 (2023).
Lv, W., Chen, Y., Wen, S., Ba, D. & Cheng, G. Modular and stereoselective synthesis of C-aryl glycosides via catellani reaction. J. Am. Chem. Soc. 142, 14864–14870 (2020).
Sun, Q. et al. Stereoselective synthesis of C-vinyl glycosides via palladium-catalyzed C−H glycosylation of alkenes. Angew. Chem. Int. Ed. 60, 19620–19625 (2021).
An, Y. et al. Palladium-catalyzed C–H glycosylation and retro Diels–Alder tandem reaction via structurally modified norbornadienes (smNBDs). Chem. Sci. 12, 13144–13150 (2021).
Giguere, D., Cloutier, P. & Roy, R. Domino Heck/lactonization-catalyzed synthesis of 3-C-linked mannopyranosyl coumarins. J. Org. Chem. 74, 8480–8483 (2009).
Chen, D. et al. Biocatalytic C-glucosylation of coumarins using an engineered C-glycosyltransferase. Org. Lett. 20, 1634–163 (2018).
Wang, Z. et al. Forming coumarin C-glycosides via biocatalysis: Characterization of a C-glycosyltransferase from Angelica decursiva. Biochem. Biophys. Res. Commun. 614, 85–91 (2022).
Saha, N. N., Desai, V. N. & Dhavale, D. D. A synthesis of new coumarin C-glycosyl derivatives. J. Org. Chem. 64, 1715–1719 (1999).
Tasior, M. et al. π-Expanded coumarins: synthesis, optical properties and applications. J. Mater. Chem. C. 3, 1421–1446 (2015).
Abe, H., Shuto, S. & Matsuda, A. Highly α- and β-selective radical C-glycosylation reactions using a controlling anomeric effect based on the conformational restriction strategy. A study on the conformation−anomeric effect−stereoselectivity relationship in anomeric radical reactions. J. Am. Chem. Soc. 123, 11870–11882 (2001).
Shi, Q. et al. Visible-light mediated catalytic asymmetric radical deuteration at non-benzylic positions. Nat. Commun. 13, 4453 (2022).
Ramanathan, D. et al. Catalytic asymmetric deuterosilylation of exocyclic olefins with mannose-derived thiols and deuterium oxide. Org. Chem. Front. 10, 1182–1190 (2023).
Xia, L., Fan, W., Yuan, X.-A. & Yu, S. Photoredox-catalyzed stereoselective synthesis of C-nucleoside analogues from glycosyl bromides and heteroarenes. ACS Catal. 11, 9397–9406 (2021).
Lasso, J. D. et al. A general platform for visible light sulfonylation reactions enabled by catalytic triarylamine EDA complexes. J. Am. Chem. Soc. 146, 2583–2592 (2024).
Nelsen, S. F. & Hintz, P. J. Electrochemical oxidation of tertiary amines. Effect of structure upon reversibility. J. Am. Chem. Soc. 94, 7114–7117 (1972).
Acknowledgements
We thank the National Natural Science Foundation of China (Grant No. 22377123 to X.M.), the China Postdoctoral Science Foundation (Grant No. 2023M743414 to D.X.), the Postdoctoral Fellowship Program of CPSF (Grant No. GZC20232572 to X.M.), the Biological Resources Program (Grant KFJ-BRP-008-002 to X.M.) from the Chinese Academy of Sciences (CAS), and the CAS Pioneer Hundred Talents Program (to X.M.) for financial support.
Author information
Authors and Affiliations
Contributions
X.M. guided the project. D.X. and W.Z. carried out reaction conditions optimization, substrate screening experiments, and mechanistic studies. J.Y. performed some of the substrate screening experiments. X.M. and D.X. wrote the manuscript with feedback from the other authors. D.X. and W.Z. contributed equally.
Corresponding author
Ethics declarations
Competing interests
The authors (D.X., W.Z., and X.M.) declare the following competing interests that two patents have been registered (202410607176.7 and 202410441139.3). The remaining author (J.Y.) declares no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, D., Zeng, W., Yang, J. et al. Visible-light-promoted direct desulfurization of glycosyl thiols to access C-glycosides. Nat Commun 15, 9187 (2024). https://doi.org/10.1038/s41467-024-53563-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53563-0
