
Abstract
The selective incorporation of a deuterium atom into small molecules with high selectivity is highly valuable for medical and chemical research. Unfortunately, this remains challenging due to the complete deuteration caused by commonly used hydrogen isotope exchange strategies. We report the development of a photocatalytic selective monodeuteration protocol utilizing C–C bond as the unconventional functional handle. The synergistic combination of radical-mediated C–C bond scission and deuterium atom transfer processes enables the effective constructions of benzylic CDH moieties with high selectivity for monodeuteration. The combinational use of a bisphosphonium photocatalyst, thiol catalyst, and CH3OD deuteration agent provides operationally simple conditions for photocatalytic monodeuteration. Moreover, the photoinduced electron transfer process of the bisphosphonium photocatalyst is elucidated through a series of spectroscopy experiments, identifying a peculiar back electron transfer process that can be regulated by subsequent nucleophilic additions.
Similar content being viewed by others
Introduction
The selective incorporation of deuterium atoms into small molecules is of great importance in medicinal and chemical research1,2,3,4. Compared to their nondeuterated analogues, deuterium-labeled compounds exhibit unique metabolism and pharmacokinetic properties, which have been frequently exploited to increase the bioavailability of pharmaceutical candidates5,6. Deuterated compounds have also been used as metabolic tracers and mass spectrometry analytic standards7. In organic chemistry, kinetic isotope effect (KIE) experiments with mono-deuterated or fully-deuterated compounds have been widely used in mechanistic investigations to elucidate reaction pathways8,9. Despite widespread uses, the preparation of deuterium-labeled molecules with high selectivity oftentimes are rather challenging tasks, as the separation of unlabeled or partially labeled materials from the desired deuterated product has been impeded by the trivial difference in physical properties.
With the advancement of transition metal catalysis, tremendous progress has been made in H/D exchange reactions, enabling various environmentally benign and operationally simple protocols2,10,11,12,13,14,15,16,17,18. In this manifold, the selectivity problem can be addressed through the complete deuteration of an aliphatic carbon which can be easily achieved by thermodynamic effect19, while the selective incorporation of a single deuterium into methylene and methyl groups is deemed impractical due to over-deuteration (Fig. 1)20. Thusly, the preparation of monodeuterated molecules heavily relies on the use of prefunctionalized substrates such as alkyl halides. The necessities of expensive deuteration reagents as well as multistep conversions has raised urgent needs to develop operationally simple and sustainable methods21,22,23,24,25,26,27,28,29,30. Recently, selective monodeuteration of C(sp3)–H bonds have been realized through elegantly designed intramolecular 1,5-HAT processes by Studer, Xie, and Zhu groups31,32. With the combinational uses of radical precursors with AIBN, or amide substrates with Ir photocatalyst, monodeuterated amides can be obtained with high selectivity. Considering the peculiar uses of mono-deuterated compounds in KIE studies and pharmaceutical investigations5,9, the development of innovative strategies and catalytic protocols remains in high demand.

a Selective deuteration: an empowering tool in medicinal and chemical research and a challenging task in organic synthesis. b Previous innovative strategies for monodeuteration: Intramolecular HAT. c. Photocatalytic ring opening protocol for selective monodeuteration.
Radical-mediated C–C bond cleavage and functionalizations, with emerging photoredox catalysis as the enabling platform to generate open-shell radical intermediates, have provided a synthetically valuable strategy to exploit ubiquitous C(sp3)–C(sp3) bonds as functional handles33,34. Under ambient temperature, a high-energy radical species such as alkoxy radical or benzene radical cation could promote the homolytic cleavage of the adjacent C–C single bond to generate a more stabilized carbon-centered radical35,36. Importantly, the resultant alkyl radical intermediate could undergo diversified conversions which have provided intriguing opportunities for functional group installations. As such, a series of ring-opening transformations have been developed over the last decade, demonstrating various intriguing applications, including oxidations, halogenation, alkylation, and arylation for the syntheses of distally functionalized carbonyl compounds37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56. We recently wondered whether a radical-mediated deuterium transfer event could be incorporated in the photocatalytic ring opening process for the selective mondeuteration, providing an intriguing strategy to utilize inert and ubiquitous C(sp3)–C(sp3) bonds as functional handles for deuterium incorporation. Compared to radical-mediated H/D exchange where conversion rates suffer from the small driving force (difference in zero-point energy, ~1 kcal/mol), ring-opening deuterations take advantage of the substantial energy gain in strain release (strain energies of 3- to 5- membered aliphatic rings, 6-27 kcal/mol) to enhance efficient deuteration.
Given the prevalence of benzylic C–H bonds in pharmaceutical candidates, we opt to first investigate the ring-opening deuteration protocol for the selective constructions of benzylic CDH moieties. In the deuteration of aromatic hydrocarbons, the relatively weakened benzylic C–H bonds can facilitate H/D exchange but have posed severe challenges for the selective monodeuteration2. We envisioned that the well-established β-bond cleavage of aromatic radical cations could render a facile approach to achieve benzylic monodeuteration, via selective ring openings of cyclic hydroaromatics and the intermediacy of a benzylic radical intermediate35,57,58,59,60,61,62. Through the use of efficient HAT catalysts, inexpensive and operationally handy deuteration agents such as CH3OD can be employed to enforce the desired deuterium atom transfer. Critically, the selective and irreversible C–C bond scission process would only allow the installation of one deuterium atom. Regarding the challenges raised by the high oxidation potential of aromatic hydrocarbons, we firstly looked into the development of highly oxidizing photocatalysts.
Organophosphorus compounds have been extensively investigated in organic electronics, optical sensing, and imaging applications63,64,65,66,67,68. Nevertheless, their unique photophysical and electrochemical properties remain largely underexploited in photoredox catalysis. Recently, we found that a bisphosphonium (BPP) compound easily prepared from the oxidative cyclization of 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), can act as a strongly oxidizing organophotocatalyst to promote the intramolecular hydroetherification of alkenols. The remarkable capacity of this photoredox catalyst, including high oxidation potential (E *1/2 = 2.17 V vs. SCE), strong absorption of visible light (λmax = 413 nm), and long excited life time, has not been fully explored for synthetic transformations69,70,71,72.
Herein, we disclose a practical and selective monodeuteration protocol by bisphosphonium photocatalysis which can enable the facile constructions of benzylic CDH moieties from easily accessed materials. The photoexcitation and photoinduced electron transfer events of bisphosphonium catalyst are elu cidated in its premiere instance by spectroscopy experiments, paving the way for future applications.
Results and discussion
Reaction optimization
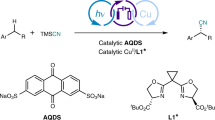
To validate our hypothesis, phenylcyclopropane (E1/2 = 1.7 V vs. SCE) was employed as the template substrate, readily available mono-deuteromethanol CH3OD was chosen as the deuteration reagent. In combination with commonly used atom transfer catalyst (TRIPS)2, a variety of oxidizing photocatalysts were evaluated under the irradiation of high intensive LED light (see supporting information for the detailed description of set-up for the parallel reactions). As depicted in Fig. 2, commonly utilized oxidizing photocatalysts Ru(bpz)32+ and triphenylpryrlium were found ineffective, while mesitylacridinium photocatalyst could afford product 2 with 68% yield and 95% D-incorporation. Among the P-containing conjugated arenes evaluated, BPP photocatalyst rendered the optimal condition, delivering the monodeuterated product with high efficiency and selectivity (83% yield, 96% D-incorporation). A seemingly positive correlation between the oxidation capacity and catalytic efficiency can be found in this series of organophotocatalyst, as phosphonium salt 3 (E *1/2 = 1.54 V vs. SCE) was found inactive and bisphosphapyrenium 4 (E *1/2 = 1.69 V vs. SCE) enabled moderate efficiency (73% yield, 96% D-incorporation). Furthermore, control experiments have indicated the essential role of BPP photocatalyst, thiol catalyst and LED light. Notably, the control experiment with H2O in entry 4 has also revealed the critical importance of anhydrous conditions to achieve high deuteration selectivity.

Reactions were performed in a parallel reactor, with 0.2 mmol 1, 2 mol% photocatalyst, 10 mol% (TRIPS)2, CH3CN (0.6 ml) and MeOD (0.3 ml). aThe yields were determined with GC-FID, the D-incorporation (D-inc) was determined by HR-MS. The recovery of 1 was presented in parentheses.
Selective monodeuteration of cyclic aromatic substrates
With the optimal condition in hand, we next explored the scope of cyclic aromatic substrates and were delighted to find that this operationally simple and inexpensive catalytic system could be adapted to 3-membered to 7-membered cyclic starting materials. Importantly, a high degree of monodeuteration selectivity was achieved across the board, delivering the desired products with more than 95% D-incorporation. Even in the presence of weakened benzylic C–H bonds and acidic α-carbonyl C–H bonds which are prone to undergo H/D exchange to induce multiple deuterium incorporation, only the desired monodeuterated product was obtained. As shown in Fig. 3, this organophotocatalytic monodeuteration protocol can be successfully applied to a variety of substituted arylcyclopropanes, generating monodeuterated 3-methoxypropylbenzenes in high D-incorporation rate. Arylbornic ester (7, 8, 14) and aryl bromide (9, 10, 26), commonly used functional handles in transition metal catalysis, could be well tolerated under the current photocatalytic condition. Owing to the high oxidation capacity of BPP catalyst, electron deficient arenes which are more challenging to activate in SET oxidations can be employed, although p-CF3 and m-Br substitutes rendered somewhat declined efficiencies. Regarding unsymmetrical arylcyclobutanones, the radical cation-mediated ring-opening process proved to be highly selective, as the C–C bond between the carbonyl and benzylic carbon terminus were selectively cleaved to generate monodeuterated 4-phenylbutanoates (16-27). For less strained 5-, 6-, 7-membered cyclic substrates, we noticed that this photocatalytic ring-opening deuteration can be effectively promoted by stabilizing the partially formed positive charge at the β-carbon terminus73. The introduction of a methoxy group at the β-carbon led to the facile cleavage of these less strained C–C bonds, rendering monodeuterated products with a distal hemiacetal functionality for further functionality manipulations (see Figs. S19–S23 for the conversions into amine, alcohol, alkene and tetrahydroquinoline moites). A variety of N-heterocyclic compounds, including pyridine and quinoline derivatives, have been effectively incorporated into monodeuterated products with yields ranging from excellent to moderate, without a significant impact on deuterium incorporation efficiency (33-36). Deuteration using D2O as the agent under standard conditions has yielded satisfactory outcomes (37, 38). Furthermore, 18O labeling is readily accomplished with H218O as the nucleophile under standard conditions (39). Importantly, the cyclic hydroaromatic moieties embedded in complex scaffolds such as steroid ring systems can be selectively cleaved, enabling the selective incorporation of benzylic CHD moieties. The implementation of a continuous-flow synthesis method has facilitated scaled-up production of 2, achieving an 87% yield and a space-time yield of 17.4 g/L•h under optimized conditions, while significantly reducing the disulfide loading to 5 mol%.

Reactions were performed in a parallel reactor, with 0.2 mmol substrate, 2 mol% photocatalyst, 10 mol% (TRIPS)2, CH3CN (0.6 ml) and MeOD (0.3 ml). Deuterium incorporation was determined by HR-MS analysis.
Spectroscopy experiments
Spectroscopy experiments were then conducted to probe the critical photoinduced electron transfer (PET) event between phenylcyclopropane 1 and BPP catalyst. Unexpectedly, Stern–Volmer quenching studies revealed an insignificant fluorescence quenching effect of phenylcyclopropane even at relatively high concentrations (Fig. 4A), reminding us that the electron transfer between excited BPP and phenylcyclopropane might be sluggish. This was in striking contrast to the observation made in the same set of experiments with triethylamine as the reductant (see Fig. S4 for the quenching experiment with triethylamine). As demonstrated in Fig. 4B, the reduced BPP ([BPP]•–) generated from the PET process with triethylamine can be clearly identified in the 580 nm region in the UV-vis absorption spectrum, while the irradiation of the solution containing BPP and phenylcyclopropane did not cause any observable changes in the absorption spectrum. From the redox potential perspective, both PET processes are thermodynamically favorable, but only the PET process with triethylamine led to the net generation of [BPP]•–. Considering that the only difference lies in the oxidative nature of the resultant radical cations, we posited that highly oxidizing aromatic radical cations might result in an overwhelming back electron transfer, leading to declined efficiency74,75. These preliminary findings, contradicting the efficient ring-opening deuteration we obtained, promoted us to elucidate the PET events between BPP photocatalyst and the phenylcyclopropane substrate.

A Stern-Volmer quenching experiment. B Photolysis experiment. C Transient absorption spectra. D ESA decay signals in the presence of oxygen. E ESA decay signals in the presence of 1. F ESA decay signals in the presence of 1 and MeOH. G proposed photoexcitation and monodeuteration pathways.
As the excitation pattern of bisphosphonium photocatalyst is currently unknown, we decided to first probe the photoexcitation process via ultrafast transient absorption (TA) spectroscopy. As depicted in Fig. 4C, the ultrafast TA spectra of BPP in acetonitrile solution (c = 0.5 mM) following 400 nm excitation (power intensity, 20 nJ per pulse) exhibit abundant excited state spectra features, including a ground state bleaching (GSB) at 420–430 nm, a simulated emission (SE) band at 430-500 nm, and two excited state absorption bands (ESA 1 and ESA 2) in the range of 500-650 nm (see Fig. S8 for details). The ESA 1 band at 540 nm shows single exponential decay of ~849 ps which is accompanied by recovery of the SE band at 466-nm region indicating the decay dynamics of the ESA 1 and SE bands originate from the same singlet excited state (S1 state). The SE negative signal coincides with the fluorescence emission spectrum of BPP measured by fluorescence spectroscopy, and we identify it as the fluorescence signal generated when the S1 state returns to the ground state after BPP excitation. The lifetime (τf = 819 ps measured by TCSPC) is consistent with the decay lifetime (τ1 = 849 ps) obtained by transient spectral dynamics analysis. The ESA 2 band at 625 nm showcases two exponential dynamics with a rise of 849 ps followed by an infinity-long lifetime decay. The rise component of 849 ps is reasonably ascribed to the intersystem crossing relaxation (ISC) time from the S1 state to the triplet state (T1) state. The latter component corresponds to the decay lifetime of T1 state.
To obtain a more complete dynamic process of the ESA 2 band, we carried out transient absorption spectroscopy in ns-μs timescale in the region of 525–700 nm. As shown in Fig. 4D, the ESA 2 band exhibits exponential decay with time constants of 29 μs. Monitoring whether the lifetime of the photoreaction system has a rapid reduction after the introduction of oxygen can be used to determine whether the triplet state exists. As demonstrated in Fig. 4D, oxygen can quickly quench 640 nm region signal, thus identifying the signal of ESA 2 band as the triplet signal generated by ISC after BPP excitation.
After the identification of the triplet state which is responsible for the desired electron transfer event, we next monitored the TA spectrum of BPP catalyst in the presence of phenylcyclopropane 1 (BPP: 1 = 1:100). With the addition of phenylcyclopropane, an accelerated decay of triplet BPP absorption can be observed (Fig. 4E). At 20 μs, the absorption centered of ESA 2 band at 625 nm returned to baseline, indicating a nearly complete consumption of triplet BPP by the SET with 1. But in the 580 nm region, the characteristic absorption of [BPP]•– was too weak to be distinguished. This peculiar observation could be explained by the fast and predominant back electron transfer between [BPP]•– and radical cation of phenylcyclopropane [1•+] that has converted [BPP]•– into ground state BPP. Thusly, no net electron transfer takes place and this gives rise to no [BPP]•–, in accordance with the observation made in the photolysis experiment. Nevertheless, in the presence of phenylcyclopropane and methanol, the characteristic absorption of [BPP]•– can be clearly observed as the triple absorption returned to baseline (Fig. 4F). The nucleophilic attack of [1•+] with methanol generates of a more stable and less oxidizing benzylic radical through a ring-opening process. Thusly, the net formation of [BPP]•– has indicated that the back-electron transfer event is suppressed by the nucleophilic attack of [1•+].
Proposed reaction mechanism
Based on these findings, the photoinduced electron transfer processes in this ring-opening deuteration have been established and the proposed reaction pathway was depicted in Fig. 4G. Upon visible-light irradiation, a transient singlet BPP can be formed, most of which undergo a fast ISC process (τISC = 849 ps) to generate a long-lived triplet state (τ1 = 29 us). The single electron oxidation of 1 by 3BPP is thermodynamically favorable and generates [1•+] and reduced BPP. Nevertheless, bimolecular back electron transfer from the resultant [1•+] to [BPP]•– regenerates ground state catalyst, rendering a fully reversible PET process. The BET process could be suppressed by the well-established nucleophilic attack of [1•+] with methanol, forming a benzylic radical intermediate. A subsequent deuterium atom transfer mediated by TRIPSD would deliver the desired monodeuteration product. Lastly, a single electron transfer event would regenerate BPP and thiol catalysts.
In summary, we show a practical and selective monodeuteration protocol utilizing radical-mediated C–C bond scission and deuterium atom transfer processes. Under LED irradiation, in the presence of bisphosphonium photocatalyst, thiol catalyst, and CH3OD, easily accessible cyclic hydroaromatics can be efficiently converted into mono-deuterated products equipped with distal functionalities. The redox-neutral and operationally simple reaction conditions provides a synthetically appealing approach for the constructions of benzylic CDH moieties. Through spectroscopy experiments including transient absorptions, the photoexcitation and photoinduced electron transfer events of bisphosphonium catalyst are established, elucidating a peculiar back-electron transfer process which can be regulated by subsequent nucleophilic additions.
Methods
General procedure for batch reactions
An 8 mL vial was charged with substrates (1.0 equiv.), BPP (0.02 equiv.), (TRIPS)2 (0.1 equiv.) and 1 mL MeCN/MeOD (v/v = 2:1). The vial was sealed with a Teflon®-lined cap, the reaction mixture was degassed by argon sparging for 10 minutes. The mixture was then irradiated with LED (λmax = 400 nm photon flux, 2.5 W) and stirred under irradiation at ambient temperature for 16 hours. After the reaction, the mixture was evaporated in vacuo and the residue was purified by flash chromatography.
Data availability
Data are available in the manuscript and supplementary materials. Data supporting the findings of this manuscript are also available from the authors upon request.
References
Kop, S. et al. Recent developments for the deuterium and tritium labeling of organic molecules. Chem. Rev. 122, 6634–6718 (2022).
Prakash, G., Paul, N., Oliver, G. A., Werz, D. B. & Maiti, D. C–H deuteration of organic compounds and potential drug candidates. Chem. Soc. Rev. 51, 3123–3163 (2022).
Li, N., Li, Y., Wu, X., Zhu, C. & Xie, J. Radical deuteration. Chem. Soc. Rev. 51, 6291–6306 (2022).
Zhou, R., Ma, L., Yang, X. & Cao, J. Recent advances in visible-light photocatalytic deuteration reactions. Org. Chem. Front. 8, 426–444 (2021).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. Deuterium- and tritium-labelled compounds: applications in the life sciences. Angew. Chem. Int. Ed. 57, 1758–1784 (2018).
Pirali, T., Serafini, M., Cargnin, S. & Genazzani, A. A. Applications of deuterium in medicinal chemistry. J. Med. Chem. 62, 5276–5297 (2019).
Mutlib, A. E. Application of stable isotope-labeled compounds in metabolism and in metabolism-mediated toxicity studies. Chem. Res. Toxicol. 21, 1672–1689 (2008).
Gómez-Gallego, M. & Sierra, M. A. Kinetic isotope effects in the study of organometallic reaction mechanisms. Chem. Rev. 111, 4857–4963 (2011).
Simmons, E. M. & Hartwig, J. F. On the interpretation of deuterium kinetic isotope effects in C–H bond functionalizations by transition-metal complexes. Angew. Chem. Int. Ed. 51, 3066–3072 (2012).
Atzrodt, J., Derdau, V., Fey, T. & Zimmermann, J. The renaissance of H/D exchange. Angew. Chem. Int. Ed. 46, 7744–7765 (2007).
Kurita, T. et al. Efficient and convenient heterogeneous palladium-catalyzed regioselective deuteration at the benzylic position. Chem. Eur. J. 14, 664–673 (2008).
Khaskin, E. & Milstein, D. Simple and efficient catalytic reaction for the selective deuteration of alcohols. ACS Catal. 3, 448–452 (2013).
Kerr, W. J., Reid, M. & Tuttle, T. Iridium-catalyzed formyl-selective deuteration of aldehydes. Angew. Chem. Int. Ed. 56, 7808–7812 (2017).
Palmer, W. N. & Chirik, P. J. Cobalt-catalyzed stereoretentive hydrogen isotope exchange of C(sp3)–H Bonds. ACS Catal. 7, 5674–5678 (2017).
Pfeifer, V. et al. Palladium nanoparticles for the deuteration and tritiation of benzylic positions on complex molecules. Angew. Chem. Int. Ed. 60, 26671–26676 (2021).
Puleo, T. R., Strong, A. J. & Bandar, J. S. Catalytic α-selective deuteration of styrene derivatives. J. Am. Chem. Soc. 141, 1467–1472 (2019).
Li, W. et al. Scalable and selective deuteration of (hetero)arenes. Nat. Chem. 14, 334–341 (2022).
Du, H.-Z. et al. Cesium amide-catalyzed selective deuteration of benzylic C-H bonds with D2 and application for tritiation of pharmaceuticals. Angew. Chem. Int. Ed. 62, e202214461 (2023).
Junk, T. & Catallo, W. J. Hydrogen isotope exchange reactions involving C–H (D, T) bonds. Chem. Soc. Rev. 26, 401–406 (1997).
Bhadra, P. K. et al. Enhancement of the properties of a drug by mono-deuteriation: reduction of acid-catalysed formation of a gut-motilide enol ether from 8-deuterio-erythromycin B. Org. Biomol. Chem. 14, 6289–6296 (2016).
Loh, Y. Y. et al. Photoredox-catalyzed deuteration and tritiation of pharmaceutical compounds. Science 358, 1182–1187 (2017).
Soulard, V., Villa, G., Vollmar, D. P. & Renaud, P. Radical Deuteration with D2O: catalysis and mechanistic insights. J. Am. Chem. Soc. 140, 155–158 (2018).
Zhang, M., Yuan, X.-A., Zhu, C. & Xie, J. Deoxygenative deuteration of carboxylic acids with D2O. Angew. Chem. Int. Ed. 58, 312–316 (2019).
Li, Y. et al. Organophotocatalytic selective deuterodehalogenation of aryl or alkyl chlorides. Nat. Commun. 12, 2894 (2021).
Li, N. et al. A highly selective decarboxylative deuteration of carboxylic acids. Chem. Sci. 12, 5505–5510 (2021).
Zhou, X., Yu, T. & Dong, G. Site-specific and degree-controlled alkyl deuteration via Cu-catalyzed redox-neutral deacylation. J. Am. Chem. Soc. 144, 9570–9575 (2022).
Zhao, G., Yao, W., Mauro, J. N. & Ngai, M.-Y. Excited-state palladium-catalyzed 1,2-spin-center shift enables selective C-2 reduction, deuteration, and iodination of carbohydrates. J. Am. Chem. Soc. 143, 1728–1734 (2021).
Zhang, Y., Ji, P., Dong, Y., Wei, Y. & Wang, W. Deuteration of formyl groups via a catalytic radical H/D exchange approach. ACS Catal. 10, 2226–2230 (2020).
Kuang, Y. et al. Visible light driven deuteration of formyl C–H and hydridic C(sp3)–H bonds in feedstock chemicals and pharmaceutical molecules. Chem. Sci. 11, 8912–8918 (2020).
Shi, Q. et al. Visible-light mediated catalytic asymmetric radical deuteration at non-benzylic positions. Nat. Commun. 13 (2022).
Wang, L., Xia, Y., Derdau, V. & Studer, A. Remote site-selective radical C(sp3)−H monodeuteration of amides using D2O. Angew. Chem. Int. Ed. 60, 18645–18650 (2021).
Li, N. et al. Highly selective single and multiple deuteration of unactivated C(sp3)-H bonds. Nat. Commun. 13, 4224 (2022).
Yu, X.-Y., Chen, J.-R. & Xiao, W.-J. Visible light-driven radical-mediated C–C bond cleavage/functionalization in organic synthesis. Chem. Rev. 121, 506–561 (2021).
Fumagalli, G., Stanton, S. & Bower, J. F. Recent methodologies that exploit C–C single-bond cleavage of strained ring systems by transition metal complexes. Chem. Rev. 117, 9404–9432 (2017).
Baciocchi, E., Bietti, M. & Lanzalunga, O. Mechanistic aspects of β-bond-cleavage reactions of aromatic radical cations. Acc. Chem. Res. 33, 243–251 (2000).
Chang, L., An, Q., Duan, L., Feng, K. & Zuo, Z. Alkoxy radicals see the light: new paradigms of photochemical synthesis. Chem. Rev. 122, 2429–2486 (2022).
Pitts, C. R., Ling, B., Snyder, J. A., Bragg, A. E. & Lectka, T. Aminofluorination of cyclopropanes: a multifold approach through a common, catalytically generated intermediate. J. Am. Chem. Soc. 138, 6598–6609 (2016).
Yayla, H. G., Wang, H., Tarantino, K. T., Orbe, H. S. & Knowles, R. R. Catalytic ring-opening of cyclic alcohols enabled by PCET activation of strong O–H bonds. J. Am. Chem. Soc. 138, 10794–10797 (2016).
Guo, J.-J. et al. Photocatalytic C−C bond cleavage and amination of cycloalkanols by cerium(III) chloride complex. Angew. Chem. Int. Ed. 55, 15319–15322 (2016).
Wang, D., Mao, J. & Zhu, C. Visible light-promoted ring-opening functionalization of unstrained cycloalkanols via inert C–C bond scission. Chem. Sci. 9, 5805–5809 (2018).
Hu, A. et al. Cerium-catalyzed formal cycloaddition of cycloalkanols with alkenes through dual photoexcitation. J. Am. Chem. Soc. 140, 13580–13585 (2018).
Petzold, D., Singh, P., Almqvist, F. & König, B. Visible-light-mediated synthesis of β-chloro ketones from aryl cyclopropanes. Angew. Chem. Int. Ed. 58, 8577–8580 (2019).
Zuo, Z., Daniliuc, C. G. & Studer, A. Cooperative NHC/photoredox catalyzed ring-opening of aryl cyclopropanes to 1-aroyloxylated-3-acylated alkanes. Angew. Chem. Int. Ed. 60, 25252–25257 (2021).
Zuo, Z. & Studer, A. 1,3-Oxyalkynylation of aryl cyclopropanes with ethylnylbenziodoxolones using photoredox catalysis. Org. Lett. 24, 949–954 (2022).
Xin, H., Duan, X.-H., Yang, M., Zhang, Y. & Guo, L.-N. Visible light-driven, copper-catalyzed aerobic oxidative cleavage of cycloalkanones. J. Org. Chem. 86, 8263–8273 (2021).
Chen, Y. G., Wang, X., He, X., An, Q. & Zuo, Z. W. Photocatalytic dehydroxymethylative arylation by synergisti cerium and nickel catalysis. J. Am. Chem. Soc. 143, 4896–4902 (2021).
Huang, L. et al. Bioinspired desaturation of alcohols enabled by photoredox proton-coupled electron transfer and cobalt dual catalysis. Nat. Commun. 13, 809 (2022).
Wang, X., Li, Y. & Wu, X. Photoredox/cobalt dual catalysis dnabled regiospecific synthesis of distally unsaturated ketones with hydrogen evolution. ACS Catal. 12, 3710–3718 (2022).
Ge, L. et al. Photoredox-catalyzed C–C bond cleavage of cyclopropanes for the formation of C(sp3)–heteroatom bonds. Nat. Commun. 13, 5938 (2022).
Ge, L. et al. Photoredox-catalyzed oxo-amination of aryl cyclopropanes. Nat. Commun. 10, 4367 (2019).
Liu, H., Li, Y., Wang, D. X., Sun, M. M. & Feng, C. Visible-light-promoted regioselective 1,3-fluoroallylation of gem-difluorocyclopropanes. Org. Lett. 22, 8681–8686 (2020).
Wang, Y. et al. Visible-light-promoted site-specific and diverse functionalization of a C(sp3)–C(sp3) bond adjacent to an arene. ACS Catal. 10, 6603–6612 (2020).
Xin, H., Duan, X. H., Liu, L. & Guo, L. N. Metal-free, visible-light-induced selective C-C bond cleavage of cycloalkanones with molecular oxygen. Chem. Eur. J. 26, 11690–11694 (2020).
Nguyen, T. V. T., Wodrich, M. D. & Waser, J. Substrate-controlled C-H or C-C alkynylation of cyclopropanes: generation of aryl radical cations by direct light activation of hypervalent iodine reagents. Chem. Sci. 13, 12831–12839 (2022).
Xu, Y. et al. Stereoselective photoredox catalyzed (3+3) dipolar cycloaddition of nitrone with aryl cyclopropane. Angew. Chem. Int. Ed. Engl. 62, e202310671 (2023).
Zhao, Y. R. et al. Synthesis of alpha-difluoromethylene ethers via photoredox-induced hyperconjugative ring opening of gem-difluorocyclopropanes. J. Org. Chem. 88, 3787–3793 (2023).
Rao, V. R. & Hixson, S. S. Arylcyclopropane photochemistry. electron-transfer-mediated photochemical addition of methanol to arylcyclopropanes. J. Am. Chem. Soc. 101, 6458–6459 (1979).
Mizuno, K., Ichinose, N. & Otsuji, Y. Photochemistry of 9, 10-dicyanoanthracene-1, 2-diarylcyclopropane systems. Photocycloaddition and photoisomerization. J. Org. Chem. 57, 1855–1860 (1992).
Taily, I. M., Saha, D. & Banerjee, P. Arylcyclopropane yet in its infancy: the challenges and recent advances in its functionalization. Org. Biomol. Chem. 19, 8627–8645 (2021).
Peng, P. et al. Electrochemical C−C bond cleavage of cyclopropanes towards the synthesis of 1,3-difunctionalized molecules. Nat. Commun. 12, 3075 (2021).
Kolb, S. et al. Electrocatalytic activation of donor–acceptor cyclopropanes and cyclobutanes: an alternative C(sp3)−C(sp3) cleavage mode. Angew. Chem. Int. Ed. 60, 15928–15934 (2021).
Liao, K. et al. Photoredox cleavage of a Csp3–Csp3 bond in aromatic hydrocarbons. J. Am. Chem. Soc. 145, 12284–12292 (2023).
Duffy, M. P., Delaunay, W., Bouit, P. A. & Hissler, M. π-Conjugated phospholes and their incorporation into devices: components with a great deal of potential. Chem. Soc. Rev. 45, 5296–5310 (2016).
Jeon, S. O. & Lee, J. Y. Phosphine oxide derivatives for organic light emitting diodes. J. Mater. Chem. 22, 4233–4243 (2012).
Federmann, P. et al. P-protected diphosphadibenzo[a, e]pentalenes and their mono- and dicationic P-bridged ladder stilbenes. Org. Lett. 21, 2033–2038 (2019).
Belyaev, A., Chou, P. T. & Koshevoy, I. O. Cationic organophosphorus chromophores: a diamond in the rough among ionic dyes. Chem. Eur. J. 27, 537–552 (2020).
Delouche, T. et al. Luminescent molecular switches based on dicationic P-doped polycyclic aromatic hydrocarbons. Mater. Adv. 1, 3369–3377 (2020).
Regulska, E. & Romero-Nieto, C. Design of organophosphorus materials for organic electronics and bio-applications. Mater. Today Chem. 22 (2021).
Cheng, H. et al. Bisphosphonium salt: an effective photocatalyst for the intramolecular hydroalkoxylation of olefins. Sci. Bull. 64, 1896–1901 (2019).
Yang, Z., Chen, J. & Liao, S. Monophosphoniums as effective photoredox oganocatalysts for visible light-regulated cationic RAFT polymerization. ACS Macro. Lett. 11, 1073–1078 (2022).
Zhang, X., Jiang, Y., Ma, Q., Hu, S. & Liao, S. Metal-free cationic polymerization of vinyl ethers with strict temporal control by employing an organophotocatalyst. J. Am. Chem. Soc. 143, 6357–6362 (2021).
Ding, J. et al. Selective oxidation of benzylic alcohols via synergistic bisphosphonium and cobalt catalysis. Chem. Commun. 59, 4055–4058 (2023).
Dinnocenzo, J. P., Zuilhof, H., Lieberman, D. R., Simpson, T. R. & McKechney, M. W. Three-electron SN2 reactions of arylcyclopropane cation radicals. 2. steric and electronic effects of substitution1. J. Am. Chem. Soc. 119, 994–1004 (1997).
Ohkubo, K., Kobayashi, T. & Fukuzumi, S. Direct oxygenation of benzene to phenol using quinolinium ions as homogeneous photocatalysts. Angew. Chem. Int. Ed. 50, 8652–8655 (2011).
Ohkubo, K., Fujimoto, A. & Fukuzumi, S. Photocatalytic monofluorination of benzene by fluoride via photoinduced electron transfer with 3-cyano-1-methylquinolinium. J. Phys. Chem. A 117, 10719–10725 (2013).
Acknowledgements
This work was supported by the National Key R&D Program of China (No. 2021YFA1500100), the National Natural Science Foundation of China (no. 22125111, 21971163), the Natural Science Foundation of Shanghai (21XD1424600), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0610000), and the Shanghai Pilot Program for Basic Research—Chinese Academy of Science, Shanghai Branch.
Author information
Authors and Affiliations
Contributions
Y.X., W.C. contributed equally to this work. Z.Z. conceived and directed the research; Z.Z., Y.X., W.C., R.P., J.D., Q.A., W.L., and Y.Y.designed the experiments. Y.X., W.C., R.P., J.D., and Q.A. performed and analyzed the reactions; Z.Z., Y.X., and W.C. prepared the manuscript, which was approved by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, Y., Chen, W., Pu, R. et al. Selective monodeuteration enabled by bisphosphonium catalyzed ring opening processes. Nat Commun 15, 9366 (2024). https://doi.org/10.1038/s41467-024-53728-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53728-x
This article is cited by
-
Photocatalyst-free photochemical deuteration via H/D exchange with D2O
Nature Communications (2025)
