
Abstract
Glass, a diverse family of amorphous materials, has significantly advanced human society across various fields. The demand for flexible ultrathin glass, driven by modern optical displays and portable optoelectronics, presents challenges in energy consumption, fabrication complexity, and recycling. Here, we demonstrate flexibility and full-color luminescence in large-scale ultrathin glasses derived from readily available natural resources, specifically egg albumen (EA) and gelatin (GEL), via an evaporation-driven self-assembly process. The dynamic crosslinked networks formed through hydrogen bonding between EA and GEL impart both high hardness and flexibility to the glasses, with hardness and flexural strength values comparable to state-of-the-art inorganic and organic glasses. Additionally, the EA–GEL-based glasses exhibit excitation-dependent and time-gated chiral ultralong phosphorescence with color from blue and red, and a lifetime of up to 180.4 ms. With their easy processability and full-color emission, these biogenic glasses can be fabricated into anti-counterfeiting patterns and optical information codes.
Similar content being viewed by others


Introduction
Glass plays a vital role in modern life due to its exceptional optical, electronic, mechanical, and thermal properties, along with its stability and affordability1,2. However, its brittleness and inability to withstand significant deformations limit its applications. Recent advancements in optical displays, wearable devices, and portable optoelectronics have created a demand for flexible ultrathin glass, typically between tens and hundreds of micrometers thick3. Unlike traditional systems, flexible glass can bend and twist without losing structural integrity or optical performance. This innovation could transform displays, semiconductors, and sensors by making them lighter, thinner, and more durable4. Despite these benefits, several challenges impede the widespread adoption of flexible glass5,6. These include: (1) the need for high-temperature fabrication (>1000 °C), (2) energy-consuming preparation methods (e.g., overflow downdraw and chemical thinning), (3) limited chemical compositions (e.g., alkali-aluminosilicate and borosilicate), and (4) difficulties in recycling and regeneration. Additionally, achieving sustainability remains challenging due to the difficulty of using abundant, environmentally friendly resources in glass production.
Self-assembly technologies have recently provided a bottom-up approach for fabricating functional molecule-based glasses7,8. However, the diversity of building blocks for these glasses remains limited, and most require complex chemical synthesis9. Currently, constructing large-size and flexible glasses through simple and effective methods is still highly desirable. Various biomacromolecules10,11, such as natural lignin, cellulose, phenolics, starch, and proteins, are rich in functional groups like –C=O, –NH2, and –OH. These groups can engage in multiple non-covalent interactions12, resulting in a wide range of bonding strengths. Under mechanical stresses like bending and twisting, the weaker bonds dissociate to dissipate energy and mitigate damage, while the stronger bonds maintain structural integrity13,14. We propose that this dynamic self-assembly capability, combined with inherent structural flexibility and mechanical strength, makes these biomolecules promising candidates for large-scale fabrication of flexible glass.
Biogenic materials could possess excellent optical and luminescent properties15. For example, ultralong phosphorescence, which continues to emit light after excitation ceases16,17,18, has been observed in several natural resources. This phenomenon can be explained by the clustering-triggered emission (CTE) mechanism, attributed to the synergistic effect of unsaturated subgroups and heteroatom clustering in a rigid biomolecular environment19. Room temperature phosphorescence (RTP) with a 34 ms lifetime was observed in bovine serum albumin solids attributed to the chain aggregation20, which facilitated clustering with π and n electrons, generating an effective intersystem crossing (ISC) process. Although RTP materials have been paid much attention to and exhibited great potential in environmental sensing, bioimaging, and color displays15,16,17, current systems are typically developed in crystalline states, which limit their practical applications due to brittleness and lack of flexibility. Assembling RTP molecules into ultrathin glasses could potentially combine mechanical flexibility with persistent luminescence, benefiting multifunctional optoelectronics and advanced information storage, although this remains speculative.
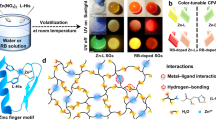
Egg albumen (EA) and gelatin (GEL) are abundant, non-toxic, biocompatible, biodegradable, and low-cost biomaterials. Here, we use EA and GEL as precursors to fabricate a type of flexible bulk molecular glass (E–G) via an environmentally friendly self-assembly strategy (Fig. 1 and Supplementary Fig. 1). The proteins in EA form dynamic hydrogen bonds with GEL oligomers (Fig. 1c), enhancing the flexibility and toughness of the molecular glass. Notably, E–G glass exhibits excitation-dependent ultralong RTP with a lifetime of up to 180.4 ms. This is due to the formation of chromophore clustering, which is restricted by multiple non-covalent interactions, such as hydrogen bonding, electrostatic interactions, and hydrophobic interactions between EA and GEL. Doping with 1-pyrenecarboxylic acid ammonium salt (PCA) results in large-scale PCA-doped glasses that exhibit excitation- and time-responsive full-color persistent luminescence from blue to red (Supplementary Fig. 2). Additionally, chiroptical properties are observed in glasses containing chiral amino acid molecules. These flexible glasses, with adjustable afterglows, excellent processability, and biodegradability, are suitable for disposable information codes and anti-counterfeiting patterns. This work thus provides a facile method for fabricating flexible and ultrathin molecular glasses, and represents an instance of biogenic molecular glasses exhibiting intelligent ultralong RTP emissions for photonic applications. It is expected that bio-based materials offer renewability and degradability, which hold promise for fabricating flexible glass with color-tunable and time-gated luminescence.

a Preparation of E–G and PCA-doped glasses at room temperature (scale bar: 3 mm) and proposed mechanism for long-lived phosphorescence of the glasses. b The photographs of glasses after exposure to UV light (scale bar: 3 mm). c Docking simulation results of ovalbumin (OVA) in EA and the partial peptide chain (PPC) in GEL were conducted with AutoDockTools.
Results
Preparation, recyclability, and biodegradability of the bio-based glasses
Air-dried EA samples, sourced from various eggs—including commercial chicken, free-range chicken, black-bone chicken, duck, goose, and pigeon eggs—yielded light yellow, brittle glassy solids (Supplementary Fig. 3). The primary solid components of EA are proteins, predominantly OVA. Other proteins, such as immunoglobulin G, gamma globulin, albumin, and chymotrypsin, can also achieve a glassy state upon drying21. For this study, EA was primarily sourced from commercially available chicken eggs. A dried aqueous solution of GEL, derived from collagen degradation, formed a transparent brittle film. The EA and GEL contain amino acids that feature functional groups, such as –OH, –NH2, and –COOH, capable of participating in hydrogen bonding. In addition, electrostatic interactions occur between the charged amino acids, specifically the positively charged groups of lysine and arginine in GEL, and the negatively charged groups of aspartic acid and glutamic acid in EA. These interactions between EA and GEL, along with hydrophobic interactions and van der Waals force, likely work in concert to facilitate the formation of a continuous glass matrix22.
Considering these factors, we fabricated flexible glasses using EA and GEL as raw materials, employing an evaporation-induced self-assembly strategy. EA and GEL were dissolved in water to create a mixed solution, which was gradually evaporated at room temperature. This process allowed biomacromolecules in EA to assemble with peptide chains in GEL, producing a compressible gel state (Supplementary Fig. 4), followed by vitrification of the dynamic networks. Similar glass materials were successfully achieved using EA from other egg types (Supplementary Fig. 5), indicating that minor compositional variations in EA did not significantly impact glass formation23. The PCA-doped glasses (denoted as E–G–Pn, where n represents 0.2, 0.5, 0.8, and 1.0) were prepared using the same method as the E–G glass, with the addition of PCA at varying mass ratios (the details can be found in the “Synthesis” section).
After immersing these glasses in various solvents (acetone, acetonitrile, N,N-dimethylformamide, cyclohexane, dichloromethane, and ethyl acetate) for 3 days, their shape and transparency remained unchanged (Supplementary Fig. 6), demonstrating their high stability. The E–G and PCA-doped glasses also exhibited exceptional recyclability, as they could be redissolved in water and reformed through solvent evaporation (Supplementary Fig. 7). Their dissolution was significantly accelerated when exposed to proteinase K solution (0.1 mg mL−1) and simulated gastric fluid containing pepsin (Supplementary Fig. 8). Within just 2 min under these conditions, the glasses underwent near-complete dissolution, indicating the considerable potential for applications in living organisms24. To assess the biodegradability of the E–G and PCA-doped glasses, we conducted a simulated biodegradation experiment by exposing them to soil. Within 1 month, the surfaces of the glasses became rough, significant disintegration was observed after 3 months, and after 8 months, the glasses had essentially degraded completely (Supplementary Fig. 9). This biodegradability could substantially reduce waste disposal costs and environmental pollution, aligning with the goals of sustainable development.
Structural characterizations
Fourier transform infrared (FT-IR) spectra were used to confirm the formation of the supramolecular glasses: amide bands I, II, and III present in the spectra of E–G and E–G–P0.5 glasses25, indicating their main components are protein macromolecules (Fig. 2a). The N–H stretching bands (amide band II, 3000–3750 cm−1) in E–G and E–G–P0.5 glasses, broader and shifted compared to solid EA and GEL, suggest stronger intra- and intermolecular hydrogen bonding involving amino groups. Differential scanning calorimetry (DSC) curves show distinct heat capacity steps at 335 K, 343 K, and 338 K for solid EA, E–G, and E–G–P0.5, respectively, indicating their glass transition temperatures (Tgs) (Fig. 2b). The higher Tg of E–G glass compared to that of solid EA may result from stronger noncovalent interactions in the polymeric networks. Powder X-ray diffraction (PXRD) patterns show broad diffraction bands for solid EA, E–G, and E–G–P0.5. These findings collectively indicate that they can be classified as glassy materials (Fig. 2c). Small-angle X-ray scattering (SAXS) curves reveal phase separation in solid EA but not in the E–G glass26 (Fig. 2d). This is possibly due to stronger interactions, such as hydrogen bonding, electrostatic interactions, and hydrophobic interactions, between EA and GEL in the glass matrix, leading to the formation of a homogeneous mixture at a molecular level. Similar vitrification behaviors are observed in other PCA-doped glasses, as evidenced by their DSC and PXRD measurements (Supplementary Figs. 10 and 11).

a FT-IR spectra of solid EA, E–G, E–G–P0.5, and GEL. b DSC curves and c PXRD patterns of EA, E–G, E–G–P0.5 glasses. d SAXS curves of glassy EA and E–G. e SEM images of cross-section (upper, scale bar: 3.7 μm) and plane (below, scale bar: 0.5 μm) for E–G glass. f UV-Vis-NIR transmittance spectra of E–G and E–G–P0.5 glasses. Two bands at approximately 1730 nm and 1500 nm, and one band at 348 nm originate from water and PCA absorption in PCA-doped glass, respectively.
Scanning electron microscope (SEM) and fluorescence microscopy images reveal the ultrathin nature of E–G and PCA-doped glasses with smooth and continuous surfaces. The thickness of glasses can be highly tuned by modulation of the initial height of the mixed solution in the mold, with a value as low as 7.0 μm (Fig. 2e and Supplementary Figs. 12–14). Elemental distribution mapping of SEM, including C, N, O, P, and S maps, indicates a homogeneous distribution of elements in the E–G sample (Supplementary Fig. 15). Freeze-dried EA and E–G dilute solutions exhibit mesoscopic protein-rich aggregates27, visible in the SEM images (Supplementary Figs. 16 and 17), which could significantly restrict the irregular motion of the chromophores, promoting efficient RTP emissions28. UV-Vis-NIR transmittance spectra show that EA, E–G, and PCA-doped glasses exhibit high transmittance of approximately 90% in the range of 500–1300 nm (Fig. 2f and Supplementary Fig. 18), confirming their high optical activities.
Flexibility and hardness characterizations
Conventional ultralong RTP materials, including purely inorganic and organic phosphors, organic-inorganic crystals, and carbon dots, often lack flexibility due to their strong covalent or ionic bonds and small sizes29,30. This limits their use in macroscopic optoelectronic applications. In contrast, E–G and PCA-doped glasses are exceptionally flexible, withstanding multiple bends and twists without structural damage (Fig. 3a, Supplementary Figs. 19 and 20, and Supplementary Movies 1 and 2). Three-point bending tests show that E–G and E–G–P0.5 glasses can endure bending strains of approximately 2.0%, achieving flexural strength values of 121.1 and 114.1 MPa, respectively (Fig. 3b). These values are comparable to or higher than those of many inorganic and organic glasses, such as polymethyl methacrylate (PMMA) and PMMA-dopant systems (Fig. 3c)31,32,33,34. Moreover, repeated bending tests reveal that the E–G glass maintains its flexural strength after ten cycles (Fig. 3d).

a Photographs of compression experiments with E–G and E–G–P0.5 glasses. b Strain-stress curves of three-point bending tests for E–G and E–G–P0.5 glasses. c Comparison of flexural strength between this work and previously reported glasses. d Repeated bending tests of E–G glass. e Nanoindentation tests of glassy films with loading-unloading curves.
Compression tests highlight the exceptional hardness of ultrathin molecular glasses. The E–G specimen supports weights of 1.5 kg, a 20 kg bucket, and even a 50 kg person for 10 min (Fig. 3e and Supplementary Fig. 21). Nanoindentation studies reveal hardness values of 33.6 kgf mm–2 for E–G glass and 34.1 kgf mm−2 for E–G–P0.5 glass (Fig. 3e), comparable to traditional metallic and inorganic glasses that lack flexibility35,36,37,38,39. In contrast, GEL in film form, used as a starting material, achieves a flexural strength value of 40.5 MPa and a hardness value of 32.8 kgf mm−2 (Supplementary Fig. 22), both lower than those of assembled E–G and PCA-doped glasses. The exceptional flexibility of these glasses stems from the dynamic hydrogen bonds between EA and GEL, which readily reform after disruption, facilitating shape recovery. Meanwhile, the increased hardness is attributed to the dense entanglements of these components, forming an intricate and cross-linked network within the supramolecular structure that effectively resists deformation40,41,42. These mechanical properties make bulk ultrathin glasses highly promising for flexible integrated photonics.
Dynamic and full-color ultralong phosphorescence
To develop promising applications in flexible optical displays, the luminescence properties of E–G and PCA-doped glasses were studied. Under UV irradiation at 295 or 395 nm, these flexible glasses exhibit blue or yellow–green emission that gradually fades but remains notably persistent at ambient conditions (Fig. 4a, b). When the excitation source is shifted to 365 nm, E–G glass shows persistent green luminescence (Supplementary Movie 3). Notably, the afterglow colors of PCA-doped glasses evolve over time once the 365 nm irradiation ceases. For instance, the afterglow of glassy E–G–P0.5 transitions from red (delayed time (td) = 0 s) to orange–red (td = 0.2 s), then to orange (td = 0.3 s), yellowish-green (td = 0.6 s), and finally to green (td = 0.9 s) (Supplementary Movie 4). To the best of our knowledge, such wide-range time-gated color-changing afterglow is still rather limited among state-of-the-art RTP materials43,44,45, which ensures its further information coding application. Similarly, the afterglow of E–G–P1.0 glass shifts from red (td = 0.1 s) to orange (td = 0.9 s), while E–G–P0.2 glass changes from orange (td = 0.1 s) to green (td = 0.3 s) (Supplementary Fig. 23). Therefore, by adjusting the PCA content, both excitation-dependent and time-gated persistent luminescence can be achieved.

Photographs of a E–G and b E–G–P0.5 glasses before and after exposure to 295 nm, 365 nm, and 395 nm UV light at ambient conditions. c Excitation-phosphorescence emission mapping of E–G glass. d Time-dependent phosphorescence emission mapping of E–G–P0.5 glass. e Time-resolved emission decay curves at 495 nm, 545 nm, and 560 nm of E–G glass under 295 nm, 365 nm, and 395 nm excitations. f Time-resolved emission decay curves at 610 nm and 670 nm of E–G–P0.5 glass under 365 nm excitation. g Intensity ratios between the 510 nm and 610/670 nm bands of E–G–P0.5 glass under 365 nm excitation. CPL spectra of h E–G and i E–G–P0.5 glasses.
We studied the photophysical properties of E–G glass using photoluminescence (PL) spectral tests. As shown in Fig. 4c, varying the excitation wavelength from 295 to 365 nm and 395 nm causes a noticeable bathochromic shift in the delayed emission, with main peaks at 495 nm, 545 nm, and 560 nm, respectively. This phenomenon is also observed in other molecular glasses with EA extracted from different eggs (Supplementary Figs. 5, 24, and 25). The afterglow hues on the Commission International de l’Eclairage (CIE) chromaticity diagram align with those perceived by the naked eye (Supplementary Fig. 26). The persistent luminescence of E–G glass is attributed to ultralong RTP, indicated by long PL lifetimes of 104.2 ms, 180.4 ms, and 137.9 ms (Fig. 4e). The intensity and lifetime decrease as the temperature increases from 77 K to 297 K in the delayed mode (Supplementary Fig. 27). Additionally, both the delayed PL spectra at 77 K and the prompt PL spectra at ambient temperature show that E–G glass displays color variations from blue to yellow and from blue–green to yellow–green, respectively, in response to different excitation wavelengths (Supplementary Figs. 28 and 29). These results highlight the broad tunability of the emission colors of E–G glass.
We investigated the luminescent properties of PCA-doped glasses. In the delayed PL spectra (td = 0.1 s) (Supplementary Fig. 30), three emission bands are observed in all PCA-doped glasses. The first band, ranging from 450 nm to 580 nm, corresponds to the phosphorescent band of E–G glass, suggesting it originates from the luminous centers in the E–G component. To explore the origin of the emission bands at 610 nm and 670 nm in PCA-doped glasses, we have incorporated PCA into a poly(vinyl alcohol) (PVA) film, known for its negligible PL properties. Upon doping with PCA, the PVA film exhibits emission peaks at ca. 610 nm and 670 nm in both the prompt and delayed PL spectra (Supplementary Fig. 31 and Supplementary Note 1). Comparing the PL spectra of glasses with varying PCA concentrations provides further insight into these emissive bands. As the doped PCA increases, the emission intensities at 610 nm and 670 nm intensify, while the short-wavelength bands decrease, as observed in both prompt and delayed PL spectra (Supplementary Fig. 32 and Supplementary Note 2). These findings demonstrate that the peaks at 610 nm and 670 nm in the PCA-doped glasses originate from the PCA itself.
Examination of the temperature-dependent delayed PL spectra and time-resolved decay profiles of PCA-doped glass (E–G–P0.5) shows that both emission intensity and lifetime decrease as the temperature rises from 77 K to 297 K (Supplementary Fig. 33), confirming that the delayed emissions are ascribed to ultralong phosphorescence. Notably, the RTP lifetimes at 610 and 670 nm significantly increase with higher PCA doping, reaching 146.6 ms and 126.5 ms, respectively, for E–G–P1.0 glass (Supplementary Fig. 30c). Thus, by adjusting the PCA doping level in E–G glass, red-shifted phosphorescence can be further obtained.
The PCA-doped glasses exhibit both excitation-dependent and time-gated afterglows. For instance, E–G–P0.5 glass, when exposed to 295 nm, 365 nm, and 395 nm UV lights, shows delayed PL spectra (td = 0.1 s) with emission features: a band at 495 nm, two bands at 610 nm and 670 nm with a shoulder from 500 nm to 580 nm, and a broad band at 545 nm (Supplementary Fig. 34). The bands at 610 nm and 670 nm (λex = 365 nm) have long RTP lifetimes of 104.4 and 93.3 ms, respectively (Fig. 4f). After irradiation ceases, these bands independently generate blue, red, and yellow–green persistent luminescence (Fig. 4a). This behavior is due to the optimal excitation wavelength for PCA being 365 nm, resulting in low luminous intensities when excited at 295 and 395 nm (Supplementary Fig. 35). In contrast, the E–G component can be excited by all three wavelengths, though its luminous intensity is relatively weak under 365 nm light. Notably, the E–G–P0.2 glass, with a balanced combination of blue, green, and red emissions, produces white-light phosphorescence under 325 nm UV light (Supplementary Fig. 36).
The time-dependent afterglows of PCA-doped glasses were further investigated by analyzing changes in the relative intensity of emissive bands corresponding to the E–G (495–560 nm) and PCA (610 nm and 670 nm) components at various delayed times. In the case of the E–G–P0.5 glass, as observed over an extended period, the intensity ratio of the peaks at 610 nm and 670 nm to the emission intensity at 510 nm gradually decreases (λex = 365 nm) (Fig. 4d, g). This shift in the intensity of these three emission bands results in time-responsive phosphorescent emissions that transition from red to orange to green in the E–G–P0.5 glass. A similar phenomenon is also observed in the time-dependent afterglows of other PCA-doped glasses (Fig. 5b, c). Collectively, the PCA-doped glasses distinguish themselves significantly by harnessing natural raw materials and exhibiting a combination of excitation- and time-dependent ultralong phosphorescence, along with flexibility—traits not commonly found in recently reported RTP molecular glasses (Supplementary Table 1)7,46,47,48,49,50,51,52,53. These exceptional characteristics could broaden their applicability in phosphorescent materials.

a Schematic of clustering and hydrogen bonding in E–G glass, and mechanism for its excitation-dependent ultralong RTP. b Delayed PL spectra (delayed time: 0.1 s, 0.2 s, 0.3 s, 0.9 s) of E–G–P0.2 and E–G–P1.0 glasses. c Demonstration of advanced multi-coding data encryption using E–G and PCA-doped glasses with excitation-dependent or/and time-dependent afterglows.
Chiro-optical properties
The chiral activity of the precursors imparts chiro-optical properties to the E–G and PCA-doped ultrathin glasses. To examine these properties, we first analyzed the circular dichroism (CD) spectrum, which reveals the ground states of the glasses. As shown in Supplementary Fig. 37, the CD spectrum of E–G glass displays absorption bands in the range from 200 nm to 225 nm. In contrast, the CD spectrum of glassy E–G–P0.5 exhibits an additional band around 300 nm, attributed to the distinctive absorption of PCA. Clear chiral signals are also evident in the circularly polarized luminescence (CPL) spectra for both E–G and E–G–P0.5 (Fig. 4h, i). Their absolute luminescence dissymmetry factor (|glum|) reaches up to −2.3 × 10−3 (Supplementary Fig. 38), comparable to many reported molecular CPL-active films54,55,56. These results highlight the potential of these glasses for developing flexible chiral photonic devices57.
Mechanisms underlying flexibility and dynamic RTP
The mechanism behind the color-tunable ultralong RTP of flexible molecular glasses was detected using both spectral tests and theoretical simulations. Delayed PL spectra for solid EA and GEL show weak red-shifted emissions with lifetimes of only 81.0 μs and 14.3 μs at room temperature, respectively (Supplementary Figs. 39–41 and Supplementary Note 3), distinct from the considerably longer lifetime (180.4 ms) observed in the glassy E–G (Fig. 4e). Additionally, the extent of red-shifted emission in EA and GEL is less pronounced compared to that of the E–G glass. Similar luminescent properties are observed in EA samples extracted from other egg resources (Supplementary Figs. 3, 42, and 43). At 77 K, delayed mode emissions show lifetimes of 668.9 ms for EA in water and 769.4 ms for the EA–GEL mixed solution (Supplementary Fig. 44). The increased lifetimes observed at 77 K, as compared to those at room temperature, are attributed to the restricted vibrations of chromophores in the frozen state. These findings indicate that the persistent RTP of the E–G glass originates from EA and GEL. Their combination in the glass matrix significantly enhances the luminescence performance, demonstrating a synergistic “1 + 1 > 2” effect.
Further investigation into the synergistic luminescence within the glassy matrix reveals that increasing the concentration of EA in water, GEL in EA solution, or EA–GEL in water leads to higher absorption and luminescence intensities (Supplementary Figs. 45 and 46). This increase is likely due to the enhanced intra- and intermolecular hydrogen bonding. Additionally, increasing the EA doping concentration in PVA film from 1 wt% to 5 wt% results in noticeable afterglow, with the RTP lifetime reaching 3.4 ms (Supplementary Fig. 47 and 48). In contrast, a polyvinylpyrrolidone (PVP) film doped with 5 wt% EA exhibits a shorter RTP lifetime of only 11.6 μs (Supplementary Fig. 49), likely due to the absence of strong hydrogen bonds to suppress nonradiative transitions.
The interactions between EA and GEL were further investigated using molecular docking (MD) simulations (Fig. 1c). The primary solid component in EA, OVA, exhibits photophysical properties nearly identical to solid EA, as shown by PL spectral tests (Supplementary Figs. 50 and 51). Therefore, OVA and a partial peptide chain in GEL (Supplementary Fig. 52) are selected to elucidate the interactions between EA and GEL. The most favorable OVA-PPC binding mode, with the lowest binding energy, is identified. MD simulations reveal an intermolecular energy of −5.57 kcal mol−1, comprising hydrogen bond and van der Waals force energy of −3.62 kcal mol−1 and electrostatic energy of −1.95 kcal mol−1. These results underscore the significant role of multiple hydrogen bonds among –C=O, –OH, and –NH2 in forming stable and dynamic networks in the glasses58.
The flexibility and full-color ultralong RTP of the molecular glasses sharply contrast with the brittleness and weak phosphorescence of their raw materials (EA and GEL). Based on experimental and theoretical investigations, several mechanisms explain the favorable features of the glasses: (1) Dynamic hydrogen bonds between EA and GEL act as sacrificial bonds, dissipating applied energy and improving flexibility. The dense entanglements form cross-linked network structures, enhancing mechanical strength. (2) Within the glassy matrix, chromophores are embedded in a more rigid molecular environment, which is maintained by the non-covalent interactions between EA and GEL molecules. This reduces the probability of non-radiative decay pathways, leading to the stabilization of triplet states and thereby resulting in a prolonged RTP lifetime in E–G glass, as compared to the pristine materials EA and GEL. (3) Clustering of functional groups such as –C=O, –OH, and –NH2, along with other subunits containing π and n electrons, creates diversified luminescent centers. Thus, the excitation-dependent long-lived RTP of E–G glass can be attributed to the CTE mechanism (Fig. 5a). (4) In PCA-doped glasses, dynamic afterglow colors, responsive to excitation wavelength and time, result from variations in the intensity of emissive bands associated with E–G and PCA components. Collectively, the synergistic effect of strong and weak hydrogen bonds, a dense network of entanglements, along multiple luminous centers, imparts extreme flexibility, toughness, and color-tunable afterglows to the biogenic glasses.
Applications of luminescent glasses
The E–G and PCA-doped glasses, with their dynamic afterglow colors and excellent processability, are highly versatile for anti-counterfeiting and encryption applications (Fig. 5b, c and Supplementary Fig. 53). The glasses, including E–G, E–G–P0.2, E–G–P0.5, and E–G–P1.0, were shaped into alphabet patterns “U,” “K,” “Z,” and “J,” respectively. Each pattern emits distinct afterglow colors corresponding to specific outputs: red (R), orange (O), yellow–green (Y), green (G), and blue (B). When the 295 nm and 395 nm UV lights are turned off, these patterns display vibrant blue and yellow–green afterglows, conveying information like “BBBB” and “YYYY.” Turning off the 365 nm UV light reveals time-responsive afterglow colors, creating diverse combinations at different intervals: “RROG” (td = 0.1 s), “RRGG” (td = 0.3 s), and “OGGG” (td = 0.9 s). This allows for the retrieval of a broad range of afterglow information. Additionally, the biodegradability of these bio-based ultrathin glasses ensures minimal environmental impact, even when discarded after use.
Discussion
In conclusion, we have developed types of large-scale flexible ultrathin glasses from natural resources EA and GEL using a facile bottom-up solution process. Our combined experimental and theoretical results highlight the crucial role of synergistic interactions of strong and weak hydrogen bonds in EA and GEL, contributing to the flexibility and toughness of the glasses. These interactions also create a rigid microenvironment for the chromophores, delivering ultralong RTP. Furthermore, doping PCA into the E–G glass enables dynamic and full-color afterglow, facilitated by diverse electron-rich emissive clusters. The excitation-dependent and time-gated afterglows observed in the PCA-doped glasses are due to variations in the intensity of emissive bands associated with E–G and PCA components. The color-adjustable circularly polarized afterglows, coupled with excellent biodegradability and processibility, make these bio-based ultrathin glasses promising for various applications, including chiro-optics, information encryption, and anti-counterfeiting. By utilizing readily available raw materials and a straightforward preparation method, this work not only provides a sustainable approach for producing flexible bulk glasses with smart-responsive ultralong RTP, but also paves the way for utilizing these eco-friendly materials as a versatile platform for advanced photonic applications.
Methods
Materials
All eggs used in this work were purchased from local food marts. GEL (Innochem, from porcine skin), 1-pyrenecarboxylic acid (PA) (Adamas, 99%), polyvinyl alcohol (PVA) (Innochem, MW ≈ 20,000), polyvinylpyrrolidone (PVP) (Innochem, K30), proteinase K solution (PK, Targetmol), and simulated gastric fluid (SGF, J.t.baker) were purchased as indicated and used without further purification. Ammonium hydroxide (25% solution in water), acetone (AC, 99+ %), acetonitrile (ACN, 99.9%), N,N-dimethylformamide (DMF, 99.9%), cyclohexane (CYH, 99.5+ %), dichloromethane (DCM, 99.9%), and ethyl acetate (EAC, 99.8%) were purchased from Beijing Oriental Shibo Fine Chemical Co., LTD and used without further purification. Deionized water was used throughout the whole experimental process.
Synthesis
Preparation of E–G glass
Firstly, 0.6 g GEL powder was dissolved into 10 mL deionized water in a glass bottle, which was then put in an oven at 50 °C for 20 min to promote the dissolution. Then, 2 mL EA taken from a fresh egg was added to the aqueous solution of GEL. Next, the mixture was thoroughly stirred and then put into plastic models. The model was left at ambient conditions overnight to allow the solvent to evaporate, forming E–G glass.
Preparation of E–G–Pn (n = 0.2, 0.5, 0.8, and 1.0) glass
0.04 g PA powder was added into a beaker containing 50 mL deionized water with 13 μL ammonium hydroxide. The mixture was stirred for 1 h to obtain a clear PCA solution. 200/500/800/1000 μL PCA solution was added into the aqueous solution of 2 mL EA and 0.6 g GEL. The mixed solution was stirred thoroughly and transferred into the plastic model. The model was left at ambient conditions overnight to allow the solvent to evaporate, forming E–G–P0.2/E–G–P0.5/E–G–P0.8/E–G–P1.0 glass. The mass fraction of PCA in the E–G–P0.2/E–G–P0.5/E–G–P0.8/E–G–P1.0 glass was ca. 0.02%, 0.05%, 0.08% and 0.10%, respectively.
Preparation of EA glass
The EA was extracted from a fresh egg and placed in a plastic container to dry overnight at ambient conditions, resulting in the formation of EA glass. The sample was transferred into a vacuum oven at 40 °C, and the dry sample was weighted every 5 h until the weight loss was less than 0.1%. It could be concluded that 2 mL of egg white had a dry weight of about 2.6 g.
Preparation of GEL film
First, 0.6 g of GEL was added to 10 mL of deionized water in a plastic bottle and stirred for 5 min. The film was then formed after leaving the bottle at ambient conditions overnight.
Preparation of PCA-doped PVA and EA-doped PVA/PVP films
Initially, EA or PCA solution was added to PVA or PVP aqueous solution and thoroughly mixed. The resulting mixture was then left at ambient conditions to allow the solvent to evaporate. This led to the formation of various films: PCA-doped PVA film (PVA-P0.5%) with a PCA mass fraction of 0.5%, EA-doped PVA films (PVA-E1%, PVA-E2%, PVA-E3%, PVA-E4% and PVA-E5%) with EA mass fractions of 1%, 2%, 3%, 4% and 5%, and EA doped PVP film (PVP-E5%) with an EA mass fraction of 5%.
Inclusion and ethics
All authors follow the recommendations set out in the Global Code of Conduct for Research in Resource-Poor Settings when designing, executing, and reporting the research.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are presented in the main article and the Supplementary Information. All other additional data can be obtained from the corresponding author upon request.
References
Toombs, J. T. et al. Volumetric additive manufacturing of silica glass with microscale computed axial lithography. Science 376, 308–312 (2022).
Zhang, N., Lin, Z. & Man, S. Fabrication and spectral properties of Tm3+-doped novel bismuthate glasses. J. Chin. Rare Earth Soc. 40, 54–59 (2022).
Macrelli, G., Varshneya, A. K. & Mauro, J. C. Ultra-thin glass as a substrate for flexible photonics. Opt. Mater. 106, 109994 (2020).
Dai, X. et al. Scalable fabrication of efficient perovskite solar modules on flexible glass substrates. Adv. Energy Mater. 10, 1903108 (2020).
Hoang, A. T. et al. Low-temperature growth of MoS2 on polymer and thin glass substrates for flexible electronics. Nat. Nanotechnol. 18, 1439–1447 (2023).
Lee, J., Lee, H., Kim, U., Chung, W. J. & Im, W. B. Flexible remote phosphor color converter based on ultra-thin glass and CsPbBr3 perovskite nanocrystal- embedded glass for a wide-color-gamut white LED. J. Mater. Chem. C 11, 898–902 (2023).
Nie, F., Wang, K.-Z. & Yan, D. Supramolecular glasses with color-tunable circularly polarized afterglow through evaporation-induced self-assembly of chiral metal–organic complexes. Nat. Commun. 14, 1654 (2023).
Sun, K. et al. Three-dimensional direct lithography of stable perovskite nanocrystals in glass. Science 375, 307–310 (2022).
Zhou, B. & Yan, D. Glassy inorganic–organic hybrid materials for photonic applications. Matter 7, 1950–1976 (2024).
Xia, Q. et al. A strong, biodegradable and recyclable lignocellulosic bioplastic. Nat. Sustain. 4, 627–635 (2021).
Jena, S. et al. Noncovalent interactions in proteins and nucleic acids: beyond hydrogen bonding and π-stacking. Chem. Soc. Rev. 51, 4261–4286 (2022).
Jones, C. D. et al. Braiding, branching and chiral amplification of nanofibres in supramolecular gels. Nat. Chem. 11, 375–381 (2019).
Huang, Z. et al. Highly compressible glass-like supramolecular polymer networks. Nat. Mater. 21, 103–109 (2022).
Liu, Y. et al. Multiple hydrogen bonding driven supramolecular architectures and their biomedical applications. Chem. Soc. Rev. 53, 1592–1623 (2024).
Luo, X. et al. Room-temperature phosphorescent materials derived from natural resources. Nat. Rev. Chem. 7, 800–812 (2023).
Dai, X.-Y., Huo, M. & Liu, Y. Phosphorescence resonance energy transfer from purely organic supramolecular assembly. Nat. Rev. Chem. 7, 854–874 (2023).
Lin, C. et al. Charge trapping for controllable persistent luminescence in organics. Nat. Photonics 18, 350–356 (2024).
El-Naggar, M. E. et al. Preparation of epoxy resin/rare earth doped aluminate nanocomposite toward photoluminescent and superhydrophobic transparent woods. J. Rare Earths 41, 397–405 (2023).
Tang, S. et al. Nonconventional luminophores: characteristics, advancements and perspectives. Chem. Soc. Rev. 50, 12616–12655 (2021).
Wang, Q. et al. Reevaluating protein photoluminescence: remarkable visible luminescence upon concentration and insight into the emission mechanism. Angew. Chem. Int. Ed. 58, 12667–12673 (2019).
Nakauchi, Y., Nishinami, S. & Shiraki, K. Glass-like protein condensate for the long-term storage of proteins. Int. J. Biol. Macromol. 182, 162–167 (2021).
Modell, A. E., Blosser, S. L. & Arora, P. S. Systematic targeting of protein–protein interactions. Trends Pharmacol. Sci. 37, 702–713 (2016).
Sun, C., Liu, J., Yang, N. & Xu, G. Egg quality and egg albumen property of domestic chicken, duck, goose, turkey, quail, and pigeon. Poult. Sci. 98, 4516–4521 (2019).
Xing, R., Yuan, C., Fan, W., Ren, X. & Yan, X. Biomolecular glass with amino acid and peptide nanoarchitectonics. Sci. Adv. 9, eadd8105 (2023).
Xu, K. et al. Egg albumen as a fast and strong medical adhesive glue. Adv. Healthc. Mater. 6, 1700132 (2017).
Ahmad, M., Ritzoulis, C., Pan, W. & Chen, J. Biologically-relevant interactions, phase separations and thermodynamics of chitosan–mucin binary systems. Process Biochem. 94, 152–163 (2020).
Safari, M. S., Byington, M. C., Conrad, J. C. & Vekilov, P. G. Polymorphism of lysozyme condensates. J. Phys. Chem. B 121, 9091–9101 (2017).
Zhang, H. et al. Aggregate science: from structures to properties. Adv. Mater. 32, 2001457 (2020).
Gao, R., Kodaimati, M. S. & Yan, D. Recent advances in persistent luminescence based on molecular hybrid materials. Chem. Soc. Rev. 50, 5564–5589 (2021).
Xing, C., Zhou, B., Yan, D. & Fang, W.-H. Dynamic photo-responsive ultralong phosphorescence from 1D halide microrods towards multi-level information storage. CCS Chem. 5, 2866–2876 (2023).
He, S. et al. Semiconductor glass with superior flexibility and high room temperature thermoelectric performance. Sci. Adv. 6, eaaz8423 (2020).
Hou, Y., Zhang, G.-H. & Chou, K.-C. Mixed alkali effect in SiO2–CaO–Al2O3–TiO2–R2O (R = Li, Na) glass ceramics. J. Alloy. Compd. 856, 158239 (2021).
Zhao, Y. et al. Optimized structural and mechanical properties of borophosphate glass. Ceram. Int. 46, 9025–9029 (2020).
Amini, A., Khavari, A., Barthelat, F. & Ehrlicher, A. J. Centrifugation and index matching yield a strong and transparent bioinspired nacreous composite. Science 373, 1229–1234 (2021).
Fan, J. T., Zhang, Z. F., Jiang, F., Sun, J. & Mao, S. X. Ductile to brittle transition of Cu46Zr47Al7 metallic glass composites. Mater. Sci. Eng. A 487, 144–151 (2008).
Qiu, X. B. et al. Tunable rejuvenation behavior of a metallic glass by residual stress modulation. J. Mater. Res. Technol. 26, 8263–8271 (2023).
Henao, J. et al. Novel Al-based metallic glass coatings by cold gas spray. Mater. Des. 94, 253–261 (2016).
Watanabe, T., Benino, Y. & Komatsu, T. Change in Vickers hardness at the glass transition region for fragile and strong glasses. J. Non-Cryst. Solids 286, 141–145 (2001).
Weeks, W. P. & Flores, K. M. Improving the precision of Vickers indentation measurements in soda-lime glass with increased dwell time. J. Non-Cryst. Solids 605, 122174 (2023).
Cai, C. et al. Bulk transparent supramolecular glass enabled by host–guest molecular recognition. Nat. Commun. 15, 3929 (2024).
Kim, J., Zhang, G., Shi, M. & Suo, Z. Fracture, fatigue, and friction of polymers in which entanglements greatly outnumber cross-links. Science 374, 212–216 (2021).
Yao, G., Pan, Y., Li, F. and Dong, S. Macrocyclic supramolecular glass: new type of supramolecular transparent material. Small https://doi.org/10.1002/smll.202405337 (2024).
Shi, M. et al. Confinement-modulated clusterization-triggered time-dependent phosphorescence color from xylan-carbonized polymer dots. J. Am. Chem. Soc. 146, 1294–1304 (2024).
Ma, H. et al. Boosting organic phosphorescence in adaptive host-guest materials by hyperconjugation. Nat. Commun. 15, 3660 (2024).
Kang, C. et al. Enabling carbonized polymer dots with color-tunable time-dependent room temperature phosphorescence through confining carboxyl dimer association. Angew. Chem. Int. Ed. 63, e202316527 (2024).
Nie, F. & Yan, D. Zero-dimensional halide hybrid bulk glass exhibiting reversible photochromic ultralong phosphorescence. Nat. Commun. 15, 5519 (2024).
Gong, Y. et al. Spectral and temporal manipulation of ultralong phosphorescence based on melt-quenched glassy metal–organic complexes for multi-mode photonic functions. Adv. Funct. Mater. 34, 2312491 (2024).
Zhou, B., Qi, Z. & Yan, D. Highly efficient and direct ultralong all-phosphorescence from metal–organic framework photonic glasses. Angew. Chem. Int. Ed. 61, e202208735 (2022).
Chen, T. et al. The trade-off anionic modulation in metal–organic glasses showing color-tunable persistent luminescence. Mater. Horiz. 11, 4951–4960 (2024).
Nie, F. & Yan, D. Macroscopic assembly of chiral hydrogen-bonded metal-free supramolecular glasses for enhanced color-tunable ultralong room temperature phosphorescence. Angew. Chem. Int. Ed. 62, e202302751 (2023).
Luo, J.-B., Wei, J.-H., Zhang, Z.-Z., He, Z.-L. & Kuang, D.-B. A melt-quenched luminescent glass of an organic-inorganic manganese halide as a large-area scintillator for radiation detection. Angew. Chem. Int. Ed. 62, e202216504 (2023).
Zhang, Z.-Z. et al. Organic–inorganic hybrid mn-based transparent glass for curved X-ray scintillation imaging. Adv. Opt. Mater. 12, 2302434 (2023).
Wang, X., Zhang, X., Liu, Y. & Zhang, Y. Shape-on-demand synthesis of luminescent (ETP)2MnBr4 glass scintillator. Chem. Eng. J. 483, 149239 (2024).
Li, H. et al. Stimuli-responsive circularly polarized organic ultralong room temperature phosphorescence. Angew. Chem. Int. Ed. 59, 4756–4762 (2020).
Wang, T., Sun, S., Jiang, X. & Ma, X. A universal strategy for modular tunable full-color circularly polarized luminescent materials with afterglow. Adv. Opt. Mater. 12, 2301770 (2024).
Yu, L. et al. Time-dependent colorful circularly polarized organic ultralong room temperature phosphorescence from a single-component chiral molecule. Small 19, 2303579 (2023).
Pacchioni, G. A chiral glass delivers colour-tunable ultralong room-temperature phosphorescence. Nat. Rev. Mater. 8, 362 (2023).
Zhang, C. et al. Molecular interaction of soybean protein and piperine by computational docking analyses. Food Hydrocoll. 146, 109249 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 22275021, D.Y.), the Newton Advanced Fellowship award (NAF\R1\201285, D.Y.), the Fok Ying-Tong Education Foundation (grant no. 171008, D.Y.), the Beijing Municipal Natural Science Foundation (grant no. L234064, D.Y.), the Beijing Nova Program (grant no. 20230484414, D.Y.), and the Fundamental Research Funds for the Central Universities (D.Y.).
Author information
Authors and Affiliations
Contributions
D.Y. and F.N. conceived the experiments. F.N. conducted and analyzed the experiments. D.Y. supervised the project. Both authors prepared and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zaijin Fang and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nie, F., Yan, D. Bio-sourced flexible supramolecular glasses for dynamic and full-color phosphorescence. Nat Commun 15, 9491 (2024). https://doi.org/10.1038/s41467-024-53963-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53963-2
This article is cited by
-
Full-color processible afterglow organic small molecular glass
Nature Communications (2025)
-
Solvent-free processing of lignin into robust room temperature phosphorescent materials
Nature Communications (2025)
-
Color-tunable supramolecular luminescent materials for information anticounterfeiting
Science China Materials (2025)
-
Lignin-derived room temperature phosphorescent materials
Science China Chemistry (2025)
-
Oxygen-independent photoactivation of kinetically trapped persistent room-temperature phosphorescence state for smart cold-chain monitoring
Science China Materials (2025)
