
Abstract
Human respiratory syncytial virus (RSV) is a major cause of acute respiratory infection. In 2020, RSV was eliminated from New Zealand due to non-pharmaceutical interventions (NPI) used to control the spread of SARS-CoV-2. However, in 2021, following a brief quarantine-free travel agreement with Australia, there was a large-scale nationwide outbreak of RSV that led to reported cases more than five-times higher than typical seasonal patterns. We generated 1470 viral genomes of both RSV-A and RSV-B sampled between 2015–2022 from across New Zealand. Using a phylodynamics approach, we used these data to better understand RSV transmission patterns in New Zealand prior to 2020, and how RSV became re-established in the community following the relaxation of COVID-19 restrictions. We found that in 2021, there was a large epidemic of RSV due to an increase in importations, leading to several large genomic clusters of both RSV-A ON1 and RSV-B BA9 genotypes. However, while a number of viral importations were detected, there was also a major reduction in RSV genetic diversity compared to pre-pandemic years. These data reveal the impact of NPI used during the COVID-19 pandemic on other respiratory infections and highlight the important insights that can be gained from viral genomes.
Similar content being viewed by others


Introduction
Respiratory syncytial virus (RSV) is a major cause of acute respiratory infection (ARI), mainly affecting children, older people, and those with comorbidities, resulting in a considerable hospitalisation burden among children in particular1,2. This RNA virus is part of the Orthopneumoviruses, a genus of viruses in the Pneumoviradae family, that generally cause upper respiratory tract infections in humans and some other vertebrates3. RSV is known to cause severe disease in children, particularly infants4, and in older adults. In 2019, for instance, there were an estimated 33 million RSV-associated ARIs globally, and over 100,000 deaths in children under 5 years old5. While serological studies have found that RSV infection is common in infants4, the virus induces poor immunity, and thus, reinfection can be frequent throughout life6. RSV infection is less severe in adults, typically causing upper respiratory infections with mild to moderate symptoms7. However, in older people, RSV can be more severe and is reported to have morbidity and mortality rates comparable with influenza virus8.
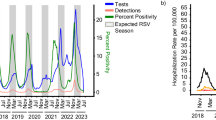
The global circulation of RSV is characterised by distinct seasonality9,10,11,12. In New Zealand, a geographically isolated country in the southern hemisphere with a temperate climate, RSV cases usually increase in autumn (i.e., at the end of April) and peak in mid-winter (late June)13. However, in 2020, the regular circulation of RSV was disrupted due to the introduction of non-pharmaceutical interventions (NPI) used to curb the spread of SARS-CoV-2, including a stay-at-home order for all non-essential workers and closure of all public venues, non-essential business, and educational facilities14. The most stringent of these NPI measures was the closure of international borders and the introduction of a 14-day stay in managed quarantine for returning citizens and residents14. These NPI were effective in eliminating the circulation and re-introduction of SARS-CoV-2 in New Zealand15,16,17, and consequently, appeared to eliminate the introduction and spread of other respiratory viruses too15. During the winter of 2020, reported cases of RSV were reduced by 98% when compared to the average annual cases recorded from 2015 to 201915. Indeed, compared to annual averages, New Zealand also saw reductions in cases of influenza virus (99.9%), metapneumovirus (92%), enterovirus (82%), adenovirus (81%), parainfluenza virus (80%), and rhinovirus (75%)15. A decrease in the circulation of RSV during 2020 was also observed in other countries that used NPI including in the USA1, Australia1, China18, Singapore19, South Africa20, UK and Ireland21.
In April 2021, following over a year of restricted travel into New Zealand, a brief quarantine-free travel agreement was initiated between New Zealand, Australia, and other countries in the South Pacific22. This relaxation of travel restrictions coincided with an increase in the circulation of respiratory viruses in New Zealand23. Specifically, New Zealand reported RSV cases five times higher, and hospitalisations three times higher, than those typically seen in the pre-pandemic phase24. Australia experienced similar epidemic surges of RSV following the relaxation of COVID-19 restrictions leading to extensive off-season outbreaks and hospitalisations during the summer of 2020/2021 from cryptically circulating lineages11. By August 2021, New Zealand had suspended quarantine-free travel and once again closed the border with Australia. New Zealand then entered its second nationwide lockdown in order to control the spread of the highly transmissible SARS-CoV-2 Delta variant25. This, in turn, was met with a sharp decline in the number of reported RSV cases26. Beginning in early 2022, the New Zealand border became unrestricted, and with the increased migration of people into the country there has been widespread community transmission of respiratory viruses once again, including influenza virus, RSV and the SARS-CoV-2 Omicron variant26,27.
Viral genomic data offers valuable insights into the pattern of viral evolution, and, therefore, enhances our understanding of outbreak epidemiology by providing a high level of detail and context to transmission patterns. When combined with geographic and population information, these data can reveal pathways of viral spread that can be used to inform effective national and local interventions16,28. Temporal and geographic information can be integrated into phylodynamic analyses to detect the spread of a pathogen in time and space, develop predictions about an outbreak’s trajectory, and identify clusters of similar sequences as an indication of infection sources and superspreading28,29,30. Herein, we describe the first genome-scale analysis of RSV in New Zealand, spanning eight years from 2015 to 2022. This timeframe incorporates several significant time points, including the New Zealand border closure in early 2020, nationwide lockdowns during the COVID-19 pandemic, the quarantine-free travel period with Australia and the South Pacific in 2021, and later, the easing of travel restrictions as New Zealand reconnected with the rest of the world in 2022. In this study, we investigate the typical patterns and dynamics of RSV transmission in New Zealand prior to 2020, and how RSV became re-established in the community following its elimination.
Results and discussion
The elimination and re-introduction of RSV during the COVID-19 pandemic
In 2021, New Zealand experienced a large epidemic of RSV following a brief period of quarantine-free travel with Australia and other countries in the South Pacific (Fig. 1a). To better understand the transmission dynamics of RSV before, during, and after this outbreak, we generated RSV genomes from patients in New Zealand over eight years. In total, 1,470 viral genomes, including 755 RSV-A and 715 RSV-B sequences, were generated from New Zealand between 2015 and 2022. Both RSV-A and RSV-B co-circulated each year in relatively even prevalence, based on the genomes generated in this study. Genomes were generated from the male and female sexes in equal proportions, and samples were mostly collected from young children with an overall median age of 1 (range 0–99) (Fig. 1e, f, Supplementary Table 3). The severity of RSV infections can be inferred by the surveillance platform from which the sample originated. Among samples from a known origin included in this study, 83% were referred from hospital-based surveillance, including outpatients and ICU, meaning that the vast majority of genomes were most likely generated from more severe infections compared to those seen in the community (Fig. 1d).

a Number of RSV-A (orange) and RSV-B (green) genomes generated over time based on sample collection date. The reflected grey bars below show the number of reported RSV-positive cases in New Zealand over the same timeframe68. b, c Map of New Zealand, coloured by the number of positive RSV-A and -B cases in each District Health Board. d The number of RSV genomes generated from cases reported in the community, hospital, intensive care unit (ICU), and those from unknown sources. e The number of RSV genomes generated from female and male patients. f The age distribution of patients from which RSV genomes were generated. A box plot indicates the mean (red), lower and upper quartiles (black), and whiskers (black lines), and the scatter points show the raw data. Source data are provided as a Source Data file.
Between 2015 and 2019, annual outbreaks of RSV were typically seasonal, with cases peaking in late June, coinciding with mid-winter in New Zealand (Fig. 1a). In 2020, however, reported RSV cases were reduced by 98%, and no community transmission of RSV was reported as a result of NPI used to control the spread of SARS-CoV-2 (see26 for percentage positivity data). Schools and childcare centers were closed during a four-week nationwide lockdown in April 2020, which effectively eliminated SARS-CoV-2 and other respiratory infections from the New Zealand community15,26,31. The following year, in April 2021, and for the first time since restrictions were placed on the New Zealand border due to the COVID-19 pandemic, people could travel to and from Australia without having to enter managed quarantine at the border. Within weeks of lifting border restrictions, there was an exceptionally large RSV epidemic in New Zealand, with a sharp surge in reported cases of both RSV-A and -B (Fig. 1a).
The 2021 RSV surge affected a wider age distribution compared to previous years (Supplementary Fig. 2). Among people who were sampled in this study, we found a 5% increase in infections among 5–18-year-olds and a 15% increase in infections among 19–65-year olds compared to previous years (p < 0.001 when comparing all groups to 2021 using a Chi-squared test) (Supplementary Table 3). This surge cannot be explained by weakened immunity due to COVID-1932, as there were fewer than 3000 reported COVID-19 cases (in a population of ~five million) before August 2021. The majority of these COVID-19 cases were associated with returning travellers, which had limited community transmission16. Rather it was more likely the result of waned immunity due to prolonged lack of viral exposure, as seen in other countries33,34,35. However, other factors related to sample collection may have also contributed to the apparent surge. One factor was a change in diagnostic viral testing regimes that were implemented during the COVID-19 pandemic, such as the increased use of point-of-care testing with an expanded respiratory viral diagnostic assay that included RSV36, as well as wider testing in hospitals as part of the admission process, in the community as part of the pandemic response, and for returning travellers as part of the new acute respiratory illness surveillance system. These new testing regimens included older children and adults15. Increased testing not only inflates case counts but can also skew patient age distribution by incorporating less severe cases37. Nevertheless, due to the associated rise in RSV-related hospitalisations26 and the return to normal levels in 2022, it is likely that the 2021 epidemic reflected a period of reduced exposure to RSV and an increase in the immunologically naïve population.
Quarantine-free travel with Australia was brought to an abrupt end in July 2021 due to the uncontrolled spread of the SARS-CoV-2 Delta Variant of Concern in Australia38. Weeks later, New Zealand entered its second nationwide lockdown due to the incursion of Delta SARS-CoV-2, which was genomically linked to cases in Australia25. The lockdown caused the number of reported RSV cases to dramatically reduce (Fig. 1).
Inferring the sources of the 2021 RSV outbreaks using phylodynamics
Phylogenomic analysis of RSV-A and RSV-B genomes, together with genomes sampled from Australia and the rest of the world, showed that in the pre-pandemic phase, genomes sampled from New Zealand were widely distributed among globally sampled genomes, falling among a range of genomic lineages. In 2021, however, genomes sampled from New Zealand formed only a small number of tightly grouped clusters. In particular, RSV-A genomes sampled in 2021 fell into only two distinct lineages with well-supported nodes (Fig. 2d–f). One clade of RSV-A genomes, predominantly sampled from Auckland and surrounding districts, formed a monophyletic clade with no close-in-time sampled genomic ancestors, and unrelated to previously circulating New Zealand lineages (Fig. 2e). Due to stringent testing regimes for all respiratory infections coupled with managed quarantine at the border for all arrivals besides those from Australia and the South Pacific, and that RSV genomes are sparsely sampled at the global scale, this lineage is unlikely to represent undetected transmission in New Zealand. Rather, it is more likely that this lineage is related to unsampled genomes, most probably in Australia.

a Map of New Zealand (where regions are categorised by District Health Board (DHB)), Australia (dark grey), and the rest of the world (light grey). b Number of RSV-A genomes sampled per region, where colours correspond to those in (a). c Root-to-tip regression analysis of RSV-A genomes versus sampling time. d Maximum likelihood time-scaled phylogenetic tree showing 755 RSV-A genomes sampled from New Zealand (coloured circles based on DHB region), 662 RSV-A genomes sampled from Australia (dark grey), and 2913 RSV-A genomes sampled from the rest of the world (light grey). A yellow vertical bar highlights the year 2021 and a dotted box shows the major New Zealand clades sampled. e, f Maximum likelihood time-scaled phylogenetic trees showing the major clades sampled in New Zealand during 2021 and their closest sampled genetic relatives, showing percentage bootstrap support from 1000 replicates for the nodes of the New Zealand clusters. Source data are provided as a Source Data file.
A second cluster of New Zealand 2021 genomes were grouped with those sampled from Australia during the months prior (Fig. 2f). This 2021 New Zealand cluster was due to multiple introductions, forming several monophyletic clades among Australian samples, and included genomes from across a wide geographic region including the North and South Islands of New Zealand. Both the highlighted clusters of RSV-A genomes were determined to be the ON1-like genotype based on their G-gene sequences (genomic clade A.3 based on Chen et al.39), which has become globally dominant in recent years40,41. This genotype, which evolved from the previous dominant RSV-A genotype NA-1, has a characteristic 72-nucleotide duplication in the G-gene42. All RSV-A genomes sampled in New Zealand conformed to a root-to-tip linear regression reflecting a temporal signal in these data (Pearson correlation coefficient, r = 0.88, p < 10−4).
Similarly, RSV-B genomes sampled in 2021 were clustered tightly with those sampled in Australia in the months leading up to the outbreak in New Zealand (Fig. 3). These genomes formed several large monophyletic clades among Australian samples and largely clustered according to geographic location in New Zealand. We again found a temporal signal in the New Zealand RSV-B data, illustrated by a root-to-tip linear regression (Pearson correlation coefficient, r = 0.93, p < 10−4). These genomes were classified as the BA9 genotype based on their G-gene sequences (or the B.5.8 genomic clade39). BA9 is one of 15 genotypes within the BA lineage reported since its emergence circa 199843. Genotype BA9 was first identified in Japan in 200644 and, like all BA genotypes, is characterised by a 60-nucleotide duplication in the G-gene45. Over the last decade, BA9 has become globally dominant and is now found worldwide40,41,46,47,48. Overall, the RSV-A and RSV-B genomes sampled in 2021 were most likely from new introductions into New Zealand rather than from cryptically circulating lineages that remained undetected in 2020. Indeed, our data support that RSV transmission was eliminated in 2020, and both subtypes were reintroduced in 2021, most likely from Australia following the lifting of border restrictions, including quarantine.

a Map of New Zealand (where regions are categorised by District Health Board (DHB)), Australia (dark grey), and the rest of the world (light grey). b Number of RSV-B genomes sampled per region, where colours correspond to those in a. c Root-to-tip regression analysis of RSV-B genomes versus sampling time. d Maximum likelihood time-scaled phylogenetic tree showing 715 RSV-B genomes sampled from New Zealand (coloured circles based on DHB region), 518 RSV-B genomes sampled from Australia (dark grey), and 2577 RSV-B genomes sampled from the rest of the world (light grey). A yellow vertical bar highlights the year 2021 and a dotted box shows the major New Zealand clade sampled. e Maximum likelihood time-scaled phylogenetic tree showing the major clades sampled in New Zealand during 2021 and their closest sampled genetic relatives, showing percentage bootstrap support from 1000 replicates for the nodes of the New Zealand clusters. Source data are provided as a Source Data file.
Examining the spatial transmission dynamics of RSV over time
We estimated the spatial transmission dynamics of RSV over time to elucidate how the number of viral introductions changed as a result of NPI, particularly border restrictions, used during the COVID-19 pandemic. While the number of people arriving in New Zealand peaked in early 2020 following an announcement that the border would be closed, a nationwide lockdown meant that any potential new arrivals of RSV did not lead to onward transmission in the community (Fig. 4). We inferred the number of introductions of both RSV-A and RSV-B into New Zealand between 2015 and 2022 using genomes sampled from both New Zealand and abroad. Although genomic sequences were too sparse to capture seasonal patterns between 2015 and 2019, introductions of both RSV-A and RSV-B decreased in 2020, resulting in near-zero introductions of either subtype (Fig. 4). In interpreting this analysis, we note that the number of introductions on any given day was approximated by counting the lineages on that day that changed from Global to New Zealand. This approach means that the introduction dates on long branches (such as those that led to the 2021 outbreaks) are likely to be underestimated. With this limitation noted, these results corroborate that the number of introductions dramatically increased the following year in 2021, particularly for RSV-A, coinciding with quarantine-free travel to and from Australia.

a Number of people arriving ino New Zealand (blue), number of people departing New Zealand (green), and the overall net migration of people into New Zealand (red)69. b, c Estimated migration rates of RSV-A and-B into New Zealand over time (mean estimates are shown in white, and 95% credible intervals are coloured). A grey-shaded area shows the period in which New Zealand underwent border restrictions during the COVID-19 pandemic. A purple-shaded area shows the quarantine-free travel period with Australia. Source data are provided as a Source Data file.
In 2022, RSV-B showed a larger number of introductions, which is consistent with the multiple different lineages that were circulating in 2022. Although both subtypes were introduced many times throughout the eight years (around 80 for RSV-A and 100 for RSV-B), only a small number of these introductions eventuated in large, detected outbreaks in 2021 (Figs. 2 and 3). We estimate that the 689 genomes and 2818 cases in 2021 were linked to just 10 (95% credible interval, CI: 6.6, 14) introductions of RSV-A and 6.7 (CI: 3.8, 10) for RSV-B. This means that each introduction in 2021 produced an average of 41 genomes and 169 cases. In contrast, the 63 (51, 76) RSV-A introductions and 76 (61, 92) B introductions between 2014 and 2019 each resulted in an average of three genomes and 90 cases. This reflects the immunologically naive population of 2021 combined with the highly stochastic nature of disease transmission, which often occurs through superspreader events49,50. In contrast, very few export events were identified. Our analysis showed that RSV-B mutated slightly faster than RSV-A (Table 1) and at a comparable rate with previous studies51. These experiments proved robust to genome subsampling, with each replicate yielding similar clock rates and migration counts.
Genomic diversity of RSV before and after the COVID-19 pandemic
Despite the border-opening associated increase in both viral introductions and genomic surveillance in New Zealand, the surge of RSV introductions in 2021 had very little genomic diversity compared to the pre-pandemic phase, as shown by the limited number of New Zealand genomic clusters in our phylogenetic analysis. Indeed, a Bayesian coalescent skyline analysis estimating the effective population size of New Zealand RSV-A and RSV-B genomes over time illustrated that the diversity in genomes decreased in 2020 and 2021 (Supplementary Fig. 3). Similar genomic bottlenecks have been reported in Europe52 and Australia11, where major lineages of RSV were collapsed following NPI during the COVID-19 pandemic. The resurgence of RSV in 2021 led to very little genomic diversity in circulating lineages, with only two major clades of RSV-A and one clade of RSV-B being reintroduced into New Zealand. Nevertheless, this reduced genetic diversity was seemingly short-lived lived with more widespread lineages reappearing by 2022 when border restrictions were removed (Figs. 2 and 3). Overall, the extensive NPI used to control SARS-CoV-2 in New Zealand significantly impacted the epidemiology of other respiratory viral infections, including the near-elimination and diminished genetic diversity of typically seasonal viruses.
Methods
Ethics
The New Zealand Northern A Health and Disability Ethics Committee approved the Southern Hemisphere Influenza Vaccine Effectiveness Research and Surveillance (SHIVERS) study (NTX/11/11/102). Ethics approval was not required for the primary care, laboratory, or hospital-based surveillance platforms as they were conducted in accordance with the Public Health Act 1956.
Sample collection
Respiratory samples (nasal, throat or nasopharyngeal swabs) were obtained from several surveillance and research platforms, each serving a different purpose. Community-based samples were obtained from general practitioners (GPs) across New Zealand from patients with influenza-like illness (ILI), for which the surveillance period typically began in late April and ended in late September. Hospital-based samples were obtained from patients with severe acute respiratory infection (SARI) from two major Auckland-based hospitals year-round. Laboratory-based surveillance samples were referred to the Institute for Environmental Science and Research (ESR) from both community and hospital diagnostic laboratories throughout the year in several major centres: Auckland, Bay of Plenty, Waikato, Hawkes Bay, Wellington, Nelson, Christchurch and Dunedin. Samples were also collected and tested at ESR for the Southern Hemisphere Influenza Vaccine Effectiveness Research and Surveillance (SHIVERS) study. This study included three Wellington-based community cohorts, where samples were obtained from participants who experienced ARI. The surveillance period for these cohorts matched that of the community-based surveillance. In 2021, a fifth iteration of SHIVERS commenced in which samples from diagnostic and hospital laboratories throughout New Zealand referred positive RSV samples to ESR for further testing and analysis. Collection dates of the genomes used in this study were collected from July 2015 up to and including October 2022.
Viral RNA extraction and qRT-PCR
Samples were received by the Clinical Virology Department at ESR from 2015 to 2022. Viral RNA was extracted using the Thermo Fisher Scientific™ MagMax Viral/Pathogen Nucleic Acid Isolation Kit (A48310) as per manufacturer’s instructions on either the ZiXpress or the Kingfisher Flex automated extraction instruments. An RSV singleplex qRT-PCR was performed on RNA using primer and probes synthesised by biosearch using sequences published by Kim et al.53, coupled with the AgPath-ID™ One step RT-PCR buffer and enzyme (4387424). Positive RSV RNA was genotyped (RSV-A, RSV-B) using primers and probes synthesised by biosearch using sequences published by Hu et al.54, coupled with the AgPath-ID™ One step RT-PCR buffer and enzyme (4387424). Samples collected between 2015 and 2021 with a cycle threshold (Ct.) below 25, as well as samples from 2022 with a Ct. below 30, were referred to the genomic sequencing department at ESR.
Generating viral genomes from RSV samples
In brief, sequencing was performed on the Illumina Nextseq platform using the respiratory virus oligo panel v2 (20044311), RNA prep enrichment kit (20040537), and the 500/550 midoutput kit (20024905) and used as per the manufacturer’s instructions. Consensus-based assembly was performed using a standardised pipeline, based on the Seattle Flu assembly pipeline, and modified for use by ESR, (https://github.com/seattleflu/assembly). Two additional references were used for consensus calling: EPI_ISL_2543807 and EPI_ISL_2543850, available from GISAID55. Consensus viral genomes were generated, and subject to quality testing, and those with fewer than 50% ambiguities were selected for further analysis (median coverage was 99.95% of the assembled genomes, with 80% of all samples having >99% or more of the genome covered, meaning that the vast majority of genomes were of high quality, see Supplementary Fig. 1). The genomes generated are available on GISAID under two blocks of consecutive accession numbers (EPI_ISL_16959469 - EPI_ISL_16960152 and EPI_ISL_19206151 - EPI_ISL_19207488) and on NCBI’s GenBank database under BioProject PRJNA939717.
Genotyping RSV genomes
Reference G glycoprotein gene (G-gene) sequences for each known genotype for both RSV-A and RSV-B were obtained from GenBank (see Supplementary Table 1 for a list of accession numbers and their genotypes). G-gene sequences were first annotated and extracted from the New Zealand genomes using Geneious Prime 2022.1.1 (https://www.geneious.com) and aligned with the reference sequences using MAFFT (v7)56 using the FFT-NS-2 algorithm. A maximum likelihood phylogenetic tree for each of RSV-A and RSV-B was estimated using IQ-TREE (v 1.6.8)57, using the Hasegawa-Kishino-Yano nucleotide substitution model with a gamma-distributed rate variation among sites (HKY + Γ)58 (determined to be the best-fit model by ModelFinder)59. Using the topology of the reference sequences, the genotypes of the New Zealand RSV G-genes were determined. Genotyping across whole genome sequences was also performed using LABEL60 based on genotypes proposed by Chen et al.39 following alignment with MAFFT (v7)56.
Inferring genomic diversity of RSV using phylodynamic analysis
Newly acquired New Zealand RSV genomes were aligned with globally sourced RSV genomes with MAFFT (v7)56 using the FFT-NS-2 algorithm (see Supplementary Table 2 for global genome accession numbers, or GISAID Epi set ID: EPI_SET_241014ew or https://doi.org/10.55876/gis8.241014ew). These global genomes (3487 RSV-A and 3002 RSV-B) were sampled uniformly at random without replacement from ~50,000 international sequences on GISAID55 between 2015 and 2022 (as of March 2024). A maximum likelihood phylogenetic tree was estimated using IQ-TREE (v 1.6.8)57 using the HKY + Γ58 nucleotide substitution model. We integrated the in-build Least Squares Dating method to estimate a time-scaled phylogenetic tree using the day of sampling. Phylogenetic branch support was assessed using the ultrafast bootstrap61 approximation. We used TempEst (v 1.5.3)62 to analyse the root-to-tip genetic regression against sampling dates to investigate the temporal signal of RSV-A and RSV-B genomes.
To measure relative genetic diversity over time (inferred through the effective population size), up to 50 New Zealand RSV genomes for each year (2015–2022) were uniformly selected at random without replacement and aligned using MAFFT (v7)56. These genomes were subject to Bayesian phylodynamic analysis using BEAST2 (v2.7)63 with the Bayesian Integrated Coalescent Epoch Plot using a structured coalescent tree prior64 and a strict clock rate estimated. We repeated this analysis three times for each RSV-A and RSV-B. We sampled the posterior distribution using Markov chain Monte Carlo (MCMC), running each analysis for 20 million steps, sampling states every 20,000 steps, and discarding the first 10% as burn-in. Convergence was assessed in Tracer v 1.665, ensuring that all analyses had over 200 effective samples for all reported parameters.
Estimating introductions of RSV using Bayesian phylodynamic analysis
To infer the migration dynamics of RSV into New Zealand over time, we treated geographic location as a discrete trait with two demes where genomes were either sampled as being ‘local’ (i.e., sampled from New Zealand) or ‘global’ (i.e., sampled from the rest of the world). Due to the size of the dataset, local and global RSV-A and RSV-B genomes were randomly split into six subsamples, making sure that near equal numbers of genomes were selected for each deme for each year. For each of RSV-A and RSV-B, each subsample comprised approximately 400 local and 400 global genomes. To ensure the geographical representativeness of subsamples, global genomes were subsampled by iteratively sampling a country and then a genome from that country. Each subsample was aligned using MAFFT (v7)56 and subject to Bayesian phylodynamic analysis using BEAST2 (v2.7)63 with the MASCOT structured coalescent tree prior66. We again used the HKY + Γ58 nucleotide substitution and a strict clock LogNormal(mean = 0.001, sigma = 0.1) clock rate prior (units: substitutions per site per year). Introduction (i.e., the transition from global to local) and export (local to global) events were assumed to be rare occurrences, and introductions were assumed much more common than exports, reflecting prior knowledge about epidemics originating overseas and then coming to New Zealand. The prior introductions were Exponential (mean = 0.1) migrations per year and Exponential (mean = 0.001) for exports. The (constant) effective population sizes of the two demes had independent LogNormal(mean = 0.2, sigma = 0.4) priors. We sampled the posterior distribution using MCMC running each analysis for 100 million steps, sampling states every 10,000 steps, and discarding the first 10% as burn-in. Convergence was assessed in Tracer v 1.665, ensuring that all analyses had over 200 effective samples for all reported parameters. All 6/6 RSV-A analyses and 2/6 RSV-B analyses were completed to this standard (the failed analyses are not reported here). The number of introductions of RSV-A and RSV-B over time was estimated using the Babel package in BEAST2, and the results were summarised in R67.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The genomes generated are available on GISAID under two blocks of consecutive accession numbers (EPI_ISL_16959469 - EPI_ISL_16960152 and EPI_ISL_19206151 - EPI_ISL_19207488) and on NCBI’s GenBank database under BioProject PRJNA939717. GISAID accession numbers of global genomes used in the analysis are available in Supplementary Table 2, accessed via GISAID Epi set ID: EPI_SET_241014ew or https://doi.org/10.55876/gis8.241014ew. Metadata included with the genomes comprises sampling date and location. Patient age and sample source data (e.g. hospital, ICU, etc.) are subject to controlled access due to privacy and ethical reasons. Data can be requested via the Institute of ESR and maybe subject to ethics approvals, and thus a timeframe for response cannot be provided. Source data are provided in this paper.
Code availability
All code used in this study is from open-source software described in the Methods.
References
Prasad, N. et al. The health and economic burden of respiratory syncytial virus associated hospitalizations in adults. PLoS One 15, e0234235 (2020).
Falsey, A. R., Hennessey, P. A., Formica, M. A., Cox, C. & Walsh, E. E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 352, 1749–1759 (2005).
Rima, B. et al. ICTV virus taxonomy profile: Pneumoviridae. J. Gen. Virol. 98, 2912–2913 (2017).
Griffiths, C., Drews, S. J. & Marchant, D. J. Respiratory syncytial virus: infection, detection, and new options for prevention and treatment. Clin. Microbiol. Rev. 30, 277–319 (2017).
Li, Y. et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in children younger than 5 years in 2019: a systematic analysis. Lancet 399, 2047–2064 (2022).
Müller-Pebody, B., Edmunds, W. J., Zambon, M. C., Gay, N. J. & Crowcroft, N. S. Contribution of RSV to bronchiolitis and pneumonia-associated hospitalizations in English children, April 1995–March 1998. Epidemiol. Infect. 129, 99–106 (2002).
Falsey, A. R. & Walsh, E. E. Respiratory syncytial virus infection in adults. Clin. Microbiol. Rev. 13, 371–384 (2000).
Haber, N. Respiratory syncytial virus infection in elderly adults. Med. Mal. Infect. 48, 377–382 (2018).
Robertson, M. et al. The spatial-temporal dynamics of respiratory syncytial virus infections across the east-west coasts of Australia during 2016-17. Virus Evol. 7, veab068 (2021).
Di Giallonardo, F. et al. Evolution of human respiratory syncytial virus (RSV) over multiple seasons in New South Wales, Australia. Viruses 10, 476 (2018).
Eden, J.-S. et al. Off-season RSV epidemics in Australia after easing of COVID-19 restrictions. Nat. Commun. 13, 2884 (2022).
Fall, A. et al. Epidemiology and molecular characterization of human respiratory syncytial virus in Senegal after four consecutive years of surveillance, 2012–2015. PLoS One 11, e0157163 (2016).
Prasad, N. et al. Respiratory syncytial virus hospitalisations among young children: a data linkage study. Epidemiol. Infect. 147, e246 (2019).
Jefferies, S. et al. COVID-19 in New Zealand and the impact of the national response: a descriptive epidemiological study. Lancet Public Health 5, e612–e623 (2020).
Huang, Q. S. et al. Impact of the COVID-19 nonpharmaceutical interventions on influenza and other respiratory viral infections in New Zealand. Nat. Commun. 12, 1001 (2021).
Geoghegan, J. L. et al. Genomic epidemiology reveals transmission patterns and dynamics of SARS-CoV-2 in Aotearoa New Zealand. Nat. Commun. 11, 6351 (2020).
Baker, R. E. et al. The impact of COVID-19 nonpharmaceutical interventions on the future dynamics of endemic infections. Proc. Natl Acad. Sci. USA 117, 30547–30553 (2020).
Liu, P. et al. Impact of COVID-19 pandemic on the prevalence of respiratory viruses in children with lower respiratory tract infections in China. Virol. J. 18, 159 (2021).
Wan, W. Y. et al. Trends in respiratory virus infections during the COVID-19 pandemic in Singapore, 2020. JAMA Netw. Open 4, e2115973 (2021).
Tempia, S. et al. Decline of influenza and respiratory syncytial virus detection in facility-based surveillance during the COVID-19 pandemic, South Africa, January to October 2020. Eurosurveillance 26, 2001600 (2021).
Williams, T. C., Sinha, I., Barr, I. G. & Zambon, M. Transmission of paediatric respiratory syncytial virus and influenza in the wake of the COVID-19 pandemic. Eurosurveillance 26, 2100186 (2021).
Department of Prime Minister and Cabinet. Timeline of significant COVID-19 events. (https://www.dpmc.govt.nz/sites/default/files/2023-10/pr-timeline-significant-events-activities.pdf) (2024).
Hatter, L., Eathorne, A., Hills, T., Bruce, P. & Beasley, R. Respiratory syncytial virus: paying the immunity debt with interest. Lancet Child Adolesc. Health 5, e44–e45 (2021).
Grant, C. C., Huang, Q. S., Trenholme, A., Taylor, S. & Wood, T. What can we learn from our 2021 respiratory syncytial virus experience? N. Z. Med. J. 134, 7–12 (2021).
Jelley, L. et al. Genomic epidemiology of Delta SARS-CoV-2 during transition from elimination to suppression in Aotearoa New Zealand. Nat. Commun. 13, 4035 (2022).
Huang, Q. S. et al. Impact of the COVID‐19 related border restrictions on influenza and other common respiratory viral infections in New Zealand. Influenza Other Respir. Viruses 18, e13247 (2024).
Douglas, J. et al. Tracing the international arrivals of SARS-CoV-2 Omicron variants after Aotearoa New Zealand reopened its border. Nat. Commun. 13, 6484 (2022).
Douglas, J. et al. Real-time genomics for tracking severe acute respiratory syndrome coronavirus 2 border incursions after virus elimination, New Zealand. Emerg. Infect. Dis. 27, 2361–2368 (2021).
Da Silva Filipe, A. et al. Genomic epidemiology reveals multiple introductions of SARS-CoV-2 from mainland Europe into Scotland. Nat. Microbiol. 6, 112–122 (2020).
Park, D. et al. Ebola virus epidemiology, transmission, and evolution during seven months in Sierra Leone. Cell 161, 1516–1526 (2015).
Cumming, J. Going hard and early: Aotearoa New Zealand’s response to Covid-19. Health Econ. Policy Law 17, 107–119 (2022).
Wang, L. et al. Association of COVID-19 with respiratory syncytial virus (RSV) infections in children aged 0–5 years in the USA in 2022: a multicentre retrospective cohort study. Fam. Med. Commun. Health 11, e002456 (2023).
Den Hartog, G. et al. Decline of RSV-specific antibodies during the COVID-19 pandemic. Lancet Infect. Dis. 23, 23–25 (2023).
Reicherz, F. et al. Waning immunity against respiratory syncytial virus during the coronavirus disease 2019 pandemic. J. Infect. Dis. 226, 2064–2068 (2022).
Rios-Guzman, E. et al. Deviations in RSV epidemiological patterns and population structures in the United States following the COVID-19 pandemic. Nat. Commun. 15, 3374 (2024).
2024 Cepheid. Available from https://www.cepheid.com/en-NZ/tests/respiratory/xpert-xpress-cov-2-flu-rsv-plus.html (2024).
Petros, B. A., Milliren, C. E., Sabeti, P. C. & Ozonoff, A. Increased pediatric respiratory syncytial virus case counts following the emergence of SARS-CoV-2 can be attributed to changes in testing. Clin. Infect. Dis. 78, 1707–1717 (2024).
Office of the Minister for COVID-19 Response. (https://covid19.govt.nz/assets/Proactive-Releases/Border/19-Jul-2022/Quarantine-Free-Travel-With-Australia_-Further-Suspension.pdf) (2024).
Chen, J. et al. Novel and extendable genotyping system for human respiratory syncytial virus based on whole‐genome sequence analysis. Influenza Other Respir. Viruses 16, 492–500 (2022).
Umar, S. et al. Molecular epidemiology and characteristics of respiratory syncytial virus among hospitalized children in Guangzhou, China. Virol. J. 20, 272 (2023).
Tsukagoshi, H. et al. Genetic analysis of attachment glycoprotein (G) gene in new genotype ON1 of human respiratory syncytial virus detected in Japan. Microbiol. Immunol. 57, 655–659 (2013).
Eshaghi, A. et al. Genetic variability of human respiratory syncytial virus A strains circulating in Ontario: a novel genotype with a 72 nucleotide G gene duplication. PLoS One 7, e32807 (2012).
Muñoz-Escalante, J. C., Comas-García, A., Bernal-Silva, S. & Noyola, D. E. Respiratory syncytial virus B sequence analysis reveals a novel early genotype. Sci. Rep. 11, 3452 (2021).
Song, J. et al. Emergence of BA9 genotype of human respiratory syncytial virus subgroup B in China from 2006 to 2014. Sci. Rep. 7, 16765 (2017).
Trento, A. et al. Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J. Gen. Virol. 84, 3115–3120 (2003).
Tabatabai, J. et al. Molecular epidemiology of respiratory syncytial virus in hospitalised children in Heidelberg, Southern Germany, 2014-2017. Infect. Genet. Evol. 98, 105209 (2022).
Razanajatovo Rahombanjanahary, N. H., Rybkina, K., Randriambolamanantsoa, T. H., Razafimanjato, H. & Heraud, J. M. Genetic diversity and molecular epidemiology of respiratory syncytial virus circulated in Antananarivo, Madagascar, from 2011 to 2017: Predominance of ON1 and BA9 genotypes. J. Clin. Virol. 129, 104506 (2020).
Yun, K. W., Choi, E. H. & Lee, H. J. Molecular epidemiology of respiratory syncytial virus for 28 consecutive seasons (1990-2018) and genetic variability of the duplication region in the G gene of genotypes ON1 and BA in South Korea. Arch. Virol. 165, 1069–1077 (2020).
Stein, R. A. Super-spreaders in infectious diseases. Int. J. Infect. Dis. 15, e510–e513 (2011).
Lloyd-Smith, J. O., Schreiber, S. J., Kopp, P. E. & Getz, W. M. Superspreading and the effect of individual variation on disease emergence. Nature 438, 355–359 (2005).
Pangesti, K. N. A., Abd El Ghany, M., Walsh, M. G., Kesson, A. M. & Hill‐Cawthorne, G. A. Molecular epidemiology of respiratory syncytial virus. Rev. Med. Virol. 28, e1968 (2018).
Chow, E. J., Uyeki, T. M. & Chu, H. Y. The effects of the COVID-19 pandemic on community respiratory virus activity. Nat. Rev. Microbiol. 21, 195–210 (2023).
Kim, C. et al. Comparison of nasopharyngeal and oropharyngeal swabs for the diagnosis of eight respiratory viruses by real-time reverse transcription-PCR assays. PLoS One 6, e21610 (2011).
Hu, A., Colella, M., Tam, J. S., Rappaport, R. & Cheng, S.-M. Simultaneous detection, subgrouping, and quantitation of respiratory syncytial virus A and B by real-time PCR. J. Clin. Microbiol. 41, 149–154 (2003).
Elbe, S. & Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 1, 33–46 (2017).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Hasegawa, M., Kishino, H. & Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22, 160–174 (1985).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Shepard, S. S. et al. LABEL: Fast and accurate lineage assignment with assessment of H5N1 and H9N2 influenza A hemagglutinins. PLoS One 9, e86921 (2014).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2, vew007 (2016).
Bouckaert, R. et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15, e1006650 (2019).
Remco R. Bouckaert. An efficient coalescent epoch model for bayesian phylogenetic inference, Syst. Biol. 71, 1549–1560 (2022).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Müller, N. F., Rasmussen, D. & Stadler, T. MASCOT: parameter and state inference under the marginal structured coalescent approximation. Bioinformatics 34, 3843–3848 (2018).
R. Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, https://www.R-project.org/. (2021).
Institute of Environmental Science and Research Limited; Available from https://www.esr.cri.nz/digital-library (2024).
Stats New Zealand. Available from https://www.stats.govt.nz/topics/migration (2024).
Acknowledgements
We gratefully acknowledge all data contributors, i.e., the Authors and their originating laboratories, responsible for obtaining the specimens and their submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based. We would like to thank the participants of the SHIVERS WellKiwi cohorts for their continued enrolment and support in our research. Second, we would like to thank the WellKiwi Clinical and Study teams for SHIVERS research, the organization, and collection of participant samples, and the metadata used in this study. We would also like to thank the clinical virology and genomic sequencing teams at the Institute of ESR for processing, testing, and sequencing all of the samples collected. We would like to acknowledge staff from all of the diagnostic laboratories and hospital laboratories in New Zealand who take the time and effort to collect, aliquot, and refer samples to ESR. A special thanks to Nicolas Zacchi from Pathlab in Bay of Plenty, Radhika Nagappan from Auckland City Hospital, Fifi Tse from Middlemore Hospital, and Samantha Hutton from Awanui Laboratory in Wellington for helping collate retrospective data for the sample analysis. Thank you to former ESR staff who contributed to this study. L.J. is funded by a University of Otago Doctoral Scholarship. J.L.G. is funded by a New Zealand Royal Society Rutherford Discovery Fellowship (RDF-20-UOO-007). This study was supported by a New Zealand Health Research Council Grant (22/138) awarded to J.L.G., J.dL., D.W., S.H., L.J., N.F., and D.W. The SHIVERS/WellKiwis cohort is funded by the US National Institute of Allergy and Infectious Diseases (NIAID): SHIVERS-III infant funded by the US-NIAID (U01 AI 144616); SHIVERS-IV household funded by the US-NIAID (CEIRR contract: 75N93021C00016).
Author information
Authors and Affiliations
Consortia
Contributions
J.L.G., J.D., L.J., N.F., Q.S.H. and D.W. conceived the ideas for the study and designed the analysis. L.J., M.O., K.B., A.C., J.W., S.U., H.J., J.B., M.A., J.d.L., C.E.T., R.S., T.W., N.A., T.J., N.T., P.M., T.D., A.T., C.B. and D.W. collected data and generated RSV genomes. J.L.G., J.D., L.J., D.W., R.W., N.F., Q.S.H. and D.W. participated in the scientific discussion and data analysis. L.J., J.D. and J.G. wrote the manuscript. All authors have discussed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jelley, L., Douglas, J., O’Neill, M. et al. Spatial and temporal transmission dynamics of respiratory syncytial virus in New Zealand before and after the COVID-19 pandemic. Nat Commun 15, 9758 (2024). https://doi.org/10.1038/s41467-024-53998-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53998-5
