
Abstract
Ruthenium (Ru) is widely recognized as a low-cost alternative to iridium as anode electrocatalyst in proton-exchange membrane water electrolyzers (PEMWE). However, the reported Ru-based catalysts usually only operate within tens of hours in PEMWE because of their intrinsically high reactivity of lattice oxygen that leads to irrepressible Ru leaching and structural collapse. Herein, we report a design concept by employing large-sized and acid-resistant lattice lead (Pb) as a second element to induce a pinning effect for effectively narrowing the moving channels of oxygen atoms, thereby lowering the reactivity of lattice oxygen in Ru oxides. The Pb-RuO2 catalyst presents a low overpotential of 188 ± 2 mV at 10 mA cm−2 and can sustain for over 1100 h in an acid medium with a negligible degradation rate of 19 μV h−1. Particularly, the Pb-RuO2-based PEMWE can operate for more than 250 h at 500 mA cm−2 with a low degradation rate of only 17 μV h−1. Experimental and theoretical calculation results reveal that Ru-O covalency is reduced due to the unique 6s−2p−4d orbital hybridization, which increases the loss energy of lattice oxygen and suppresses the over-oxidation of Ru for improved long-term stability in PEMWE.
Similar content being viewed by others

Introduction
Proton exchange membrane water electrolysis (PEMWE) technology attracts great interest due to its excellent intermittent matching, high H2 purity, low Ohmic resistance, and large current density1,2,3,4,5. Nevertheless, the large-scale application of PEMWE is severely restricted owing to the lack of high-efficiency and low-cost catalysts for acidic oxygen evolution reaction (OER). The biggest bottleneck lies in how to strengthen the durability of catalysts under strong acidic and oxidative conditions during the high-current-density operation6,7. Till now, iridium (Ir)-based materials with robust stability and high activity are regarded as the best acid OER catalysts8,9. However, the usage of Ir in commercial PEMWE is usually quite high (2–5 mg cm−2), which limits their large-scale applications10,11. Recently, ruthenium (Ru)-based catalysts are of great interest due to the higher OER activity and lower cost compared to Ir-based catalysts12,13,14,15,16, which renders the Ru-based catalysts as attractive alternatives to Ir-based catalysts. However, Ru-based catalysts always exhibit unsatisfactory stability even operating at a low current density17,18, which is far below the practical requirements of PEMWEs (>500 mA cm−2). The main reason can be attributed to the high Ru-O covalency and inevitable lattice oxygen mechanism (LOM) of Ru-based catalysts, which increases the participation of lattice oxygen during the reaction, thereby leading to the overoxidation of Ru sites and irrepressible structural collapse of catalysts. Therefore, developing an effective strategy to reduce the reactivity of lattice oxygen is quite significant for improving the durability of catalysts, however, implementing this target remains a grand challenge.
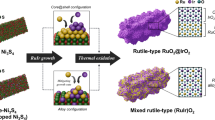
Herein, we propose a design concept to strengthen the stability of RuO2 by introducing large-sized and acid-resistant lead (Pb) into the lattice of RuO2 to pin the moving channels of oxygen atoms for increasing the diffusion activation energies of lattice oxygen, thereby reducing the reactivity of lattice oxygen (Fig. 1). The Pb-RuO2 catalyst possesses an overpotential of 188 ± 2 mV and can sustain for over 1100 h at 10 mA cm−2, longer than the Ru-based catalysts in previous works. Moreover, the Pb-RuO2-based PEMWE requires a cell voltage of 1.688 V to achieve 1000 mA cm−2 and show long-term stability at 500 mA cm−2 for more than 250 h with a low degradation rate of only 17 μV h−1. XAFS, in situ DEMS, and DFT calculations collectively elucidate that the Ru-O covalency is weakened due to the reduced Ru charge derived from the hybridization between Pb 6s, O 2p, and Ru 4d orbits, which enhances the energy barrier of lattice oxygen loss and suppresses the leaching of Ru for promoted long-term acid OER durability in PEMWE.

The design concept of using large-sized Pb to narrow the moving channel of lattice oxygen atoms for reduced lattice oxygen reactivity of RuO2.
Results
Materials synthesis and characterization
The Pb-RuO2 catalyst was prepared by a one-pot glucose-blowing method. Briefly, a porous foam consisting of glucose and homogeneously dispersed Ru and Pb ions was first obtained, followed by calcinating in air to form the Pb-RuO2 catalyst (Supplementary Fig. 1). As a comparison, pure RuO2 (Hm-RuO2) was also synthesized via a similar procedure without the addition of Pb ions.
X-ray diffraction (XRD) pattern of Pb-RuO2 shows that it has the rutile-type structure without the appearance of peaks related to PbOx (Supplementary Fig. 2). The characteristic peaks of Pb-RuO2 shift to lower angles compared to those of RuO2, suggesting the lattice expansion due to the incorporation of large-sized Pb atoms into the lattice of RuO2 crystal. Transmission electron microscopy (TEM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and annular bright field (ABF) images of the Pb-RuO2 show that it is composed of small crystals with a size of around 6.5 nm, corresponding to the (110) plane of RuO2 (Supplementary Figs. 3–5). Atomic-revolution HAADF-STEM images clearly reveal that the metal atoms of Pb-RuO2 display order arrangement along the crystal lattice of RuO2 at [001] direction, which is highly consistent with the structural model (Fig. 2a, b). The absence of the PbOx crystal lattice indicates the successful involvement of Pb atoms into the lattice of RuO2. The (110) lattice spacing of Pb-RuO2 (0.332 nm) is larger than that of standard RuO2 (0.320 nm), further demonstrating the lattice expansion due to the substitution of Pb to Ru. Elemental mapping images confirm the coexistence of Ru, Pb, and O within the crystal (Fig. 2c). Inductively coupled Plasma-optical emission spectrometer (ICP-OES) shows that the relative mass fraction of Pb and Ru is 12.8 and 87.2 wt%, respectively, further supported by the EDS results (Supplementary Table 1).

a HAADF-STEM image and FFT pattern (inset) of the Pb-RuO2 catalyst. b Atomic-resolution HAADF-STEM image and structural model of the Pb-RuO2 crystal from the [001] direction. c Elemental mappings of Pb, Ru, and O in the Pb-RuO2 catalyst. d Pb L3-edge XANES spectra of Pb-RuO2 and PbO2. e Ru K-edge XANES spectra of Pb-RuO2, RuO2, and Ru foil. The inset is the valence state of Ru. f The Fourier-transformed Ru K-edge EXAFS spectra of Ru foil, RuO2, and Pb-RuO2. g–i The Wavelet transforms the Ru K-edge EXAFS signals of Ru foil, RuO2, and Pb-RuO2.
X-ray photoelectronic spectroscopy (XPS) was performed to characterize the chemical state of the catalysts. As shown in the Pb 4f spectra, the valence state of Pb in Pb-RuO2 is lower than +4 due to its higher binding energy than that of PbO219 (Supplementary Fig. 6). The high-resolution Ru 3p spectra display that the Ru3+ ratio in Pb-RuO2 is higher than that in RuO2, indicating the lower oxidation state of Ru in Pb-RuO2 (Supplementary Fig. 7). X-ray absorption fine structure spectroscopy (XAFS) was conducted to elucidate the electronic structure and coordination environment of Pb and Ru in Pb-RuO2. The Pb L3-edge X-ray absorption near edge structure (XANES) spectrum of PbO2 shows an obvious shoulder at 13,030.0 eV, which is related to the dipole-allowed transition from the 2p3/2 core level to the unoccupied 6s state19,20 (Fig. 2d). However, the shoulder could not be observed for Pb-RuO2, indicating that 6s electrons exist in Pb-RuO2 yet is absent in PbO2. The Ru K-edge XANES spectra display that the edge of Pb-RuO2 is close to that of RuO2, suggesting the oxidized feature of Ru (Fig. 2e). The negative shift of the edge for Pb-RuO2 relative to that of RuO2 reveals its lowered Ru oxidation state, consistent with the XPS analysis. The Ru valence state is determined to be +3.7 by linearly fitting the first derivative position (E0) of Ru K-edge XANES (inset of Fig. 2e and Supplementary Fig. 8). The Ru K-edge extended X-ray absorption fine structure (EXAFS) spectra show two distinct peaks at 1.47 and 3.16 Å, corresponding to the Ru-O and Ru-Ru bonds, respectively, further confirmed by the wavelet transform (WT) analysis (Fig. 2f-i). The EXAFS fitting results reveal that the coordination number (CN) of Ru-O and Ru-Ru in Pb-RuO2 is lower than that of RuO2, mainly ascribed to the assistance of Pb for breaking the structural symmetry of RuO2 (Supplementary Figs. 9–11, Supplementary Table 2). Further analysis of the bond length suggests that the Ru-O distance of Pb-RuO2 is stretched compared to the pure RuO2 (Supplementary Table 2), demonstrating its weakened Ru-O covalency21.
Electrocatalytic activity
The acid OER performances of Pb-RuO2 and reference catalysts were evaluated in the O2-saturated 0.5 M H2SO4 electrolyte. Pb-RuO2 with proper Pb proportion exhibits an overpotential of only 188 ± 2 and 255 ± 4 mV to achieve 10 and 100 mA cm−2, respectively, smaller than those of Hm-RuO2 (245 ± 5 and 363 ± 6 mV at 10 and 100 mA cm−2) and commercial (Com)-RuO2 (280 ± 7 and 446 ± 6 mV at 10 and 100 mA cm−2) (Fig. 3a and Supplementary Figs. 12, 13). The reduced Tafel slope of Pb-RuO2 relative to those of Hm-RuO2 and Com-RuO2 suggests its enhanced OER reaction kinetics (Fig. 3b, c), further verified by the lowered charge-transfer resistance in electrochemical impedance spectroscopy (EIS) measurement (Supplementary Fig. 14). Pb-RuO2 also delivers a much higher mass activity (MA) than those of Hm-RuO2 and Com-RuO2 (Supplementary Figs. 15,16), demonstrating its improved intrinsic OER activity. Electrochemical surface area (ECSA) measurement reveals that Pb-RuO2 has the highest active area among different reference catalysts (Supplementary Fig. 17).

a The iR-corrected polarization curves (R was 1.15 ± 0.05 Ω) tested at a scan rate of 5 mV s−1, b Tafel slope, and c the comparison of overpotential at 10 mA cm-2 and Tafel slope of Pb-RuO2, home-made (Hm-RuO2) and commercial (Com-RuO2). d The iR-corrected polarization curves of Pb-RuO2, Hm-RuO2, and Com-RuO2 before and after stability cycling tests. e Chronopotentiometry test of Pb-RuO2 at 10 mA cm-2.
The Pb-RuO2 exhibits negligible variation of overpotential after 100,000 cycling tests in acid electrolyte, while the Hm-RuO2 and Com-RuO2 catalysts show obvious performance decay after only 10,000 cycling tests (Fig. 3d and Supplementary Fig. 18). The chronopotentiometry tests display that Pb-RuO2 presents a quite stable potential at 10 mA cm−2 for more than 1100 h, superior to that of Com-RuO2 with distinct potential increase during the 56 h operation (Fig. 3e and Supplementary Fig. 19). The notable durability of Pb-RuO2 with long operation time and relatively low overpotential degradation rate surpasses the reported state-of-the-art Ru, Ir-based electrocatalysts (Supplementary Fig. 20 and Supplementary Table 3). Moreover, the Pb-RuO2 can even operate at a quite high current density of 100 mA cm-2 for 250 h with only 19 mV decay of potential, superior to the reported Ru, Ir-based electrocatalysts and Com-RuO2 (Supplementary Figs. 21–23).
Catalytic mechanism
We first conducted a methanol-assisted method to probe the *OH intermediate22, since the activity of RuO2 is closely related to the binding strength of *OH. The larger current difference between the methanol oxidation reaction (MOR) and OER of Pb-RuO2 compared to that of RuO2 (Fig. 4a and Supplementary Fig. 24) indicates the enhanced *OH adsorption on Pb-RuO2, since the bigger current difference suggests stronger MOR competition reaction that is more active on the surfaces with stronger *OH adsorption. In situ electrochemical Raman spectra were also performed to investigate the adsorption behavior of the intermediate species, in which Pb-RuO2 displays a negative shift of the Raman frequency for *OH adsorption (~700 cm−1) relative to that of RuO2 (Fig. 4b, Supplementary Fig. 25), further indicating the enhanced binding with *OH species23,24. According to previous researches18,25, the promoted *OH binding strength of Pb-RuO2 supplies near-optimal free energies for each intermediate, which leads to reduced theoretical OER overpotential.

a The iR-corrected polarization curves of Pb-RuO2 and RuO2 in 0.5 M H2SO4 solution with (dashed lines) and without (solid lines) 1 M methanol. b In situ Raman spectra of Pb-RuO2 and RuO2 obtained under various applied potentials. c Schematic illustration of the in situ DEMS. DEMS signals of 32O2, 34O2, and 36O2 from the gaseous products for 18O-labeled d Pb-RuO2 and e RuO2 catalysts in H216O aqueous H2SO4 electrolyte during three times of cycles. f The mass spectroscopy peak area ratio of 34O2/32O2 of Pb-RuO2 and RuO2.
In situ differential electrochemical mass spectroscopy (DEMS) with isotope 18O labeling was conducted to reveal the lattice oxygen participation of different catalysts (Fig. 4c, Supplementary Fig. 26), since the lattice oxygen mechanism (LOM) path always results in a rapid decline of performance in acid medium. The surface of catalysts was first labeled with 18O by operating at 1.6 V vs. RHE for 600 s in 0.5 M H2SO4/H218O solution. Then, the generated O2 from 18O labeled catalysts during electrochemical OER in 0.5 M H2SO4/H216O was detected by mass spectroscopy. Most signals of the evolved O2 from Pb-RuO2 and RuO2 are 32O2 (Fig. 4d, e and Supplementary Fig. 27), indicating that the main reaction path of Pb-RuO2 and RuO2 is adsorbate evolution mechanism (AEM). However, RuO2 shows more distinct 36O2 signals compared to Pb-RuO2, demonstrating the inhibited LOM process of Pb-RuO2. The 34O2/32O2 percentage of RuO2 (3.38%) and Pb-RuO2 (0.87%) is higher than 0.2% in natural abundance of H218O11,18, suggesting the participation of lattice oxygen in both catalysts (Fig. 4f). While Pb-RuO2 shows significantly reduced 34O2/32O2 percentage compared to RuO2, demonstrating the crucial role of Pb for suppressing the lattice oxygen participation for promoting the long-term stability of Pb-RuO2 in acid OER.
We further employed density functional theory (DFT) calculations to reveal the reason for the improved OER performance of the Pb-RuO2 catalyst by establishing the Pb-RuO2 and RuO2 models (Fig. 5a, Supplementary Fig. 28, and Supplementary Data 1, 2). The Gibbs free energy of oxygen intermediates on the AEM path was calculated to investigate the origin of performance enhancement of Pb-RuO2 based on the DEMS results. As shown in Fig. 5b and Supplementary Figs. 29–32, the potential determining step (PDS) was *O to *OOH for both Pb-RuO2 and RuO2. Pb-RuO2 displays a reduced energy barrier from 1.832 to 1.817 V relative to RuO2, indicating improved activity with the assistance of Pb. The crystal orbital Hamilton populations (COHP) of the Ru active site with *OOH adsorbed (Ru-OOH) further elucidate the enhanced *OOH adsorption, which leads to decreased energy barrier (*O-*OOH) for reduced reaction overpotential (Supplementary Fig. 33).

a Atomic structure of the Pb-RuO2 and RuO2 (110) surface. b The reaction paths of Pb-RuO2 at 0 V. c Projected DOS plots of Ru (d), O (p), and Pb (s) of Pb-RuO2. Calculated COHP of (d) Pb-RuO2 and (e) RuO2. f -ICOHP of Pb-RuO2 and RuO2 with varied Ru charge. Inset is the Ru charge of Pb-RuO2 and RuO2 based on Bader charge analysis. g Lattice oxygen loss energy of Pb-RuO2 and RuO2. h The dissolved Ru ions of Pb-RuO2 and RuO2 as a function of reaction time at 10 mA cm−2. i Comparison of the Ru 3d spectra of Pb-RuO2 and RuO2 before and after OER electrolysis.
Projected density of states (pDOS) plots were collected to further reveal the effect of Pb on the electronic structure of RuO2. The coupling between Pb 6s, Ru 4d, and O 2p orbits (6s-2p-4d) leads to the changed electronic environment around the Ru active sites for balanced adsorption of the oxygen intermediates (Fig. 5c, Supplementary Fig. 34). Moreover, the O p band center of Pb-RuO2 shows a downshift relative to that of RuO2 (Supplementary Table 4), leading to the weakened Ru-O covalency25. The integrated COHP (ICOHP) calculations further indicate that the involvement of Pb can reduce the Ru-O covalency (Fig. 5d, e). The Barder charge analysis reveals that the 6s-2p-4d orbital hybridization of Pb-RuO2 leads to charge redistribution for reduced Ru charge (Fig. 5f), which not only optimizes the adsorption of the oxygen intermediates for decreased OER overpotential, but also weakens the Ru-O covalency and inhibits the lattice oxygen participation for promoted long-term stability of Pb-RuO2 in acid OER.
To elucidate the unique effect of large-sized Pb on the stability of RuO2, we further calculated the energy barrier of lattice oxygen loss, which shows that Pb-RuO2 presents an increased barrier relative to that of RuO2 (Fig. 5g), indicating the decreased reactivity of lattice oxygen. ICP-MS analysis displays that the pure RuO2 shows accelerated Ru dissolution, revealing the irrepressible structural collapse due to the high activity of lattice oxygen. While the leaching of Ru can be suppressed with the assistance of large-sized lattice Pb due to the unique pinning effect for reducing the reactivity of lattice oxygen (Fig. 5h). Besides, the dissolution of Pb was also controllable as displayed in Supplementary Fig. 35. XPS spectra after electrolysis exhibit that the Ru valence state of Pb-RuO2 is more stable than that of pure RuO2, demonstrating that the over-oxidation of Ru atoms can be suppressed due to the inhibition of lattice oxygen participation (Fig. 5i). TEM images after electrolysis also show the almost maintained structure of the Pb-RuO2 catalyst, further indicating the enhanced stability of RuO2 due to the assistance of Pb (Supplementary Figs. 36, 37).
Performance of PEMWE devices
A PEM electrolyzer using Pb-RuO2 and Pt/C as anode and cathode catalysts were constructed to evaluate the potential of our catalyst in industrial applications (Supplementary Fig. 38). Pb-RuO2-based PEMWE can achieve industrial current densities of 500, 1000, and 2000 mA cm−2 at 1.582, 1.688, and 1.848 V, respectively, better than those of Hm-RuO2-based PEMWE (1.623, 1.736, and 1.947 V) and Com-RuO2-based PEMWE (1.676, 1.817, and 2.125 V) (Fig. 6a, b). Impressively, the Pb-RuO2-based PEMWE can sustain for over 250 h at an industrial current density of 500 mA cm−2. The degradation rate was determined to be only 17 μV h−1, the lowest value among the reported Ru-based catalysts (Fig. 6c, Supplementary Fig. 39, and Supplementary Table 5). When operating at a larger current density of 1000 mA cm−2, the Pb-RuO2-based PEMWE can also work for more than 100 h with a degradation rate of only 87 μV h−1, which is much smaller than that of Hm-RuO2-based PEMWE (3.2 mV h−1 of degradation rate during 38 h operation). The durability of Pb-RuO2 at both 500 and 1000 mA cm-2 in PEMWE is well-placed among select recently reported Ru-based catalysts (Supplementary Fig. 40 and Supplementary Table 5).

a Polarization curves of PEMWEs using Pb-RuO2, Hm-RuO2, and Com-RuO2 as anode catalysts without iR-correction. b Comparison of the cell voltages of Pb-RuO2, Hm-RuO2, and Com-RuO2-based PEMWEs at 500, 1000, and 2000 mA cm−2. c Chronopotentiometry curves of the Pb-RuO2 and Hm-RuO2-based PEMWEs.
Discussion
We reported a class of quite stable Pb-RuO2 catalysts for PEMWE by introducing large-sized and acid-resistant Pb into the lattice of RuO2 to induce the pinning effect, and narrow the moving channels of oxygen atoms for increasing the diffusion activation energies of lattice oxygen. XAFS, in situ DEMS, and DFT calculation results reveal that the involvement of Pb can weaken the reactivity of lattice oxygen owing to the reduced Ru-O covalency derived from the 6s-2p-4d orbital hybridization, which accordingly suppresses the over-oxidation of Ru for well-maintained crystal structure. The Pb-RuO2 catalyst presents an impressively low overpotential of 188 ± 2 mV, and can sustain for over 1100 h at 10 mA cm-2 with a negligible performance decay, surpassing the reported Ru-based catalysts. Moreover, the Pb-RuO2-based PEMWE requires a voltage of 1.688 V to reach 1000 mA cm−2, and operates for more than 250 h at 500 mA cm-2 with a small degradation rate of only 17 μV h−1. This finding provides valuable insights into stabilizing RuO2 for practical application in PEMWE.
Methods
Materials
Ruthenium (III) chloride (RuCl3) with a purity of 99.5% was purchased from Innochem. Lead nitrate (Pb(NO3)2) with a purity of 99% was obtained from Shanghai Aladdin Biochemical Co., Ltd. Glucose (C6H12O6·H2O) with a purity of 99% and urea (H2NCONH2) with a purity of 99% were purchased from Beijing Tong Guang Fine Chemicals Company. Nafion 212 membrane with a thickness of 0.05 mm was obtained from DuPont Co. No further purification was conducted for all reagents.
Characterization
The phase composition of the catalyst was determined by X-ray diffraction (XRD) on a Rigaku D/max-2500 powder diffractometer (Cu Kα source with λ of 0.15406 nm). The morphology of the catalyst was characterized by transmission electron microscope (TEM) on JEOL JEM-2010F at 200 kV. Spherical-aberration corrected transmission electron microscope (JEM-ARM200F) was conducted to investigate the atomic structure of catalyst by HAADF-STEM, and element composition and distribution by energy dispersive X-ray spectroscopy (EDS) element mappings. We further performed X-ray photoelectron spectroscopy (XPS) to characterize the valence state of the catalyst on Thermo Scientific K-Alpha spectrometer equipped with an Al Kα X-ray source (1.4866 keV). The composition of the catalyst was determined by inductively coupled plasma atomic emission spectrometer (ICP-OES) on Thermo Fisher iCAP PRO. Raman spectra were tested on Renishaw inVia with an excitation laser of 532 nm. Differential electrochemical mass spectroscopy (DEMS) was carried out on PM-DEMS. We further applied X-ray absorption near-edge structure (XAFS) spectra to reveal the valence state and coordination structure of the catalyst by the Shanghai (BL14W1) and Beijing Synchrotron Radiation Facility (BL1W1B). Athena and Artemis were conducted to analyze the relevant XAFS data according to the standard procedure.
Synthesis of Pb-RuO2 and RuO2
Typically, RuCl3 (40.1 mg) and Pb(NO3)2 (4.6 mg) were added into deionized water (5 mL) containing glucose (5 g) and urea (1 g). The mixture was stirred for 30 min, subsequently transferred into a breaker and treated at 140 °C for 8 h in an oven to form a porous foam, followed by annealing at 500 °C for 10 h in the air to obtain the Pb-RuO2 catalyst. To investigate the effect of Pb concentration on catalytic performance, Pb-RuO2-L (RuCl3 of 41.2 mg and Pb(NO3)2 of 2.6 mg) and Pb-RuO2-H (RuCl3 of 36.5 mg and Pb(NO3)2 of 9.8 mg) were also prepared. As a comparison, pure RuO2 (Hm-RuO2) was also synthesized without the addition of Pb(NO3)2.
Electrochemical measurements in three-electrode cell
Electrochemical performances of the catalysts were evaluated in a three-electrode cell connected to the electrochemical workstation (CHI 760E). Hg/Hg2SO4, as the reference electrode, was calibrated by cyclic voltammetry in H2-saturated 0.5 M H2SO4 electrolyte using a purified Pt wire as the working and counter electrode26. The thermodynamic potential corresponding to Hg/Hg2SO4 was determined by the average potential where the current approaches zero27. The RHE potentials were obtained by the equation of E (vs. RHE) = E (vs. Hg/Hg2SO4) + 0.65 V + 0.059 pH. Graphite rod was used as the counter electrode, and 0.5 M H2SO4 solution was conducted as electrolyte. The electrolyte was freshly prepared and promptly utilized (pH is 0.3 ± 0.1), which was prepared by adding 27.2 mL of sulfuric acid (98%) into a container with 500 mL of deionized water, followed by adjusting the volume to 1000 mL in a volumetric flask. The catalysts coated carbon paper (CP) was used as working electrode, which was prepared by dropping 250 μL of catalyst ink onto the CP (1 cm × 1 cm) to obtain a catalyst loading of 0.5 mg cm−2 determined by the mass of catalyst on carbon paper and geometric area of the electrode. The catalyst ink was obtained by dispersing 2 mg of catalyst into 1 mL solution containing 990 μL of isopropanol and 10 μL of Nafion D521.
For comparison, home-made (Hm) RuO2 or commercial (Com) RuO2 with the same loading was prepared by a similar procedure. When testing the linear sweep voltammetry (LSV) polarization curves, three independent curves were measured with a scan rate of 5 mV s−1. Accelerated durability tests were conducted to assess the stability of the catalyst, which was achieved by cycling for 100,000 cycles within 1.3 to 1.5 V vs. RHE at room temperature. We further evaluate the durability of the catalysts by the chronopotentiometry measurement at a current density of 10 mA cm-2 for 1100 h. Electrochemical impedance spectroscopy (EIS) was tested between a frequency range (100,000 to 0.1 Hz) with an overpotential (270 mV) and amplitude (5 mV). The double-layer capacitance (Cdl) was calculated by CVs at various scan rates from 20 to 100 mV s−1. The electrochemical active surface area (ECSA) of the catalyst was determined by the equation of ECSA = Cdl/Cs, where Cs is 0.035 mF cm-2 28,29.
Electrochemical measurements in PEMWE device
The MEA was prepared through a modified procedure in our previous work26. Briefly, the anode (Pb-RuO2) and cathode catalysts (commercial 70 wt% Pt/C) were air sprayed onto the Nafion 212 membrane (treated with H2O2 (80 °C, 1 h) and 0.5 M H2SO4 (80 °C, 1 h)) to obtain the catalyst coated membrane (CCM) with a Ru and Pt loading of about 1.0 and 0.4 mg cm−2, respectively. The MEA was assembled by sandwiching the Ti bipolar plate, Pt-coated Ti fiber, CCM, carbon paper, and Ti bipolar plate with a torque (10 N m). DI water was circulated through an ion exchange filter (XK-CR-003P, XUANKE HYDROGEN) to purify the cations from the slight catalyst dissolution during the operation, and flow into the anodic side of the electrolyzer with a flow rate (20 mL min−1) by a peristaltic pump. A battery test system (CT-4008T-5V12A-S1-F, NEWARE) was used to evaluate the performance of Pb-RuO2. The polarization curves were tested within a range (0–2000 mA cm−2), and the stability was evaluated by chronopotentiometry tests at the current densities of 500 and 1000 mA cm−2. The degradation rate was calculated by ΔE/Δt, where ΔE represents the potential difference before and after stability tests and Δt represents the testing time. Commercial RuO2 and home-made RuO2 were also air sprayed onto the membrane with the same Ru loading for comparison. The polarization curves and stability tests in PEMWE were obtained without iR-correction.
Differential electrochemical mass spectroscopy (DEMS) measurements
The involvement of lattice oxygen was identified by in situ DEMS on PM-DEMS through a modified procedure in previous works21,30. Briefly, the working electrodes were obtained by dipping the catalysts on the Au electrode with a mass loading of 0.3 mg cm−2. The reference and counter electrodes were Ag/AgCl electrode and Pt wire, respectively. First, we labeled the catalysts with 18O isotopes by operating at 1.6 V vs. RHE for 600 s in 0.5 M H2SO4/H218O solution. Afterward, H216O was utilized to eliminate the redundant H218O on the 18O-labeled electrodes. Finally, CV cycles were performed in 0.5 M H2SO4/H216O solution at a scan rate of 5 mV s−1 within the potential range (1.2–1.6 V vs. RHE), and the gas products generated during the reaction were tested by the real-time mass spectroscopy.
Calculation details
The DFT calculations were conducted by Vienna ab initio simulation package (VASP) through a modified procedure in previous work31. The generalized gradient approximation (GGA), as parameterized by Perdew–Burke–Ernzerhof (PBE), was employed to describe the electronic exchange and correlations32,33. A cut-off energy of 450 eV was applied for the plane wave basis. For the slab, a 2 × 2 × 2 supercell that contains 24 Ru atoms and 52 O atoms was modeled as RuO2 (110) surface. For the Pb-RuO2, two Ru atoms in the second layer of the RuO2 (110) surface were replaced by Pb atoms. A vacuum spacing of 20 Å was introduced along the z-direction to avoid interactions between the slab and its periodic motif. The terminations of RuO2 and Pb-RuO2 (110) feature half of the Ru sites occupied by oxygen, with the remaining coordinatively unsaturated Ru sites unoccupied, thereby acting as active sites for intermediate adsorption. Geometry relaxation and electronic structure convergence criteria were set to 0.05 eV/Å and 5 × 10−5 eV, respectively. The Brillouin zone was sampled using the Monkhorst-Pack method with a 3 × 3 × 1 mesh. The free energies of the reaction were calculated using the equation of \({\varDelta G}_{{ads}}=\,{\varDelta E}_{{ads}}+\,{\varDelta E}_{{ZPE}}-T\varDelta S\), where ΔEads represents the adsorption energy of intermediates, T is the temperature, ΔEZPE and ΔS account for the zero-point energy and entropy contributions, respectively. The formation energy of O vacancies was calculated, assuming that the lost oxygen atoms are located around the unsaturated Ru coordination sites at the surface for both Pb-RuO2 and RuO2 (110).
Data availability
The data that support the conclusions of this study are available within the paper and Supplementary information. Source data are provided with this paper.
References
Shi, Z. et al. Phase-dependent growth of Pt on MoS2 for highly efficient H2 evolution. Nature 621, 300–305 (2023).
Chong, L. et al. La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science 380, 609–616 (2023).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2023).
Liu, R.-T. et al. Recent advances in proton exchange membrane water electrolysis. Chem. Soc. Rev. 52, 5652–5683 (2023).
Zhang, G. et al. Porous flow field for next-generation proton exchange membrane fuel cells: Materials, characterization, design, and challenges. Chem. Rev. 123, 989–1039 (2023).
Chen, F.-Y., Wu, Z.-Y., Adler, Z. & Wang, H. Stability challenges of electrocatalytic oxygen evolution reaction: from mechanistic understanding to reactor design. Joule 5, 1704–1731 (2021).
Seitz, L. C. et al. A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction. Science 353, 1011–1014 (2016).
Zheng, Y.-R. et al. Monitoring oxygen production on mass-selected iridium-tantalum oxide electrocatalysts. Nat. Energy 7, 55–64 (2022).
Hao, S. et al. Torsion strained iridium oxide for efficient acidic water oxidation in proton exchange membrane electrolyzers. Nat. Nanotechnol. 16, 1371–1377 (2021).
Chen, Z. et al. Advances in oxygen evolution electrocatalysts for proton exchange membrane water electrolyzers. Adv. Energy Mater. 12, 2103670 (2022).
Xu, J. et al. IrOx·nH2O with lattice water-assisted oxygen exchange for high-performance proton exchange membrane water electrolyzers. Sci. Adv. 9, eadh1718 (2023).
Hao, S. et al. Dopants fixation of Ruthenium for boosting acidic oxygen evolution stability and activity. Nat. Commun. 11, 5368 (2020).
Zhou, L. et al. Stabilizing non-iridium active sites by non-stoichiometric oxide for acidic water oxidation at high current density. Nat. Commun. 14, 7644 (2023).
Lin, Y., Dong, Y., Wang, X. & Chen, L. Electrocatalysts for the oxygen evolution reaction in acidic media. Adv. Mater. 35, 2210565 (2023).
Yao, Y. et al. Engineering the electronic structure of single-atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304–313 (2019).
Jin, H. et al. Safeguarding the RuO2 phase against lattice oxygen oxidation during acidic water electrooxidation. Energy Environ. Sci. 15, 1119–1130 (2022).
Xu, Y. et al. Strain-modulated Ru-O covalency in Ru-Sn oxide enabling efficient and stable water oxidation in acidic solution. Angew. Chem. Int. Ed. 63, e202316029 (2024).
Wang, Y. et al. Breaking the Ru-O-Ru symmetry of a RuO2 catalyst for sustainable acidic water oxidation. Angew. Chem. Int. Ed. 63, e202316903 (2024).
Ye, X. et al. Observation of novel charge ordering and spin reorientation in perovskite oxide PbFeO3. Nat. Commun. 12, 1917 (2021).
Chen, K. et al. Valence state of Pb in transition metal perovskites PbTMO3 (TM = Ti, Ni) determined from X-ray absorption near-edge spectroscopy. Phys. Status Solidi B. 255, 1800014 (2018).
Ping, X. et al. Locking the lattice oxygen in RuO2 to stabilize highly active Ru sites in acidic water oxidation. Nat. Commun. 15, 2501 (2024).
Tao, H. B. et al. A general method to probe oxygen evolution intermediates at operating conditions. Joule 3, 1498–1509 (2019).
Chen, X. et al. Revealing the role of interfacial water and key intermediates at ruthenium surfaces in the alkaline hydrogen evolution reaction. Nat. Commun. 14, 5289 (2023).
Chen, H.-Q. et al. Unmasking the critical role of the ordering degree of bimetallic nanocatalysts on oxygen reduction reaction by in situ raman spectroscopy. Angew. Chem. Int. Ed. 61, e202117834 (2022).
Shi, Z. et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 14, 843 (2023).
Zhou, C. et al. Oxophilic gallium single atoms bridged ruthenium clusters for practical anion-exchange membrane electrolyzer. Nat. Commun. 15, 6741 (2024).
Li, L. et al. Optimizing the electronic structure of ruthenium oxide by neodymium doping for enhanced acidic oxygen evolution catalysis. Adv. Funct. Mater. 33, 2213304 (2023).
McCrory, C. C. L., Jung, S., Peters, J. C. & Jaramillo, T. F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977–16987 (2013).
Su, H. et al. Tensile straining of iridium sites in manganese oxides for proton-exchange membrane water electrolysers. Nat. Commun. 15, 95 (2024).
Lu, M. et al. Artificially steering electrocatalytic oxygen evolution reaction mechanism by regulating oxygen defect contents in perovskites. Sci. Adv. 8, eabq3563 (2022).
Li, L. et al. Lanthanide-regulating Ru-O covalency optimizes acidic oxygen evolution electrocatalysis. Nat. Commun. 15, 4974 (2024).
Hammer, B., Hansen, L. B. & Nørskov, J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Acknowledgements
This study was financially supported by National Natural Science Foundation of China (Nos. 52333010 (S.J.G.), 52025133 (S.J.G.), 22305010 (C.H.Z.), 52302207 (L.L.), 22102003 (K.W.)), National Key R&D Program of China (No. 2022YFE0128500 (S.J.G.)), China National Petroleum Corporation-Peking University Strategic Cooperation Project of Fundamental Research, the Beijing Natural Science Foundation (No. Z220020 (S.J.G.)), Tencent Foundation through the XPLORER PRIZE, CNPC Innovation Found (No. 2021DQ02-1002 (S.J.G.)) and China Postdoctoral Science Foundation (No. 2023M730051 (C.H.Z.)). The authors thank the photoemission photoendstations BL14W1, BL11B, and BL20U in the Shanghai Synchrotron Radiation Facility (SSRF) and the 1W1B beamline at the Beijing Synchrotron Radiation Facility (BSRF) for the help with XAFS characterizations. The authors also thank Shanghai Pro-tech Limited Company for their help with DEMS tests.
Author information
Authors and Affiliations
Contributions
S.J.G. conceived the project. C.H.Z. designed the research and performed the material synthesis, characterization, electrochemical tests, and the assembly and tests of PEMWE. L.L. conducted the DFT calculations. Z.Q.D., F.L., H.Y.G., K.W., M.G.L., Z.Y.Q., N.Y., and Z.L. participated in part of basic experiments. C.H.Z. wrote the paper. S.J.G. and M.C.L. guided the paper writing. All authors participated in the project discussions and production of the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Huanyu Jin, Porun Liu and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, C., Li, L., Dong, Z. et al. Pinning effect of lattice Pb suppressing lattice oxygen reactivity of Pb-RuO2 enables stable industrial-level electrolysis. Nat Commun 15, 9774 (2024). https://doi.org/10.1038/s41467-024-53905-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53905-y
This article is cited by
-
Grain boundary in RuO2 to boost acidic oxygen evolution activity and stability
Science China Chemistry (2025)
