
Abstract
Metabolic disorder significantly contributes to diabetic vascular complications, including diabetic retinopathy, the leading cause of blindness in the working-age population. However, the molecular mechanisms by which disturbed metabolic homeostasis causes vascular dysfunction in diabetic retinopathy remain unclear. O-GlcNAcylation modification acts as a nutrient sensor particularly sensitive to ambient glucose. Here, we observe pronounced O-GlcNAc elevation in retina endothelial cells of diabetic retinopathy patients and mouse models. Endothelial-specific depletion or pharmacological inhibition of O-GlcNAc transferase effectively mitigates vascular dysfunction. Mechanistically, we find that Yes-associated protein (YAP) and Transcriptional co-activator with PDZ-binding motif (TAZ), key effectors of the Hippo pathway, are O-GlcNAcylated in diabetic retinopathy. We identify threonine 383 as an O-GlcNAc site on YAP, which inhibits its phosphorylation at serine 397, leading to its stabilization and activation, thereby promoting vascular dysfunction by inducing a pro-angiogenic and glucose metabolic transcriptional program. This work emphasizes the critical role of the O-GlcNAc-Hippo axis in the pathogenesis of diabetic retinopathy and suggests its potential as a therapeutic target.
Similar content being viewed by others
Introduction
The vascular complications of diabetes, including atherosclerosis, diabetic nephropathy and retinopathy are among the most severe manifestations of the disease. Among these complications, diabetic retinopathy (DR) stands out as a primary cause of global vision loss and blindness1. DR is characterized by early microvascular abnormalities such as pericyte loss, microaneurysms, and intraretinal hemorrhages in its non-proliferative stage (non-proliferative diabetic retinopathy, NPDR)2,3. Impaired blood-retinal barrier (BRB) function in this stage can lead to diabetic macular edema (DME), a major cause of visual impairment and blindness in DR. The disease can progress to a proliferative stage (proliferative diabetic retinopathy, PDR), characterized by neovascularization, vitreal contraction, and retinal detachment2. While laser photocoagulation and vascular endothelial growth factor (VEGF)-neutralizing therapies are common approaches to treating DR, they have limitations. Laser photocoagulation therapies can destroy retinal tissue, leading to scotomas, and the effectiveness of anti-VEGF therapy in proliferative retinopathy is limited by the need for frequent interventions due to the short-lived effects of these agents4. Additionally, some patients may exhibit poor or no response to VEGF-targeting drugs, and in certain cases, VEGF neutralization can accelerate the atrophy of photoreceptors5. Thus, it is imperative to deepen our understanding of the pathogenesis of diabetic retinopathy in order to develop more effective and sustainable therapeutic strategies.
Endothelial cells (ECs) form the innermost layer of blood vessels, which places them in direct contact with the bloodstream and enables them to sense the nutrient levels in the blood. ECs exhibit high glycolytic activity, consuming glucose at rates similar to many cancer cells, and primarily rely on glycolysis for generating around 85% of their ATP6. Several key regulators of glycolysis, including PFKFB3, ADORA2A, and HK2, have been shown to play a critical role in regulating ECs energy homeostasis and angiogenesis6,7,8. Yet, how these metabolic signals transduce to protein modifications and gene transcription remains to be clarified.
The O-GlcNAcylation modification of proteins acts as a sensor of nutrients, particularly the glucose levels in the environment. Glucose can be shunted into the hexosamine biosynthesis pathway (HBP), a side branch of glycolysis, to generate UDP-GlcNAc, which serves as a donor molecule for O-GlcNAcylation9,10. O-GlcNAcylation is an important post-translational modification, reversibly catalyzed at protein serine/threonine residues by O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), a pair of enzymes responsible for adding and removing O-GlcNAc moieties from proteins. The amount of glucose that enters the cell directly affects the availability of UDP-GlcNAc, thus, O-GlcNAcylation is tightly regulated by ambient glucose. Dysregulation of O-GlcNAc homeostasis has been associated with several chronic human diseases, including cardiovascular diseases, Alzheimer’s disease, and cancer9. Despite this, the role of O-GlcNAc signaling in regulating ECs, particularly in the context of diabetic retinopathy, remains elusive. Here, we studied the role of O-GlcNAcylation in vascular disorders during diabetic retinopathy and assessed the anti-angiogenic therapeutic potential.
Results
Elevated O-GlcNAcylation level in diabetic retinopathy
To evaluate the O-GlcNAcylation level in DR, we first analyzed the abundance of UDP-GlcNAc, a final product of HBP and the substrate for protein O-GlcNAcylation in the vitreous fluid of PDR patients. Enzyme-linked immunosorbent assay (ELISA) showed that the UDP-GlcNAc levels were significantly elevated in the vitreous fluid of PDR patients compared with non-diabetic patients (Fig. 1A).

A ELISA analysis of UDP-GlcNAc levels in the vitreous fluid of proliferative diabetic retinopathy (PDR) and non-diabetic patients (n = 7/11 patients). Values are shown as mean ± SEM, with statistical significance at p < 0.0001 (two-tailed Student’s t-test). B Schematic of the streptozotocin (STZ)-induced diabetic model. The upper timeframe represents postnatal weeks, while the lower depicts the duration of diabetes induction by STZ. C Schematic diagram illustrating the oxygen-induced retinopathy (OIR) mouse model. RA: room air. D Western blot and quantification showing elevated O-GlcNAc-modified protein levels in the retina of diabetic mice compared to controls (n = 5/5 mice). Values as mean ± SEM, p = 0.0478 (two-tailed Student’s t-test). E Western blot and quantification showing O-GlcNAc-modified proteins in OIR pups compared to controls (n = 6/6 pups). Values as mean ± SEM, p = 0.046 (two-tailed Student’s t-test). F, G Flat-mounted retinas stained for O-GlcNAc (green) and CD31/IsoB4 (red), illustrating O-GlcNAc levels in control and diabetic/OIR mice. Representative images of O-GlcNAc (green), CD31 (red), and DAPI (blue) staining in frozen eye sections (H) and quantification of O-GlcNAc-modified protein levels in retinal vessels (CD31+ area) (I) from diabetic and control mice at different time points (n = 6/5, 6/5, 6/6 mice). Mean ± SEM, p-values < 0.0001 (two-tailed Student’s t-test). Representative images (J) and quantification (K) of O-GlcNAc-modified proteins in retinal vessels (IsoB4+ area) from OIR pups and controls (n = 6/6 mice). Mean ± SEM, p = 0.0071 (two-tailed Student’s t-test). L Western blot and quantification of O-GlcNAc-modified proteins in HRCECs exposed to varying glucose concentrations (n = 3 independent experiments). Mean ± SEM, p = 0.0054, p = 0.0007 (one-way ANOVA with Dunnett’s test). M Western blot and quantification of O-GlcNAc-modified proteins in HRCECs under hypoxia for 0, 12, and 24 h (n = 3 independent experiments). Mean ± SEM, p = 0.0051, p = 0.0007 (one-way ANOVA with Dunnett’s test). *p < 0.05; **p < 0.01; ***p < 0.001; scale bars: 100 µm (F, G), 30 µm (H, J). Source data provided as a Source Data file.
Next, we determined the levels of O-GlcNAc-modified proteins in two different models for DR: a streptozocin (STZ)-induced mouse DR model (Fig. 1B, Supplementary Fig. 1A), which develops retinal vascular leakage without neovascularization11, and oxygen-induced retinopathy (OIR), an animal model of PDR and retinopathy of prematurity with neovascularization and vascular leakage12 (Fig. 1C). Western blot analysis on whole retina tissue lysates showed that the overall O-GlcNAcylation levels were significantly elevated in the STZ and OIR mice, compared with the age-matched control (Fig. 1D, E). To further determine the cell type with a high level of O-GlcNAc-modified proteins, we performed immunofluorescence staining and found that O-GlcNAc signal intensity was enriched in CD31 or IsoB4-labeled blood vessels and was significantly higher in STZ and OIR mice as compared with control mice (Fig. 1F–K). Moreover, in the STZ model, O-GlcNAc intensity in blood vessels increased with the duration of diabetes (Fig. 1H, I).
To further elucidate whether the levels of O-GlcNAc-modified proteins is also changed during physiological retinal angiogenesis, we performed immunofluorescence staining in mouse retinas at different stages. While O-GlcNAc was highly enriched in IsoB4-labeled blood vessels during retina vasculature development (postnatal day 3 and 6, P3 and P6), adult mice exhibited significantly lower levels of O-GlcNAc in ECs (Supplementary Fig. 1B–E). Interestingly, O-GlcNAc intensity was higher in the angiogenic front compared with the vascular plexus in mouse retina at P3 and P6 (Supplementary Fig. 1B, C), suggesting EC tip cells might have a higher level of O-GlcNAc-modified proteins.
Given that glucose is the primary source of O-GlcNAcylation and the STZ model induces hyperglycemia, we treated human retinal capillary endothelial cells (HRCECs) with different concentrations of glucose to assess its effect on O-GlcNAcylation. We observed that glucose levels dose-dependently increased the overall O-GlcNAcylation levels in HRCECs (Fig. 1L). Previous study indicated that hypoxia-induced metabolic changes can lead to increased O-GlcNAcylation levels13,14. As the OIR model is a hypoxia-driven angiogenesis model15, we exposed HRCECs to hypoxia and found that hypoxia also increased the level of O-GlcNAc-modified proteins in HRCECs (Fig. 1M). Collectively, these findings suggest that both glucose and hypoxia contribute to elevated O-GlcNAcylation levels in retina ECs.
Increased O-GlcNAcylation promotes vascular dysfunction
Afterwards, we aimed to investigate the functional implications of O-GlcNAcylation by conducting a series of angiogenesis assays. To assess whether O-GlcNAcylation affects EC proliferation, we treated HRCECs and Human Retinal Microvascular Endothelial Cells (HRMECs), a primary retinal microvascular EC type with PUGNAc, an OGA inhibitor that increases protein O-GlcNAcylation, and performed a BrdU incorporation assay. The results revealed that PUGNAc treatment significantly increased EC proliferation (Fig. 2A, B, Supplementary Fig. 2A, B). In addition to utilizing inhibitors, we also employed siRNA to knock down OGT in HRCECs and conducted BrdU assay. Our findings revealed reduced EC proliferation in HRCECs transfected with siOGT (Supplementary Fig. 2C, D).

A BrdU incorporation in HRCECs treated with DMSO or 25 μM PUGNAc for 24 h. B Quantification of BrdU+ cells (red) as a percentage of total cell number in (A) (n = 3/3 independent experiments). Mean ± SEM, p = 0.0249 (two-tailed Student’s t-test). C Representative images of mouse aortic rings treated with DMSO or 25 μM PUGNAc for 5 days, stained with Phalloidin (red). D Quantification of microvascular sprouting area / total ring area in (C) (n = 6/7 aortic rings). Mean ± SEM, p = 0.0271 (two-tailed Student’s t-test). E Paracellular resistance in HRCECs treated with DMSO or 25 μM PUGNAc for 12 h. (n = 4/4 independent experiments). Multiple unpaired t-tests using two-stage step-up (Benjamini, Krieger, and Yekutieli) method. F Schematic of STZ and 4-OHT injections in OgtWT and OgtiΔEC mice. G Desmin (Cyan) and CD31 (Magenta) staining of retinas 9 weeks after STZ onset. Arrowheads indicate vessels without mural cell coverage. H Quantification of mural cell coverage (%) in (G) (n = 6/6 mice). Mean ± SEM, p = 0.0038 (two-tailed Student’s t-test). I Retinal trypsin digestion showing acellular capillaries (Ac-Cap) in STZ mice. Black arrowheads indicate acellular capillaries. J Quantification of Ac-Cap in (I). Ac-Caps were quantified in 3 random fields per retina (n = 6/6 retinas). Mean ± SEM, p = 0.0018 (two-tailed Student’s t-test). K Collagen-lV (green) and CD31 (red) staining of retinas from (G). Arrowheads indicate CollV+CD31- sleeves. L Quantification of CollV+CD31- sleeves per field of retina (n = 3/3 mice, 15 fields per retina). Mean ± SEM, p = 0.0311 (two-tailed Student’s t-test). M Schematic of tamoxifen injections in the OIR model. N Representative images of retinal vasculature stained with IsoB4 (red) in OIR pups at P17. Quantification of retinal neovascularization (O) and avascular area (P) in (N) (n = 13/8 pups). Mean ± SEM, p = 0.0489 (two-tailed Student’s t-test). *p < 0.05; **p < 0.01. Scale bars: 200 µm in (A), (C), 100 µm in (G), 50 µm in (I), (K), and 500 µm in (N). Source data are provided as a Source Data file.
We also evaluated the role of O-GlcNAcylation in vessel sprouting using an ex vivo aortic ring assay and found that treatment with PUGNAc significantly increased vascularization and sprouting in this model (Fig. 2C, D). A similar effect was observed in a choroid sprouting model (Supplementary Fig. 2E, F). As vascular mural cells and EC sprouts cannot be distinguished by Phalloidin staining in aortic ring and choroidal sprouting assays, we conducted the bead-sprouting assay, a well-established model for studying EC sprouting16. The results indicated a significant increase in sprout length with PUGNAc treatment, whereas siOGT led to decreased sprouting (Supplementary Fig. 2G–J).
In addition, we performed a transendothelial electrical resistance (TER) assay to evaluate junctional integrity and barrier function. Our results demonstrated that PUGNAc compromised the junctional integrity of retinal ECs (Fig. 2E), suggesting that increased protein O-GlcNAc modification activates ECs and negatively impacts their barrier function.
Ogt loss in ECs impairs development but reduces dysfunction
Our data revealed that O-GlcNAc levels are elevated in ECs during diabetic retinopathy, which promotes angiogenesis and EC activation. To investigate the physiological and pathological significance of protein O-GlcNAcylation in ECs, we generated OgtΔEC mice by crossing Ogtfl/fl mice with Cdh5-Cre mice (Supplementary Fig. 3A). Ogt mRNA level was significantly reduced in isolated brain ECs from OgtΔEC mice (Supplementary Fig. 3B, C), confirming the knockout efficiency. OgtΔEC mice were born at expected Mendelian ratios, however, OgtΔEC pups weighted significantly less than OgtWT littermates (Supplementary Fig. 3D).
We then analyzed the postnatal angiogenesis of the retina. Quantification of postnatal day (P6) whole-mount retinas showed a significant reduction of vascular area, number of sprouts and radial outgrowth in OgtΔEC pups (Supplementary Fig. 3E–I). Thus, these results indicated that OGT in ECs serves as an important regulator of postnatal retinal angiogenesis.
As loss of Ogt in ECs affects physiological retinal angiogenesis, we then generated inducible EC-specific Ogt knockout mice (OgtiΔEC) by crossing Ogtfl/fl mice with Cdh5-(PAC)-CreERT2 mice (Supplementary Fig. 3J). Cre recombination in newborn pups was induced as previously described (Supplementary Fig. 3K)16, and the knockout efficiency was confirmed in isolated brain ECs (Supplementary Fig. 3L, M). O-GlcNAc-modified proteins in blood vessels were also reduced by Ogt knockout (Supplementary Fig. 3N, O). OgtiΔEC pups also showed impaired developmental angiogenesis in P6 pups, similar to OgtΔEC pups (Supplementary Fig. 3P–S).
Next, we explored whether the loss of Ogt in ECs could also affect the vasculature in pathological conditions. We induced diabetes and then intravitreally injected 4-hydroxytamoxifen (4-OHT) (Fig. 2F). The levels of O-GlcNAc-modified proteins in blood vessels were significantly reduced by Ogt knockout, while the blood glucose was not altered (Supplementary Fig. 4A–C). Nine weeks after the onset of STZ-induced diabetes, we assessed the retina vascular permeability by injecting the Cadaverine-conjugated Alexa Fluor-555 tracer. The extravasations of the fluorescent tracers were observed in OgtWT STZ mice, but significantly reduced in OgtiΔEC STZ mice (Supplementary Fig. 4D, E). In addition, OgtiΔEC mice exhibited notable improvement in vascular abnormalities associated with DR, including increased mural cell coverage and a reduced number of acellular capillaries (Fig. 2G–J). The regressing vessel branches leave collagen type IV (ColIV+) empty sleeves behind17. Therefore, we employ CollV and CD31 staining to analyze vessel regression by quantifying the number of CollV+CD31- sleeves. The results showed reduced number of CollV+CD31- sleeves, indicative decreased vessel regression in OgtiΔEC mice (Fig. 2K, L).
We also applied the OIR model and analyzed the retina vasculature (Fig. 2M). Ogt expression was significantly reduced in brain ECs, and O-GlcNAc-modified proteins in blood vessels were significantly reduced by Ogt knockout (Supplementary Fig. 4F–I). We observed a significant reduction in pathological neovascularization, as quantified by measuring the neovascular tufts area in OgtiΔEC compared to OgtWT pups, while the avascular area remained comparable between the two groups (Fig. 2N–P). This reduction in neovascularization may be due to reduced EC proliferation, as demonstrated by the significantly lower number of EdU+ IsoB4+ cells in OgtiΔEC pups (Supplementary Fig. 4J, K). Additionally, the abnormal vascular growth observed in the OIR model leads to a breakdown of the BRB, resulting in severe retinal hemorrhages in OgtWT OIR pups. In contrast, OgtiΔEC pups showed significantly less retinal hemorrhage, as quantified by the area of blood islands (Supplementary Fig. 4L, M). Furthermore, we injected FITC-Dextran to determine vessel leakage and found that extravasation of FITC-Dextran was markedly reduced in OgtiΔEC OIR pups (Supplementary Fig. 4N, O).
O-GlcNAcylation regulates Hippo pathway in ECs
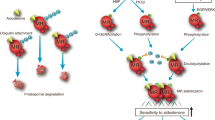
To determine the mechanism of O-GlcNAcylation in regulating the function of retinal ECs, we profiled transcriptome from PUGNAc or vehicle-treated HRCECs. 193 genes were upregulated, while 138 genes were downregulated upon PUGNAc treatment. Pathway enrichment analysis of downregulated transcripts in PUGNAc-treated HRCECs revealed pronounced alterations in the Hippo signaling pathway (Fig. 3A). We and others have previously shown that Hippo pathway and its effectors YAP (Yes-associated protein) and TAZ (transcriptional coactivator with PDZ-binding motif) as crucial regulators of developmental angiogenesis and tumor angiogenesis16,18,19. Thus, we first tested whether YAP and TAZ can be O-GlcNAcylated in retinal ECs. HRCECs were infected with Ad-Flag-YAP and Ad-Flag-TAZ respectively, and then exposed to 5.5 mM or 25 mM glucose. Afterwards, YAP and TAZ were immunoprecipitated with anti-Flag beads from cell lysates for blotting O-GlcNAc. We found that O-GlcNAcylation of YAP and TAZ can be detected already in low (5.5 mM) glucose, and increased under high (25 mM) glucose exposure (Fig. 3B, C).

A HRCECs were treated with DMSO or 25 μM PUGNAc for 24 h, and total RNA was harvested for RNA-seq. The enriched pathways for significantly upregulated genes under PUGNAc treatment are shown. The p-value for the Hippo-Yap signaling pathway (WP4537) is 0.0005, computed using the Fisher exact test. B, C HRCECs were infected with Ad-Flag-YAP and Ad-Flag-TAZ for 24 h, followed by treatment with 5.5 mM or 25 mM glucose for an additional 24 h. YAP and TAZ O-GlcNAcylation was analyzed via immunoprecipitation using anti-Flag beads and western blot (n = 3 independent experiments). D, E YAP was purified from HRCECs and analyzed by MS to identify the O-GlcNAcylation sites. Two different O-GlcNAcylation sites in YAP are shown (T358 and T383). F HEK 293T cells were transfected with Flag-YAP-T358A, Flag-YAP-T383A, or Flag-YAP-2TA (T358A and T383A) plasmids. YAP O-GlcNAcylation was analyzed via immunoprecipitation followed by western blot (n = 3 independent experiments). Mean ± SEM, p-values = 0.4728, 0.0488, and 0.0221 (one-way ANOVA with Dunnett’s multiple comparisons test). G Identification of the O-GlcNAcylation modification sites of YAP by in vitro O-GlcNAcylation assay. Purified wild-type or mutant YAP was used as substrates. Coomassie blue staining shows the YAP protein used in the assay. Three independent experiments were performed. *p < 0.05. Source data are provided as a Source Data file.
Next, we performed mass spectrometry and identified T358 and T383 as candidate O-GlcNAc-modified sites of YAP (Fig. 3D, E). To validate this, we generated adenoviruses expressing Flag-tagged YAP mutants with alanine substitutions at T358 (T358A), T383 (T383A), or both sites (2TA). Notably, the level of O-GlcNAcylation markedly decreased in YAP-2TA mutants and the T383A mutant, but no significant change was observed in the T358A mutant (Fig. 3F). To further confirm that OGT directly induces O-GlcNAcylation of YAP at T383 and T358, we conducted an in vitro O-GlcNAcylation assay. Recombinant proteins were generated for YAP-WT, YAP-T358A, and YAP-T383A for this assay. The results revealed a significant reduction in O-GlcNAcylation levels in the YAP-T383A mutant compared to YAP-WT, while marginal changes were observed in the T358A mutant. This finding suggests that T383 is the primary O-GlcNAcylation site on YAP (Fig. 3G). YAP threonine 383 is conserved in human, chimpanzee, rhesus monkey, dog, mouse and other species (Supplementary Fig. 5A). Moreover, YAP-2TA still showed O-GlcNAcylation, suggesting the presence of other potential O-GlcNAcylation sites on YAP (Fig. 3F).
O-GlcNAcylation regulates YAP phosphorylation and stability
O-GlcNAcylation can affect phosphorylation by influencing the accessibility or recognition of nearby phosphorylation sites on target proteins10,20,21. YAP activity is tightly regulated by its phosphorylation status, with YAP-S397 being an important phosphorylation site. Phosphorylation of YAP at S397 (S381) by LATS1/2 promotes YAP ubiquitination-dependent degradation and reduces its nuclear localization22. As S397 and T383 have high proximity, we employed AlphaFold2-multimer to analyze the potential impact of O-GlcNAcylation at T383 on its binding interface with LATS1/2 (Supplementary Fig. 5B, C). The predicted structure showed a binding interface of LATS1/2 and YAP at S397. Furthermore, both S397 and T383 are closely located and are situated in the loop region of YAP. Therefore, based on the predicted structure, it is possible that O-GlcNAcylation at T383 affects its binding interface with LATS1/2 at S397. Based on this result, we tested whether O-GlcNAcylation of T383 affects phosphorylation at S397. Indeed, we found that YAP-T383A as well as YAP-2TA showed a higher phosphorylation level of S397, while YAP T358A had no obvious effect (Fig. 4A).

A HEK 293T cells transfected with indicated plasmids. pYAP-S397 was analyzed via immunoprecipitation followed by western blot (n = 3 independent experiments). Mean ± SEM, p-values = 0.0068, 0.004 (one-way ANOVA, Dunnett’s test). B Western blot and quantification of pYAP-S397 in HRCECs treated with DMSO or 25 μM PUGNAc for 24 h (n = 3 independent experiments). Mean ± SEM, p = 0.0002 (two-tailed Student’s t-test). C pYAP-S397 in HRCECs treated with DMSO or 15 μM OSMI-1 for 24 h in 25 mM glucose medium (n = 3 independent experiments). Mean ± SEM, p = 0.0053 (two-tailed Student’s t-test). D, E YAP O-GlcNAcylation in HRCECs infected with Ad-Flag-YAP, transfected with siCtrl or siOGT/siOGA under 25 mM glucose (n = 3 independent experiments). Mean ± SEM, p-values: (D) = 0.0091, 0.0318; (E) = 0.0336, 0.0339 (two-tailed Student’s t-test). F, G Relative CTGF and CYR61 mRNA in HRCECs treated as in (D) analyzed by qRT-PCR (n = 3 independent experiments). Mean ± SEM, p-values: (F) = 0.0191, 0.0458; (G) = 0.0298, 0.0443 (two-tailed Student’s t-test). H HRCECs infected with indicated adenovirus, treated with DMSO or 25 μM PUGNAc, and analyzed for pYAP-S397 (n = 3 independent experiments). Mean ± SEM, p = 0.0012 (one-way ANOVA, Dunnett’s test). I Ubiquitination and β-TRCP levels in HRCECs infected with indicated adenovirus and treated with MG132 or vehicle (n = 3 independent experiments). Mean ± SEM, p-values = 0.0011, 0.0002, 0.0196, 0.0125 (one-way ANOVA, Tukey’s test). J YAP stability in HRCECs infected with indicated adenovirus and treated with 50 µg/ml CHX for western blot. Flag-YAP protein levels were quantified as Flag-YAP/β-Actin (n = 3 independent experiments). Mean ± SEM, p-values: 0.0064, <0.0001 (two-way ANOVA, Šídák’s test). K–N Images and quantification of YAP/TAZ (green) within vessels (CD31+ or IsoB4+ areas, red) from OgtWT and OgtiΔEC STZ or OIR mice (n = 4/4 mice in L, 6/6 mice in N). Mean ± SEM, p-values: (L) = 0.0443; (N) = 0.0002 (two-tailed Student’s t-test). *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars: 30 µm in (K, M). Source data provided as a Source Data file.
To investigate whether O-GlcNAcylation can regulate YAP phosphorylation at S397 and in turn affect YAP activity, we treated HRCECs with PUGNAc and the OGT inhibitor OSMI-1. We observed that PUGNAc significantly decreased pYAP-S397, whereas OSMI-1 increased it (Fig. 4B, C). We also measured the expression of YAP/TAZ target genes CTGF and CYR61 and found that OSMI-1 treatment decreased their expression (Supplementary Fig. 5D). Similar results were observed when using HRMECs (Supplementary Fig. 5E–H). In addition to utilizing inhibitors, we also employed siRNA to knock down OGT and OGA in HRCECs cells (Fig. S5I, J). Knocking down OGT decreased YAP O-GlcNAcylation and increased pYAP-S397, whereas OGA knockdown had the opposite effect (Fig. 4D, E). Additionally, the expression of CTGF and CYR61 decreased with siOGT but increased with siOGA (Fig. 4F, G). To overexpress OGT/OGA, we constructed adenovirus expressing OGT and OGA, respectively (Supplementary Fig. 5K, L). OGT overexpression increased YAP O-GlcNAcylation, decreased pYAP-S397, and increased target gene expression, while OGA overexpression showed the opposite effects (Supplementary Fig. 5M–P).
To further confirm that T383 is the key O-GlcNAcylation site of YAP that modulates phosphorylation of S397, we treated the HRCECs with PUGNAc and found that pYAP-S397 was significantly reduced in cells expressing YAP-WT, but not in those expressing YAP-T383A mutants (Fig. 4H). To explore whether YAP-T383A influence the mRNA levels of CTGF and CYR61, we knocked down endogenous YAP using siRNA targeting the 3’UTR of YAP (Supplementary Fig. 5Q) and infected HRCECs with adenovirus expressing YAP-WT and YAP-T383A. CTGF and CYR61 expression were significantly reduced in YAP-T383A expressing cells, further indicating that T383A expression reduced YAP activity (Supplementary Fig. 5R). Collectively, these results further support the notion that O-GlcNAcylation of YAP plays a pivotal role in the regulation of YAP phosphorylation and downstream target gene expression.
Additionally, we determined the effect of O-GlcNAcylation on YAP stability using a cycloheximide (CHX) chase assay. We found that PUGNAc treatment significantly promoted YAP stabilization (Supplementary Fig. 6A). Moreover, YAP was relatively stable in HRCECs cultured in 25 mM glucose, but OSMI-1 treatment significantly accelerated its degradation (Supplementary Fig. 6B). YAP-S397 phosphorylation also affect its subcellular localization23. We observed increased nuclear translocation of YAP/TAZ in HRCECs upon PUGNAc stimulation, but increased cytosol accumulation after OSMI-1 treatment (Supplementary Fig. 6C–F). YAP-S397 phosphorylation generates phosphodegron motifs that are recognized by the SCFβ-TRCP E3 ligase24. We determined whether the binding of YAP and SCFβ-TRCP is affected by knockdown/overexpressing of OGT or YAP O-GlcNAcylation deficient mutant through Co-IP experiments. The result indicated that OGT knockdown increased the association of YAP and β-TRCP (Supplementary Fig. 6G), while overexpression of OGT reduced this association (Supplementary Fig. 6H). Additionally, the YAP O-GlcNAcylation-deficient mutant (YAP-T383A) showed an increased level of ubiquitination, which could be inhibited by MG132 treatment (Fig. 4I). CHX chase assay also showed that YAP-T383A mutants were less stable compared with YAP-WT (Fig. 4J).
Next, investigate whether the O-GlcNAcylation of YAP mediates its nuclear translocation and downstream target gene expression, specifically requiring S397. In HRCECs transfected with YAP 3’UTR siRNA to knockdown endogenous YAP, we expressed Flag-YAP-WT or Flag-YAP-S397A by adenovirus and transfected siCtrl or siOGT. The YAP-S397A mutant exhibited increased nuclear translocation, consistent with previous findings (Supplementary Fig. 6I, J)23. Furthermore, knockdown of OGT resulted in reduced nuclear localization and target gene expression in YAP-WT expressing cells but not in YAP-S397A, suggesting that nuclear translocation by O-GlcNAcylation depends on the S397 site of YAP (Supplementary Fig. 6I, J). Similar results were observed in the expression levels of CTGF and CYR61 (Supplementary Fig. 6K). We further conducted ChIP-qPCR analyses to determine whether knockdown of OGT affect YAP-S397A binding to the promoter region of CTGF and CYR61. The results indicated that the YAP-S397A mutant showed increased binding to the promoter regions of CTGF and CYR61. Interestingly, OGT knockdown reduced the binding in YAP-WT expressing cells but not in YAP-S397A expressing cells (Supplementary Fig. 6L).
Furthermore, co-staining of IsoB4 and YAP/TAZ showed lower expression of YAP/TAZ in OgtiΔEC mice compared with OgtWT mice in both STZ model and OIR model, suggesting O-GlcNAcylation also regulates YAP/TAZ protein level in vivo (Fig. 4K–N).
YAP/TAZ are highly expressed and activated in ECs of DR
As we observed that O-GlcNAc-modified proteins were increased in the blood vessel in the retinas of STZ and OIR mice, we then tested whether YAP/TAZ O-GlcNAcylation is also increased. Therefore, we utilized the proximity ligation assay (PLA), a technique for detecting protein–protein interactions in situ at endogenous protein levels, to assess the regulation of O-GlcNAcylation by targeting a substrate protein to OGT25. The results revealed a significant increase in interactions between OGT and YAP/TAZ in ECs in both models compared to age-matched controls (Fig. 5A–D), suggesting the upregulation of endogenous YAP/TAZ O-GlcNAcylation in ECs of DR models.

A Proximity ligation assay (PLA) of YAP/TAZ and OGT interaction in frozen eyeball sections from control and STZ-induced diabetic mice. CD31 was stained for blood vessels. B Quantification of PLA counts (red) in CD31+ areas (green) (n = 3/3 mice). Mean ± SEM, p = 0.016 (two-tailed Student’s t-test). C PLA of YAP/TAZ and OGT interaction in frozen eyeball sections from control and OIR model pups. CD31 was stained for blood vessels. D Quantification of PLA counts in CD31+ areas (n = 3/3 mice). Mean ± SEM, p = 0.0033 (two-tailed Student’s t-test). E Immunostaining of YAP/TAZ (green), UEA1 (red), and DAPI (blue) in paraffin-embedded eyeball sections from non-diabetic control and PDR patients. Arrowheads indicate YAP/TAZ localization in EC nuclei. F Quantification of YAP/TAZ fluorescence intensity within vessels (UEA1+ areas) from non-diabetic controls and PDR patients (n = 4/4 individuals). Mean ± SEM, p = 0.008 (two-tailed Student’s t-test). G Relative expression of CTGF and CYR61 in retinal microvascular ECs from PDR patients compared to non-diabetic controls, analyzed from the Omnibus Database (GSE94019) (n = 4/9 individuals). Mean ± SEM, p = 0.0268 (two-tailed Student’s t-test). H Immunostaining of YAP/TAZ (green) and CD31 (red) in sagittal retinal sections from STZ-induced diabetic mice at different stages and age-matched controls. I Quantification of YAP/TAZ fluorescence intensity in CD31+ areas (n = 3/3 mice per time point). Mean ± SEM, p-values = 0.0175, 0.0008, 0.0007 (two-tailed Student’s t-test). J Immunostaining of YAP/TAZ (green) and IsoB4 (red) in sagittal retinal sections from OIR model mice and age-matched controls. K Quantification of YAP/TAZ fluorescence intensity in IsoB4+ areas (n = 6/6 mice). Mean ± SEM, p < 0.0001 (two-tailed Student’s t-test). *p < 0.05; **p < 0.01. Scale bars: 5 µm in (A, C); 30 µm in (E, H, J). Source data are provided as a Source Data file.
To further determine whether YAP/TAZ levels are affected in DR endothelium, we analyzed their abundance and activity in both human patients and mouse models. In retinas from patients with PDR, we observed increased levels of YAP/TAZ in ECs (labeled with UEA1) compared to controls (Fig. 5E, F). Moreover, we noted an increased expression of CTGF and CYR61 in the retinal microvascular ECs of PDR patients, with CTGF showing statistical significance (analyzed from Gene Expression Omnibus Database dataset GSE94019; Lam et al., 2017) (Fig. 5G).
Using mouse models, we found that the levels of YAP/TAZ were significantly higher in retinal ECs in the STZ and OIR model, with the STZ model showing a time-dependent increase in YAP/TAZ abundance (Fig. 5H–K). Additionally, we found that YAP/TAZ were highly enriched in the nucleus of ECs in both models, indicating their activation (Fig. 5H, J).
As we have proved that glucose and hypoxia increased O-GlcNAc-modified proteins in HRCECs, we then tested whether they can also affect YAP activation. Treatment with glucose or hypoxia resulted in YAP/TAZ activation, which was demonstrated by upregulation of CTGF and CYR61 (Supplementary Fig. 7A, B). These findings suggest that glucose and hypoxia promote YAP/TAZ activation in HRCECs.
Previous studies conducted by our team and other researchers have highlighted VEGF as a potent activator of YAP/TAZ16,18. Therefore, we analyzed the timing of VEGFA elevation and O-GlcNAcylation increase in the OIR model. Our observations revealed that retinal VEGFA levels increased on P15, whereas the elevation in O-GlcNAcylation occurred earlier, on P12 (Supplementary Fig. 7C, D). This discrepancy suggests that O-GlcNAcylation is a rapid response process and may contribute to vascular dysfunction at an earlier stage. This relationship was observed not only in the OIR model but also in the early stages of diabetes. In STZ-treated mice, Vegfa levels remained comparable to those in vehicle-treated littermates until week 14, as previously reported11. However, O-GlcNAcylation exhibited a notable increase at 4 weeks post diabetes onset (Fig. 1H, I). These findings suggest that O-GlcNAcylation may serve as an early response factor contributing to vascular dysfunction in DR.
Loss of Yap/Taz in ECs attenuates vascular dysfunction in DR
To investigate the role of YAP/TAZ in vascular disorders in DR, we utilized Cdh5-(PAC)-CreERT2; Yapfl/flTazfl/fl mice (Yap/TaziΔEC mice) in which Yap/Taz can be specifically depleted in ECs upon tamoxifen treatment (Fig. 6A). We first applied the STZ model, in which Cre recombination was induced via intravitreal injection of 10 µg 4-OHT one week after the onset of STZ-induced diabetes (Fig. 6B). Immunostaining of YAP/TAZ confirmed the successful knockout efficiency in this model, while the blood glucose was not altered between the two groups (Supplementary Fig. 8A–C). To assess the impact of YAP/TAZ on the process of DR in the STZ model, we injected Cadaverine-conjugated AlexaFluor-555 tracer in Yap/TazWT and Yap/TaziΔEC STZ mice and found that endothelial-specific depletion of Yap/Taz significantly reduced the accumulation of the tracer (Supplementary Fig. 8D, E). Furthermore, Yap/TaziΔEC mice exhibited increased mural cell coverage and reduced number of acellular capillaries (Fig. 6C–F). Quantification of CollV+CD31- sleeves showed decreased vessel regression in Yap/TaziΔEC mice (Fig. 6G, H). In the OIR model, we induced Cre recombination through intraperitoneal administration of Tamoxifen (Fig. 6I). The mRNA levels of Yap and Taz was significantly reduced in the isolated brain ECs from Yap/TaziΔEC mice, validating the efficacy of the knockout approach (Supplementary Fig. 8F, G). Immunostaining also showed a significant reduction of YAP/TAZ in the retina blood vessels in Yap/TaziΔEC mice (Supplementary Fig. 8H, I). We found that Yap/TaziΔEC pups exhibited significantly less neovascularization compared to Yap/TazWT pups, with no significant differences in the avascular area (Fig. 6J–L). Further analysis using EdU incorporation with IsoB4 co-staining demonstrated a reduction in EC proliferation in the absence of YAP/TAZ (Supplementary Fig. 8J, K). In addition, Yap/TaziΔEC pups showed significantly less retinal hemorrhage (Fig. 6M, N). This was further confirmed by immunohistological analysis for TER119 (an erythrocyte marker) and IsoB4, as Yap/TaziΔEC pups showed significantly less erythrocyte extravasation, determined by quantifying TER119+ IsoB4– area in the whole-mount retina (Fig. 6O, P). Furthermore, Yap/TaziΔEC retinas exhibited markedly reduced extravasation of FITC-dextran, as demonstrated by FITC-dextran leakage assays (Fig. 6Q, R).

A Schematic showing the generation of EC-specific Yap/Taz knockout mice. B Intravitreal 4-OHT injection and STZ administration in EC-specific Yap/Taz knockout (Yap/TaziΔEC) and littermate control (Yap/TazWT) mice. C Desmin (Cyan) and CD31 (magenta) staining in retinas of Yap/TazWT and Yap/TaziΔEC mice, 9 weeks post-STZ onset. Arrowheads indicate vessels lacking mural cell coverage. D Quantification of mural cell coverage (%) within CD31+ vessel areas from (C) (n = 6/6 mice). Mean ± SEM, p = 0.0484 (two-tailed Student’s t-test). E Retinal trypsin digestion showing acellular capillaries in Yap/TazWT and Yap/TaziΔEC STZ mice. Black arrowheads indicating acellular capillaries. F Acellular capillaries (Ac-Cap) in (E), based on 3 random fields per retina (n = 6/6 retinas). Mean ± SEM, p = 0.0484 (two-tailed Student’s t-test). G Collagen lV (green) and CD31 (red) staining in retinas, 9 weeks post-STZ-induced diabetes. Arrowheads indicate Ac-Caps. H Quantification of CollV+CD31- sleeves in (G) (15 random fields per retina, n = 3/3 mice). Mean ± SEM, p = 0.0479 (two-tailed Student’s t-test). I Schematic of Tamoxifen injection in the OIR model. J IsoB4 staining of retinal vasculature in P17 OIR retinas from Yap/TazWT and Yap/TaziΔEC pups. K, L Quantification of neovascularization and avascular areas in Yap/TazWT and Yap/TaziΔEC OIR retinas (n = 7/8 pups). Mean ± SEM, p < 0.0001 in (K), p = 0.9361 in (L) (two-tailed Student’s t-test). M, N Images and quantification of blood island areas in P17 OIR retinas (n = 10/14 pups). Mean ± SEM, p = 0.0013 (two-tailed Student’s t-test). O, P Images and quantification of TER119+ red blood cell (RBC) (Cyan) leakage in flat-mounted retinas of OIR pups (n = 8/9 pups). Mean ± SEM, p = 0.0003 (two-tailed Student’s t-test). Q, R Images and quantification of extravasated FITC-dextran (70 kDa) (green) in IsoB4-stained (red) flat-mounted retinas (n = 5/8 pups). Mean ± SEM, p = 0.001 (two-tailed Student’s t-test). *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars: 100 µm in (C), 50 µm in (G), 500 µm in (J, O, Q), and 1 mm in (M). Source data are provided as a Source Data file.
YAP-T383/S397 axis mediates O-GlcNAc-mediated DR pathology
Next, we aimed to evaluate the functionality of YAP-T383/S397 axis. By using a recently developed AAV-BR1 system to specifically target CNS vasculature (including retina blood vessels) for gene delivery26, we constructed wild-type YAP (AAV-YAP-WT) or a mutant YAP-T383A (AAV-YAP-T383A). We first analyzed physiological angiogenesis by retro-orbitally injected AAV-EGFP, AAV-YAP-WT, AAV-YAP-T383A, and AAV-YAP-S397A in P3 pups and analyzed the outcomes at P13. GFP expression was detected in both retinal and brain vasculature, indicating efficient and specific gene delivery (Supplementary Fig. 9A). The injection of AAV constructs encoding YAP-WT, YAP-T383A, and YAP-S397A significantly elevated Yap mRNA expression levels in isolated brain ECs, indicating successful gene delivery (Supplementary Fig. 9B). In mice, the retinal vasculature develops as ECs migrate from the optic nerve to the retinal surface at birth, progressing radially to form the superficial plexus. By P7, sprouting vessels extend into the outer plexus layer (OPL), establishing the deep plexus. Between P11 and P12, vessels from the deep plexus ascend into the inner plexus layer (IPL), forming the intermediate plexus, which completes vascularization around P1527 (Supplementary Fig. 9C). We separately assessed the vascular coverage in each layer. The results revealed no statistical difference between the four groups in the superficial layer and the deep layer. Interestingly, when analyzing the intermediate layer, we found that the AAV-YAP-WT group increased vascular density compared to the AAV-EGFP group, while the AAV-YAP-T383A group showed a similar density to that in the AAV-EGFP group. The AAV-YAP-S397A group exhibited the highest vascular coverage in the intermediate layer (Supplementary Fig. 9D–G). These results suggest that while YAP-WT and YAP-S397A promote developmental angiogenesis, YAP-T383A fails to do so.
To further prove that O-GlcNAc modification promotes retinal vascular dysfunctions in DR conditions via the YAP-T383/S397 axis, we performed a series of rescue experiments. Firstly, we investigated the necessity of YAP-T383 in retinal neovascularization in the OIR model. We expressed AAV-YAP-WT or AAV-YAP-T383A in Yap/TaziΔEC pups and subjected them to the OIR model (Fig. 7A). Mice injected with AAV- EGFP served as controls. The results demonstrated that expression of YAP-WT significantly promoted neovascularization. In contrast, the AAV-YAP-T383A group exhibits comparable levels to the control group, suggesting the inability to undergo O-GlcNAc modification prevents YAP-T383A from stimulating pathological angiogenesis in the retina (Fig. 7B, C). Similarly, in the STZ model, our results demonstrated that the expression of wild-type YAP significantly decreased Desmin-positive mural cell coverage, while the AAV-YAP-T383A group exhibited levels comparable to the control group (Fig. 7D–F).

A Schematic showing AAV-BR1 injection in the OIR model. AAV-BR1 was injected retro-orbitally at P7, followed by tamoxifen intraperitoneal injections at P12, P13, P14, and P16 in Yap/TaziΔEC pups. B IsoB4-stained images of retinal vasculature in OIR retinas from AAV-BR1-injected Yap/TaziΔEC pups at P17. C Quantification of retinal neovascularization area in (B) (n = 6/6/7 pups). Values are mean ± SEM, p = 0.0023, p = 0.0097 (one-way ANOVA with Tukey’s multiple comparisons test). D Schematic of AAV-BR1 injection in the STZ model. One week after STZ onset, 4-OHT was injected intravitreally into Yap/TaziΔEC mice, with AAV-BR1 injected into the tail vein. Retinas were harvested after 8 weeks for analysis. E Desmin (Cyan) and CD31 (magenta) staining of retinas from AAV-BR1-injected Yap/TaziΔEC mice. Arrowheads show vessels lacking mural cell coverage. F Quantification of mural cell coverage (%) in CD31+ vessel areas from (E) (n = 6/6/5 mice). Values are mean ± SEM, p = 0.003, p = 0.0424 (one-way ANOVA with Tukey’s test). G Schematic of AAV-BR1 injection in STZ model. 4-OHT was intravitreally injected into OgtWT and OgtiΔEC mice one week after STZ onset, with AAV-BR1 injected intravenously. Retinas were harvested after 8 weeks. H Desmin (Cyan) and CD31 (magenta) staining of retinas from AAV-BR1-injected OgtWT and OgtiΔEC mice. Arrowheads indicate vessels lacking mural cell coverage. I Quantification of mural cell coverage (%) in CD31+ vessel areas from (H) (n = 8/7/7/9/8 mice). Values are mean ± SEM, p = 0.0051, p = 0.0002, p < 0.0001 (one-way ANOVA with Tukey’s test). J Schematic of AAV-BR1 injection in OIR model, with AAV-BR1 injected retro-orbitally at P7 into OgtWT and OgtiΔEC pups. K Representative IsoB4-stained images of retinal vasculature from AAV-BR1-injected OgtWT and OgtiΔEC OIR pups. L Quantification of retinal neovascularization in AAV-BR1-injected OgtWT and OgtiΔEC pups (n = 5/6/6/6/5 pups). Values are mean ± SEM, p = 0.0059, p = 0.0018, p < 0.0001 (one-way ANOVA with Tukey’s test). *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars: 500 µm in (B, K), 100 µm in (E, H). Source data are provided as a Source Data file.
Next, we investigated whether re-expressing YAP-WT or YAP-S397A could mitigate the protective effect of Ogt depletion. In the STZ model, we intravitreally injected 4-OHT and subsequently injected AAV- EGFP, AAV-YAP-WT, and AAV-YAP-S397A (Fig. 7G). In OgtWT mice, the overexpression of YAP-WT significantly decreased the mural cell coverage compared with EGFP group. Conversely, in OgtiΔEC mice, overexpression of YAP-WT resulted in only a slight reduction of mural cell coverage. These results suggest that in the STZ model, the effect of YAP in promoting mural cell loss depend on OGT. However, the expression of YAP-S397A could nullify the protective effect of OgtiΔEC, suggesting that the effect of S397A is not dependent on OGT (Fig. 7H, I). Furthermore, we retro-orbitally injected AAV-EGFP, AAV-YAP-WT, and AAV-YAP-S397A, and subjected OgtWT and OgtiΔEC pups to the OIR model, similar results were observed as in the STZ model (Fig. 7J–L). Taken together, these results suggest that O-GlcNAcylation at YAP-T383 promotes vascular dysfunction by inhibiting YAP-S397 phosphorylation and stabilizing/activating YAP. Our findings indicate that the T383/S397 regulatory axis is crucial for O-GlcNAc modification-mediated vascular abnormalities in DR models.
Inhibition O-GlcNAcylation and YAP/TAZ reduced DR pathology
We next investigated the therapeutic potential of two inhibitors, OSMI-1 and Super-TDU, for reducing DR pathology. OSMI-1 is a small molecule inhibitor of OGT that was previously optimized from a high-throughput screen hit28. Our results showed that OSMI-1 treatment significantly enhanced mural cell coverage in the STZ model (Supplementary Fig. 10A–C). Moreover, it effectively reduced neovascularization, blood island areas, and FITC-dextran extravasation in the OIR model (Supplementary Fig. 10D–K), indicating its potential as a promising therapeutic agent for DR.
Super-TDU is an inhibitor peptide that targets YAP-TEADs interaction, inhibiting YAP-mediated TEAD transactivation and YAP function29. To efficiently target ECs in the retina, we used a method we previously established by loading therapeutic peptides on exosomes12. By linking Super-TDU to EC-derived exosomes via an anchoring peptide, CP05, we generated EXOSuper-TDU (Fig. 8A). The efficacy of both Super-TDU and EXOSuper-TDU was tested in the STZ and OIR models. In the STZ model, both Super-TDU and EXOSuper-TDU exhibited a protective effect by increasing mural cell coverage and reducing the number of acellular capillaries (Fig. 8B–F). Notably, EXOSuper-TDU demonstrated superior efficacy compared to Super-TDU (Fig. 8C–F). In the OIR model, both Super-TDU and EXOSuper-TDU effectively inhibited neovascularization, retinal hemorrhage, and FITC-dextran extravasation (Fig. 8G–N). Furthermore, EXOSuper-TDU exhibited better efficacy compared to Super-TDU (Fig. 8H–N). These results suggest that pharmacological targeting of OGT or YAP/TAZ could be a promising therapeutic approach for reducing DR pathology.

A Schematic showing the preparation of EXOSuper-TDU, where Super-TDU is anchored to EC-derived exosomes using the CP05 peptide. B Schematic of Super-TDU and EXOSuper-TDU administration in the STZ model. PBS, Super-TDU, or EXOSuper-TDU were injected retro-orbitally one week after STZ onset, with vascular phenotypes analyzed 8 weeks later. C Desmin (Cyan) and CD31 (magenta) staining of retinas from STZ mice treated as indicated. Arrowheads indicate vessels lacking mural cell coverage. D Quantification of mural cell coverage (%) in CD31+ vessels from (C) (n = 6 mice per group). Mean ± SEM, p = 0.0319, p < 0.0001, p = 0.0217 (one-way ANOVA with Tukey’s test). E Retinal trypsin digestion showing acellular capillaries in STZ mice. Black arrowheads indicate acellular capillaries. F Quantification of acellular capillaries (Ac-Cap) in (E). Three random fields per retina were quantified (n = 6 mice per group). Mean ± SEM, p = 0.0108, p < 0.0001, p = 0.0177 (one-way ANOVA with Tukey’s test). G Schematic of Super-TDU treatment in the OIR model. PBS, Super-TDU, or EXOSuper-TDU were injected retro-orbitally at P12, and retinas were harvested at P17. H IsoB4-stained images of retinal vasculature in OIR retinas from treated pups. I, J Quantification of neovascularization and avascular areas in OIR retinas (n = 6/6 pups). Mean ± SEM, p = 0.0349, p = 0.0002, p = 0.0499 (one-way ANOVA with Tukey’s test). K, L Images of the retinal cup inner surface and quantification of blood island area (n = 8/7/7 pups). Mean ± SEM, p = 0.003, p < 0.0001, p = 0.0374 (one-way ANOVA with Tukey’s test). M, N Confocal images and quantification of extravasated FITC-dextran and IsoB4 in flat-mounted OIR retinas. FITC-dextran+ and IsoB4− areas indicate vessel leakage (n = 6 pups per group). Mean ± SEM, p = 0.0487, p < 0.0001, p = 0.0074 (one-way ANOVA with Tukey’s test). *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars: 100 µm (C), 50 µm (E), 1 mm (K), 500 µm (H, M). Source data are provided as a Source Data file.
YAP/TAZ regulates glucose metabolism and O-GlcNAcylation in ECs
To investigate the downstream effects of YAP/TAZ on regulating DR, we profiled transcriptome from siYAP/TAZ or siCtrl transfected HRCECs cultured in high (25 mM) glucose medium (Fig. 9A). Our analysis identified a total of 1107 differentially expressed genes, including 774 downregulated genes and 333 upregulated genes. Gene Ontology (GO) analysis followed by network visualization of enriched GO terms using BiNGO revealed that the downregulated genes are associated with angiogenesis and metabolism (Fig. 9B), which is consistent with previous research18,30. To validate these findings, we examined the expression of key enzymes in the HBP and O-GlcNAcylation (Fig. 9C). We found that HK1, HK2, GPI, UAP1 and OGT were downregulated in YAP/TAZ knockdown HRCECs (Fig. 9D). These results align with the observation that retinal microvascular ECs from patients with PDR exhibited upregulation of HK1, HK2, GPI, PGM3 and OGT genes compared to those from normal subjects, with two of them showing statistical significance (Fig. 9E). These results suggest that YAP/TAZ may be a critical regulator of the HBP and O-GlcNAcylation. Indeed, western blot analysis showed that overall O-GlcNAc-modified protein levels were significantly reduced in YAP/TAZ knockdown HRCECs, and restoring YAP/TAZ can rescue O-GlcNAc levels (Fig. 9F). Consistent with these findings, we observed that the protein O-GlcNAcylation levels were significantly decreased in the blood vessels of Yap/TaziΔEC mice in both the STZ and OIR models (Fig. 9G–J). Together, these results indicate that the activation of YAP/TAZ through O-GlcNAcylation plays a key role in promoting vascular dysfunction by inducing a pro-angiogenic and glucose metabolic transcriptional program. This regulatory mechanism may contribute to the formation of a positive feedback loop driving the progression of vascular dysfunction in DR.

A HRCECs were transfected with siCtrl or siYAP/TAZ and cultured in 25 mM glucose medium for 48 h. Total RNA was then harvested for RNA-seq. B Network visualization of GO terms for genes downregulated by YAP/TAZ knockdown. C Schematic representation of the hexosamine biosynthetic pathway (HBP), with key enzymes and O-GlcNAcylation processes highlighted in yellow boxes. D Heatmap of RNA-seq data from (A), displaying expression levels of key enzymes involved in HBP and O-GlcNAcylation. E Relative expression levels of key enzymes in the HBP and O-GlcNAcylation in retinal microvascular ECs from PDR patients compared to non-diabetic controls. Data from GSE94019 (n = 4/9 individuals). Mean ± SEM, p = 0.0466, p = 0.0234 (multiple unpaired Student’s t-tests). F Western blot and quantification of overall O-GlcNAc-modified proteins in HRCECs transfected with siCtrl or siYAP/TAZ, followed by infection with Ad-Flag-YAP/TAZ and cultured in 25 mM glucose medium (n = 3 independent experiments). Mean ± SEM, p = 0.036, p = 0.0341 (one-way ANOVA with Tukey’s test). G, H O-GlcNAc (green) and CD31 (red) staining with DAPI (blue) in frozen sections of eyeballs from Yap/TazWT and Yap/TaziΔEC mice 9 weeks after the onset of STZ-induced diabetes (n = 8/6 mice). Mean ± SEM, p = 0.002 (two-tailed Student’s t-test). I, J O-GlcNAc and IsoB4 (red) staining with DAPI (blue) in frozen sections of eyeballs from Yap/TazWT and Yap/TaziΔEC OIR pups at P17 (n = 4/5 pups). Mean ± SEM, p = 0.0084 (two-tailed Student’s t-test). K The proposed working model demonstrates the role of O-GlcNAcylation in regulating vascular dysfunction in DR through the modulation of YAP/TAZ. Exposure to high glucose or hypoxia results in O-GlcNAcylation of YAP/TAZ at site T383, which impedes phosphorylation at S397, leading to the stabilization and activation of YAP/TAZ. This modification ultimately contributes to vascular dysfunction in DR. Additionally, YAP/TAZ also exerts regulatory control over EC metabolism and protein O-GlcNAcylation, establishing a reciprocal interplay between the Hippo signaling pathway and protein O-GlcNAcylation. *p < 0.05; **p < 0.01. Scale bars: 30 µm in (G) and (I).
Discussion
Although diabetic retinopathy continues to be a prominent cause of vision impairment globally, the exact underlying factors of metabolic imbalance that contribute to vascular disorders and the decline of retinal functions are yet to be fully understood. In this study, we provide evidence that protein O-GlcNAcylation in response to high glucose or hypoxia exposure play a key role in mediating endothelial activation and pathological angiogenesis in the retinal vasculature. The hyper-O-GlcNAcylation level leads to the activation of a key angiogenesis regulatory pathway, the Hippo pathway via altering YAP phosphorylation and protein stability. Targeting O-GlcNAcylation-Hippo regulatory axis by genetic and pharmacological approaches attenuates mural cell loss and vascular leakage in an STZ model as well as alleviates pathologic retinal neovascularization in an OIR model. Therefore, our study provided mechanistic insight into the metabolic regulation in DR, and reveals the therapeutic potential of targeting the O-GlcNAcylation-Hippo signaling pathway in the treatment of DR (Fig. 9K).
ECs are the first-in-line cell type that is exposed to blood-borne metabolites. Recent studies have recognized the metabolism of ECs as a driving force of angiogenesis31. Metabolic pathways including glycolysis, fatty acid oxidation, and glutamine metabolism play distinct and essential roles in the process of vessel formation31. Furthermore, pathological conditions such as cancer and diabetes significantly disrupt EC metabolism32. The metabolism of ECs differs from many other cell types. For example, inhibiting CPT1A, the key enzyme for fatty acid oxidation in ECs, does not lead to energy depletion or disruption of redox homeostasis. However, it does impair the de novo nucleotide synthesis required for DNA replication33. Additionally, ECs have a unique reliance on the serine biosynthesis pathway. Unlike other cells, ECs do not heavily depend on this pathway for protein synthesis or DNA methylation. Inhibiting PHGDH, the key enzyme in the serine synthesis pathway, hampers heme synthesis, reduces purine and pyrimidine synthesis, and leads to mitochondrial respiration defects and oxidative stress34. ECs exhibit a pronounced reliance on glycolysis as their primary energy source, surpassing not only other healthy cell types but also several cancer cell types6. ECs also use alternative glucose metabolism pathways in the context of angiogenesis35. HBP was suggested to function as a nutrient sensor that modulates angiogenesis. Interestingly, during retinal development, N-glycosylation modulate tip cell selection and physiological angiogenesis via Fringe Glycosyltransferases-mediated modification of Notch36. N-glycosylation in ECs also regulates endothelial-to-hematopoietic transition during embryogenesis within the aorta-gonad-mesonephros region of the dorsal aorta37. Here, we found that in DR conditions, the O-GlcNAcylation level is particularly high in ECs and it is essential for regulating pathological angiogenesis. Although we didn’t focus on the role of N-glycosylation in DR, it would be interesting to investigate how N-glycosylation regulates DR pathogenesis.
Vascular disorders including pathological angiogenesis, pericyte loss, and BRB breakdown are hallmarks of DR. The use of anti-angiogenesis therapies targeting VEGF has revolutionized ocular angiogenesis treatment. The anti-VEGF agents are the first-line of therapies for DR, but their efficacy is limited, and non- or poor response to treatment poses a therapeutic challenge4,5. Previous studies have identified other factors, including Sema3A, Sema4D, angiopoietin-2 and netrin contribute to neovascularization and vascular barrier dysfunction11,38,39,40. Here, we identified O-GlcNAcylation targeting two transcriptional co-factors YAP and TAZ for regulating endothelial behaviors. Previous research has highlighted the Hippo pathway and its effectors YAP and TAZ as crucial regulators of cell behavior and organ growth. Upon Hippo pathway activation, YAP/TAZ are phosphorylated and inhibited, while in their non-phosphorylated state, they translocate into the nucleus and bind to different transcription factors to regulate downstream gene expression41. In ECs, YAP/TAZ regulate a transcription program including cell proliferation, inflammation, cytoskeleton organization, angiogenesis, vesicle trafficking and EC metabolism16,30,42,43. Furthermore, YAP/TAZ also controls VEGF signaling and angiopoietin-2 signaling to modulate angiogenesis16,44.
The activity of YAP/TAZ in ECs is highly dynamic. During development, YAP/TAZ are highly activated in ECs16. Once the vasculature is established, YAP/TAZ are present in ECs but at low levels, primarily localized in the cytoplasm, indicating an inactive state16. Depleting YAP/TAZ in adult mice’s ECs does not lead to observable vascular defects18, further underscoring the nonessential role of YAP/TAZ in quiescent ECs. During tumor angiogenesis, YAP/TAZ is again activated and supports tumor growth and dissemination19. Here, in this study we found that YAP/TAZ is also involved in vascular disorders during the pathogenesis of DR. Remarkably, while YAP/TAZ can be reactivated and contribute to the pathogenesis in these pathological angiogenesis conditions, the regulation of YAP/TAZ activity may vary across distinct physiological and pathological contexts. During retinal and spinal cord development, neuronal-derived VEGF triggers the activation of YAP/TAZ in ECs via RhoA and cytoskeleton rearrangement-dependent pathways16. In fibrotic conditions, YAP/TAZ activity can be modulated by sensing the stiffness of the extracellular matrix, thereby promoting angiogenesis and activating ECs45. Additionally, in tumor microenvironments characterized by high levels of inflammatory cytokines, VEGF and TNFα stimulate YAP/TAZ activation through STAT3-mediated nuclear translocation19. In this study, we identified in diabetes conditions, glucose, and hypoxia potently activate YAP/TAZ via adding O-GlcNAc moieties to YAP/TAZ. We further demonstrated that hyper-activated YAP/TAZ promote vascular leakage in an STZ model and pathologic retinal neovascularization in an OIR model. These results further highlight that YAP/TAZ is a master regulator of EC function and angiogenesis which activity could be modulated in response to different stimuli in different circumstances.
The activity of YAP/TAZ is regulated through posttranscriptional modifications. Phosphorylation of YAP at Ser 127 (or Ser 89 of TAZ) by LATS leads to their cytoplasmic localization by facilitating binding with 14-3-3 proteins. LATS phosphorylation also controls YAP and TAZ activity by promoting their degradation via phosphorylation at YAP Ser 397 (or TAZ Ser 311). This phosphorylation event generates phosphodegron motifs that are recognized by the SCFβ-TRCP E3 ligase. In addition to these phosphorylation events, other modifications, such as ubiquitination, sumoylation, methylation, and glycosylation, have also been identified to influence YAP activity41,46. Previous research has characterized Ser109 and Thr241 as O-GlcNAcylation sites on YAP, which inhibits Ser127 phosphorylation and activates YAP47,48. However, in our mass spec screening using retinal ECs, we did not detect these specific sites. These findings suggest that O-GlcNAcylation modification sites may differ across cell types, as Ser109 and Thr241 were identified in HEK293T cells and tumor cells, respectively47,48. Interestingly, a previous study indicated that TAZ does not undergo direct O-GlcNAcylation in HEK 293T cells47. However, our data from human retinal ECs clearly indicate the presence of O-GlcNAcylation modification of TAZ. While our study did not aim to identify the O-GlcNAcylation site of TAZ in ECs, it is intriguing to speculate that O-GlcNAcylation modification may vary across different cell types to regulate distinct cellular functions and behaviors.
Interestingly, in addition to being regulated by metabolic signaling, YAP/TAZ also play a role in regulating the metabolism of ECs. Previous studies have demonstrated that YAP/TAZ regulate MYC and glycolysis in ECs to support vascular growth18. Furthermore, YAP/TAZ-TEAD signaling regulates mTORC1 in ECs, linking endothelial nutrient acquisition to angiogenic growth30. In our study, we discovered that YAP/TAZ exhibit additional regulatory functions by modulating a set of genes involved in the HBP pathway and consequently impact protein O-GlcNAcylation. This reciprocal regulation forms a positive feedback loop that promotes EC activation and angiogenesis.
The dysregulation of O-GlcNAcylation is implicated in numerous chronic diseases, including cancer, neurodegenerative disorders, and diabetes, highlighting the therapeutic potential of targeting O-GlcNAc enzymes. Three OGA inhibitors (MK-8719 from Merck/Alectos, ASN-120290 from Asceneuron S.A., LY-3372689 from Eli Lilly) have entered clinical trials to treat neurodegenerative disorders49. Additionally, several potent OGT inhibitors, such as Alloxan, Ac4-5SGlcNAc, and OSMI-1, have been developed for cancer therapy28,50. Given the widespread expression of both OGA and OGT in various tissues, therapeutic inhibition of these enzymes may lead to global alterations in cellular biological processes. Therefore, an effective approach for therapeutic intervention lies in obtaining specific inhibitors targeting key O-GlcNAcylated proteins in specific disease conditions.
While endothelial hyperactivation of YAP/TAZ in DR has detrimental effects, YAP/TAZ activity is crucial for retinal regeneration in certain situations such as retinal injury51. Although anti-angiogenic therapy is considered safe according to some studies, concerns arise regarding the potential risks of long-term therapy, including the need for repeated invasive intravitreal injections that may lead to complications such as endophthalmitis and retinal detachment52,53. Hence, there is a need to develop safer therapeutic methods that specifically target ECs and effectively treat proliferative retinopathy with reduced invasiveness. In our study, we utilized an established exosome-mediated drug delivery system to load a therapeutic amount of the YAP/TAZ inhibitor, Super-TDU29, via a previously identified anchor peptide12,54. This delivery system demonstrated efficient and specific targeting of retinal blood vessels12, presenting an effective approach to selectively target YAP/TAZ in ECs and establishing the feasibility of this approach for future clinical translation in therapy development.
The limitation of this study should be acknowledged: the O-GlcNAc-modification of YAP/TAZ in retinal vessels was only analyzed in mouse models. Further investigation is needed to examine this in patient-derived samples.
In summary, through the utilization of clinical specimens and animal models, we have identified a critical role for O-GlcNAc modification in DR. We have further demonstrated that the Hippo-YAP/TAZ pathway, a key regulator of angiogenesis, is targeted by O-GlcNAcylation. Additionally, we have discovered a previously unrecognized O-GlcNAcylation site, YAP-T383, which plays a crucial role in regulating the stability and activation of YAP in retinal ECs. Notably, our findings unveil a reciprocal interplay between the Hippo signaling pathway and protein O-GlcNAcylation, whereby the Hippo pathway exerts regulatory control over protein O-GlcNAcylation. This reciprocal regulation significantly contributes to pathological angiogenesis. Overall, these insights underscore the potential therapeutic value of targeting the O-GlcNAcylation-Hippo signaling axis in diabetic retinopathy.
Methods
Study approval
Our study complied with all relevant ethical regulations and the Declaration of Helsinki. The Ethical Committee of Tianjin Medical University General Hospital approved the protocol for patient sample usage in ELISA (Approval No. IRB2019-WZ-099). Eye samples from PDR patients and age-matched controls for immunohistochemistry were obtained from the National Disease Research Interchange, with storage in accordance with the UK Human Tissue Act (2004). Ethical approval for the research was also obtained from the Ethical Review Boards of Queen’s University Belfast. Informed consent was obtained from all participants, who did not receive compensation. Sex and/or gender were not considered in the study design as they were not relevant to the findings, and participants were not selected based on self-report or assignment. All animal study protocols were approved by the Institutional Animal Care and Use Committee of Tianjin Medical University (Approval No. TMUaMEC 2020006), following the ARVO guidelines for the use of animals in vision research. Male mice were used in the STZ model based on previous evidence that only 50% of female mice responded to streptozotocin treatment55. In the OIR model and P6 mice experiments, both male and female mice were included.
Cell culture
HRCECs were purchased from Jennio Biotechnology Co. Ltd (Guangzhou, China) and HRMECs were purchased from Procell Co. Ltd. HRCECs and HRMECs were cultured in Endothelial Cell Medium (ScienCell, #1001), containing 5% FBS, 1% endothelial cell growth supplements and 1% antibiotic solution (P/S). Cells were maintained in a humidified incubator at 37 °C with 5% CO2. HRCECs and HRMECs from passages 1 to 6 were used. HEK 293T cell line (ATCC CRL-3216, female) was used in the study. HEK 293T cells were cultured in Dulbecco’s modified Eagle’s medium (HyClone, #SH30243.01), supplemented with 10% FBS and 1% antibiotic solution (P/S). To evaluate protein O-GlcNAcylation levels in HRCECs in response to high glucose or hypoxia (1% O2), and to assess the activation of YAP/TAZ in HRCECs upon PUGNAc (Sigma Aldrich, #A7229) treatment, cells were pretreated overnight in 1% FBS Endothelial Cell Medium. To analyze YAP/TAZ activity in HRCECs in response to OSMI-1 (Sigma Aldrich, #SML1621), cells were pretreated with 25 mM Glucose for 12 h in 1% FBS Endothelial Cell Medium.
Mice
Mice were housed under a 12-h light-dark cycle at 21–25 °C, with 30–70% humidity, and provided unrestricted access to food and water. Ogtfl/fl mice (Jackson Laboratory 004860) were crossed with Cdh5-Cre mice56 or Cdh5-(PAC)-CreERT2 mice57 to generate OgtΔEC line or OgtiΔEC line to specifically deleted Ogt in the endothelium. Yapfl/fl;Tazfl/fl mice58 were crossed with Cdh5-(PAC)-CreERT2 mice57 to specifically delete YAP/TAZ in the endothelium. For analysis at P6, Cre recombination was induced by intragastric injection of 50 μl tamoxifen (1 mg/ml) per day from P1 to P3. In the OIR model, Cre recombination was induced by intraperitoneal injection of 70 μl tamoxifen (1 mg/ml) at P12, P13, P14, and P16. For the STZ mice, Cre recombination was induced by intravitreal injection of 10 µg 4-OHT in 1 µl DMSO one week after STZ onset. For therapeutic experiments, mice were randomly assigned to experimental groups and were anesthetized with 5% isoflurane and intravitreally injected with 1 μl OGT inhibitor OSMI-1 (30 μM) 5 weeks after the onset of the STZ model and at P14 in OIR model. All mouse lines were C57BL/6 J background. In our study design, male mice were selected in the STZ model based on the previous study that only 50% of female mice were responding to the streptozotocin treatment55. While in OIR model and P6 mice, Sex are not relevant for any findings, therefore male and female mice were both involved.
Oxygen-induced retinopathy (OIR) in mice
Neonatal C57BL/6J mouse pups with the nursing mother on day P7 were exposed to hyperoxia (75% O2) for 5 days. Mice were returned to room air on P12. On P17, the mice were sacrificed and the retinas were harvested for analysis15. Four corners of the retinas were cut with spring scissors to flatten the whole-mount retinas, which were then photographed using a confocal microscope (LSM 800, Carl Zeiss). The retinal neovascularization area was quantified using the SWIFT_NV59 plugin in NIH ImageJ.
Streptozotocin (STZ) mouse model
Hyperglycemia was induced in male adult (8-weeks-old) mice via 5-consecutive-day intraperitoneal injection of STZ (50 mg/kg, in trisodium citrate buffer, pH 4.5; Sigma Aldrich, #S0130). Control animals were administered with an equivalent volume of the vehicle.
ELISA analysis
Vitreous samples were obtained from 11 patients undergoing vitrectomy for PDR and 7 patients with macular disease not related to diabetes (mean ± SD age at recruitment: 62.1 ± 7.5 years, 10 males and 8 females). All samples were collected by pars plana vitrectomy, centrifuged at 16,000 × g at 4 °C for 15 min and stored at −80 °C until further use. This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of Tianjin Medical University General Hospital (NO. IRB2019-WZ-099). ELISA tests for detection of human UDP-GlcNAc was performed following the manufacturer’s instructions (Jiangsu Jingmei Biological Technology Co., Ltd, China).
Human tissue processing for immunohistochemistry
The eyes were maintained in formalin. Upon arrival, the eyes were dissected and embedded in paraffin and sectioned at 5 µm thickness for follow-up immunohistochemistry analysis (mean ± SD age at recruitment: 67.8 ± 14.6 years, 4 males and 4 females). Antigen retrieval was carried out in Trilogy buffer (Sigma-Aldrich, #920P-06) for 15 min at 95 °C in a pressure cooker. Following antigen retrieval, retinal sections were blocked using 10% FCS for 1 h at room temperature (RT) and permeabilized with 0.3% Triton X-100. The samples were then incubated overnight in primary antibody at (anti-YAP/TAZ, Cell Signaling Technology, #8418, 1:50) 4 °C, followed by incubations with corresponding secondary antibodies (Jackson ImmunoResearch, 1:300) and Fluorescein conjugated Ulex Europaeus Agglutinin I (UEA I) (Vector Labs, # FL-1061-2, 1:100) at RT for 1 h. The sections were mounted in Vectashield with 4′, 6-diamidino-2-phenylindole (DAPI; Vector Laboratories, USA, H-1200). The sections were examined under Leica DMi8 epifluorescence microscope.
Immunoprecipitation and immunoblotting
Coimmunoprecipitation assays were performed as previously described19. Cells were lysed using mild lysis buffer [20 mM tris at pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 10% glycerol, 1×protease inhibitor cocktail, and 1× phosphatase inhibitor (Roche)]. Cell lysates were centrifuged for 10 min, and supernatants were used for immunoprecipitation. To immunoprecipitate Flag-tagged proteins, supernatants were collected and incubated with anti-Flag M2 magnetic beads (Sigma-Aldrich, #M8823). The beads were washed and 3×Flag peptide (Absin, bulk-peptide) was applied to elute the Flag-protein complex. The antibodies used for immunoblotting were anti-O-GlcNAc (Abcam, #2739, 1:1000), anti-pYAP-S397 (Cell Signaling Technology, #13619, 1:1000), anti- Flag (Proteintech, # 66008-4ig, 1:1000), anti-OGT (Ptm-biolab, #PTM-5497, 1:1000), anti-OGA (Cell Signaling Technology, #60406, 1:1000), anti-GFPT1 (Abcam, #125069, 1:1000), anti-β-TRCP (Cell Signaling Technology, #4394, 1:1000), anti-Ubiquitin (Cell Signaling Technology, #3936, 1:1000) and anti-β-Actin (Abclonel, #AC004, 1:1000).
Immunofluorescence
Eyeballs were embedded in cutting temperature compound (OCT, Sakura) for cryosections. For staining of O-GlcNAc, mice were perfused with warm phosphate-buffered saline (PBS), and then eyeballs were harvested and fixed in 4% paraformaldehyde (PFA)/PBS for 1 h at 4 °C, and dehydrated in 30% sucrose at 4 °C overnight60. Cryosections were washed, permeabilized in PBS containing 0.3% TritonX-100 then blocked with Mouse Polymer IHC Kit (Abcam, #ab269452) for 1 h, and incubated with anti-O-GlcNAc (Abcam, #2739, 1:200) and anti-CD31 (BD Biosciences, #557355, 1:150). After washing, sections were incubated with corresponding secondary antibody for 2 h. For staining of YAP/TAZ, cryosections were washed, permeabilized in 0.3% TritonX-100 PBS, blocked in 2% bovine serum albumin (BSA) / 0.3 % TritonX-100 PBS for 1 h, and incubated with anti-YAP/TAZ (Cell Signaling Technology, #8418, 1:200) and anti-CD31 (BD Biosciences, #557355, 1:150), After washing, sections were incubated with corresponding secondary antibody for 2 h. For flat-mounted retinas, eyes were enucleated from mice and fixed in 4% PFA for 1 h at 4 °C. Retinas were dissected, washed with PBS and permeabilized with PBS containing 1% TritonX-100 overnight at 4 °C then blocked in PBS containing 2% BSA,0.3% TritonX-100 for 12 h at 4 °C. After blocking, for visualization of retinal vasculature in OIR models, flat-mounted retinas were stained with Alexa Fluor 594-conjugated isolectinGS-IB4 (Invitrogen, #I21413, 1:100) for 2 h at RT. For immunofluorescence staining of flat-mounted retinas, retinas were incubated in blocking solution with anti-Desmin (Abcam, #15200, 1:200), anti-CD31 (BD Biosciences, #557355, 1:150) or anti-TER119 (R&D, #MAB1125, 1:200) at 4 °C overnight. After washing, the retinas were incubated with corresponding secondary antibodies (Jackson ImmunoResearch, 1:300) and Alexa Fluor 594-conjugated isolectinGS-IB4 for 2 h at RT. Images were collected on a confocal microscope (LSM 800, Carl Zeiss).
EdU injection in OIR pups
EdU (US EVERBRIGHT INC., #E6032) was administrated to pups in the OIR model at a concentration of 50 μg/g body weight, 2.5 h prior to sacrifice at P17. The eyes were enucleated and fixed in 4% PFA at 4 °C for 1 h. Afterwards, they were transferred to 30% sucrose/PBS solution and kept at 4 °C overnight. Subsequently, the eyes were embedded in OCT and frozen at −80 °C. Cryosections were prepared in a sagittal orientation. EdU+ cells were detected by using YF® 488 Click-iT EdU Stain Kits (US EVERBRIGHT INC., #C6033), according to the manufacturer’s instructions. Retinas were counterstained with isolectinGS-IB4 (1:200) and DAPI (Invitrogen, #D1306, 1:1000). The numbers of EdU+IsoB4+ cells were counted from five sagittal eye sections per eye. To ensure consistent quantification, sections intersecting the optic nerve area were used for each eye.
Analyses of vascular leakage
In OIR model, vascular leakage was analyzed by intracardial injection of FITC-conjugated dextran61. In brief, a 31-G needle was positioned above the heart 2 mm parasternal to the left at a virtual line connecting both armpits, in caudal and lateral angles of 30° and 10°. A total of 50 μl of warm PBS containing FITC-conjugated dextran (70 kDa, Sigma-Aldrich, #FD70S, 25 mg/ml) were injected. After 10 min of circulation, the eyes were enucleated and fixed in 4% PFA at 4 °C for 1 h. Retinas were then dissected, washed with PBS, permeabilized overnight at 4 °C with PBS containing 1% TritonX-100 and stained with isolectinGS-IB4 (1:100) for 2 h at RT. Subsequently, the retinas were flat-mounted in fluorescent mounting medium (Fluoromount-G®, SouthernBiotech, #0100-01) after several washes. In the STZ model, 50 μl of Alexa Fluor 555 cadaverine (Invitrogen, #A30677, 1 mg/ml) was intravenously injected into the STZ mice as described62. After allowing the cadaverine to circulate for 2 h, the eyes were fixed in 4% PFA for 2 h, and the retinas were isolated. Following permeabilization and blocking, the retinas were incubated overnight at 4 °C with the primary antibody anti-CD31 (BD Biosciences, #557355, 1:150). Subsequently, a corresponding secondary antibody was applied for 2 h at RT. Vascular leakage was observed and imaged using a confocal fluorescence microscope (LSM800, Carl Zeiss). The leakage of FITC-conjugated dextran and cadaverine was quantified by measuring the area of extravasation outside the vessels divided by the total measured area of the retina using NIH ImageJ software.
Immunofluorescence of cultured cells
For YAP/TAZ staining, HRCECs were seeded on coverslips. Following the respective treatment, cells were fixed with 4% PFA/PBS for 15 min at RT, permeabilized with 0.2% Triton X-100 PBS, blocked in 2% BSA/0.2% Triton X-100 PBS, and incubated with YAP/TAZ antibody (Cell Signaling Technology, #8418, 1:200). After washing, cells were incubated with secondary antibody. For each group, at least 4 random fields each group from three independent experiments were chosen. The counting of cells exhibiting mainly nuclear, equal nuclear and cytoplasmic, or mainly cytoplasmic YAP/TAZ localization was performed in a blinded manner.
Mouse aortic ring assay
1-mm long segments of the thoracic aorta were cultured in Opti-MEMTM (Thermo Scientific, #31985070) with 100 μ/ml penicillin and 100 μg/ml streptomycin overnight. The aortic rings were embedded in growth factor reduced Matrigel (Corning, #354230) in 24-well plates. Subsequently, the aortic ring was cultured in Opti-MEMTM supplemented with 2.5% FBS, 25 μM PUGNAc in a humidified 37 °C, 5% CO2 incubator for 5 days63. Images were acquired by using a Nikon TI2-U microscope and analyzed with NIH ImageJ software.
Choroid sprouting assay
The choroid-RPE-sclera complex was dissected and cut into ~1 mm × 1 mm pieces. The choroid explants were then embedded in growth factor-reduced Matrigel (Corning, #354230) and cultured in 0.5 ml M199 medium (Corning, NY, #11150067) in a 24-well plate. On day 2, the culture medium was supplemented with DMSO or 25 μM PUGNAc. On day 4, the sprouting choroidal explants were fixed and stained with Phallodin (Solarbio, #CA1610, 1:300)64. Images were acquired by using a confocal fluorescence microscope (LSM 800; Carl Zeiss). The sprouting area was quantified with NIH ImageJ.
Fibrin gel bead sprouting assay
The Fibrin Gel Bead Sprouting Assay was conducted following the protocol outlined (Choi et al., 2015). In summary, cytodex 3 microcarrier beads (GE Healthcare) were coated with HRCECs (or siRNA-transfected HRCECs) (mixed at a concentration of 200 cells per bead) and then embedded in 2 mg/mL fibrin gels in 24-well plates. This embedding process involved combining 2 mg/mL fibrinogen (Calbiochem) in PBS with 0.625 Units/mL thrombin (Sigma-Aldrich) and 0.15 Units/mL aprotinin (Sigma-Aldrich). The HRCECs-coated beads were cultured in ECM under the indicated stimulation conditions. After 24 h, these embedded beads were fixed with 4% PFA for 15 min, followed by blocking in 1% BSA and 0.2% Triton X-100 in PBS. Subsequently, they were incubated with 1 mg/mL Phalloidin (Sigma-Aldrich) for 2 h. Confocal images were acquired using the Zeiss LSM 800 microscope, and sprout length was analyzed using NIH ImageJ.
Transendothelial electrical resistance (TER) assay
The iCelligence machine utilized in this study was obtained from Acea Biosciences Inc, San Diego, CA. To analyze the TER, HRCECs were cultured until reaching confluence over gold microelectrodes connected to a phase-sensitive lock-in amplifier. Prior to cell growth on gold microelectrode-embedded plates, 200 μl of complete ECM were added to each well to record baseline TER Cell Index values. As the cells proliferated and spread across the microelectrode surface, the TER cell index values increased, while loss of junctional adhesion was monitored by a decrease in TER cell index values. Cell Index values were analyzed following manufacturer’s instructions65. The comparisons of TER values were conducted by analyzing the maximal barrier disruption recorded values and expressing them as a percentage of the barrier disruption observed for PUGNAc versus DMSO.
Brain ECs isolation
Brain ECs were isolated by using a magnetic cell separation method (Miltenyi Biotec, Germany) according to the manufacturer’s instructions. Briefly, freshly collected brains from mice were transported in Hanks’ balanced salt solution (HBSS; Gibco, #14025134), cut into small pieces, and then digested with 1 mg/ml Collagenase/Dispase (Roche, #10269638001) and 60 μ/ml DNase I (Roche, #4716728001) in 2% FBS endothelial cell medium at 37 °C for 5 min. The single-cell suspension was strained through a 40 μm cell strainer and washed with PBS containing 2 % FBS and 2 mM EDTA. Cells were then incubated with Myelin Removal Beads (Miltenyi, #130-096-733) to deplete myelin. Afterwards, the cells were incubated with CD31 MicroBeads (Miltenyi, #130-097-418) to obtain the ECs. Brain ECs were then used for RNA extraction and quantitative real-time PCR analysis to assess genes knockout or over-expression efficiency.
RNA extraction and quantitative real-time PCR analysis
Cells were harvested for RNA extraction using TRIzol reagent (SIGMA, #101254514). RNA concentration was determined by spectrophotometry using a Nanodrop ND-1000 (Thermo Fisher Scientific, USA) and RNA samples were reverse-transcribed to complementary DNA (cDNA) using an mRNA Reverse Transcription Kit (Thermo Fisher Scientific, #K1622) according to the protocol instructions. Quantitative reverse transcription PCR (qRT-PCR) was performed using SYBR™ Green Master Mix (TRANS, #AQ601) and the Real-Time PCR 384-well system or a Real-Time PCR 96-well system (Applied biosystems QuantStudio 3, USA). Primers used in this study are listed in Supplemental Table 1. β-Actin was used as the internal control for mouse RNA samples. All qRT-PCR results were obtained from at least three biological replicates.
Proximity ligation assay
The experiments were conducted using the Duolink® In Situ Red Starter Kit, with frozen sections of eyeballs from STZ, OIR model mice. Sections were washed with PBS containing glycine and then permeabilized with PBS containing 0.1% Triton X-100 for 20 min. After blocking, cells were incubated overnight at 4 °C with a mixture of anti-YAP/TAZ, anti-OGT, and anti-CD31 antibodies in blocking solution. Subsequently, the pre-diluted anti-rabbit plus and anti-mouse minus probes were incubated with the cells for 1 h at 37 °C. Following incubation, the cells were incubated at 37 °C with ligation enzyme for 30 min and polymerase for 100 min before mounting and imaging.
In vitro O-GlcNAcylation assay
1 mg of recombinant OGT protein (DIA·AN) was incubated with 2 mg of recombinant wild-type YAP or its mutant in 50 mL reaction volumes containing 50 mM Tris-HCl, 12.5 mM MgCl2, 2 mM UDP-GlcNAc, 1 mM DTT, and adjusted to pH 7.5. The incubation was carried out for 4 h at 37 °C. Subsequently, the reaction mixtures were resolved using SDS-PAGE and subjected to western blotting analysis employing an anti-O-GlcNAc (RL2) antibody.
ChIP-qPCR
For chromatin immunoprecipitation (ChIP) followed by qRT-PCR, HRCECs were subjected to treatment with indicated siRNA and adenovirus. Following treatment, cells were fixed in 1% formaldehyde for 10 min at room temperature (RT), followed by quenching of the reaction with 125 mM glycine for 5 min at RT. The cells were then washed twice with PBS and collected in ice-cold PBS. The cell pellet was obtained by centrifugation at 900 × g for 3 min at 4 °C, resuspended in lysis buffer, and incubated on ice for 10 min. The lysates were sonicated using a sonication system for 10 cycles at high power, with 30 s on and 30 s off. Subsequently, the samples were centrifuged at 14,000 × g for 10 min at 4 °C, and the supernatant was collected in a new tube. A portion (20 μl) of the supernatant was reserved as Input, while the remaining soluble chromatin was diluted in IP dilution buffer to a final concentration of 4 ml (1:10) and subjected to immunoprecipitation with flag-beads rotating for 2 h at 4 °C. The precipitated complexes were washed several times with washing buffer, and the protein-DNA complexes were then de-crosslinked overnight with 250 mM NaCl at 65 °C. Subsequently, RNA and proteins were digested using 80 mg RNase A and proteinase K, respectively. The precipitated DNA was extracted using a kit (TIANGEN DP204-03), and quantification of the precipitated DNA was performed by qRT-PCR. The results were normalized to both input and flag-vector adenovirus-infected control.
LC-MS/MS analysis
We infected HRCECs with FLAG-YAP adenovirus, followed by 25 mM glucose treatment for 24 h. Flag-tagged YAP protein was pulled down using anti-Flag-beads. The experiment was conducted using samples pooled from three biological repeats. After gel electrophoresis and Coomassie Brilliant Blue staining, the protein bands corresponding to the expected molecular weight were excised and subjected to tryptic digestion. The resulting tryptic peptides were dissolved in 0.1% formic acid (solvent A) and directly loaded onto a reversed-phase analytical column (15 cm length, 75 μm i.d.). The gradient consisted of an increase from 6% to 23% solvent B (0.1% formic acid in 98% acetonitrile) over 16 min, 23% to 35% in 8 min, and climbing to 80% in 3 min, holding at 80% for the last 3 min, all at a constant flow rate of 400 nl/min using an EASY-nLC 1000 UPLC system. Peptides were subjected to NSI source followed by tandem mass spectrometry (MS/MS) using a Q Exactive™ Plus (Thermo) instrument coupled online to the UPLC. The electrospray voltage was 2.0 kV. The m/z scan range was 350 to 1800 for full scan, with intact peptides detected in the Orbitrap at a resolution of 70,000. Peptides were selected for MS/MS using a normalized collision energy (NCE) setting of 28, and the fragments were detected in the Orbitrap at a resolution of 17,500. The data-dependent procedure alternated between one MS scan followed by 20 MS/MS scans with a 15.0 s dynamic exclusion. Automatic gain control (AGC) was set at 5 × 104.
Mass spectra data processing
The resulting MS/MS data were analyzed using Proteome Discoverer 2.4 software66. Tandem mass spectra were searched against the human YAP1 sequence. Trypsin/P was specified as the cleavage enzyme, allowing up to two missed cleavages. Mass error was set to 10 ppm for precursor ions and 0.02 Da for fragment ions. Fixed modification was set for carbamidomethylation of cysteine, while variable modifications included methionine oxidation, N-terminal protein acetylation, and O-GlcNAcylation of serine/threonine residues. Peptide confidence was set to high, and peptide ion scores greater than 20 were considered significant. This analysis identified two O-GlcNAcylation modification sites, T358 and T383. Detailed results are provided in the Supplementary Data file and source data files.
Retinal digest preparation
Retinal vascular preparations were performed using a pepsin–trypsin digestion method67. Briefly, enucleated eyes were fixed in 4% PFA (pH 7.4) for two days, followed by a 1-h wash in distilled water at 37 °C. The retinal vasculature was isolated using a combined pepsin (5% pepsin in 0.2% HCl for 1 h) and trypsin (2.5% in 0.2 M Tris pH 7.4 for 30 min) digestion. The samples were air-dried and stained with periodic acid schiff and hematoxylin to visualize basement membranes and capillary nuclei. The number of acellular capillaries was counted in 3 randomly selected fields per retina and normalized to the relative capillary area. All evaluations were performed in a blinded manner.
Recombinant adenovirus construction and infection
Recombinant adenovirus expressing wild type YAP gene (Isotype 9) (GenBank accession no. NM_001282101) and Ad-OGT, Ad-OGA, Flag-YAP-T383A mutant, Flag-YAP-S397A mutant, Flag-YAP-2TA mutant were generated as previously described68. HRCECs were infected with adenovirus at multiplicity of infection of ~100. 36 h after infection, cells were used for analysis.
Recombinant AAV vector production
Brain microvasculature endothelial cell-specific AAV-BR1 was prepared as follow steps26: HEK293T cells were transfected with transfer plasmids (pAAV-CMV EGFP, pAAV- CMV -YAP-WT, pAAV-CMV-YAP-T383A, or pAAV-CMV-YAP-S397A), along with pCapsid NRGTEWD (BR1) and pHelper, to package the vectors. The cells were cultured in DMEM supplemented with 1% P/S and 10% fetal bovine serum at 37 °C with 5% CO2. Transfection was performed using linear polyethylenimine (Polysciences). After five days, the cells were harvested and lysed using salt-active nuclease. The virus was then purified through iodixanol density-gradient ultracentrifugation, and the vector copy number was determined using qRT-PCR.
In vivo administration of rAAV vectors
In developmental mice, recombinant AAV vectors were retro-orbitally injected into P3 mice at a dose of 5 × 1010 gp per pup. In STZ model, recombinant AAV vectors were injected into the tail vein at a dose of 1 × 1011 genomic particles (gp) per mouse after 1 week of STZ administration. In the OIR model, recombinant AAV vectors were retro-orbitally injected at a dose of 5 × 1010 gp per pup at P7. Mice in each experimental groups were of similar age and weight and randomly allocated to AAV treatment groups.
Exosome isolation, peptides conjugation and in vivo administration
The HRCECs culture medium was centrifuged at 1000 × g for 10 min, followed by 10,000 × g for 30 min. The supernatant was collected and filtered through a 0.22 µm filter, and then subjected to ultracentrifugation at 100,000 × g for 1 h. The exosome pellets were washed with a large volume of PBS and recovered by centrifugation at 100,000 × g for 1 h. They were then resuspended in sterile PBS. The protein concentration of the isolated exosomes was quantified using the Bradford assay kit (Sangon Biotech, China). The Super-TDU sequence was SVDDHFAKSLGDTWLQIGGSGNPKTANVPQTVPMRLRKLPDSFFKPPE; the Super-TDU-CP05 sequence was SVDDHFAKSLGDTWLQIGGSGNPKTANVPQTVPMRLRKLPDSFFKPPECRHSQMTVSRL. Super-TDU and Super-TDU-CP05 were synthesized by ChinaPeptides (Shanghai, China) with a purity of 96.4%. For exosome treatment, 25 μg of exosomes, 50 μg of Super-TDU and EXOSuper-TDU (50 μg Super-TDU-CP05 and 25 μg EXO were incubated for 6 h at 4 °C before injection) were dissolved in sterile PBS and injected retro-orbitally at P12 in OIR pups and 1 week after the onset of STZ in STZ model.
siRNA transfection
Sixteen hours prior to transfection, cells were seeded in a six-well plate and allowed to reach 70 to 80% confluency. Transfection was performed using Lipofectamine™ RNAiMAX (Invitrogen, #13778150) and either control siRNA or siRNA targeting YAP/TAZ (Dharmacon Research, #M-012200-00-0005 and #M-016083-00-0005) following the manufacturer’s instructions. Human OGT siRNA: UUUAUCAGGAUUGUGCAUG dTdT, Human OGA siRNA: GGGAUAUCAAGAGUAUAAU dTdT. The siRNA solution was used at a final concentration of 160 nM.
RNA sequencing and data analyses
HRCECs were treated with DMSO or 25 μM PUGNAc for 24 h or transfected with siCtrl or siYAP/TAZ and subsequently cultured in Endothelial Cell Medium containing 25 mM glucose for 48 h. Total RNA was harvested for RNA-seq. The quality of RNA samples was analyzed using an Agilent 2100 bioanalyzer (Agilent technologies). Sequencing was performed by Novogene facility (Beijing, China) on Illumina Hiseq platform according to the manufacturer’s guide (Illumina, San Diego, CA, USA). Expression changes between groups were calculated with DESeq2 package.
Gene ontology terms network visualization
774 downregulated and 333 upregulated genes (fold-change < 0.5 or >1.5, P < 0.05) by YAP/TAZ knockdown. And the downregulated genes were applied in Cytoscape 3.9.169 using BiNGO plugin70,71 to perform the network generation.
Bioinformatic analysis
In this study, Pathway enrichment analysis was carried out by Enrichr (https://maayanlab.cloud/Enrichr/) including sources from WikiPathway 2021 Human and Gene Ontology (GO) analysis followed by network visualization of enriched GO terms16.
Statistics & reproducibility
Statistical analyses were performed using GraphPad Prism version 9.0. All data are expressed as the mean ± SEM across a minimum of three independent experiments. To calculate statistical significance, a two-tailed Student’s t-test, multiple unpaired Student’s t-tests, one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons or Tukey’s multiple comparisons, or two-way ANOVA with Šídák’s multiple comparisons test were used. No statistical method was used to predetermine sample size. Sample size was determined according to previous publications where at least three animals per group were analyzed. The experimenters were blinded to animal genotype and grouping information and all data were derived from biological replicates as indicated. P-values less than 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under accession code GSE266071 and GSE266072. The processed RNA-seq data are available at GEO database (GSE266071 and GSE266072). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE72 partner repository with the dataset identifier PXD056603.The remaining data are available within the Article, Supplementary Information or Source Data file. The cited data used in this study are available in the GEO database under accession code GSE94019. Source data are provided with this paper.
References
Ogurtsova, K. et al. IDF diabetes atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 128, 40–50 (2017).
Antonetti, D. A., Klein, R. & Gardner, T. W. Diabetic retinopathy. N. Engl. J. Med. 366, 1227–1239 (2012).
Duh, E. J., Sun, J. K. & Stitt, A. W. Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight 2, e93751 (2017).
Borrelli, E. et al. Long-term visual outcomes and morphologic biomarkers of vision loss in eyes with diabetic macular edema treated with anti-VEGF Ttherapy. Am. J. Ophthalmol. 235, 80–89 (2022).
Grunwald, J. E. et al. Risk of geographic atrophy in the comparison of age-related macular degeneration treatments trials. Ophthalmology 121, 150–161 (2014).
De Bock, K. et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154, 651–663 (2013).
Yu, P. et al. FGF-dependent metabolic control of vascular development. Nature 545, 224–228 (2017).
Liu, Z. et al. Endothelial adenosine A2a receptor-mediated glycolysis is essential for pathological retinal angiogenesis. Nat. Commun. 8, 584 (2017).
Yang, X. & Qian, K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 18, 452–465 (2017).
Wang, T. et al. O-GlcNAcylation of fumarase maintains tumour growth under glucose deficiency. Nat. Cell Biol. 19, 833–843 (2017).
Cerani, A. et al. Neuron-derived semaphorin 3A is an early inducer of vascular permeability in diabetic retinopathy via neuropilin-1. Cell Metab. 18, 505–518 (2013).
Dong, X. et al. Exosome-mediated delivery of an anti-angiogenic peptide inhibits pathological retinal angiogenesis. Theranostics 11, 5107–5126 (2021).
Ferrer, C. M. et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol. Cell 54, 820–831 (2014).
Yi, W. et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 337, 975–980 (2012).
Connor, K. M. et al. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat. Protoc. 4, 1565–1573 (2009).
Wang, X. et al. YAP/TAZ orchestrate VEGF signaling during developmental angiogenesis. Dev. Cell 42, 462–478.e7 (2017).
Baffert, F. et al. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am. J. Physiol. Heart Circ. Physiol. 290, H547–H559 (2006).
Kim, J. et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J. Clin. Invest. 127, 3441–3461 (2017).
Shen, Y. et al. STAT3-YAP/TAZ signaling in endothelial cells promotes tumor angiogenesis. Sci. Signal 14, eabj8393 (2021).
Yang, Y. et al. O-GlcNAc transferase inhibits visceral fat lipolysis and promotes diet-induced obesity. Nat. Commun. 11, 181 (2020).
Leney, A. C., El Atmioui, D., Wu, W., Ovaa, H. & Heck, A. J. R. Elucidating crosstalk mechanisms between phosphorylation and O-GlcNAcylation. Proc. Natl Acad. Sci. USA 114, E7255–E7261 (2017).
Zhao, B., Li, L., Tumaneng, K., Wang, C. Y. & Guan, K. L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF beta-TRCP. Gene Dev. 24, 72–85 (2010).
Hu, J. K. et al. An FAK-YAP-mTOR signaling axis regulates stem cell-based tissue renewal in mice. Cell Stem Cell 21, 91–106.e106 (2017).
Zhao, B., Li, L., Tumaneng, K., Wang, C. Y. & Guan, K. L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 24, 72–85 (2010).
Zhu, Y. & Hart, G. W. Dual-specificity RNA aptamers enable manipulation of target-specific O-GlcNAcylation and unveil functions of O-GlcNAc on beta-catenin. Cell 186, 428–445.e427 (2023).
Korbelin, J. et al. A brain microvasculature endothelial cell-specific viral vector with the potential to treat neurovascular and neurological diseases. EMBO Mol. Med. 8, 609–625 (2016).
Chen, D. Y. et al. Endothelium-derived semaphorin 3G attenuates ischemic retinopathy by coordinating beta-catenin-dependent vascular remodeling. J. Clin. Invest. 131, e135296 (2021).
Ortiz-Meoz, R. F. et al. A small molecule that inhibits OGT activity in cells. ACS Chem. Biol. 10, 1392–1397 (2015).
Jiao, S. et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 25, 166–180 (2014).
Ong, Y. T. et al. A YAP/TAZ-TEAD signalling module links endothelial nutrient acquisition to angiogenic growth. Nat. Metab. 4, 672–682 (2022).
Li, X., Sun, X. & Carmeliet, P. Hallmarks of endothelial cell metabolism in health and disease. Cell Metab. 30, 414–433 (2019).
Eelen, G., de Zeeuw, P., Simons, M. & Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 116, 1231–1244 (2015).
Schoors, S. et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520, 192–197 (2015).
Vandekeere, S. et al. Serine synthesis via PHGDH is essential for heme production in endothelial cells. Cell Metab. 28, 573–587.e513 (2018).
Rohlenova, K., Veys, K., Miranda-Santos, I., De Bock, K. & Carmeliet, P. Endothelial cell metabolism in health and disease. Trends Cell Biol. 28, 224–236 (2018).
Benedito, R. et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137, 1124–1135 (2009).
Kasper, D. M. et al. The N-glycome regulates the endothelial-to-hematopoietic transition. Science 370, 1186–1191 (2020).
Hammes, H. P. et al. Angiopoietin-2 causes pericyte dropout in the normal retina: evidence for involvement in diabetic retinopathy. Diabetes 53, 1104–1110 (2004).
Wu, J. H. et al. Inhibition of Sema4D/PlexinB1 signaling alleviates vascular dysfunction in diabetic retinopathy. EMBO Mol. Med. 12, e10154 (2020).
Miloudi, K. et al. Truncated netrin-1 contributes to pathological vascular permeability in diabetic retinopathy. J. Clin. Invest. 126, 3006–3022 (2016).
Ma, S., Meng, Z., Chen, R. & Guan, K. L. The Hippo pathway: biology and pathophysiology. Annu. Rev. Biochem. 88, 577–604 (2019).
Wang, L. et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540, 579–582 (2016).
Sakabe, M. et al. YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc. Natl Acad. Sci. USA 114, 10918–10923 (2017).
He, J. et al. Yes-associated protein promotes angiogenesis via signal transducer and activator of transcription 3 in endothelial cells. Circ. Res. 122, 591–605 (2018).
Shen, Y. et al. Reduction of liver metastasis stiffness improves response to bevacizumab in metastatic colorectal cancer. Cancer Cell 37, 800–817.e807 (2020).
Varelas, X. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 141, 1614–1626 (2014).
Peng, C. et al. Regulation of the Hippo-YAP pathway by glucose sensor O-GlcNAcylation. Mol. Cell 68, 591–604.e595 (2017).
Zhang, X. et al. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat. Commun. 8, 15280 (2017).
Bartolomé-Nebreda, J. M., Trabanco, A. A., Velter, A. I. & Buijnsters, P. O-GlcNAcase inhibitors as potential therapeutics for the treatment of Alzheimer’s disease and related tauopathies: analysis of the patent literature. Expert Opin. Ther. Pat. 31, 1117–1154 (2021).
Gloster, T. M. et al. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat. Chem. Biol. 7, 174–181 (2011).
Rueda, E. M. et al. The Hippo pathway blocks mammalian retinal muller glial cell reprogramming. Cell Rep. 27, 1637–1649.e1636 (2019).
Brown, D. M. et al. Long-term outcomes of ranibizumab therapy for diabetic macular edema: the 36-month results from two phase III trials: RISE and RIDE. Ophthalmology 120, 2013–2022 (2013).
van Wijngaarden, P., Coster, D. J. & Williams, K. A. Inhibitors of ocular neovascularization: promises and potential problems. JAMA 293, 1509–1513 (2005).
Gao, X. et al. Anchor peptide captures, targets, and loads exosomes of diverse origins for diagnostics and therapy. Sci. Transl. Med. 10, eaat0195 (2018).
Martinez, N. et al. Glycerol contributes to tuberculosis susceptibility in male mice with type 2 diabetes. Nat. Commun. 14, 5840 (2023).
Zhao, Y. et al. PGF(2alpha) facilitates pathological retinal angiogenesis by modulating endothelial FOS-driven ELR(+) CXC chemokine expression. EMBO Mol. Med. 15, e16373 (2023).
Wang, Y. et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486 (2010).
Xin, M. et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl Acad. Sci. 110, 13839–13844 (2013).
Stahl, A. et al. Computer-aided quantification of retinal neovascularization. Angiogenesis 12, 297–301 (2009).
Dong, X. et al. Natural killer cells promote neutrophil extracellular traps and restrain macular degeneration in mice. Sci. Transl. Med. 16, eadi6626 (2024).
Walchli, T. et al. Quantitative assessment of angiogenesis, perfused blood vessels and endothelial tip cells in the postnatal mouse brain. Nat. Protoc. 10, 53–74 (2015).
Segarra, M. et al. Endothelial Dab1 signaling orchestrates neuro-glia-vessel communication in the central nervous system. Science 361, eaao2861 (2018).
Baker, M. et al. Use of the mouse aortic ring assay to study angiogenesis. Nat. Protoc. 7, 89–104 (2011).
Tomita, Y. et al. An ex vivo choroid sprouting assay of ocular microvascular angiogenesis. J. Vis. Exp. 162, 10.3791/61677 (2020).
Sinha, R. K. et al. Apolipoprotein E receptor 2 mediates activated protein C-induced endothelial Akt activation and endothelial barrier stabilization. Arterioscler. Thromb. Vasc. Biol. 36, 518–524 (2016).
Klykov, O. et al. Efficient and robust proteome-wide approaches for cross-linking mass spectrometry. Nat. Protoc. 13, 2964–2990 (2018).
Jiang, Q. et al. Circular RNA-ZNF532 regulates diabetes-induced retinal pericyte degeneration and vascular dysfunction. J. Clin. Invest. 130, 3833–3847 (2020).
Liu, J. et al. Heterologous prime-boost immunizations with chimpanzee adenoviral vectors elicit potent and protective immunity against SARS-CoV-2 infection. Cell Discov. 7, 123 (2021).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Xue, J. et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288 (2014).
Maere, S., Heymans, K. & Kuiper, M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449 (2005).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–d552 (2022).
Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2020YFA0803703, 2021YFC2401404), National Natural Science Foundation of China (Grant Numbers 82122018, 82330031, 82020108007, 32171101 and 8230041151), Natural Science Foundation of Tianjin (23JCJQJC00050), Postdoctoral Fellowship of CPSF (GZB20230516 and 2024T170653) and International Student Exchange Program Award from Belfast Association for the Blind. The authors thank Prof. Dr. Andreas Stahl for providing SWIFT_NV macros, Prof. Martin Trepel and Dr. Jakob Körbelin for providing the AAV-BR1 system. We thank the Core Facility of Research Center of Basic Medical Sciences at Tianjin Medical University for technical support.
Author information
Authors and Affiliations
Contributions
H.Y. and X.W. designed the study and acquired the funding. H.Z., M.D., J.Z., T.W., J.H., Q.L., H.X., M.C., H.Y., and X.W. supervised the research. TJ. L and M.C. provided the resource. Y.L., Q.L., B.C., F.W., Y.L., X.D., N.M., Z.W., L.W., Y.F., Y.Z., Y.L., and Y.S. performed the research and analyzed the data. X.W. and Y.L. wrote the manuscript. All authors reviewed the paper and approved the final draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jian-xing Ma, Lance Wells and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lei, Y., Liu, Q., Chen, B. et al. Protein O-GlcNAcylation coupled to Hippo signaling drives vascular dysfunction in diabetic retinopathy. Nat Commun 15, 9334 (2024). https://doi.org/10.1038/s41467-024-53601-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53601-x
This article is cited by
-
MSCs-EXOs containing miR-21a-5p promoted cell proliferation in the rat retinal Müller cell rMC-1 via targeting LATS1
Stem Cell Research & Therapy (2025)
-
Posttranslational modifications of YAP/TAZ: molecular mechanisms and therapeutic opportunities
Cellular & Molecular Biology Letters (2025)
-
Crosstalk between O-GlcNAcylation and phosphorylation in metabolism: regulation and mechanism
Cell Death & Differentiation (2025)
