
Abstract
The direct utilization of solar energy for the artificial photosynthesis of hydrogen peroxide (H2O2) provides a reliable approach for producing this high-value green oxidant. Here we report on the utility of high-entropy oxide (HEO) semiconductor as an all-in-one photocatalyst for visible light-driven H2O2 production directly from H2O and atmospheric O2 without the need of any additional cocatalysts or sacrificial agents. This high-entropy photocatalyst contains eight earth-abundant metal elements (Ti/V/Cr/Nb/Mo/W/Al/Cu) homogeneously arranged within a single rutile phase, and the intrinsic chemical complexity along with the presence of a high density of oxygen vacancies endow high-entropy photocatalyst with distinct broadband light harvesting capability. An efficient H2O2 production rate with an apparent quantum yield of 38.8% at 550 nm can be achieved. The high-entropy photocatalyst can be readily assembled into floating artificial leaves for sustained on-site production of H2O2 from open water resources under natural sunlight irradiation.
Similar content being viewed by others
Introduction
Nature photosynthesis process within green plants converts sunlight energy directly into chemical energy. For decades, the use of artificial photocatalytic systems to achieve photosynthetic production of fuels and high-value chemicals has long been explored for research into, for instance, the photocatalytic water splitting, carbon dioxide reduction, and nitrogen fixation to ammonia1,2,3. In more recent years, there has been increasing interest in studies of the artificial photosynthesis of hydrogen peroxide from water and oxygen4,5. H2O2 is an important green chemical oxidizer with widespread applications both in large-scale industrial processes, such as chemical synthesis and environmental remediation, as well as in small-scale on-site use including household disinfection. However, the industrial production of H2O2 relies heavily on the costly anthraquinone process that suffers from high energy consumption and substantial toxic by-products6. From a sustainability perspective, the solar-driven photosynthetic process provides a blueprint towards the renewable and sustainable production of H2O2 especially for fulfilling on-site use, but unfortunately, the realization of such a process is still hindered by its limited efficiency. Currently, a key challenge in this regard has been the quest for innovative high-performance photocatalytic materials that can efficiently absorb and transform light into photoexcited charge carriers for driving the catalytic chemical conversion of H2O and/or O2 into H2O2 at sufficiently high efficiencies.
In general, the photocatalytic and electrochemical synthesis of H2O2 involves two possible reaction pathways (Supplementary Figure 1): the two-electron oxygen reduction reaction (2e− ORR) and the two-electron water oxidation reaction (2e− WOR)7,8. The elementary steps for oxygen reduction are associated with the interaction between the catalyst surface and a serial of chemical intermediates, namely ·OOH, ·OH, and ·O, respectively. As far as the 2e− ORR pathway is concerned, only the ·OOH intermediate is preferentially involved9,10; otherwise, if the ·OH and ·O intermediates are also activated due to the dissociation of O–O bond, then the competing 4e− ORR process will be kinetically favored, which will lead to the formation of H2O instead of H2O2. Due to the uphill thermodynamics, the WOR half-reaction is intrinsically more difficult to occur than the ORR process, and similarly, there also exists competition between the 4e−and 2e− pathways in WOR11. It is only the favorable binding of ·OH intermediate with catalyst surface that will selectively generate H2O2 via 2e− pathway12.To accomplish the high-efficiency photosynthesis of H2O2, a straightforward but challenging strategy is to couple the countervailing ORR/WOR half-reactions together to constitute a full-reaction cycle that can maximize the utilization of photo-generated electrons and holes. In this way, the simultaneous light-driven generation of H2O2 via both 2e− ORR and 2e− WOR will be achieved without the need of any external sacrificial agents13,14. Here in this work, we demonstrate the unique utility of high-entropy oxide (HEO) material as such a full-reaction photocatalyst that can simultaneously catalyze 2e− ORR and 2e− WOR for highly efficient visible-light-driven H2O2 generation. This innovative HEO photocatalyst contains eight earth-abundant metal elements with a nominal stoichiometry of (Ti0.2V0.2Cr0.2 Nb0.2Mo0.05W0.05Cu0.05Al0.05) O2 (TVCNMWCA-HEO) that are stabilized in a single rutile phase (Fig. 1a), and importantly, it can operate in an all-in-one manner with neither sacrificial agents nor any external photosensitizer and cocatalysts.

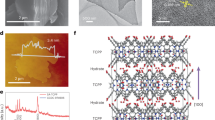
a Schematic illustration of the rutile crystal structure of TVCNMWCA-HEO. b XRD pattern with related Rietveld refinement result. c SEM image. d Representative HAADF-STEM image and inset of the corresponding FFT showing a typical rutile structure along the <001> zone axis. e Atomic-resolution HAADF-STEM images along the <001> zone axis. The line scan profile shows the variation of intensities of atomic column rows from the red box. f Schematic lattice structure of cation positions for TVCNMWCA-HEO (yellow dots) and ideal rutile phase (pink dots). The expanded views taken from the selected area highlighted by the yellow box in (e). g Geometric phase analysis based on the HAADF image (e). The results of the normal strain components \({\varepsilon }_{{xx}}\) and \({\varepsilon }_{{yy}}\) indicate local distortion. h Atomic EDS elemental mapping of element distributions of the selected area highlighted by the magenta box in (e). These source data are provided as a Source Data file.
Over the past few years, the concept of high-entropy stabilization has been emerging as a fruitful paradigm for the design and discovery of catalyst materials for use especially in renewable energy conversion reactions through either electrocatalysis or photocatalysis15,16. For high-entropy materials in general, the chemically homogeneous distribution of multiple constituent elements in a single-phase solid solution gives rise to ultra-large configurational entropy in conjugation with severe local lattice distortion and inherent surface complexity17. By leveraging these unique traits, the high-entropy catalysts can normally provide a near-continuous distribution of adsorption energies, which is of crucial importance for complicated catalytic reactions requiring diverse adsorption energy levels18. Meanwhile, a huge number of possible atom arrangements on the high-entropy stabilized surfaces will offer abundant active catalytic sites and variable adsorption modes for target reactants and key intermediates19,20. For high-entropy oxides in particular, as compared to conventional simple metal oxides, they are much more prone to the formation of higher density of oxygen vacancies (Ovs) due to severe lattice distortion and unbalanced bonding states for oxygen atoms21,22,23. Surface oxygen vacancies are well-documented as active sites in various catalytic processes, thereby endowing high-entropy oxides with an even richer array of active sites.24. As semiconductor materials, high-entropy oxides effectively serve as light-harvesting antennas in photocatalytic systems. The abundant oxygen vacancies enhance the absorption of low-energy photons by creating intermediate states, making these materials promising candidates for visible light-active photocatalysts. Moreover, the complex electronic interactions among different metal cations can broaden the d-band and reduce the band gap, further improving optical absorption capabilities25,26. These intrinsic properties allow high-entropy oxides to integrate both light absorption and catalytic functions—two essential components of photocatalytic systems—positioning them as promising all-in-one photocatalysts. The potential of high-entropy photocatalysts was initially demonstrated by Edalati and colleagues in 202027. Since then, high-entropy materials have been explored for various roles in photocatalytic systems, functioning as optical antennas to enhance low-energy photon capture28,29, as co-catalysts to provide active sites30, and in certain few cases, as both light absorbers and catalytic sites31. Despite these promising advancements, research on all-in-one high-entropy photocatalysts remains in its early stages32,33.
In our present work, such an all-in-one high-entropy photocatalyst has been designed to achieve highly efficient photocatalytic H2O2 production under visible light irradiation. It is the combination of surface compositional variety and exposed surface oxygen vacancies that affords the different sets of active sites for both 2e− ORR and 2e− WOR, thereby obviating the need for any coupled co-catalysts. Apart from this, the presence of a high density of oxygen vacancies also plays a vital role in mediating the electronic and optical absorption characteristics of TVCNMWCA-HEO. To be specific, oxygen vacancies induce an intermediate band within the bandgap for harvesting low-energy photons in the visible and near-infrared regions, thereby enabling the full-spectrum solar energy utilization of TVCNMWCA-HEO. The TVCNMWCA-HEO photocatalyst exhibited exceptional activity in producing H2O2 from only H2O and O2, achieving an apparent quantum yield of 38.8% at 550 nm and a solar-to-chemical conversion efficiency of 1.72%. Owing to the inherent simplicity of the all-in-one operation manner, this high-entropy photocatalyst is practically scalable and easy for implementation in versatile application scenarios.
Results
Synthesis and structural characterization of TVCNMWCA-HEO
For the synthesis of high-entropy photocatalytic materials, it is essential to consider both structural stability and catalytic performance. In our high-entropy oxide, we select elements with suitable sizes and electronegativities to construct the crystal structure, while additionally incorporating catalytic active components to enhance the material’s catalytic properties. Figure 1a illustrates the crystal structure of our high-entropy oxide, in which eight cations (Ti/V/Nb/Cr/Mo/W/Cu/Al) randomly occupy the sub-lattice position, forming an entropy-stabilized oxide with a single-phase tetragonal structure (P42/mnm). In general, the presence of eight metallic elements with diverse cationic radii, coordination and charges poses a tendency to form multiphase structures. In this work, a well-modified Pechini method was utilized to synthesize a homogeneous metal precursor, followed by calcination at 600 ˚C in air to obtain the TVCNMWCA-HEO powder. Figure 1b shows the powder X-ray diffraction (XRD) profile along with the Rietveld refinement, directly confirming the phase purity of the rutile-type TVCNMWCA-HEO (Supplementary Table 1). In contrast, when the material is synthesized at 450 °C, two phases are observed (Supplementary Fig. 2). This indicates that a higher synthesis temperature is necessary to enhance the entropic contribution and lower the Gibbs free energy of the system to realize the stability of the single-phase rutile structure34. Moreover, the exact stoichiometries of the TVCNMWCA-HEO were confirmed by inductively coupled plasma-atomic emission spectroscopy (ICP-OES) in Supplementary Table 2, showing results similar to the nominal composition. Figure 1c displays a representative scanning electron microscope (SEM) image of the ellipsoidal-shaped TVCNMWCA-HEO nanoparticles, which are evenly dispersed, with an individual particle size of ~70 nm (Supplementary Fig. 3). During the synthesis process, we observed that the incorporation of the aluminum element significantly enhanced the anti-sintering ability of these nanoparticles, thereby preserving their optimal morphology and increasing the surface area, as shown in Supplementary Fig. 4. This endows the TVCNMWCA-HEO nanoparticles to expose more active sites on the surface, resulting in excellent catalytic performance.
High-entropy materials are recognized for their stable structures with long-range crystal periodicity, but at the atomic scale, the compositional disorder can naturally lead to complex chemical environments35. The unique atomic structure of TVCNMWCA-HEO material has been confirmed through atomic-resolution imaging. Shown in Fig. 1d is a typical high-angle annular dark-field (HAADF) image recorded by scanning transmission electron microscopy (STEM) of an individual TVCNMWCA-HEO nanoparticle. The corresponding fast Fourier transform (FFT) pattern reveals a tetragonal structure along the <001> orientation, consistent with the XRD results. The sharp diffraction spots indicate the high crystallinity of the TVCNMWCA-HEO sample. Further selecting the direction of the crystal in Fig. 1e enables direct observation of different metal atoms, with white spots corresponding to the columns of metal ions. The brightness of each spot represents the abundance of relatively heavy atoms in the metal column and then the intensity variations of these white dots along the row of the red area are clearly observed, which is great and irregular. This obvious contrast difference highlights the intense chemistry disorder of these eight metal atoms in the lattice, leading to random fluctuations in the atomic fraction of Ti, V, Cr, Nb, Mo, W, Al or Cu in each atomic column. In addition, the yellow boxed region is magnified to compare the atomic structure of the standard tetragonal model with that of TVCNMWCA-HEO, as shown in Fig. 1f. The metal ions (yellow dots) are observed to deviate slightly from the standard positions, which are located at the center of the oxygen octahedron (pink dots). Furthermore, the atomic strain distribution pattern of Fig. 1e based on geometric phase analysis (GPA), clearly indicates severe lattice distortion across whole region due to the variation in ionic radius (Fig. 1g). This TVCNMWCA-HEO material characterized by chemical disorder and local lattice distortion, facilitates the formation of abundant defects in its structure, including cation vacancies and oxygen vacancies, which can efficiently tune the band structure21,36. Figure 1h and Supplementary Fig. 5 reveal the elemental mapping of Ti, V, Cr, Nb, Mo, W, Al, and Cu at low and high magnifications. Although some elements may concentrate locally in nanometer-sized regions, there is no evident preference for specific neighbors. This indicates long-range chemical disorder and homogeneous mixture in the sample (Supplementary Table 3). Especially in Fig. 1h, the atomic-resolution STEM-EDS images were recorded to show the atom species at each lattice site, substantiating the randomness of these eight metal elements within metal atom columns. Such local chemical complexity and lattice distortion of TVCNMWCA-HEO were also revealed in Raman spectroscopy analysis. The Raman spectrum of TVCNMWCA-HEO shows a broad and featureless peak ranging from 600-1000 cm−1, which results from severe chemical disorder, the tilts and twists of the octahedra and the existence of defects (Supplementary Fig. 6). Besides, the inherently present mass and force constant fluctuations in high-entropy materials can induce significant phonon scattering and broadening37. We employed the widely recognized methods to calculate the configurational entropy of the TVCNMWCA-HEO, which shows an ideal configuration of 1.86 R per the specific cation sublattice, supporting the stabilization of its entropy-driven single-phase structure34,38.
Band structure, optical properties and charge carrier dynamics
We employed X-ray absorption spectroscopy (XAS) to investigate the chemical states and local coordination structures of transition metal elements in TVCNMWCA-HEO. Shown in Fig. 2a–d are the X-ray absorption near-edge structure (XANES) spectra of Ti, V, Cr, and Nb K-edges, which are the four main constituents of TVCNMWCA-HEO. It is noted that the Ti K-edge of TVCNMWCA-HEO shows a similar shape to that of pristine rutile TiO2, meaning a typical octahedral coordination structure (Fig. 2a). However, differences in shape and intensity can still be observed in the Ti K pre-edge peak of TVCNMWCA-HEO, which should be attributed to the local lattice distortions around Ti atoms. Additionally, the Ti K-edge of TVCNMWCA-HEO shifted to lower energies with respect to pristine rutile TiO2, suggesting the partial reduction of Ti species in the high-entropy lattice. Derivative linear combination fitting (LCF) reveals that Ti mainly exists as Ti⁴+ with some Ti³⁺ (Supplementary Fig. 7), resulting in an average valence state of approximately + 3.76. Similarly, other transition metal elements also exhibit mixed valence states, with Nb at + 4.05, Cr at + 3.83, and V at + 4.03 (Fig. 2b–d, Supplementary Table 4). Apart from these four main constituents, we also investigated the XANES spectra of W L-edge and Cu K-edge, as shown in Supplementary Fig. 7. All the valence state characteristics have confirmed the electron transfer and complex charge compensation in TVCNMWCA-HEO induced by the chemical disorder, which is also the primary factor for the formation of oxygen vacancies.

a The Ti K-edge XANES spectra of the TVCNMWCA-HEO and the corresponding reference materials (Ti foil, TiO2 and Ti2O3). b The V K-edge XANES spectra of the TVCNMWCA-HEO and the corresponding reference materials (V foil, VO2, V2O5). c The Cr K-edge XANES spectra of the TVCNMWCA-HEO and the corresponding reference materials (Cr foil, CrO2, and Cr2O3). d The Nb K-edge XANES spectra of the TVCNMWCA-HEO and the corresponding reference materials (Nb foil, NbO2, and Nb2O5). e The FT-EXAFS spectra of Ti, V, Cr, and Nb K-edge of the TVCNMWCA-HEO and FT-EXAFS spectra of Ti foil, TiO2, and Ti2O3. f The O 1 s XPS spectra of TVCNMWCA-HEO. The inset is the EPR spectra. These source data are provided as a Source Data file.
EXAFS was then employed to analyze coordination number, distance between neighboring atoms, and disorder of neighboring atoms (Supplementary Fig. 8 and 9). In Fig. 2e, the peak around 1.47 Å in the Ti K-edge EXAFS spectrum corresponds to the first shell of the Ti-O path, with a bond length of 1.96 Å close to that of pristine rutile TiO2 (Supplementary Table 5). Similarly, the bond lengths of the first shell for Ti, V, Nb, Cr, W, and Cu are very close, indicating that all metals occupy the same lattice positions in TVCNMWCA-HEO, i.e., the cation sites in the rutile structure. Although the coordination environment of Ti in the TVCNMWCA-HEO configuration remains unchanged, the coordination number (C. N.) of Ti is estimated to be 3.7, suggesting the presence of oxygen vacancies. Straightforwardly, the existence of oxygen vacancies is evidenced by the O 1 s XPS spectra and EPR results (Fig. 2f). The O 1 s spectra reveal a prominent peak at 531.9 eV corresponding to oxygen adsorption species on surface oxygen vacancies24,39. This signal intensity accounts for 38% of the total oxygen signal, reflecting the significant amount of surface oxygen vacancies. Also, the EPR spectrum of the HEO exhibits a distinct signal at g = 2.003, clearly evidencing the presence of single-electron-trapped oxygen vacancies22,40. There is virtually no direct and precise method to measure the amount of oxygen vacancies in such a complex metal oxide. To address this, we compared the oxygen occupancy between TVCNMWCA-HEO and pristine TiO2 by Rietveld refinement to estimate its total concentration of oxygen vacancies to be around 8%, including both bulk and surface oxygen vacancies (Supplementary Fig. 10). The entropy-driven local structural disorder and abundant oxygen vacancies are typical features of HEOs, even in high-entropy oxides with fewer elementary components (Supplementary Fig. 11), and these characteristics are particularly prominent in our TVCNMWCA-HEO composed of eight elements.
Oxygen vacancies are ubiquitous in metal oxides and can effectively modify the electronic structure and optical properties41,42. The high density of oxygen vacancies in the TVCNMWCA-HEO could result in the formation of intermediate band (IB) states within the bandgap, therefore harvesting low-energy photons and impacting photo-carrier dynamics43. As shown in Fig. 3a, the presence of intermediate band states between the Fermi level and the valence band in TVCNMWCA-HEO, has been demonstrated by both the XPS valence band spectrum and the UPS valence band spectrum. The presence of IB enables the efficient absorption of sub-bandgap photons, making the materials promising for use in solar energy conversion applications43,44. The electronic structure of the TVCNMWCA-HEO was quantitatively determined by employing XPS, UPS, and Mott-Schottky analysis (Fig. 3a, b). Shown in Fig. 3c is the energy band diagram of TVCNMWCA-HEO, with a band gap of ~3.05–3.34 eV. The intermediate band is inferred to be located about 1 eV below the conduction band (CB). According to the above analysis, the TVCNMWCA-HEO possesses a sufficiently large bandgap and suitable valence band (VB) and conduction band edge to generate H2O2 through WOR and ORR processes simultaneously.

a Valence band spectra of TVCNMWCA-HEO acquired by XPS and UPS. The red regions mark the intermediate band states formed by abundant oxygen vacancies. b Mott–Schottky plots at different frequencies of TVCNMWCA-HEO. c Schematic band structure diagram of TVCNMWCA-HEO determined from XPS, UPS, and M-S analysis. d Diffuse reflectance UV–vis spectra of the TVCNMWCA-HEO (inset: the digital photo of TVCNMWCA-HEO powder) e Transient absorption spectra at various time delays of TVCNMWCA-HEO powder after pumping at 550 nm. f Corresponding decay kinetics probed at 700 nm showing a slow recombination process because of the presence of IB. g The photoluminescence spectra of TVCNMWCA-HEO excited at 325 nm and fitted by Gaussian function, suggesting various possible transitions of TVCNMWCA-HEO; The black circles is the experiment result; the red line is the fitting result. h Three typical time-resolved photoluminescence decay curves of band-edge emission at 400 nm and visible emission at 560 nm and 700 nm. The experimentally obtained curves are fitted by two-component exponential decay curves (black solid line). These source data are provided as a Source Data file.
The optical absorption of the brown TVCNMWCA-HEO powder offers insight into its optical behavior (Fig. 3d). Benefiting from the IB, distinct absorption features appeared in a broad region from 500 to 2000 nm. It has been demonstrated that electronic transition could occur from VB to IB, from IB to CB, or within IB (Fig. 3c). As a result, the TVCNMWCA-HEO achieves full-spectrum absorption from the near-infrared to ultraviolet regions, making it an excellent candidate for solar radiation harvesting. Furthermore, Supplementary Fig. 12 shows the transient absorption (TA) spectra of TVCNMWCA-HEO material when excited by a pump laser at 380 nm. A broad positive absorption band extends across the entire spectral range and decays slowly from 10 ps to 5.5 ns. This signal is attributed to carrier transitions from the occupied IB to CB via sub-bandgap photons. For further strict comparison, the transient absorption spectrum excited by a 550 nm femtosecond laser can be seen in Fig. 3e and Supplementary Fig. 13. In this spectrum, the photon energy is unable to directly excite electrons from VB to CB. The decay dynamic at 700 nm is governed by a two-step decay with a fast component of 15.9 ps and a slower component of about 1.6 ns, which corresponds to the relaxation of carriers within the IB and the interband relaxation, respectively (Fig. 3f). These results indicate that the intermediate band states enable efficient absorption of sub-band-gap photons. Likewise, time-resolved photoluminescence (TRPL) was used as a complementary technique to elucidate the carrier dynamics in the material. The steady PL spectra of TVCNMWCA-HEO nanoparticles were tested under excitation at both 325 nm and 520 nm, as indicated in Fig. 3g and Supplementary Fig. 14. Various fluorescent signals are detected, indicating that the carrier recombination process is altered when the carriers are excited into the delocalized intermediate band and subsequently scatter or diffuse to different luminescent centers. Figure 3h shows the TRPL curves of the TVCNMWCA-HEO nanoparticles when irradiated with a 380 nm laser, illustrating the electrons excited from VB or IB to CB. The lifetime at 400 nm is ~0.88 ns, while the carriers undergo two-step recombination at 560 nm and 700 nm and exhibit a longer lifetime. All of these pump-probe techniques have provided solid evidence that the presence of intermediate bands has offered a high density of states for electron storage and slower charge-carrier recombination, thereby contributing to the efficiency of photocatalysis.
Photocatalytic performance for H2O2 generation
The photocatalytic performance of the TVCNMWCA-HEO sample was evaluated for H2O2 production without any sacrificial agents under visible light irradiation (λ ≥ 420 nm) (Supplementary Fig. 15). In Fig. 4a, the TVCNMWCA-HEO catalyst achieved exceptional H2O2 production, yielding 21 mM within the first hour. Furthermore, the TVCNMWCA-HEO catalyst exhibited sustained photocatalytic activity even under red light irradiation (Fig. 4b). Under near-infrared light at 850 nm, the H2O2 yield reaches 4.51 mM within an hour, highlighting the effective use of near-infrared light energy by TVCNMWCA-HEO. Besides, the AQY of TVCNMWCA-HEO was evaluated using other wavelengths in the visible light spectrum, as shown in Fig. 4c. The TVCNMWCA-HEO photocatalyst exhibits excellent AQY, reaching 38.8% at 550 nm, and the AQY values at 475 nm, 600 nm, 650 nm, and 700 nm consistently remained above 25%. Notably, a solar-to-chemical conversion efficiency of 1.72% is achieved on the TVCNMWCA-HEO photocatalyst in pure water and air conditions, as illustrated in Supplementary Fig. 16. These values exceed most previously reported advanced photocatalysts under the same conditions (Supplementary Table 6).

a Photocatalytic H2O2 production with TVCNMWCA-HEO, other septenary high-entropy oxides, and TiO2 in pure water under visible light irradiation (λ ≥ 420 nm, 300 mW/cm2). b Photocatalytic H2O2 production with TVCNMWCA-HEO under monochromatic irradiation at 650, 700, and 850 nm. c Wavelength-dependent apparent quantum yield (AQY) measurement for TVCNMWCA-HEO. d Stability tests of TVCNMWCA-HEO in water for photocatalytic H2O2 production under visible light irradiation. e Long Time course of H2O2 production under visible light irradiation (λ ≥ 420 nm, 300 mW/cm2) and AM1.5 irradiation (100 mW/cm2). f The H2O2 formation (Kf) and the decomposition (Kd) rate constant over TVCNMWCA-HEO under visible light irradiation (λ ≥ 420 nm, 300 mW/cm2) and AM1.5 irradiation (100 mW/cm2). g Amount of H2, O2, and H2O2 produced with TVCNMWCA-HEO under vacuum conditions and visible light irradiation (λ ≥ 420 nm, 300 mW/cm2). These source data are provided as a Source Data file.
As documented in previous studies on high-entropy catalysis, there is a strong correlation between the variation of total configuration entropy of high-entropy catalysts and their catalytic performance45. Generally, the catalytic efficiency tends to improve as the configuration entropy increases by adjusting components within a certain range. Our experimental results are consistent with this observed trend. Specifically, starting with the octonary TVCNMWCA-HEO, we systematically removed Mo, W, Al, or Cu elements one by one to form a series of septenary high-entropy oxides. All of these septenary subsystems maintain entropy-stabilized single rutile phases (Supplementary Fig. 11 and Supplementary Table 2). The H2O2 production efficiencies of these septenary subsystems exhibit a significant drop compared to octonary TVCNMWCA-HEO as illustrated in Fig. 4a. These comparative results indicate that the presence of each component is crucial for the catalytic reaction. Meanwhile, we note that the degree of decline in H2O2 production rates varies among these septenary high-entropy oxides, as the role of each component in the catalytic process is not equivalent. In addition to septenary high-entropy oxides, we selectively synthesized senary HEO with fewer components, which also displayed lower photocatalytic activity (Supplementary Fig. 17). After thorough optimization, the TVCNMWCA-HEO stands out as the most effective photocatalyst in our high-entropy oxide system, exhibiting the highest photocatalytic efficiency in generating H2O2. As depicted in Fig. 4d, the photocatalytic performance of TVCNMWCA-HEO was fully maintained after several consecutive cycles, clearly indicating the stability and reusability of this photocatalyst. Additionally, there were no significant changes in the XRD, Raman, and O 1 s spectra of TVCNMWCA-HEO before and after the photocatalytic reaction (Supplementary Fig. 18). This synthesized photocatalyst exhibited no decline in photocatalytic activity, even after three months of storage in air (Supplementary Fig. 19).
Another significant factor in the photocatalytic generation of H2O2 is its decomposition rate, as H2O2 is unstable and easily decomposes under light, high temperature, and other conditions46. The TVCNMWCA-HEO can perform photocatalysis with low light energy, utilizing visible and near-infrared light to avoid the strong decomposition effect of H2O2 caused by UV light. As shown in Fig. 4e, the long-term H2O2 production tests were performed under visible light irradiation and AM1.5 G simulated sunlight irradiation. A continuous increase in the H2O2 concentration was observed during the first 4 hours, after which it gradually became saturated. This is due to the dynamic competition between the generation and decomposition of H2O2, which eventually reaches an equilibrium state. The rate constant for H2O2 formation (Kf, × mM min−1) and decomposition (Kd, min−1) over the TVCNMWCA- HEO sample were evaluated by assuming zero-order and first-order kinetics, respectively47. The values of Kf and Kd under visible light and
AM1.5 G simulated sunlight irradiation are depicted in Fig. 4f, which are 0.580 mM min−1, 0.297 mM min−1, 0.016 min−1 and 0.08 min−1. Ultimately, the TVCNMWCA-HEO photocatalyst exhibited the H2O2 evolution yield of 37.2 mM under visible light irradiation and 35.9 mM under AM1.5 G simulated sunlight irradiation. Such a high photocatalytic yield of H2O2 is expected to be suitable for daily practical applications.
Investigation of the full reaction mechanism
We then observed that the TVCNMWCA-HEO catalyst can achieve water oxidation reactions to generate photocatalytic H2O2, even under low-oxygen and anaerobic conditions. Shown in Fig. 4g is the production of a certain amount of H2O2 and O2 under vacuum conditions, suggesting the WOR could occur via two pathways: the two-electron process (2e− WOR) producing H2O2 and the four-electron process (4e− WOR) producing O211. More importantly, as the reaction proceeds, the yield of H2O2 continuously increases while the yield of O2 decreases. This indicates that the generated O2 was eventually reduced to H2O2 through the 2e− ORR pathway. Additionally, the production of a small amount of H2 via photocatalytic water reduction also confirms that the CB level of TVCNMWCA-HEO is higher than the H2 reduction potential, fulfilling the energy requirement of 2e− ORR. Moreover, as shown in Fig. 4g, it is unlikely that such a large amount of H2O2 in the first hour was accumulated through the 4e− WOR process and the 2e− ORR process. It is inferred that the generation of H2O2 under vacuum conditions mainly occurs through the 2e− WOR process.
We further carried out the photocatalytic reaction in different atmospheres as displayed in Fig. 5a. Continuous injection of high-purity oxygen into the solution results in a higher H2O2 yield compared to the atmospheric environment. Conversely, the yield of H2O2 decreases when the dissolved oxygen is removed from the water by purging it with N2, suggesting that TVCNMWCA-HEO exhibits high activity in the 2e− ORR process. Moreover, the half-reactions were carried out in a methanol aqueous solution (CH3OH, 10% v/v) and an AgNO3 aqueous solution (1 mM), respectively (Fig. 5b). It was found that the H2O2 generation of semi-redox reaction and semi-oxide reaction was not completely suppressed. These results confirm the process of WOR and ORR for TVCNMWCA-HEO, which can generate H2O2 through the 2e− transfer process. It can be suspected that the relative contribution of ORR and WOR processes may vary in different atmospheres. Given the wide adaptability and self-regulatory capacity of high-entropy catalysts, we expect efficient photocatalysis production in diverse environments in future experiments.

a Comparison of H2O2 production catalyzed by TVCNMWCA-HEO at different atmospheres (N2 / O2 /Air / Vacuum) under visible light for 60 minutes. b Comparison of H2O2 production catalyzed by TVCNMWCA-HEO under visible light in water without any sacrificial agents, and in CH3OH aqueous (10% v/v) and in AgNO3 (1 mM), respectively (c) DMPO spin-trapping EPR spectra of TVCNMWCA-HEO for measuring ·OOH and ·OH in dark and visible light (λ ≥ 420 nm), respectively. These source data are provided as a Source Data file.
To elucidate reaction mechanisms and understand the evolution of groups generated on the catalyst surface during the photocatalytic H2O2 production process, we employed electron paramagnetic resonance (EPR) spectroscopy and in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements to monitor the photocatalytic reactions. The EPR measurements were carried out in the methanol solution with dissolved O2 for ORR testing and in the Ar-saturated anaerobic aqueous solution for WOR testing, with 5,5-dimethyl-1-pyrroline N-oxide (DMPO)) used as the free-radical spin-trapping agent. As shown in Fig. 5c, six characteristic peaks of DMPO-·OOH and four characteristic peaks of DMPO-·OH are observed under light irradiation48. Furthermore, we performed DRIFTS measurements to monitor the photocatalytic reactions, during utilizing in situ D2O exchange to refine the precision of identifying adsorbed species. Benefiting from the high activity of the TVCNMWCA-HEO surface, a weaker water dissociation adsorption signal can be observed even in the dark (Supplementary Fig. 20). Under light irradiation, typical infrared absorption signals of adsorbed oxygen intermediates appear, with their intensity increasing as the illumination time is extended (Figs. 6a and 6b). Specifically, a shoulder located at 2685 cm−1 began to form under illumination, which can be attributed to the photocatalytically generated ·OD species, implying that the hydrogen peroxide can be produced through a 2e− WOR pathway on the TVCNMWCA-HEO surface. Additionally, the gradually increasing bands appear at 1357 cm−1. assigned to ·O2−, and at 1281 cm−1, assigned to ·OOD species, are key intermediates during the 2e− ORR process. In H2O, the corresponding intermediate species were also found in Supplementary Fig. 21. The key intermediates for both 2e− WOR and 2e− ORR mechanisms were effectively observed, indicating that TVCNMWCA-HEO has dual pathways for H2O2 production, which also accounts for its high activity.

a Time-resolved DRIFTS spectroscopy of changes on TVCNMWCA-HEO surface under illumination. Main changes in spectra related to the evolution of the reaction intermediates are marked with arrows. b Details on the spectral region with characteristic oxygen intermediate species vibration bands. c Charge density difference plots for H2O adsorption on the Mo and W sites of TCVNMWCA-HEO surface. Yellow and cyan represent electron accumulation and electron depletion, respectively. The isosurface value is 0.003 e/Bohr3. d Gibbs free energy (ΔG*OH) profiles on various catalytic sites of the TVCNMWCA-HEO surface. e Charge density difference plots for O2 adsorption on the V and Cu sites of TCVNMWCA-HEO. Yellow and cyan represent electron accumulation and electron depletion, respectively. The isosurface value is 0.0015 e/Bohr3. f Gibbs free energy (ΔG*OOH) profiles on various catalytic sites of the TVCNMWCA-HEO surface. These source data are provided as a Source Data file.
Then, theoretical simulations were used to investigate the possible catalytic sites on the TVCNMWCA-HEO surface and to verify the viability of 2e− WOR and 2e− ORR mechanisms. Here, the high-entropy structure of TiVNbCrO8 was first constructed with reference to the ICP-OES characterization results (Supplementary Table 2), and the thermodynamically most stable configuration was selected as the basis for subsequent calculations. It should be noted that in TVCNMWCA-HEO, these transition metal elements (Ti/V/Cr/Nb/Mo/W/Cu) play a significant role in influencing the electronic structure, while Al primarily functions to prevent nanoparticle sintering and maintain morphological stability. In this context, the effect of low-content transition metal elements, such as Mo, W, and Cu, are considered as dopants, through local mixing near the active site to establish a reasonably simplified TVCNMWCA-HEO model (More details in the Method). Based on the experimental detection of massive oxygen vacancies, we first theoretically evaluated the rationality of the existence of oxygen vacancies in high-entropy models. As shown in Supplementary Fig. 22, the introduction of Mo, W, and Cu can significantly reduce the formation energy of oxygen vacancy. For example, the formation enthalpy of oxygen vacancies in TiVNbCrO8 is + 1.26 eV, whereas it decreases to −0.89 eV after Mo, W, and Cu doped into its lattice. This indicates that a high-entropy configuration is more conducive to the generation of oxygen vacancies, which should be an apparent manifestation of charge compensation in HEO materials and is consistent with experimental results. Thus, this resultant high-entropy structure not only contains various metal sites but also a high density of oxygen vacancies, providing a highly active surface with diverse sites for catalytic reactions.
Since our H2O2 synthesis was carried out in an aqueous medium, we first evaluated the possibility of the 2e− WOR reaction mechanism on the TVCNMWCA-HEO surface (Supplementary Tables 7 and 8). As displayed in Supplementary Fig. 23, the H2O molecule can spontaneously adsorb on diverse metal sites surrounding the oxygen vacancy, confirming the diversity and richness of active sites in TVCNMWCA-HEO materials. According to the adsorption configuration and differential charge analysis (Fig. 6c and Supplementary Fig. 24), we observed that the adsorption process of H2O is accompanied by obvious charge accumulation in the M-O (M=Mo, W, Cu, V, and Cr) bond and charge consumption near O-H bond. The charge transfers caused by H2O adsorption on the Mo and W sites are more significant than those on other sites, which means that these sites may have stronger H2O molecule activation ability. For the adsorption configuration of H2O on the Cr site, the oxygen atom of H2O actually occupies the position of the oxygen vacancy, and the H2O dissociation process is thermodynamically non-feasible, suffering an energy barrier of 0.16 eV (Supplementary Fig. 23). Although the oxygen vacancy is not active *OH formation, it enhances the catalytic activity of surrounding metal sites by modulating the material’s electronic structure. Moreover, H2O stably adsorbed on oxygen vacancy can prevent the recombination of active oxygen atoms and vacancy during the catalytic process, thereby maintaining the stability of oxygen vacancies. Similarly, H2O adsorbed on V sites requires additional energy (0.29 eV) to form *OH. In contrast, the adsorbed H2O molecules undergo the dissociation reaction and form OH species spontaneously at the Mo, W, and Cu sites, with *OH formation energies of −0.02 eV, −0.38 eV, and −1.00 eV, facilitating the excellent hydrogen peroxide production performance via 2e− WOR process (Fig. 6d).
Subsequently, we assessed the feasibility of producing hydrogen peroxide via the 2e− ORR pathway at various active sites (Supplementary Tables 9 and 10). Oxygen adsorption, the first step of the 2e− ORR process was investigated. Obvious charge accumulation at oxygen atoms and charge consumption near the O-O bond implies the effective activation of the O2 molecule at Cu, V, Mo, and W sites (Fig. 6e and Supplementary Fig. 25). Two structural adsorption configurations of O2, the Griffiths-type and the Pauling-type, were observed on the TVCNMWCA-HEO surface (Supplementary Fig. 26)11,49. O2 exhibited a Pauling-type adsorption mode at the Cu site, which is recognized as a weaker adsorption mode and inherently advantageous for hydrogen peroxide production via the 2e− ORR process. Additionally, O2 preferentially adsorbs in the form of Griffiths-type configuration at the V, Mo, and W sites, with free energy of 0.69 eV, 0.22 eV, and −0.06 eV, respectively. It is well known that the key to generating H2O2 lies in preventing O-O bond breakage during the catalytic process, which requires weak adsorption between O2 and the active sites. Thus, we conjecture that the Mo and W sites may be unfavorable for the ORR process due to their high activity. O2 adsorbed on the V site in a bridge-like manner may maintain a strong O-O bonding state due to its weak adsorption strength, thereby generating an *OOH intermediate (Supplementary Tables 11). Furthermore, the energy of the adsorbed O2 to form the *OOH intermediate through the hydrogenation step was calculated. As expected, due to the strong adsorption of O2 at the Mo and W sites, the hydrogenation process is accompanied by the breaking of O-O bonds, resulting in the generation of separated *O and *OH, which is often considered to be the typical symptom of a 4e− ORR process. In contrast, *OOH can be spontaneously formed at the Cu and V sites (Fig. 6f), with free energies of −0.18 eV and −0.29 eV, suggesting these sites can reduce O2 to generate hydrogen peroxide through the 2e− ORR pathway. The combined EPR, DRIFTS data, and calculation results demonstrate that photocatalytic H2O2 generation over the TVCNMWCA-HEO catalyst occurs via the ·OOH and ·OH radical mechanism.
Outdoor photocatalytic performance
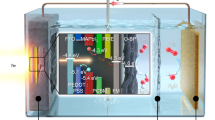
The high-level photocatalytic performance of TVCNMWCA-HEO, combined with the intrinsic simplicity of its all-in-one operation, promises great utilization of this photocatalyst in various application scenarios. In this work, we exploit the performance of TVCNMWCA-HEO by constructing simply engineered, stand-alone artificial leaves (TVCNMWCA-HEO/SA membrane) for outdoor testing of the photocatalytic H2O2 production under sunlight irradiation (Fig. 7a, b). The solar-driven photocatalytic setup just consists of pure water and TVCNMWCA-HEO/SA membranes, as shown in Fig. 7c. We observed the production of a high-concentration solution of H2O2 in the beakers outdoors under sunlight, which can be directly used in antibacterial experiments. In Fig. 7d and Supplementary Fig. 27, the result reveals a significant decrease in the survival ratio of Escherichia coli, which visualizes the generation of H2O2 through solar-driven photocatalysis. In this representative on-site application, the TVCNMWCA-HEO/SA membrane can work as a stand-alone and floatable “leaf”, capable of producing a certain concentration of H2O2 only from open water resources and oxygen under sunlight. Furthermore, the use of these artificial leaves allows for the direct production of pure aqueous H2O2 solution, obviating the need of extra energy-consuming separation processes.

a Schematic illustration of the floatable TVCNMWCA-HEO/SA membrane using a gelation method by sodium alginate, which provides excellent permeability for water molecules to freely move inside the membrane. b Digital photo of the TVCNMWCA-HEO/SA membrane with a diameter of 4 cm. c Digital photo showing the TVCNMWCA-HEO/SA membrane in water under sunlight irradiation. d Digital photos of the sterilization of Escherichia coli with a photocatalytically produced H2O2 solution in (c). These source data are provided as a Source Data file.
Discussion
The greatly expanded compositional space and high-entropy mixing effect of HEO provide a unique opportunity to tailor key chemical and physical properties of metal oxide semiconductors. A leading manifestation of this in our TVCNMWCA-HEO is the spontaneous formation of a high concentration of stable oxygen vacancies during synthesis. For metal oxides in general, oxygen vacancies are prevalent defects that can profoundly impact their photoelectrochemical and photocatalytic performance50. It has been widely proved that oxygen vacancies can effectively enhance visible light absorption by introducing electronic mid-gap states. Meanwhile, the surface oxygen vacancies can also serve as active sites to promote the chemical adsorption of reactants and initiate catalytic reactions. For conventional metal oxides with simpler chemical components, a general requirement for the generation of abundant oxygen vacancies usually involves the post-synthesis treatment of metal oxide samples in an oxygen-deficient atmosphere. And, due to their inherent instability, the as-formed oxygen vacancies in conventional oxides will easily undergo re-oxidization under certain circumstances (e.g., by air-annealing treatment). For our TVCNMWCA-HEO, by contrast, it appeared that the formation of a high-concentration of oxygen vacancies with extraordinary stability can be readily spontaneously accomplished over the course of HEO synthesis of in open air. Essentially, it is a synergistic effect involving chemical disorder, lattice distortion, and charge compensation in TVCNMWCA-HEO that effectively lowers the formation energy of oxygen vacancies and facilitates the escape of oxygen atoms from the high-entropy stabilized rutile lattice23.
A typical photocatalyst system normally consists of two essential components: the light absorber and the coupled co-catalysts. To drive a specific photocatalytic reaction, it requires the efficient transfer of photo-induced charge carriers from the light absorber to the co-catalyst components, where active sites are actually located. For our TVCNMWCA-HEO photocatalyst instead, it can not only act as a good light-absorber for near-perfect broadband solar light harvesting, but also provide an abundance of naturally occurring surface active sites for both O2-to-H2O2 conversion via the 2e− ORR pathway and H2O-to-H2O2 conversion via 2e− WOR pathway. At the high-entropy stabilized lattice surface of TVCNMWCA-HEO photocatalyst, the exposed multi-component metal cations in diverse local coordination environments, particularly those surrounding oxygen vacancies, form a diverse set of active sites that are responsible for 2e− ORR and 2e− WOR reaction pathways, respectively. The key intermediate species for photocatalytic conversion of H2O to H2O2 and O2 to H2O2 are ·OH and ·OOH radicals, respectively, whose high-rate formation is also manifested in ESR and DRIFTS tests. Given that TVCNMWCA-HEO displays not only excellent light-harvesting capability and optimized carrier dynamics but also superior catalytic functions for both 2e− ORR and 2e− WOR reaction pathways, this earth-abundant high-entropy photocatalyst can operate in an all-in-one manner for highly efficient full-reaction photosynthesis of H2O2 directly from open water resources and oxygen with neither sacrificial agents nor any external photosensitizer and cocatalysts.
Methods
Chemicals and reagents
Titanium (IV) n-butoxide (C16H36O4Ti, > 99%), vanadium chloride (VCl3, 99%), niobium chloride (NbCl5, 99.9%), chromium chloride hexahydrate (CrCl3 6H2O, > 99%), molybdenum chloride (MoCl5, 99.6%), tungsten chloride (WCl6, 99%), aluminum chloride (AlCl3, 99%), and copper chloride dihydrate (CuCl2·2H2O, > 99%) were purchased from Alfa Aesar Co., Ltd. Polyethylene glycol 20000 (PEG), citric acid (99%), ethanol (EtOH, AR) and deionized water were purchased from Beijing InnoChem Science & Technology Co., Ltd. All the chemicals were used as received.
Synthesis of high-entropy oxides
Synthesis of TVCNMWCA-HEO
The TVCNMWCA-HEO was synthesized by a two-step process. Initially, citric acid (10 mmol, 1.9 g) was added into 100 ml EtOH. Afterwards, C16H36O4Ti (1 mmol, 340.3 mg), VCl3 (1 mmol, 157.3 mg), CrCl3 6H2O (1 mmol, 266.5 mg), NbCl5 (1 mmol, 270.2 mg), MoCl5 (0.25 mmol, 68.3 mg), WCl6 (0.25 mmol, 99.1 mg), CuCl2·2H2O (0.25 mmol, 42.6 mg) and AlCl3 (0.25 mmol, 33.3 mg) were co-added into the ethanol solution. Then, this solution was stirred and PEG (0.25 mmol, 5 g) was slowly added. The mixture was heated at 120 °C for a duration of three hours to obtain a dried gel, which was introduced in a tubular furnace to 500 °C for 30 minutes. This precursor was finely pestled with some water and EtOH in an agate mortar, and then it was calcined at 600 °C for 10 h in the air to obtain the final product TVCNMWCA-HEO ( ~ 0.42 g).
Synthesis of (TiVCrNbMoAlCu)O2-x
The (TiVCrNbMoAlCu)O2-x was synthesized by a two-step process. Initially, citric acid (10 mmol, 1.9 g) was added into 100 ml EtOH. Afterwards, C16H36O4Ti (1 mmol, 340.3 mg), VCl3 (1 mmol, 157.3 mg), CrCl3 6H2O (1 mmol, 266.5 mg), NbCl5 (1 mmol, 270.2 mg), MoCl5 (0.33 mmol, 90.2 mg), CuCl2·2H2O (0.33 mmol, 56.3 mg) and AlCl3 (0.33 mmol, 44.0 mg) were co-added into the ethanol solution. Then, this solution was stirred and PEG (0.25 mmol, 5 g) was slowly added. The mixture was heated at 120 °C for a duration of three hours to obtain a dried gel, which was introduced in a tubular furnace to 500 °C for 30 minutes. This precursor was finely pestled with some water and EtOH in an agate mortar, and then it was calcined at 600 °C for 10 h in the air to obtain the final product (TiVCrNbMoAlCu)O2-x (~0.40 g).
Synthesis of (TiVCrNbMoWCu)O2-x
The (TiVCrNbMoWCu)O2-x was synthesized by a two-step process. Initially, citric acid (10 mmol, 1.9 g) was added to 100 ml EtOH. Afterwards, C16H36O4Ti (1 mmol, 340.3 mg), VCl3 (1 mmol, 157.3 mg), CrCl3 6H2O (1 mmol, 266.5 mg), NbCl5 (1 mmol, 270.2 mg), MoCl5 (0.33 mmol, 90.2 mg), WCl6 (0.33 mmol, 130.9 mg) and CuCl2·2H2O (0.33 mmol, 56.3 mg) were co-added into the ethanol solution. Then, this solution was stirred and PEG (0.25 mmol, 5 g) was slowly added. The mixture was heated at 120 °C for a duration of three hours to obtain a dried gel, which was introduced in a tubular furnace to 500 °C for 30 minutes. This precursor was finely pestled with some water and EtOH in an agate mortar, and then it was calcined at 600 °C for 10 h in air to obtain the final product (TiVCrNbMoWCu)O2-x (~ 0.44 g).
Synthesis of (TiVCrNbWAlCu)O2-x
The (TiVCrNbWAlCu)O2-x was synthesized by a two-step process. Initially, citric acid (10 mmol, 1.9 g) was added to 100 ml EtOH. Afterwards, C16H36O4Ti (1 mmol, 340.3 mg), VCl3 (1 mmol, 157.3 mg), CrCl3 6H2O (1 mmol, 266.5 mg), NbCl5 (1 mmol, 270.2 mg), WCl6 (0.33 mmol, 130.9 mg), CuCl2·2H2O (0.33 mmol, 56.3 mg) and AlCl3 (0.33 mmol, 44.0 mg) were co-added into the ethanol solution. Then, this solution was stirred and PEG (0.25 mmol, 5 g) was slowly added. The mixture was heated at 120 °C for a duration of three hours to obtain a dried gel, which was introduced in a tubular furnace to 500 °C for 30 minutes. This precursor was finely pestled with some water and EtOH in an agate mortar, and then it was calcined at 600 °C for 10 h in air to obtain the final product (TiVCrNbWAlCu)O2-x (~ 0.43 g).
Synthesis of (TiVCrNbMoWAl)O2-x
The (TiVCrNbMoWAl)O2-x was synthesized by a two-step process. Initially, citric acid (10 mmol, 1.9 g) was added into 100 ml EtOH. Afterwards, C16H36O4Ti (1 mmol, 340.3 mg), VCl3 (1 mmol, 157.3 mg), CrCl3 6H2O (1 mmol, 266.5 mg), NbCl5 (1 mmol, 270.2 mg), WCl6 (0.33 mmol, 130.9 mg), MoCl5 (0.33 mmol, 90.2 mg) and AlCl3 (0.33 mmol, 44.0 mg) were co-added into the ethanol solution. Then, this solution was stirred and PEG (0.25 mmol, 5 g) was slowly added. The mixture was heated at 120 °C for a duration of three hours to obtain a dried gel, which was introduced in a tubular furnace to 500 °C for 30 minutes. This precursor was finely pestled with some water and EtOH in an agate mortar, and then it was calcined at 600 °C for 10 h in air to obtain the final product (TiVCrNbMoWAl)O2-x (~0.44 g).
Synthesis of (TiVCrNbMoW)O2-x
The (TiVCrNbMoW)O2-x was synthesized by a two-step process. Initially, citric acid (10 mmol, 1.9 g) was added into 100 ml EtOH. Afterwards, C16H36O4Ti (1 mmol, 340.3 mg), VCl3 (1 mmol, 157.3 mg), CrCl3 6H2O (1 mmol, 266.5 mg), NbCl5 (1 mmol, 270.2 mg), MoCl5 (0.50 mmol, 136.6 mg) and WCl6 (0.50 mmol, 198.3 mg) were co-added into the ethanol solution. Then, this solution was stirred and PEG (0.25 mmol, 5 g) was slowly added. The mixture was heated at 120 °C for a duration of three hours to obtain a dried gel, which was introduced in a tubular furnace to 500 °C for 30 minutes. This precursor was finely pestled with some water and EtOH in an agate mortar, and then it was calcined at 600 °C for 10 h in air to obtain the final product (TiVCrNbMoW)O2-x powder ( ~ 0.45 g).
Statistics and reproducibility
No data were excluded from the analyzes. No statistical method was used to predetermine sample size. All synthesis and photocatalytic experiments were performed at least three times independently, ensuring the reproducibility of the results.
Characterization
The powder X-ray diffraction (XRD) patterns were recorded with a Rigaku diffractometer using Cu–Kα irradiation, operated at 40 KV and 40 mA. The XRD profiles were further analyzed by the Rietveld refinement program. The diffraction patterns were recorded in the range of 10–80°. Scanning electron microscopy (SEM) images were acquired by a Hitachi SU8220 SEM apparatus. For the scanning transmission electron microscopy (STEM observation, the samples were first dispersed into ethyl alcohol by ultrasonic dispersion and then transferred to TEM grids. All electron microscope images presented here were recorded in STEM mode using JEM-ARM300F (JEOL Co.), operating at 300 kV. The elemental mapping was also performed on an energy–dispersive X-ray analyzer (EDX) attached to the TEM apparatus. X-ray photoelectron spectroscopy (XPS) (Thermo Fisher ESCALAB Xi + ) measurement was carried out using monochromatic aluminum Kx–rays. The XPS data was analyzed with the XPS–Peak software. The valence band of the sample was also measured by UV electron spectroscopy (UPS) using a 21.2 eV monochromatic black source. Raman spectra were obtained on a Raman spectrometer (LabRAM HR Evolution, HORIBA, France) with radiation of Ar laser at 532 nm. The photoluminescence (PL) spectroscopy was measured by a Hitachi-7000 fluorescence spectrometer with an excitation wavelength of 350 nm at room temperature. The UV–vis diffuse reflectance spectrum (UV–vis DRS) was conducted in a UV-3700 spectrometer at the range of 250–800 nm. Time-resolved PL spectra at excitation of 380 nm were recorded by Edinburgh FS5 transient state fluorescence spectrometer. The femtosecond transient absorption spectrometer is composed of a regenerative-amplified Ti: sapphire laser system (Coherent) and a Helios pump-probe system (Ultrafast Systems). In our work, the transient absorption spectra were measured from 400 nm to 800 nm after band-gap excitation using 380 nm laser pulses and 550 nm laser pulses, respectively.
The metal contents in the prepared samples were determined by using a Varian 720-ES inductively coupled plasma optical emission spectrometer (ICP-OES). The Nb K-edge and Mo K-edge X-ray absorption fine structure (XAFS) spectra of the prepared samples were collected respectively at beamline BL14W1 located at the Shanghai Synchrotron Radiation Facility (SSRF) and the Ti K-edge, V K-edge, Cr K-edge, W L-edge and Cu K-edge XAFS spectra were obtained at Singapore Synchrotron Light Source center (SSLS). The obtained XAFS data was processed in Athena (version 0.9.26) for background, pre-edge line and post-edge line calibrations. Then Fourier transformed fitting was carried out in Artemis (version 0.9.26). The k3 weighting, k-range of 2−10 Å−1 and R range of 1−3 Å were used for the fitting of Ti, V, Cr, W and Cu elements and k-range of 2−7Å−1 and R range of 1−4 Å were used for the fitting of Nb element. The four parameters, coordination number, bond length, Debye-Waller factor and E0 shift (CN, R, σ2, ΔE0) were fitted without anyone being fixed, constrained, or correlated.
The electron paramagnetic resonance (EPR) spectra of as-synthesized samples were recorded at liquid nitrogen temperature with a Bruker A300 spectrometer. The EPR measurement was also performed to detect the radicals with DMPO as the radical-trapping reagent at room temperature. The DRIFTS analysis was conducted on an IRTracer-100 FTIR spectrometer equipped with a mercury–cadmium–tayuride (MCT) detector. Continuous nitrogen purging was maintained for both the interferometer and the infrared light path in the spectrometer. In the dark, the TVCNMWCA-HEO in H2O and D2O were monitored for 5 minutes, respectively. The last spectrum of the experiment in the dark was considered as the blank reference spectra for the experiment conducted under illumination. Subsequently, samples were irradiated and the sequence of spectra was recorded for real-time tracking of the reaction intermediates.
Calculation of the configurational entropy
When applying the concept of “high entropy” to HEOs where multiple sublattices exist, the value of configurational entropy (Sconfig) is correlated to the normalization reference, such as by a specific sublattice, or atoms.
For our HEO materials, the configurational entropy per cation sublattice can be calculated using the formula51:
where ms is the multiplicity of sublattice s and xi,s is the fraction of element i on sublattice s. Another method of the calculation of configurational entropy for complex HEO materials was proposed by Dippo in 2021, which introduced a new entropy measurement metric, the EM value38. The formulas can calculate the configurational entropy of the per mole of atoms in the sublattices:
where \({a}_{s}\) is the number of different sites on sublattice s, xi,s is the fraction of element i on sublattice s. As a result, the TVCNMWCA-HEO has an ideal configuration of 1.86 R per the specific cation sublattice and 1.24 R per mole of atoms in the sublattices.
Photocatalytic experimental measurement
50 mg of as-synthesized photocatalyst was dispersed in 100 ml aqueous solution. The suspension was stirred in the dark for 15 min after ultrasound treatment for 10 min to reach an adsorption-desorption equilibrium. The light intensity was adjusted to 300 mW/cm2 using a Xenon lamp (PLS-SXE 300D/ 300DUV, Beijing Perfectlight) with a 420 nm cut-off filter or 100 mW/cm2 using an AM 1.5 G solar simulator. The irradiation area is 28.26 cm−2. The bottle was kept in a water bath at 298 K. During illumination, 2 ml solution was sampled every 10 minutes and filtrated with a 0.45 μm to remove the photocatalyst. The production of H2O2 was analyzed by a colorimetric method. Typically, a 500 μL volume of 1% o-tolidine in 0.1 M HCL was added to 2 ml obtained aqueous phase product, which was kept for 30 minutes. Then the mixed solution was detected by UV-vis spectroscopy on the basis of absorbance at 453 nm, from which the quantity of generated H2O2 was estimated. Supplementary Fig. 13 shows the standard curve of H2O2 and the raw data of the UV-vis absorption spectrum of H2O2 generated by TVCNMWCA-HEO in pure water under visible light (λ ≥ 420 nm). Moreover, we also combined other methods to determine the amount of H2O2 such as using copper (II) ion and 2,9-dimethyl-1,10-phenanthroline (DMP).
The apparent quantum yield (AQY) was determined by equation:
The solar-to-chemical conversion (SCC) efficiency was determined by equation
Furthermore, according to the technology of sodium alginate–based membranes52, we synthesized the TVCNMWCA-HEO/SA membranes. Firstly, the TVCNMWCA-HEO photocatalyst powder (2 mg/ml) and 2% wt of sodium alginate aqueous solution were stirred overnight. Then the resulting homogeneous mixture was cast onto a clean glass plate, which was left to dry at room temperature for 24 hours to evaporate the solvent. The prepared membranes were used after chemical cross-linkage with calcium chloride. Every membrane approximately contains 1–2 mg powder. A few pieces of the TVCNMWCA-HEO membranes floated on the pure water in five 1000 ml glass beakers. These solutions were irradiated for 6 h under sunlight from 10:00 to 16:00. In this case, the solution was separated directly without any further separation step and 2 ml solution was sampled every 2 h to analyze the production of H2O2. Moreover, an antibacterial experiment was applied to visualize the generation of H2O2 through solar-driven photocatalysis. Gram-negative E. coli was used as model bacteria, which was cultivated on the nutrient agar plate by a sterile. With a sterile loop, touch an isolated colony on the plate and then rub the loop in the PBS on the slide to disperse cells into the buffer. Bacteria mixtures were diluted 100, 101, and 102 times with PBS. Then 1 ml solution was sampled from the beaker at different time intervals and added into 2 ml E.coli suspension and mixed thoroughly. Finally, bacteria dilution (3 mL) was added to nutrient agar (15 mL) and incubated at 37 °C for 24 h. The number of colonies forming units (CFU) was recorded and pictured.
Computational details
In this theoretical calculations, density functional theory (DFT) as implemented in the VASP code with exchange-correlation energy functional were applied, which were modeled by Perdew-Burke-Ernzerhof (PBE) functional53,54,55. The vast number of possible atomic configurations in high-entropy materials is well-known to demand great computational cost, making computational studies challenging. Recently, several studies have proposed more feasible computational approaches. For example, Zheng’s team explored the effects of high-entropy oxides through local mixing near the active site and developed a simplified high-entropy oxide model56. In this work, we adopted this approach to establish a reasonably simplified high-entropy oxide model. Firstly, a 7 × 7 × 11 k-points was selected to sample the Brillouin zone of anatase TiO2 cell. Then, the optimized TiO2 cell is expanded by a factor of 2 along the c-axis to accommodate enough metal atoms, enabling us to construct the high-entropy structure of TiVNbCrO8 within the smallest three-dimensional unit cell. Based on the ICP-MS characterization results, the proportions of the main metal elements, such as Ti, V, Cr, and Nb are approximately equal, resulting in six possible occupancy configurations in TiVNbCrO8 cell. The thermodynamically most stable TiVNbCrO8 structure and V-exposed (001) surface were selected as the basis for subsequent calculations. A (2 × 2) supercell with eight layers of TiVNbCrO8 was used to describe the TiVNbCrO8 (001) surface, with the bottom four layers fixed to simulate the bulk structure. The thickness of the vacuum in all slabs was set to 15 Å to eliminate the interactions between the layers caused by the periodic boundary condition. The cut-off energy was set to be 520 eV and structural relaxation was conducted with the criteria for energy convergence of 10−4 eV/atom and force convergence of 0.02 eV/Å, respectively. The vdW-DF2 method was applied to describe the long-range van der Waals (vdW) interaction in all the structures57,58.
Note that, Al is catalytically inactive and serves as structural support and anti-sintering agent in many cases59,60,61, so we excluded this element when focusing on the catalytic reaction. Considering the low content of Mo, W, and Cu elements, we constructed the TVCNMWCA-HEO model by treating these low-content elements as dopants for the sake of simplification. To compare the influence of different doping atoms on the formation energy of Ov in TiVNbCrO8 (001) surface, Ov was constructed on TiVCrNbO2, Cu-doped TiVCrNbO2, MoCu-doped TiVCrNbO2 and MoWCu-doped TiVCrNbO2, to form (TiVCrNb)O2-x, (TiVCrNbCu)O2-x, (TiVCrNbMoCu)O2-x and (TiVCrNbMoWCu)O2-x, respectively. The formation energy of Ov was defined as \(\triangle {{{\rm{E}}}}_{{{\rm{ov}}}}={{{\rm{E}}}}_{{{\rm{slab}}}-{{\rm{O}}}}+{{{\rm{O}}}}_{{{\rm{atom}}}}-{{{\rm{E}}}}_{{{\rm{slab}}}}\), where \({{{\rm{E}}}}_{{{\rm{slab}}}-{{\rm{O}}}}\), \({{{\rm{O}}}}_{{{\rm{atom}}}}\) and \({{{\rm{E}}}}_{{{\rm{slab}}}}\) represent computational surface with Ov, the oxygen atom and total energy of perfect surface, respectively.
To identify the ORR and WOR catalytic sites on (TiVCrNbMoWCu)O2-x, we first investigate the adsorption of H2O on Mo, W, Cu, V and Ov sites. The adsorption energies (\(\triangle {{{\rm{G}}}}_{{{\rm{ads}}}}\)) of H2O were defined as \(\triangle {{{\rm{G}}}}_{{{\rm{ads}}}}\) = \({{{\rm{G}}}}_{{{\rm{total}}}}\) – \({{{\rm{E}}}}_{{{\rm{surf}}}}\) – \({{{\rm{G}}}}_{{{\rm{water}}}}\), where \({{{\rm{G}}}}_{{{\rm{total}}}}\), \({{{\rm{E}}}}_{{{\rm{surf}}}}\), and \({{{\rm{G}}}}_{{{\rm{water}}}}\) represent the energies of adsorption configurations, computational surfaces, and water, respectively. Then, sequential deprotonation step was considered to depict the water dissociation process. The dissociation energy (\(\triangle {{{\rm{G}}}}_{1}\)) of H2O was defined as \(\triangle {{{\rm{G}}}}_{1}\) = \({{{\rm{G}}}}_{{{\rm{OH}}}}\) + \({{{\rm{G}}}}_{{{\rm{H}}}}\)–\({{{\rm{G}}}}_{{{{\rm{H}}}}_{2}{{\rm{O}}}}\), where \({{{\rm{G}}}}_{{{\rm{OH}}}}\), \({{{\rm{G}}}}_{{{\rm{H}}}}\) and \({{{\rm{G}}}}_{{{{\rm{H}}}}_{2}{{\rm{O}}}}\) represent the energies of OH adsorption configuration, hydrogen atom, and H2O adsorption configuration, respectively.
As for the ORR reaction pathway, we first considered the adsorption of O2 on different active sites and then calculated the hydrogenation energy for generating the important intermediate product *OOH. The adsorption energies (\(\triangle {{{\rm{G}}}}_{{{\rm{ads}}}}\)) of O2 were defined as \({{\triangle {{{\rm{G}}}}_{{{\rm{ads}}}}={{\rm{G}}}}_{{{\rm{total}}}}-{{{\rm{E}}}}_{{{\rm{surf}}}}-{{\rm{G}}}}_{{{{\rm{O}}}}_{2}}\), where \({{{\rm{G}}}}_{{{\rm{total}}}}\), \({{{\rm{E}}}}_{{{\rm{surf}}}}\), and \({{{\rm{G}}}}_{{{{\rm{O}}}}_{2}}\) represent the energies of adsorption configurations, computational surfaces, and O2, respectively. The hydrogenation energy (\(\triangle {{{\rm{G}}}}_{2}\)) was defined as \(\triangle {{{\rm{G}}}}_{2}\) = \({{{\rm{G}}}}_{{{\rm{OOH}}}}-{{{\rm{G}}}}_{{{\rm{H}}}}-{{{\rm{G}}}}_{{{{\rm{O}}}}_{2}}\), where \({{{\rm{G}}}}_{{{\rm{OOH}}}}\), \({{{\rm{G}}}}_{{{\rm{H}}}}\), \({{{\rm{G}}}}_{{{{\rm{O}}}}_{2}}\) represent the energies of OOH adsorption configuration, hydrogen atom, and O2 adsorption configuration, respectively. All the energies were corrected by considering the zero-point energy and Entropy corrections under the standard conditions (p0 = 1 bar and T0 = 298.15 K). Note that, the chemical potentials of H+ and e− can be described based on the computational hydrogen electrode62.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data that support the findings in this paper are available within the article and its Supplementary Information. Source data are provided with this paper.
References
Andrei, V. et al. Floating perovskite-BiVO4 devices for scalable solar fuel production. Nature 608, 518 (2022).
Nguyen, D. N., Fadel, M., Chenevier, P., Artero, V. & Tran, P. D. Water-splitting artificial leaf based on a triple-junction silicon solar cell: One-step fabrication through photoinduced deposition of catalysts and electrochemical operando monitoring. J. Am. Chem. Soc. 144, 9651–9660 (2022).
Shi, R., Zhao, Y., Waterhouse, G. I. N., Zhang, S. & Zhang, T. Defect engineering in photocatalytic nitrogen fixation. ACS Catal. 9, 9739–9750 (2019).
Fu, Y. J. et al. Photocatalytic H2O2 and H2 generation from living chlorella vulgaris and carbon micro particle comodified g-C3N4. Adv. Energy Mater. 8, 1802525 (2018).
Escobedo, S. & de Lasa, H. Synthesis and performance of photocatalysts for photocatalytic hydrogenp production: Future perspectives. Catalysts 11, 1505 (2021).
Campos-Martin, J. M., Blanco-Brieva, G. & Fierro, J. L. G. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 45, 6962–6984 (2006).
Guo, Y., Tong, X. & Yang, N. Photocatalytic and electrocatalytic generation of hydrogen peroxide: principles, catalyst design and performance. Nano-Micro Lett. 15, 77 (2023).
Hou, H., Zeng, X. & Zhang, X. Production of Hydrogen Peroxide by Photocatalytic Processes. Angew. Chem. Int. Ed. 59, 17356–17376 (2020).
Siahrostami, S. et al. Enabling direct H2O2 production through rational electrocatalyst design. Nat. Mater. 12, 1137–1143 (2013).
Verdaguer-Casadevall, A. et al. Trends in the electrochemical synthesis of H2O2: enhancing activity and selectivity by electrocatalytic site engineering. Nano Lett. 14, 1603–1608 (2014).
Siahrostami, S. et al. A review on challenges and successes in atomic-scale design of catalysts for electrochemical synthesis of hydrogen peroxide. ACS Catal. 10, 7495–7511 (2020).
Xia, C. et al. Confined local oxygen gas promotes electrochemical water oxidation to hydrogen peroxide. Nat. Catal. 3, 125–134 (2020).
Zhang, Y. et al. H2O2 generation from O2 and H2O on a near-infrared absorbing porphyrin supramolecular photocatalyst. Nat. Energy 8, 361–371 (2023).
Yan, H. et al. Spontaneous exciton dissociation in organic photocatalyst under ambient conditions for highly efficient synthesis of hydrogen peroxide. PNAS 119, e2202913119 (2022).
Jia, Z. et al. A novel multinary intermetallic as an active electrocatalyst for hydrogen evolution. Adv. Mater. 32, e2000385 (2020).
Nellaiappan, S. et al. High-entropy alloys as catalysts for the CO2 and CO reduction reactions: experimental realization. ACS Catal. 10, 3658–3663 (2020).
Sarkar, A. et al. High-entropy oxides: Fundamental aspects and electrochemical properties. Adv. Mater. 31, 1806236 (2019).
Batchelor, T. A. A. et al. High-entropy alloys as a discovery platform for electrocatalysis. Joule 3, 834–845 (2019).
Li, H., Lai, J., Li, Z. & Wang, L. Multi‐sites electrocatalysis in high‐entropy alloys. Adv. Funct. Mater. 31, (2021).
Wu, D. et al. Noble-metal high-entropy-alloy nanoparticles: atomic-level insight into the electronic structure. J. Am. Chem. Soc. 144, 3365–3369 (2022).
Chae, S., Williams, L., Lee, J., Heron, J. T. & Kioupakis, E. Effects of local compositional and structural disorder on vacancy formation in entropy-stabilized oxides from first-principles. Npj Comput. Mater. 8, 95 (2022).
Gu, K. et al. Defect-rich high-entropy oxide nanosheets for efficient 5-Hydroxymethylfurfural electrooxidation. Angew. Chem. Int. Ed. Engl. 60, 20253–20258 (2021).
Zhang, Y. et al. Stabilizing oxygen vacancy in entropy-engineered CoFe2O4-type catalysts for co-prosperity of efficiency and stability in an oxygen evolution reaction. ACS Appl. Mater. Interfaces 12, 32548–32555 (2020).
Ciftyurek, E., Li, Z. & Schierbaum, K. Adsorbed oxygen ions and oxygen vacancies: Their concentration and distribution in metal oxide chemical sensors and influencing role in sensitivity and sensing mechanisms. Sensors 23, 29 (2022).
Katzbaer, R. R., Vieira, F. M. D., Dabo, I., Mao, Z. Q. & Schaak, R. E. Band gap narrowing in a high-entropy spinel oxide semiconductor for enhanced oxygen evolution catalysis. J. Am. Chem. Soc. 145, 6753–6761 (2023).
Du, K. et al. High entropy oxides modulate atomic‐level interactions for high‐performance aqueous zinc‐ion batteries. Adv. Mater. 35, 2301538 (2023).
Edalati, P. et al. Photocatalytic hydrogen evolution on a high-entropy oxide. J. Mater. Chem. A. 8, 3814–3821 (2020).
Edalati, P. et al. High-entropy oxynitride as a low-bandgap and stable photocatalyst for hydrogen production. J. Mater. Chem. A. 9, 15076–15086 (2021).
Nundy, S. et al. Bandgap engineering in novel fluorite‐type rare earth high‐entropy oxides (RE‐HEOs) with computational and experimental validation for photocatalytic water splitting applications. Adv. Sustain. Syst. 6, 2200067 (2022).
Huang, H. et al. Noble-metal-free high-entropy alloy nanoparticles for efficient solar-driven photocatalytic CO2 reduction. Adv. Mater. 36, 2313209 (2024).
Akrami, S. et al. Significant CO2 photoreduction on a high-entropy oxynitride. Chem. Eng. J. 449, 137800 (2022).
Qi, S. et al. Water-stable high-entropy metal-organic framework nanosheets for photocatalytic hydrogen production. Adv. Mater. 36, 2403328 (2024).
Akrami, S. et al. Defective high-entropy oxide photocatalyst with high activity for CO2 conversion. Appl. Catal. B: Environ. 303, 120869 (2022).
Rost, C. M. et al. Entropy-stabilized oxides. Nat. Commun. 6, 8485 (2015).
Su, L. et al. Direct observation of elemental fluctuation and oxygen octahedral distortion-dependent charge distribution in high entropy oxides. Nat. Commun. 13, 2358 (2022).
Vásquez, G. C. et al. Oxygen vacancy related distortions in rutileTiO2nanoparticles: A combined experimental and theoretical study. Phys. Rev. B. 94, 235209 (2016).
Körmann, F., Ikeda, Y., Grabowski, B. & Sluiter, M. H. F. Phonon broadening in high entropy alloys. Npj Comput. Mater. 3, 1–9 (2017).
Dippo, O. F. & Vecchio, K. S. A universal configurational entropy metric for high-entropy materials. Scr. Mater. 201, 113974 (2021).
Su, R. et al. Utilizing the oxygen-atom trapping effect of Co3O4 with oxygen vacancies to promote chlorite activation for water decontamination. PNAS 121, e2319427121 (2024).
Feng, D. et al. Holey lamellar high-entropy oxide as an ultra-high-activity heterogeneous catalyst for solvent-free aerobic oxidation of benzyl alcohol. Angew. Chem. Int. Ed. Engl. 59, 19503–19509 (2020).
Selim, S. et al. Impact of oxygen vacancy occupancy on charge carrier dynamics in BiVO4 photoanodes. J. Am. Chem. Soc. 141, 18791–18798 (2019).
Pan, S. et al. Engineering the intermediate band states in amorphous Ti3+-doped TiO2 for hybrid dye-sensitized solar cell applications. J. Mater. Chem. A. 3, 11437–11443 (2015).
Jiang, W. et al. Role of oxygen vacancy in metal oxides for photocatalytic CO2 reduction. Appl. Catal. B: Environ. 321, 122079 (2023).
Chatzitakis, A. & Sartori, S. Recent advances in the use of black TiO2 for production of hydrogen and other solar fuels. ChemPhysChem 20, 1272–1281 (2019).
Tang, L. et al. High configuration entropy activated lattice oxygen for O2 formation on perovskite electrocatalyst. Adv. Funct. Mater. 32, 2112157 (2022).
Yu, W. et al. Photocatalytic hydrogen peroxide evolution: What is the most effective strategy? Nano Energy 104, 107906 (2022).
Chu, C. et al. Spatially separating redox centers on 2D carbon nitride with cobalt single atom for photocatalytic H2O2 production. PNAS 117, 6376–6382 (2020).
Chang, J. N. et al. Oxidation-reduction molecular junction covalent organic frameworks for full reaction photosynthesis of H2O2. Angew. Chem. Int. Ed. Engl. 62, e202218868 (2023).
Teng, Z. et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat. Catal. 4, 374–384 (2021).
Wang, P. et al. Oxygen vacancy defect engineering to promote catalytic activity toward the oxidation of VOCs: a critical review. Catal. Rev. 66, 586–639 (2022).
Mahdavi-Shakib, A., Rich, L. C., Whittaker, T. N. & Chandler, B. D. Hydrogen adsorption at the Au/TiO2 interface: quantitative determination and spectroscopic signature of the reactive interface hydroxyl groups at the active site. ACS Catal. 11, 15194–15202 (2021).
Wang, Y. & Lu, Y. Sodium alginate-based functional materials toward sustainable applications: Water treatment and energy storage. Ind. Eng. Chem. Res. 62, 11279–11304 (2023).
Kresse, G. & Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Baek, J. et al. Synergistic effects of mixing and strain in high entropy spinel oxides for oxygen evolution reaction. Nat. Commun. 14, 5936 (2023).
Dion, M., Rydberg, H., Schröder, E., Langreth, D. C. & Lundqvist, B. I. Van der Waals density functional for general geometries. Phys. Rev. Lett. 95, 109902 (2005).
Lee, K., Murray, É., Kong, L. Z., Lundqvist, B. I. & Langreth, D. C. Higher-accuracy van der Waals density functional. Phys. Rev. B. 82, 081101 (2010).
He, L. et al. Robust and coke-free Ni catalyst stabilized by 1–2 nm-thick multielement oxide for methane dry reforming. ACS Catal. 11, 12409–12416 (2021).
Wang, L., Wang, L., Meng, X. & Xiao, F.-S. New strategies for the preparation of sinter-resistant metal-nanoparticle-based catalysts. Adv. Mater. 31, 1901905 (2019).
Zhang, Z. et al. Spatial segregation of three-dimensional Al2O3 supported PtSn catalyst for improved sintering-resistant at high temperature. Appl. Catal. B-Environ. 358, 124334 (2024).
Norskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22109108, 12334001, 51991344), National Key R&D Program of China (2021YFA1400204), the Strategic Priority Research Program of Chinese Academy of Sciences (XDB30000000, XDB33030200), Guangdong Major Project of Basic and Applied Basic Research (2021B0301030002), Guangdong Basic and Applied Basic Research Foundation (2023A1515140102).
Author information
Authors and Affiliations
Contributions
H.L. and W.W. conceived the idea and designed the experiments. W.W. and X.B. supervised the research. H.L. performed the synthesis of the samples, material characterizations, photocatalysis measurements, and data analysis. L.C. and W.L. performed the DFT calculations. L.C. assisted with the discussion of the experiments. H.S., L.L., and J.W. performed the STEM analysis. L.S.L., X.Z., Y.L., and R.L. contributed to the structure characterization. J.Z. performed the antibacterial experiments; H.L. and W.W. wrote this manuscript with the input of all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yongfa Zhu and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ling, H., Sun, H., Lu, L. et al. Sustainable photocatalytic hydrogen peroxide production over octonary high-entropy oxide. Nat Commun 15, 9505 (2024). https://doi.org/10.1038/s41467-024-53896-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53896-w
This article is cited by
-
Nanoscale reduction-site-selective oxygen regulation for promoting hydrogen peroxide production
Nature Communications (2025)
-
Defect-modulated oxygen adsorption and Z-scheme charge transfer for highly selective H2O2 photosynthesis in pure water
Nature Communications (2025)
-
Photocatalytic technology in lignin: from traditional depolymerization to actively light-driven conversion
Advanced Composites and Hybrid Materials (2025)
