
Abstract
Activities of ATP binding cassette (ABC) proteins are regulated by multiple mechanisms, including protein interactions, phosphorylation, proteolytic processing, and/or oligomerization of the ABC protein itself. Here we present the structure of yeast cadmium factor 1 (Ycf1p) in its mature form following cleavage by Pep4p protease. Ycf1p, a C subfamily ABC protein (ABCC), is homologue of human multidrug resistance protein 1. Remarkably, a portion of cleaved Ycf1p forms a well-ordered dimer, alongside monomeric particles also present in solution. While numerous other ABC proteins have been proposed to dimerize, no high-resolution structures have been reported. Both phosphorylation of the regulatory (R) region and ATPase activity are lower in the Ycf1p dimer compared to the monomer, indicating that dimerization affects Ycf1p function. The interface between Ycf1p protomers features protein-protein interactions and contains bound lipids, suggesting that lipids stabilize the dimer. The Ycf1p dimer structure may inform the dimerization interfaces of other ABCC dimers.
Similar content being viewed by others

Introduction
ATP binding cassette (ABC) proteins are integral membrane proteins found in all kingdoms of life1,2,3. ABC proteins contain a core structure comprised of two half-transporters, each containing a transmembrane domain (TMD) and a cytosolic nucleotide binding domain (NBD)4,5,6. Different ABC proteins are found either with the domains TMD1-NBD1-TMD2-NBD2 arranged in sequence in a single polypeptide (Fig. 1a), with separate polypeptides forming each half-transporter, or with separate polypeptides forming the TMDs and NBDs. Binding and hydrolysis of ATP at the NBDs of most ABC proteins powers the active transport of solutes across membranes, while in other ABC proteins ATP hydrolysis allows the protein to function as a channel or to regulate the activity of interacting proteins1,2. Owing to these capabilities, ABC proteins are involved in numerous biological processes, such as nutrient import in prokaryotes, export of cytotoxic molecules in prokaryotes and eukaryotes, and peptide antigen presentation and cellular metal trafficking in eukaryotes1,2,7.

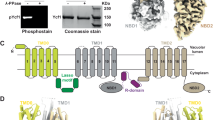
a Schematic diagram of Ycf1p showing the three transmembrane domains (TMD0, TMD1, and TMD2), the L0 linker, the nucleotide binding domains (NBD1, NBD2), the regulatory (R) region, and the coupling helices (CH1, CH2, CH3, and CH4) that are located in TMD1 and TMD2 and contact the NBDs. Phosphorylation sites in the L0 linker (S25134,121) and R region (S903, S908, T911, and S91433,35,36) are depicted with a black “P” circled in red. The loop connecting the first two transmembrane helices in TMD1 (loop-6), which contains a 17-residue insertion compared to most other ABCC proteins, is shown by a dashed curve. Cleavage of this loop by Pep4p protease (pink scissors) yields the N-terminal and C-terminal proteolytic fragments60. b 12% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) gels showing purified samples of cleaved Ycf1p-3xFLAG (left) and uncleaved Ycf1p-3xFLAG (right). The two fragments of cleaved Ycf1p (measuring at ~141 kDa and ~33 kDa from the gel on the left) are consistent with proteolytic cleavage of Ycf1p at the Pep4p site. The migration of the single band in the uncleaved Ycf1p gel is consistent with the 173.8 kDa molecular weight of Ycf1p-3×FLAG. SDS-PAGE gels shown are representative of those for 23 cleaved and 10 uncleaved Ycf1p preparations generated for this study. c Analytical gel filtration chromatography of cleaved (black trace) and uncleaved (grey trace) Ycf1p. Both cleaved and uncleaved Ycf1p samples contain dimeric forms of the protein, with cleaved Ycf1p samples containing up to ~40% dimers. d 2D class average images showing cleaved Ycf1p monomer (left) and dimer (right). e Cryo-EM maps for two different conformations of the cleaved Ycf1p monomer in which the NBDs have different degrees of separation (IFwide-α and IFwide-β). The protein domains are colored as in panel (a) and the lipids are colored in grey. f Cryo-EM map of cleaved dimeric Ycf1p, in which both protomers are in the IFwide-β conformation, with protein domains coloured as in panel (a) and lipids in grey.
Human ABC transporters are divided into seven subfamilies, designated as ABCA to ABCG, based on the amino acid sequences of the TMDs and NBDs in the core structure1,2. While many ABC proteins contain only the core structure, some subfamilies of ABC proteins possess additional domains and linkers that regulate the activity of the protein3,4,5,8. The ABCC subfamily comprises the most functionally diverse set of proteins, and includes the multidrug resistant proteins (MRPs) that function as transporters, the cystic fibrosis transmembrane conductance regulator (CFTR) Cl- channel, and the sulphonylurea receptors (SUR1, SUR2) that form the regulatory subunits in ATP sensitive K+ (KATP) channels1. All ABCC proteins contain an N-terminal extension composed of either TMD0 and an L0 linker (Fig. 1a) or just an L0 tail6. ABCC N-terminal extensions are involved in cellular trafficking, mediate protein interactions, and/or regulate ABC protein activity9,10,11,12,13,14,15. The TMD0 domain of ABC proteins is highly dynamic, and is often absent or seen only at low resolution in cryo-EM structures of isolated ABCC (and ABCB) transporters13,15,16,17,18,19,20,21,22. Many ABC proteins also possess disordered linkers, such as the one connecting NBD1 to TMD2 that is known as the regulatory (R) region in CFTR and other ABCC proteins (Fig. 1a)4,5,23,24,25,26.
Regulation of the activity of ABC proteins can occur through several different processes. Phosphorylation, which generally occurs in loops, or in the R region or other intrinsically disordered regions, has been shown to alter the structural features and interactions of the disordered region and/or affect the activity of the protein. Regulation by phosphorylation has been observed for CFTR23,24,25,27,28,29, SUR2A30,31, multidrug resistant protein 1 (MRP1)32, and Ycf1p33,34,35,36, as well as ABC proteins from other subfamilies37. ABC proteins have also been reported to oligomerize, including those from the ABCC subfamily10,11,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57. The function of oligomerization is not well understood but may provide an additional layer of regulation. Proteolytic processing is another regulatory mode and occurs in luminal loops in both human ABCA358,59 and Ycf1p60,61.
Ycf1p is a yeast ABCC protein that transports glutathione-conjugated heavy metals such as Cd2+ from the cytosol into the vacuole to detoxify the cell62. Pep4p protease-mediated cleavage of the luminal loop connecting the first two transmembrane helices in TMD1 (loop-6, Fig. 1a) yields mature Ycf1p and affects the substrate specificity of the protein60. Here we present structures of the endogenous, Pep4p-cleaved Ycf1p. Surprisingly, proteolytic cleavage promotes the formation of a well-ordered dimer of the protein, which is held together by protein-protein interactions involving TMD0, the L0 linker, and the R region, as well as by protein-lipid interactions. The dimeric Ycf1p exists alongside the monomeric protein, with ATPase activity and phosphorylation decreased in the dimer compared to the monomer. The structures reveal the molecular features of dimeric ABCC transporters, and give insight into dimerization of MRP110,42,44,53, a close homologue of Ycf1p that can functionally complement its activity63.
Results
Proteolytically cleaved Ycf1p exists in multiple well-ordered oligomeric states
To obtain the endogenous proteolytically processed (or cleaved) Ycf1p, the W303-1A yeast strain, which expresses the Pep4p protease, was modified to include a 3×FLAG tag at the 3′ end of the YCF1 gene in the chromosomal DNA. Similar to the procedure used to obtain uncleaved Ycf1p from a yeast strain lacking endogenous proteases35, the modified yeast strain was cultured and harvested, and its membranes were isolated and solubilized with the detergent dodecyl-β-D-maltopyranoside (DDM). Ycf1p was then purified with anti-FLAG affinity chromatography and exchanged into the detergent glycol-diosgenin (GDN). Because the two proteolytic fragments of Ycf1p are metabolically stable and remain associated following cleavage by Pep4p61, an intact but cleaved Ycf1p could be isolated by this procedure (Fig. 1b, left and Supplementary Fig. 1a, left). The resulting two fragments of Ycf1p have sizes consistent with proteolytic cleavage of loop-6, as described previously60. The smaller fragment corresponds in size with Ycf1p TMD0, the L0 linker, and the first TM helix of TMD1 (calculated MW of 36.7 kDa vs. determined MW from SDS-PAGE of ~33 kDa). The larger fragment is the size of the remaining TM helices 7–11 of TMD1, connecting loops, NBD1, R region, TMD2, and NBD2, as well as the 3xFLAG tag (calculated MW of 137.1 kDa vs. determined MW from SDS-PAGE of ~141 kDa). Mass spectrometry of tryptic peptides confirms that the Pep4p cleavage site is located within loop-6, and specifically in the 17-residue insertion in loop-660 (Supplementary Fig. 1b and Supplementary Data 1, Entries1316-1361 and Supplementary Data 2, Entries 84–121). However, Pep4p-mediated cleavage of Ycf1p does not occur at a single site. Rather, the mass spectrometry data indicate multiple proteolytic sites between residues D325 and N335. In contrast, endogenous Ycf1p-3×FLAG from the modified BJ2168 yeast strain35, which lacks the gene for Pep4p protease, is not proteolytically cleaved (Fig. 1b, right and Supplementary Fig. 1a, right and reference35). Size exclusion chromatography of the proteolytically cleaved Ycf1p-3×FLAG, which will herein be referred to as cleaved Ycf1p, suggests multiple oligomeric states of the protein. However, while cleaved Ycf1p exists in both monomeric and dimeric forms (Fig. 1c, black trace), Ycf1p from a ∆Pep4p strain appears as a primarily monomeric species along with some higher-order species (Fig. 1c, grey trace and reference36). The fraction of aggregated protein cannot be discerned from these data, as aggregated protein scatters light and thus appears to absorb light strongly even at low concentrations64.
Cryo-EM of the cleaved Ycf1p (Supplementary Figs. 2, 3, and 4, and Supplementary Table 1) allowed calculation of two-dimensional (2D) class average images that show both monomeric Ycf1p (Fig. 1d, left), as well as dimeric Ycf1p (Fig. 1d, right). Image analysis yielded three-dimensional (3D) maps of monomeric Ycf1p in two different conformations (Fig. 1e and Supplementary Figs. 2, 3a–d, and 4a,b) and a 3D map of a well-ordered cleaved Ycf1p dimer (Fig. 1f, and Supplementary Figs. 2, 3a,b,e, and 4c). The two maps of cleaved monomeric Ycf1p (at 3.4 Å and 3.1 Å) are in the inward-facing conformation with the substrate binding site exposed to the cytosol and the NBDs separated (Fig. 2 and Supplementary Table 1), as expected in the absence of bound nucleotide6. These two different conformations of Ycf1p differ in the separation of their NBDs. Using nomenclature introduced previously, the inward-facing conformations of cleaved Ycf1p are referred to as IFwide-α and IFwide-β on account of their similarity to the IFwide, rather than the IFnarrow, conformation of uncleaved Ycf1p in the detergent digitonin36.

a Atomic models of monomeric Ycf1p in IFwide-α (left) and IFwide-β (right) conformations, coloured as in Fig. 1. Close-up views of the interactions between lipids and each conformer are shown below. A phosphatidylethanolamine molecule (PE1) is located between TMD0 and TM helix bundle 1 on the luminal site of the protein for both the IFwide-α (lower panel) and IFwide-β (lower left panel) conformations and is shown as a stick in CPK colouring with the cryo-EM density in grey. The sidechains of interacting residues, defined as those within 4 Å of the lipid, are also shown as sticks with their cryo-EM densities. A phosphatidylethanolamine (PE2) is bound to the ABC core of the IFwide-β conformation (lower right panel). b The sequence of loop-6 and surrounding residues in TM6 and TM7 is shown on top. Below, map-in-model fit for residues in TM6, TM7, and loop-6. The backbone is shown as a blue ribbon with all modeled sidechains shown as sticks. The cryo-EM density is shown at two different thresholds, denoted by the σ values, to highlight the poor (or missing) density for loop-6. The portion of loop-6 missing in the atomic model is schematically represented by a dashed pink curve. Residues labeled in pink are from the loop-6 insertion, whereas residues labeled in blue are from TM6, loop-6, and TM7, and are found in other ABCC proteins. c Plots of the specific ATPase activities of monomeric cleaved Ycf1p (pink) and monomeric uncleaved Ycf1p (grey) in the presence of increasing concentrations of glutathione disulfide, GSSG (left), and estradiol 17β-(D)-glucuronide, E217βG (right). Bars represent the mean value with error bars showing the standard deviation derived from three independent measurements each from two separate preparations of cleaved and uncleaved Ycf1p, with the exception of assays with uncleaved Ycf1p and 0 μM, 25 μM, and 125 μM GSSG. In these cases, values are derived from three independent measurements each from three separate preparations of protein.
CryoEM of dimeric Ycf1p shows the individual Ycf1p protomers in the dimer in the IFwide-α or the IFwide-β conformations, leading to three different dimer populations: IFwide-α:IFwide-α, IFwide-β:IFwide-β, and IFwide-α:IFwide-β (Supplementary Fig. 2). However, a high-resolution structure (at 3.2 Å) could be determined only for the IFwide-β:IFwide-β Ycf1p dimer (Fig. 1f and Supplementary Fig. 2). This map allowed construction of an atomic model for dimeric Ycf1p with both protomers in the IFwide-β conformation (Fig. 3 and Supplementary Table 1). As expected for ABCC proteins8, TMD1 and TMD2 in the Ycf1p monomers and in each protomer in the Ycf1p dimer are domain-swapped such that TM bundle 1 and TM bundle 2 each possess TM helices from both halves of the ABC core (Supplementary Fig. 5).

a An atomic model of the Ycf1p dimer is shown with cylinders representing α-helices and arrows representing β-strands (centre). The protein domains are coloured as in Fig. 1. One of the protomers (on the right-hand side) is coloured in lighter shades to distinguish it from the other protomer (on the left-hand side). Bound lipids that could be modeled are shown in CPK colouring except with pink instead of grey for the hydrocarbon tails. Right, Protein interactions between protomers are shown. The protein backbone is represented by a schematic ribbon diagram and interacting residues are shown as sticks along with their cryo-EM densities. Left, Protein-lipid interactions that link the two protomers are shown. Cryo-EM densities for the lipids are shown in Supplementary Fig. 8. b The Ycf1p dimer is shown as viewed from the luminal side (right). Left, the density for additional hydrophobic molecules located at the TMD0/TMD0 interface between the two protomers is shown in purple. Cryo-EM densities for Ycf1p residues that interact with these molecules are shown in Supplementary Fig. Fig. 8. c Plots of the specific ATPase activity of monomeric and dimeric Ycf1p, without and with 100 μM GSSG. Bars represent the mean value with error bars showing the standard deviation derived from three independent measurements each from two separate preparations of cleaved Ycf1p.
As described above, size exclusion chromatography of purified uncleaved Ycf1p suggests the presence of some dimeric complexes in this sample also. In order to investigate if dimeric uncleaved Ycf1p was missed in the initial analysis of that sample35, cryo-EM data for uncleaved Ycf1p was re-processed using the same workflow that led to a high-resolution structure of the cleaved Ycf1p dimer and showed some 2D classes consistent with a Ycf1p dimer (Supplementary Fig. 6a). However, the protomers in this apparent dimer do not maintain consistent orientations relative to each other and 3D refinement of this dataset resulted in a low-resolution map with clear artifacts (Supplementary Fig. 6b–d). Therefore, it appears that monomeric Ycf1p particles can interact with each other, at least transiently, before Ycf1p is cleaved, but a well-ordered and stable dimer forms only upon cleavage of the loop-6 insertion, allowing determination of the Ycf1p dimer structure. Size exclusion data show a larger dimer peak and much smaller aggregate peak for cleaved Ycf1p compared to uncleaved Ycf1p (Fig. 1c). This observation, combined with the finding that uncleaved Ycf1p can form non-specific interactions between molecules (Supplementary Fig. 6), suggests that the uncleaved Ycf1p may be more prone to non-specific aggregation than the cleaved Ycf1p.
Nucleotide-free uncleaved and cleaved Ycf1p adopt similar conformations
Atomic models were constructed for both the IFwide-α and IFwide-β conformations of monomeric cleaved Ycf1p, comprising ~90% of the protein’s residues (Fig. 2a). Comparison of the cleaved IFwide-α and IFwide-β structures to three previously-determined structures of uncleaved Ycf1p35,36 indicates that cleavage of loop-6 (Fig. 1a) to yield the mature form of Ycf1p results in few structural changes in the protein (Supplementary Fig. 7, left and centre). The r.m.s.d. values between the cleaved IFwide-α or IFwide-β conformer and uncleaved Ycf1p determined in the same detergent system35 are both 0.49 Å when overlaying TM bundle 1, which consists of transmembrane helices 6, 7, 8, 11, 15, and 16, and coupling helices (CH) 1 and 4 (Supplementary Fig. 5). The cleaved IFwide-α and IFwide-β conformers are also very similar (Supplementary Fig. 7, right). The lack of large-scale structural changes in Ycf1p upon cleavage is consistent with the fact that loop-6 is disordered and does not interact with other regions of the protein, as evidenced by the lack density for most loop-6 residues in all inward-facing Ycf1p structures (Fig. 2b and references35,36).
Additionally, and as in the uncleaved Ycf1p structure35, the structures of cleaved Ycf1p show a phosphatidylethanolamine molecule (PE1) nestled between TMD0 and TM helix bundle 1 on the luminal side of the protein, mediating additional contacts between TMD0, TMD1, and TMD2 (Fig. 1e and Fig. 2a, lower panel for IFwide-α and lower left panel for IFwide-β). The IFwide-β conformation also contains a phosphatidylethanolamine (PE2) contacting the C-terminal end of the R region and TMD2 (Fig. 1e, right panel and Fig. 2a, lower right panel for IFwide-β). Clear density is observed for the hydrocarbon tails in these lipids (Fig. 2a, lower panels). Further, the head group density of each lipid is consistent with an ethanolamine group, leading to the assignment of these two lipids as phosphatidylethanolamine, a common lipid in yeast vacuolar membranes65,66.
Yeast viability studies with Cd2+ and As2+ metal ions indicate that proteolytic processing of loop-6 is not necessary for transport function, but enhances the activity of Ycf1p and/or alters its substrate specificity, depending on the strain used60. In order to probe the connection between proteolytic cleavage of loop-6 and substrate specificity in vitro, we measured the ATPase activity of the cleaved and uncleaved Ycf1p in the presence of glutathione disulfide (GSSG) and estradiol 17β-(D)-glucuronide (E217βG) (Fig. 2c), substrates of Ycf1p36,67, MRP168, and MRP369. These experiments were performed on cleaved and uncleaved Ycf1p monomers, which were isolated from their dimeric counterparts by gel filtration chromatography (Fig. 1c). Note that we can not perform a similar comparison for uncleaved and cleaved Ycf1p dimers on account of the low amounts of uncleaved Ycf1p dimer in our preparations (Fig. 1c) and that the uncleaved Ycf1p dimer is not stable (Supplementary Fig. 6). The ATPase activities of both cleaved and uncleaved monomeric Ycf1p increase with increasing substrate concentrations, as expected considering that substrates increase the ATPase activity of ABC transporters6. Further, the ATPase activity of cleaved monomeric Ycf1p is greater than that of the uncleaved protein across the different substrate concentrations used (Fig. 2c). However, there is no difference in the relative ATPase activities of cleaved and uncleaved Ycf1p for the different substrates. The cleaved Ycf1p monomer possesses ~2-fold greater specific activity compared to the uncleaved protein in the presence of various amounts of either GSSG or E217βG, even though these substrates have markedly different structures. Thus, these data suggest that there is no change in the substrate specificity of Ycf1p upon cleavage, at least in vitro. It is possible that the in vivo effects of loop-6 cleavage on substrate specificity seen for one yeast strain but not another requires a cellular component not in our purified Ycf1p preparations60. It is also possible that the substrate-specificity is confined to different glutathione-conjugated metals only and/or is due to different activities of cleaved and uncleaved Ycf1p at another step of transport cycle other than the ATPase step probed here.
Ycf1p dimerization involves TMD0, the L0 linker, and lipids
Examination of the Ycf1p dimer structure indicates that both protein-protein and protein-lipid interactions are critical for its formation (Fig. 3). The protein-protein interactions are formed by both membrane-embedded and cytosolic residues (Fig. 3a, right panels). Interactions within the membrane involve TMD0 residues from TM2 (e.g., W70, V73, S74, A77, A84, L88, L91) and TM5 (I171) from one protomer and TMD2 residues from TM15 (e.g., W1090′, F1094′, P1098′, F1102′, Y1105′) and TM16 (F1197′) from the opposite protomer (residues in one protomer are labeled with “ ′ ” to distinguish them from residues in the opposite protomer). Note that in the SUR proteins and in MRP3, which possess alternate orientations for TMD0 with respect to the ABC core compared to Ycf1p (and MRP1) and MRP2, either TM2 or both TM2 and TM3 of TMD0 bind TM helix bundle 135,70,71,72. Thus, the Ycf1p dimer structure, which has TM2/TM helix bundle 1 inter-protomer interactions in addition to TM4/TM helix bundle 1 intra-protomer contacts, further demonstrates the flexibility and plasticity of TMD0. Cytoplasmic loop interactions are formed between residues in the TMD0 loops (e.g., K65, R67, N69) in one protomer and residues in the L0 linker (e.g., K231′, E235′) and in the C-terminal end of the R region (e.g., E928′, E930′) in the opposite protomer. Additional cytoplasmic contacts are made between the L0 linkers of each protomer (e.g., M195, E235′). While many of the cytoplasmic loop contacts involve polar and charged amino acids, both the polar groups of side chains (e.g., the guanidino group of R67 and carboxyl group of E235′) and the non-polar groups of side chains (e.g., aliphatic tails of R67 and K231′) mediate interactions. As mentioned above, a stable dimer is only formed upon proteolytic cleavage of loop-6 (Fig. 1c, d, f and Supplementary Fig. 6). However, there are no apparent structural features in cleaved Ycf1p that would explain its propensity to form dimers. Therefore, dimerization of Ycf1p on cleavage is likely driven by interactions of the protein with the cellular environment.
Lipid molecules are also integral components of the dimer interface. As in both the monomeric cleaved Ycf1p structures presented here and a structure of uncleaved Ycf1p35, a phosphatidylethanolamine molecule (PE1) is bound between TMD0 and TM bundle 1 on the luminal side of each protomer in the dimer (Fig. 3a, upper left panels and Supplementary Fig. 8). In addition to interacting with TMD0 and TM helix bundle 1 of one protomer, also observed in monomeric Ycf1p (Fig. 2 and reference35), this phosphatidylethanolamine contacts TMD0 residues (F157′, Q167′, and I171′) in the opposite protomer in the Ycf1p dimer (Fig. 3a, upper left panels and Supplementary Fig. 8a, right). As observed for the cleaved Ycf1p IFwide-β monomer, each protomer of the Ycf1p dimer also contains a bound phosphatidylethanolamine (PE2) located where the C-terminal end of the R region meets TMD2 (Fig. 3a, centre and Supplementary Fig. 8). However, this lipid contacts the R region and TMD2 residues in one protomer only. Assignment of these lipids as phosphatidylethanolamine is based on density seen for the hydrocarbon tails and head groups, as done for the cleaved Ycf1p monomer structure. Further, and as seen in monomeric Ycf1p structures, there are additional lipids bound to TMD1 and TMD2 (Supplementary Fig. 8, light purple density).
The Ycf1p dimer includes two lipid molecules, each of which is located on the cytoplasmic side of the protein and contacts both protomers (Fig. 3a, lower left and Supplementary Fig. 8b, middle right). These lipid molecules, which are modeled as phosphatidic acid (PA) owing to clear density for the two hydrocarbon chains and the lack of observable density for their head groups, contact TMD0 residues from both protomers (W70′, S74′, T143, S182′, L189). Each phosphatidic acid also binds the L0 linker (K191, F218, S219, S224, S227, K231) and TMD2 (F1102, Y1106, R1179) of one protomer. The cryo-EM map for the cleaved Ycf1p dimer also possesses non-protein density located on both the luminal and cytoplasmic sides of the dimer interface, between TMD0 of each protomer (dark purple density in Figs. 3b and in Supplementary Fig. 8). The identities of these molecules are ambiguous, possibly because of their location on the symmetry axis, which can lead to artefacts in cryo-EM maps. However, we hypothesize that they are also lipids based on map density that is not on the symmetry axis and that resembles long hydrocarbon lipid tails. These putative lipid molecules interact with multiple TMD0 residues in TM2 (W70, I71), TM4 (F147, L150, I151, L154, H157, T158), loop-4 (G161, W163, Q167), and TM5 (F170, I171, L174, F175, V177, I178, A181, S182, L186, L189), suggesting that they are important for the stability of the Ycf1p dimer (Supplementary Fig. 8b, left panels). Notably, dimerization of Ycf1p results in only 1,613 Å2 of buried surface area and few electrostatic contacts, further suggesting that the observed lipids are needed to stabilize the dimer73.
Dimeric cleaved Ycf1p is less active than monomeric cleaved Ycf1p
In keeping with a regulatory role for Ycf1p dimerization, the cleaved monomeric and dimeric Ycf1p species exhibit different levels of ATPase activity (Fig. 3c). Cleaved monomeric Ycf1p purified by gel filtration chromatography has a specific ATPase activity of 78.2 ± 7.9 nmol Pi/mg total protein/min (mean ± s.d. from 3 independent assays from each of 2 separate protein purifications) compared to 38.2 ± 3.1 nmol Pi/mg total protein/min (mean ± s.d. from 3 independent assays from each of 2 separate protein purifications) for cleaved dimeric Ycf1p purified by gel filtration. The ~2-fold lower ATPase activity of the Ycf1p dimer compared to the Ycf1p monomer species is found both in the presence and absence of the substrate GSSG, which stimulates the ATPase activity of the monomer and dimer to the same extent. Based on this difference in ATPase activity, we checked if addition of MgATP or the non-hydrolyzable MgAMP-PNP alters the relative population of monomeric and dimeric Ycf1p species but found that it does not (Supplementary Fig. 9a), indicating that the Ycf1p dimer does not dissociate during the ATP hydrolysis reaction. Although the cleaved Ycf1p dimer is less active than the cleaved Yc1p monomer, the dimeric form constitutes a small fraction ( ~35 to 40 %) of the total Ycf1p, at least in detergent, and consequently cleavage results in an overall increase in the ATPase activity of the protein. Thus, the in vitro ATPase and size exclusion data are consistent with yeast viability studies showing that Pep4-mediated cleavage of Ycf1p enhances the activity of the protein in some yeast strains60.
One possible explanation for the difference in ATPase activities of the cleaved monomer and dimer is that the different species also possess different phosphorylation states. Mass spectrometry revealed that while S903, S908, T911, and S914 are all heavily phosphorylated in the cleaved Ycf1p monomer, only S903 is highly phosphorylated in the dimeric form of the protein (Supplementary Fig. 9b and Supplementary Data 3, Entries 3424-4042 and Supplementary Data 4, Entries 3082-3444). Phosphorylation of S908 is absolutely required for Ycf1p transport activity33,74 and phosphorylation of S903 also contributes significantly74, as indicated by yeast growth assays in the presence Cd2+. Phosphorylation of T911 can also enhance Ycf1p transport function33. Further, phosphorylation of S903, S908, and T911 are all required for Ycf1p ATPase activity, whereas phosphorylation of S914 is dispensable36,74. Thus, the higher ATPase activity of the Ycf1p monomer vs. the Ycf1p dimer is expected given the higher phosphorylation states of S903, S908, and T911 in the monomer (Supplementary Fig. 9b). Except for residues H928-K935, most of the R region is not observed in any of our cryo-EM maps of cleaved Ycf1p or is at low resolution and is modeled as poly-Ala, likely because of the flexible nature of the R region. However, regardless of the level of phosphorylation, part of the R region in both the monomeric and dimeric cleaved Ycf1p structures binds the peripheral side of NBD1, which is opposite the nucleotide binding and NBD2 dimerization interfaces (red density in Figs. 1e,f and red curve in Figs. 2a and 3a). Interactions of the R region and the peripheral side of NBD1 are also observed in structures of uncleaved, phosphorylated Ycf1p (Fig. 4ai and references35,36), albeit mediated by different R region residues in the different structures. The different R region interactions captured in the different uncleaved, phosphorylated Ycf1p structures are likely because the intrinsically disordered nature of the R region allows for multiple NBD1/R region binding events35,36.

a Schematic diagrams of the NBD1/R region interactions in the fully phosphorylated Ycf1p monomer (i), the non-phosphorylated Ycf1p monomer (ii), and the partly phosphorylated Ycf1p dimer (iii). Possible locations for unobserved portions of the R region are shown by a diffuse red density to denote its multiple possible interactions, with the most likely locations of the R region in dark red and the less likely locations in light red. The fully phosphorylated R region likely binds the peripheral face of NBD1, allowing for binding (i). ATP is shown with the adenine base and ribose in blue and the phosphates depicted as red circles. The non-phosphorylated R region can bind in the substrate binding cavity74 and also contact the ATP binding site of NBD135 (ii), preventing nucleotide binding. The partly phosphorylated R region in the Ycf1p dimer can interact with the ATP binding site or with the peripheral side of NBD1 (iii). ATP binds when the R regions binds the peripheral side of NBD1. b Schematic diagram of the cleaved Ycf1p dimer (top) and the proposed MRP1 dimer (bottom). Two orientations are shown for each protein. The Ycf1p dimer is viewed from the lumen (top left) and from within the membrane (top right). Similarly, the MRP1 dimer is shown from the extracellular side of the plasma membrane (bottom left) and from within the membrane (bottom right). Transmembrane helices of the cleaved Ycf1p dimer cryo-EM structure and the MRP1 dimer model are shown as cylinders. Transmembrane helices in TMD0 are coloured in orange for Ycf1p, as in Fig. 1a, and in coral for MRP1. As in Fig. 1a, transmembrane helices in TMD1 and TMD2 are in blue and green, respectively, for both proteins. Transmembrane helices in one protomer are labeled with “ ‘ ” to distinguish them from those in the opposite protomer. While the structure of the ABC core is similar in Ycf1p and MRP1, the orientation of TMD0 helices differ slightly between the two proteins, leading to different inter-protomer TMD0 contacts between the Ycf1p and MRP1 dimers.
Multiple interactions also occur for the R region when it is not phosphorylated (Fig. 4aii). Structures of de-phosphorylated Ycf1p74 and the related ABCC transporter MRP271 show part of the R region, including S903, S908, and T911 in Ycf1p74, in the substrate-binding cavity, thus inhibiting ABC transporter activity. In order to become phosphorylated, the R region must disengage from the substrate-binding cavity so that is accessible to cellular kinases. Thus, the non-phosphorylated R region must also sample multiple structural states. Notably, previous work on Ycf1p35 and the ABCC protein CFTR24,26,75,76,77,78 show that the non-phosphorylated R region can contact NBD1 at the ATP binding and NBD2 dimerization sites, and that these interactions are altered with R region phosphorylation24,26,35,75,78,79. At least for CFTR, this disruption leads to greater ATPase activity and ABC protein function27.
It is also possible that the unresolved portions of the singly and fully phosphorylated R region mediate different interactions with NBD1 in the cleaved Ycf1p dimer and monomer, respectively. To test this possibility, we performed Trp fluorescence quenching assays using I¯ and acrylamide for NBD1 linked to the R region with the S903D phosphomimetic mutation (NBD1-RS903D) and NBD1-R with the S903D, S908D, T911E, and S914 phosphomimetic mutations (NBD1- RS903D/S908D/T911E/S914D) (Supplementary Fig. 10a, left). Phosphomimetic mutations have been used to probe the effects of phosphorylation on Ycf1p activity74. These data are compared to previous quenching data for isolated NBD1, NBD1-R, and NBD1-RS908D/T911E 35. Because Trp residues are found only in NBD1 and in different environments (Supplementary Fig. 10a, right) these experiments can assess interactions of the different phosphorylated forms of the R region with different sites in NBD135.
The differences in I¯ and acrylamide quenching between the free and ATP-loaded states of the proteins (Supplementary Fig. 10b) are expected, as the Trp residues in NBD1 affected by I¯ and acrylamide are near regions of the protein that bind ATP6. Residue W701 is in the γ-phosphate linker and W849 is near the Walker A and B motifs. The smaller KSV values for NBD1-RS903D and NBD1-RS903D/S908D/T911E/S914D compared to NBD1 show that both singly and fully phosphorylated R region lower the surface accessibilities of W701 and W849 in NBD1, likely through direct binding of the R region to NBD1. A similar effect is seen for NBD1-R and NBD1-RS908D/T911E (Supplementary Fig. 10b and reference35). Additionally, the smaller KSV values for acrylamide quenching of NBD1-RS903D and NBD1-RS903D/S908D/T911E/S914D compared to NBD1 and NBD1-R suggest that singly and fully phosphorylated forms of R region bind W849 on the peripheral face of NBD1 (Fig. 4ai, aiii), consistent with the cryo-EM structures (Fig. 1e,f) and Trp quenching data on NBD1-RS908D/T911 (Supplementary Fig. 10b and reference35).
The KSV values for I¯ quenching, which differ between most proteins in both the nucleotide-free and MgATP-bound states, suggest that interactions between NBD1 (at W701) and the R region depend on the phosphorylation state of the R region. In particular, the KSV value for NBD1-RS903D in the MgATP-loaded state is slightly lower than that for NBD1-RS908D/T911E and NBD1-RS903D/S908D/T911/S914D. Thus, the R region in the Ycf1p dimer, which is phosphorylated primarily at S903 only, may more stably contact NBD1 in a manner that prevents NBD1/NBD2 dimerization (Fig. 4aiii), at least partly, leading to the lower ATPase activity of the Ycf1p dimer (Fig. 3c). One model to explain binding of the R region to multiple NBD1 sites is that specific R region residues contact specific NBD1 residues. Alternatively, the R region may engage NBD1 in a more dynamic complex in which multiple R region residues interact with NBD1, as seen for interactions of other disordered regions with their folded protein binding partners80,81. Such a dynamic complex may explain the small differences in binding of the singly and fully phosphorylated R region to NBD1 shown by the quenching data. The diffuse red density in Fig. 4a indicates the proposed transient interactions between the R region, in different phosphorylation states, with different parts of Ycf1p, as suggested by fluorescence quenching experiments (Supplementary Fig. 10), structures of cleaved Ycf1p (Fig. 1e) and previously published structures of uncleaved Ycf1p35,36,74.
Discussion
Functions of the Ycf1p dimer
Because of lower ATPase activity, phosphorylation levels, and abundance of the cleaved Ycf1p dimer compared to the monomer (Fig. 1c, Fig. 3c, and Supplementary Fig. 9b), it is possible that the Ycf1p dimer serves as a reservoir of protein for when more Ycf1p needed, such as in environments with large quantities of toxic metals. It is also possible that the Ycf1p dimer engages in activities other than transport. In addition to its transport function, Ycf1p recruits the soluble SNARE protein Vam7p to the vacuole during vacuolar fusion82. This Ycf1p activity depends on a functional Walker A motif in NBD1, which binds ATP6 and which is occluded, at least transiently, in the non-phosphorylated protein (Fig. 4aii, Supplementary Fig. 10b, and reference35). Notably, vacuolar fusion is also regulated by lipids83,84,85, such as phosphatidic acid, a lipid found in the cleaved Ycf1p dimer interface (Fig. 3a and Supplementary Fig. 8b). Further, the lipid composition of vacuoles changes during the fusion process83,84,85. Ycf1p has also been shown to interact with membrane and cytosolic proteins other than Vam7p86,87, and these protein-protein interactions may change depending on the phosphorylation state of the protein, as in other ABC transporters4. Thus, the oligomeric state of the protein, which may be modulated by lipids, with its different phosphorylation state, could affect Ycf1p interactions and downstream functions.
Lipids form critical interactions at interfaces of membrane protein oligomers
The presence of lipid molecules at the Ycf1p dimer interface has also been observed for other membrane protein oligomers. Studies of other α-helical transmembrane proteins indicate that the presence of lipids at protomer interfaces is inversely correlated with oligomeric stability73, as assessed by buried surface area and number of electrostatic contacts between protomers. Further, for transmembrane proteins that depend on lipids for oligomeric stability, titration of the stabilizing lipid increases the stability and population of the transmembrane protein oligomer88. The limited buried surface area and electrostatic interactions in the cleaved Ycf1p dimer also imply that the dimer interface is dynamic. Thus, it is possible that dimerization of Ycf1p will change with changes in lipid composition, which has been described for other transmembrane proteins73. Further, changes in oligomerization may regulate the function of Ycf1p, as it does for other ABC transporters11,43,46,51,52,53,55,56.
Ycf1p dimer structure is consistent with predicted dimers of other ABCC proteins
While high-resolution cryo-EM structures have been determined for MRP1 only in the monomeric state16,17,18,19, studies of purified protein samples and studies in living cells indicate that MRP1 can also dimerize10,42,44,53. The dimerization interface of MRP1 has been proposed to involve TMD010, and specifically that TM5 and the preceding extracellular loop (or loop-4 as defined in Fig. 1a) mediate the interactions between MRP1 protomers11. Experiments also indicate that the L0 linker is involved in MRP1 dimerization11, as with Ycf1p (Fig. 3a, lower right panel). Notably, co-expression of a TMD0-L0 fragment of MRP1, which can associate with full length MRP1, inhibits MRP1 transport activity10. One possible explanation for these data is that the MRP1 monomer is functional unit and that dimerization regulates MRP1 function10, which could also be the case for Ycf1p.
The Ycf1p dimer structure allows construction of a model of the MRP1 dimer that is compatible with these findings (Fig. 4b). This model of the MRP1 dimer was generated by aligning the ABC cores of two MRP1 proteins to the ABC cores of the two Ycf1p protomers in the Ycf1p dimer. Because the position of TMD0 relative to the ABC core differs slightly between these proteins35, TM5 in MRP1 is at the dimer interface instead of TM2 in Ycf1p. Notably, and consistent with our finding of reduced ATPase activity for dimeric Ycf1p (Fig. 2c), co-expression of the N-terminal extension of MRP1 with full-length MRP1 reduces its transport activity10.
Similar to MRP1, only monomeric structures of CFTR have been determined previously76,77,78,79,89,90. However there is substantial evidence that CFTR (or ABCC7) exists in both monomeric and dimeric forms in cells41,43,45,47,48,53 and that at least part of the CFTR dimerization interface is mediated by interactions between residues in the R region of each protomer47. The fact that there are some R region/R region contacts between Ycf1p protomers in the dimer further establishes that the Ycf1p dimer structure is a good model for dimers of other ABCC proteins.
Oligomerization interfaces vary between different classes of ABC transporters
ABC transporters outside the C subfamily have also been shown to oligomerize. Dimers and higher order oligomers have been detected in cells and in membrane preparations for the full-length transporters ABCA146,51,55, ABCA356, and P-glycoprotein (or ABCB1)38,39,40, as well as the dimeric half-transporter ABCG249,50,52,54. The oligomeric state of ABCA transporters can change during the transport cycle (e.g., ABCA1)51 or based on the cellular location of the protein (e.g., ABCA356). Further, as seen for Ycf1p (Supplementary Fig. 9b), the phosphorylation state of P-glycoprotein can differ in different oligomeric states40. However, as with MRP116,17,18,19 and CFTR76,77,78,79,89,90, high resolution structures have been determined only for monomeric states of ABCA191,92, ABCA359, P-glycoprotein93,94, and the dimeric form of the half transporter ABCG295. The lack of dimeric structures for these ABC transporters, for which oligomerization has been detected biochemically, further suggests that oligomerization is transient or that the oligomeric state is unstable. In addition, ABCA1, ABCA3, ABCB1, and ABCG2 lack the N-terminal extension found in ABCC proteins and consequently ABCA, ABCB, and ABCG proteins must oligomerize differently than Ycf1p. Instead, ABCG2 has been suggested to oligomerize using TM helices and loops in TMD152,54, while ABCA1 dimerization employs the large extracellular domains53 that are hallmarks of the ABCA family4,6.
Multimeric complexes of ABC proteins are also found in prokaryotes, such as the complex between six copies of the ABC half-transporter HlyB, which are arranged as three HlyB homodimers, and three copies of the single pass transmembrane protein HlyD57. HlyB and HlyD form part of a type I secretion system in Gram negative bacteria96. Similar to Ycf1p, associated lipids mediate critical interactions between the HlyB homodimers. However, and in contrast to Ycf1p, HlyB transmembrane helices do not participate in complex formation and instead protein-protein contacts are formed between HlyB and HlyD transmembrane helices. Further, and again unlike Ycf1p, the HlyB NBDs also form part of the interface between the HlyB homodimers. Because the Ycf1p dimer interface is partially formed by TMD0, the NBDs in different Ycf1p protomers are too far apart to contact one another in the inward-facing conformation seen here (Figs. 1, 3) and likely also in other states of the transporter.
The studies presented here define the molecular structure of an ABCC transporter dimer. The dimeric protein displays different activity and phosphorylation compared to the monomeric form of the protein. Protein-protein contacts in the dimer involve the variably-oriented TMD035 and the intrinsically disordered R region35,36, highlighting the dynamic nature of ABC oligomerization. The structure also shows that bound interfacial lipids are likely integral to the stability of the Ycf1p dimer. These findings emphasize the flexibility of ABC dimers and explain why both monomeric and dimeric Ycf1p species are present in our preparations and likely also in cells, as seen for other ABC transporters11,38,39,41,42,43,45,46,47,48,49,51,53,54,55. The Ycf1p dimer structure provides a model for understanding dimerization, and regulation by oligomerization, in other ABCC transporters.
Methods
Construction and growth of the Ycf1p-3×FLAG Yeast Strain, and purification of Ycf1p-3×FLAG
The S. cerevisiae strain W303-1A (MATa {leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15}) was further modified by insertion of sequence encoding a C-terminal 3×FLAG tag and a URA3 marker downstream of the YCF1 gene, as done previously in the BJ2168 strain35. The modified W303-1A is named ySCB2.
Growth of the ySCB2 yeast strain and subsequent purification of cleaved Ycf1p-3×FLAG protein followed the procedure used for purifying uncleaved Ycf1p-3×FLAG from the strain ySCB135, with minor modifications. A 10 or 11-L culture of the ySCB2 yeast strain was grown in YPD medium (1% [w/v] yeast extract, 2% [w/v] peptone, 2% [w/v] dextrose), supplemented with 100 mg/mL ampicillin and 0.02% (v/v) antifoam, at 30 °C in a New Brunswick BioFlow fermenter. The culture was grown for a total of ~48 h to an optical density at 660 nm (OD600) of 6.0–10.0. Yeast cells were pelleted by centrifugation at 4000 × g for 15 min at 4 °C and resuspended in 1 mL of lysis buffer (8 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4, 0.24 g/L KH2PO4, 80 g/L sucrose, 20 g/L D-sorbitol, 20 g/L D-glucose, 5 mM 6-aminocaproic acid, 5 mM benzamidine, 5 mM EDTA, and 10 mg/L PMSF at pH 7.4) per gram of cells. The resuspended yeast cells were lysed with a Biospec Beadbeater at 4 °C, with 0.5 mm glass beads in 5 cycles of 1 min on, 2 min off. Cellular debris was removed by centrifugation at 4000 × g for 15 min at 4 °C, after which the membranes were collected via ultracentrifugation (Beckman L-90K) at 152,947 × g for 40 min at 4 °C. Membranes were resuspended, using either a Dounce or tissue homogenizer, in 0.5 mL of lysis buffer per gram of harvested cells and then frozen in liquid nitrogen and stored at −80 °C.
Frozen membranes were thawed in a tepid water bath and solubilized with 1% (w/v) n-dodecyl-β-D-maltopyranoside (DDM; Anatrace) with mixing for ∼1 h. Solubilized membranes were ultracentrifuged at 181,078 × g for 70 min (Beckman L-90K) and the supernatant containing Ycf1p was filtered through a 0.45 μm syringe filter (Pall) and applied to a 1.5 mL M2 affinity gel column (Millipore Sigma) that was pre-equilibrated with DTBS (50 mM Tris HCl, 150 mM NaCl, 0.04% [w/v] DDM, pH 8.0). The column was washed with 12 column volumes of DTBS, followed by 8 column buffers of GTBS (50 mM Tris HCl, 150 mM NaCl, 0.006% [w/v] glyco-diosgenin [GDN; Anatrace], pH 8.0). Ycf1p was eluted with 6 column volumes of GTBS containing 150 μg/mL 3×FLAG peptide. An additional five column volumes of GTBS without 3×FLAG peptide was applied to wash residual Ycf1p from the column. The use of GDN enables acquisition of cryo-EM images of membrane proteins with a good distribution of particles with excellent image contrast97,98,99,100,101,102,103. Elution fractions containing Ycf1p were concentrated with a 100-kDa molecular weight cut off (MWCO) Amicon Ultra centrifugal filter (Millipore Sigma). Samples for cryo-EM were concentrated to ~5 mg/mL and used immediately, while samples for biochemical assays were concentrated to 1.2–2.2 mg/mL, flash-frozen in liquid nitrogen, and stored at −80 °C until use. When gel filtration chromatography was used, purified Ycf1p samples (100 μL) were loaded onto a Superose 6 Increase (10/300 GL) column that was pre-equilibrated with GTBS. The column was run at a flow rate of 0.1 mL/min at 4 °C using an ÄKTA Pure system. Where indicated, samples of purified cleaved Ycf1p were pre-incubated with 2 mM MgCl2 and either 2 mM ATP or AMP-PNP before applying the sample to the column.
Malachite Green Assay
ATPase activity of Ycf1p was measured using the Malachite Green assay104, which detects the amount of inorganic phosphate in solution. The uncleaved and cleaved Ycf1p monomer and dimer were separated by size exclusion chromatography, and either the monomer fraction or both the monomer and dimer fractions were used fresh in the ATPase assay. Samples of 20 µL that contained either 0.114 µg of the uncleaved Ycf1p monomer, the cleaved Ycf1p monomer, or the cleaved Ycf1p dimer in 50 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) pH 8.0, 3 mM MgCl2, 0.006% (w/v) GDN, and 2 mM ATP were prepared. The ATPase activity in the presence of substrate was measured by including 100 μM glutathione disulfide (GSSG), or with varying concentrations of GSSG or estradiol 17β-(D)-glucuronide (E217βG). Control samples in the reaction buffer, with and without substrate, were prepared and phosphate standard samples (from 0–40 μM) were generated from a phosphate stock (Sigma). All samples were incubated at 30 °C for 90 min, then promptly diluted 8-fold in MilliQ H2O and flash frozen in liquid nitrogen until analysis.
Samples were thawed at room temperature for ~20 min and 80 µL of each reaction were transferred to a 96-well plate. Each solution was then mixed with 20 µL of the phosphate detection reagent (0.01% [w/v] Malachite Green, 0.17% [v/v] Tween 20, 1.48% [w/v] ammonium molybdate) and incubated at room temperature for 20 min. The colour development reaction was quenched with 11 µL of a 34% (w/v) sodium citrate, the samples were mixed, incubated for 7 min at room temperature, and the absorbances measured at 620 nm with a SpectraMax i3X Multi-Mode Assay Microplate Reader. The amount of phosphate produced was determined based on a standard curved using the phosphate standards. Each measurement of phosphate production was corrected for the amount of phosphate produced in a sample that contained all components except the protein. ATPase assays were performed in triplicate for each of two independent Ycf1p preparations.
Mass spectrometry analysis of Pep4p cleaved of Ycf1p
In-gel trypsin digestion of the two SDS-PAGE bands (at ~32 kDa and ~140 kDa) from purified Ycf1p samples followed by MS/MS was performed by the SPARC BioCentre Laboratory, Sick Kids Hospital, Toronto, Canada. Briefly, 10 μl of ~0.35 mg/mL purified Ycf1p was loaded onto a 12% SDS-PAGE acrylamide gel. The gel was stained with Coomassie Brilliant Blue and destained with MilliQ. After dehydrating the gel bands with acetonitrile, the Ycf1p fragments were reduced by incubation with 10 mM dithiothreitol (DTT) at 60 °C for 1 h and further reacted with 55 mM iodoacetamide at room temperature for 20 min in the dark. The gel bands were re-dehydrated with acetonitrile, and the Ycf1p fragments were subsequently digested with trypsin (0.5 μg) at 37 °C overnight. The resulting peptides were subjected to LC MS/MS. The raw MS files were analyzed with PEAKS Studio (v10.5) software using the S. cerevisiae database and the sequence of Ycf1p-3×FLAG protein. The parent and fragment mass tolerances were set to 10 ppm and 0.02 Da, respectively, and only tryptic peptides with a maximum of three missed cleavage sites were included. The addition of a carbamidomethyl group on Cys was specified as a fixed modification, while deamidation of Asn and Gln, oxidation of Met, acetylation of the N terminus, and phosphorylation of Ser, Thr, or Tyr were set as variable modifications. Data was visualized using Scaffold (v5.3.3).
Mass spectrometry analysis of phosphorylation of Ycf1p
Similar to identification of the Pep4p cleavage site, in-gel trypsin digestion followed by MS/MS was used to analyze phosphorylation states in the cleaved Ycf1p monomer and dimer. However, in this case, purified Ycf1p was subjected to 4–16% Bis-Tris blue native PAGE. Gel bands corresponding to the cleaved Ycf1p monomer and the cleaved Ycf1p dimer were excised, dehydrated with acetonitrile, and incubated with DTT and iodoacetamide, as above. Following further dehydration of the gel bands, the in-gel cleaved Ycf1p monomer and dimer proteins were digested with trypsin and resulting peptides analyzed by LC MS/MS using the parameters described above, using Proteome Discoverer (v2.5.0.4.00) software. Phosphorylated and non-phosphorylated peptides were detected for both the cleaved Ycf1p monomer and dimer, and the ratio of the number of fragments detected from the phosphorylated and non-phosphorylated peptides, when the total number of fragments is high, approximately indicates the extent of phosphorylation35.
Cryo-EM Sample Preparation and Data Collection
Freshly purified Ycf1p from the ySCB2 strain was applied to homemade nanofabricated holey gold grids105 with ~1.2 µm holes. Grids were glow-discharged in air (Pelco easiGlow) for 2 min prior to the application of ~1.5–3 µL of sample. Samples were frozen with a Leica Automatic Grid Plunging device with 1.5 s blotting time at 4 °C with a relative humidity of ~100%. A total of 6,195 images were collected on a Titan Krios G3 electron microscope (Thermo Fischer Scientific) operating at 300 kV with a prototype Falcon 4i camera. Data collection was monitored using cryoSPARC Live106 and automated with the EPU software package. Two sets of movies were recorded. One set was recorded for 6.97 s, with a total exposure and exposure rate of 46.6 e/Å2 and 6.89 e/pixels/s, respectively. The second set of movies was recorded for 8.94 s, with a total exposure of and exposure rate of 46.0 e/Å2 and an exposure rate of 5.89 e/pixels/s.
Imaging analysis and model building/validation
CryoSPARC v2106 was used post-data collection for image processing. Patch motion correction was applied to the first 29 exposure fractions of each movie and the contrast transfer function (CTF) of the resulting aligned images were estimated with patch CTF estimation. Movies were manually curated removing exposures with undesirable ice thickness, poor CTF fits, and sample aggregation, which resulted in a total of 6195 movies used in particle analysis. Blob selection with particle dimensions between 250 and 350 Å was employed to generate initial 2D classes of monomeric and dimeric Ycf1p particles to use as templates for particle selection. Initial rounds of 3D particle sorting as well as non-uniform refinement107 were performed to remove non-Ycf1p particles and separate the monomeric and dimeric Ycf1p particles. Local motion correction108 was then applied to improve particle alignment and further rounds of 3D particle sorting and non-uniform refinement were conducted. 3D variability analysis109 was used to identify different modes of nucleotide binding domain (NBD) motion and generated templates with varying NBD distances, which were used in 3D sorting of conformations of both monomeric and dimeric Ycf1p. Final rounds of 3D particle sorting and non-uniform refinement provided five final cryo-EM maps (Supplementary Fig. 2). Cryo-EM maps for monomeric Ycf1p in the IFwide-α and IFwide-β conformations were at 3.4 Å and 3.1 Å, respectively. A cryo-EM map at 3.2 Å was obtained for a Ycf1p dimer in the IFwide-βːIFwide-β conformation. In contrast, maps of Ycf1p dimers in the Ycf1p IFwide-αːIFwide-α and IFwide-αːIFwide-β conformation are at 6.3 and 6.8 Å resolution, respectively. Models for the Ycf1p IFwide-α and IFwide-β monomers, and for the IFwide-βːIFwide-β Ycf1p dimer were built in Coot110 using PDB 7MPE35 as a starting model. On account of the lower resolution of the NBDs and part of the R region, these regions were modeled as poly-Ala. Refinement was performed in both Phenix111,112 and iSOLDE113. Lipids were added using the Coot ligand library110 or imported using the SMILES string.
Preparation of NBD1-RS903D and NBD1-RS903D/S908D/T911E/S914D proteins
Preparation of isolated NBD1-RS903D and NBD1-RS903D/S908D/T911E/S914D was accomplished as previously described for other Ycf1p NBD1 and NBD1-R proteins35. Briefly, NBD1-RS903D and NBD1-RS903D/S908D/T911E/S914D were expressed in E. coli as fusion proteins with an N-terminal His6-SUMO tag. The His6-SUMO-NBD1-RS903D and His6-SUMO-NBD1-RS903D/S908D/T911E/S914D were first isolated using Ni-NTA affinity chromatography and then subjected to dialysis to remove the high concentrations of imidazole that was used to elute the proteins from the Ni-NTA column. His6-Ulp protease was included in the dialysis bag in order to remove the His6-SUMO tag from the NBD1-R proteins. The resulting mixture containing the His6-SUMO tag and NBD1-RS903D (or NBD1-RS903D/S908D/T911E/S914D) was applied to a Co2+-agarose column. The flow through and wash fractions from the Co2+-agarose column, which contained the isolated NBD1-RS903D (or NBD1-RS903D/S908D/T911E/S914D) were concentrated and applied to a size exclusion column (Superdex 75, Cytiva) that was pre-equilibrated in 20 mM Na3PO4, 150 mM NaCl, 2% (v/v) glycerol, 2 mM DTT, 2 mM MgCl2, 2 mM ATP, 100 mM benzamidine, 100 mM ε-amino n-caproic acid, pH 7.3 to purify the protein to homogeneity. Protein concentrations were determined using the Bradford protein assay114.
Trp fluorescence quenching experiments
Fluorescence quenching experiments were performed on 2 μM NBD1-RS903D and NBD1-RS903D/S908D/T911E/S914D, as previously described for NBD1 and NBD1-R35. Briefly, the purified NBD1-RS903D and NBD1-RS903D/S908D/T911E/S914D proteins were exchanged into the fluorescence buffer (20 mM Na3PO4, 150 mM NaCl, 2% [v/v] glycerol, pH 7.3) by size exclusion chromatography. MgATP-bound and nucleotide-free states of NBD1-RS903D and NBD1-RS903D/S908D/T911E/S914D were generated by adding 2 mM MgATP and 2 mM EDTA, respectively, to the protein eluents. Fresh reductant (2 mM DTT) was also added to the eluents.
Trp fluorescence quenching experiments were performed on a Fluoromax-4 fluorimeter (Horiba Scientific) equipped with an automated titrator and a Peltier temperature control unit. Trp emission spectra were recorded from 310 nm to 450 nm using an excitation wavelength of 289 nm, and excitation and emission slit widths of 2 nm and 5 nm, respectively. All spectra were recorded at 15 °C. For I¯ quenching experiments, successive additions of 0.8 M KI in the fluorescence buffer, which was prepared fresh and contained 0.2 mM Na2SO3115, was added to the protein solutions. The change in ionic strength and sample dilution were accounted for with a parallel titration using 0.8 M KCl. The initial fluorescence (\({F}_{o}\)) was divided by the fluorescence (\(F\)) at each KI or KCl titration point to give \({({F}_{o}/F)}_{{KI}}\) or \({({F}_{o}/F)}_{{KCl}}\), respectively. A plot of the change in fluorescence, (Fo/F)KI/(Fo/F)KCl against [I-] was used to determine the Stern-Volmer constant (\({K}_{{SV}}\))116. Similarly, the acrylamide quenching experiments were performed by successive addition of a solution containing 1.6 M acrylamide in the fluorescence buffer to the protein samples. A control titration with buffer was performed to account for sample dilution. Absorbances at 298 nm (A298) and 352 nm (A352) were measured after each addition of acrylamide to correct the observed fluorescence for the inner filter effect with the equation: \({F}_{{corrected}}={F}_{{observed}}\frac{{10}^{{A}_{298}}+{10}^{{A}_{352}}}{2}\) 117,118. The A298 and A352 values at the highest acrylamide concentrations were less than 0.050. Analogous to the I¯ quenching experiments, \({F}_{{corrected}}\) was divided fluorescence for the dilution control (\({F}_{{dilution}}\)), and the value for \({F}_{{corrected}}/{F}_{{dilution}}\) was plotted against [acrylamide] to obtain the \({K}_{{SV}}\) value.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support this study are available from the corresponding authors upon request. Cryo-EM maps have been deposited in the Electron Microscopy Data Bank with accession codes EMD-40710 (cleaved Ycf1p monomer in the IFwide-α conformation), EMD-40709 (cleaved Ycf1p monomer in the IFwide-β conformation), and EMD-40692 (cleaved Ycf1p dimer in the IFwide-β conformation). The coordinates have been deposited in the Protein Data Bank with accession codes 8SQM (cleaved Ycf1p monomer in the IFwide-α conformation), 8SQL (cleaved Ycf1p monomer in the IFwide-β conformation), and 8SQ0 (cleaved Ycf1p dimer in the IFwide-β conformation). Source data for Figs. 1b, c, 2c, and 3c, and Supplementary Figs. 1a, 9a, and 10b are provided. Mass spectrometry data have been deposited, via the PRIDE partner repository119, to ProteomeXChange Consortium120 with dataset identifiers PXD057166 and PXD057187. Source data are provided with this paper.
References
Dean, M., Moitra, K. & Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Hum. Mutat. 43, 1162–1182 (2002).
Vasiliou, V., Vasiliou, K. & Nebert, D. W. Human ATP-binding cassette (ABC) transporter family. Hum. Genomics 3, 281–290 (2009).
Ford, R. C. & Beis, K. Learning the ABCs one at a time: structure and mechanism of ABC transporters. Biochem. Soc. Trans. 47, 23–36 (2019).
Bickers, S. C., Sayewich, J. S. & Kanelis, V. Intrinsically disordered regions regulate the activities of ATP binding cassette transporters. Biochim. Biophys. Acta - Biomembr 1862, 183202–183202 (2020).
Ford, R. C., Marshall-Sabey, D. & Schuetz, J. Linker domains: why ABC transporters ‘live in fragments no longer’. Trends Biochem. Sci. 45, 137–148 (2020).
Thomas, C. & Tampe, R. Structural and mechanistic principles of ABC transporters. Annu. Rev. Biochem. 89, 605–636 (2020).
Davidson, A. L., Dassa, E., Orelle, C. & Chen, J. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 72, 317–364 (2008).
Thomas, C. et al. Structural and functional diversity calls for a new classification of ABC transporters. FEBS Lett 594, 3767–3775 (2020).
Bandler, P. E., Westlake, C. J., Grant, C. E., Cole, S. P. C. & Deeley, R. G. Identification of regions required for apical membrane localization of human multidrug resistance protein 2. Mol. Pharmacol. 74, 9–19 (2008).
Yang, Y. et al. Regulation of function by dimerization through the amino-terminal membrane-spanning domain of human ABCC1/MRP1. J. Biol. Chem. 282, 8821–8830 (2007).
Yang, Y., Mo, W. & Zhang, J. T. Role of transmembrane segment 5 and extracellular loop 3 in the homodimerization of human ABCC1. Biochemistry 49, 10854–10861 (2010).
Kiss, K. et al. Role of the N-terminal transmembrane domain in the endo-lysosomal targeting and function of the human ABCB6 protein. Biochem. J. 467, 127–139 (2015).
Wang, C. et al. Cryo-electron microscopy structure of human ABCB6 transporter. Protein Sci. 12, 2363–2374 (2020).
Bakos, É. et al. Functional multidrug resistance protein (MRP1) lacking the N-terminal transmembrane domain. J. Biol. Chem. 273, 32167–32175 (1998).
Blees, A. et al. Structure of the human MHC-I peptide-loading complex. Nature 551, 525–528 (2017).
Johnson, Z. L. & Chen, J. Structural basis of substrate recognition by the multidrug resistance protein MRP1. Cell 168, 1075–1085.e9 (2017).
Johnson, Z. L. & Chen, J. ATP binding enables substrate release from multidrug resistance protein 1. Cell 172, 81–89.e10 (2018).
Wang, L. et al. Characterization of the kinetic cycle of an ABC transporter by single-molecule and cryo-EM analyses. Elife 9, e56451 (2020).
Pietz, H. L. et al. A macrocyclic peptide inhibitor traps MRP1 in a catalytically incompetent conformation. Proc. Natl. Acad. Sci. USA. 120, e2220012120 (2023).
Oldham, M. L., Grigorieff, N. & Chen, J. Structure of the transporter associated with antigen processing trapped by herpes simplex virus. Elife 5, e21829 (2016).
Oldham, M. L. et al. A mechanism of viral immune evasion revealed by cryo-EM analysis of the TAP transporter. Nature 529, 537–540 (2016).
Park, J. G. et al. The lysosomal transporter TAPL has a dual role as peptide translocator and phosphatidylserine floppase. Nat. Commun. 13, 5851 (2022).
Dulhanty, A. M. & Riordan, J. R. Phosphorylation by cAMP-dependent protein kinase causes a conformational change in the R domain of the cystic fibrosis transmembrane conductance regulator. Biochemistry 33, 4072–4079 (1994).
Baker, J. M. R. et al. CFTR regulatory region interacts with NBD1 predominantly via multiple transient helices. Nat. Struct. Mol. Biol. 14, 738–745 (2007).
Ostedgaard, L. S., Baldursson, O., Vermeer, D. W., Welsh, M. J. & Robertson, A. D. A functional R domain from cystic fibrosis transmembrane conductance regulator is predominantly unstructured in solution. Proc. Natl. Acad. Sci. USA. 97, 5657–5662 (2000).
Bozoky, Z. et al. Regulatory R region of the CFTR chloride channel is a dynamic integrator of phospho-dependent intra- and intermolecular interactions. Proc. Natl. Acad. Sci. USA. 110, 4427–4436 (2013).
Li, C., Ramjeesingh, M. & Bear, C. E. Purified cystic fibrosis transmembrane conductance regulator (CFTR) does not function as an ATP channel. J. Biol. Chem. 271, 11623–11626 (1996).
Bozoky, Z. et al. Synergy of cAMP and calcium signaling pathways in CFTR regulation. Proc. Natl. Acad. Sci. USA. 114, E2086–E2095 (2017).
Chin, S., Hung, M. & Bear, C. E. Current insights into the role of PKA phosphorylation in CFTR channel activity and the pharmacological rescue of cystic fibrosis disease-causing mutants. Cell. Mol. Life Sci. 74, 57–66 (2017).
Quinn, K. V., Giblin, J. P. & Tinker, A. Multisite phosphorylation mechanism for protein kinase A activation of the smooth muscle ATP-sensitive K+ channel. Circ. Res. 94, 1359–1366 (2004).
Sooklal, C. R., López-Alonso, J. P., Papp, N. & Kanelis, V. Phosphorylation Alters the Residual Structure and Interactions of the Regulatory L1 Linker Connecting NBD1 to the Membrane-Bound Domain in SUR2B. Biochemistry 57, 6278–6292 (2018).
Shukalek, C. B. et al. Arsenic triglutathione [As(GS)3] transport by multidrug resistance protein 1 (MRP1/ABCC1) is selectively modified by phosphorylation of Tyr920/Ser921 and glycosylation of Asn19/Asn23. Mol. Pharmacol. 90, 127–139 (2016).
Eraso, P., Martinez-Burgos, M., Falcón-Pérez, J. M., Portillo, F. & Mazón, M. J. Ycf1-dependent cadmium detoxification by yeast requires phosphorylation of residues Ser908 and Thr911. FEBS Lett 577, 322–326 (2004).
Paumi, C. M., Chuk, M., Chevelev, I., Stagljar, I. & Michaelis, S. Negative regulation of the yeast ABC transporter Ycf1p by phosphorylation within its N-terminal extension. J. Biol. Chem. 283, 27079–27088 (2008).
Bickers, S. C., Benlekbir, S., Rubinstein, J. L. & Kanelis, V. Structure of Ycf1p reveals the transmembrane domain TMD0 and the regulatory region of ABCC transporters. Proc. Natl. Acad. Sci. USA. 118, e2025853118 (2021).
Khandelwal, N. K. et al. The structural basis for regulation of the glutathione transporter Ycf1 by regulatory domain phosphorylation. Nat. Commun. 13, 1278 (2022).
Stolarczyk, E. I., Reiling, C. J. & Paumi, C. M. Regulation of ABC transporter function via phosphorylation by protein kinases. Curr. Pharm. Biotech. 12, 621–635 (2011).
Boscoboinik, D. et al. Dimerization of the P-glycoprotein in membranes. Biochim. Biophys. Acta 1027, 225–228 (1990).
Naito, M. & Tsuruo, T. Functionally active homodimer of P-glycoprotein in multidrug-resistant tumor cells. Biochem. Biophys. Res. Commun. 185, 284–290 (1992).
Poruchynsky, M. S. & Ling, V. Detection of oligomeric and monomeric forms of P-glycoprotein in multidrug resistant cells. Biochemistry 33, 4163–4174 (1994).
Eskandari, S., Wright, E. M., Kreman, M., Starace, D. M. & Zampighi, G. A. Structural analysis of cloned plasma membrane proteins by freeze-fracture electron microscopy. Proc. Natl. Acad. Sci. USA. 95, 11235–11240 (1998).
Soszynski, M., Kaluzna, A., Rychlik, B., Sokal, A. & Bartosz, G. Radiation inactivation suggests that human multidrug resistance-associated protein 1 occurs as a dimer in the human erythrocyte membrane. Arch. Biochem. Biophys. 354, 311–316 (1998).
Wang, S., Yue, H., Derin, R. B., Guggino, W. B. & Li, M. Accessory protein facilitated CFTR-CFTR interaction, a molecular mechanism to potentiate the chloride channel activity. Cell 103, 169–179 (2000).
Rosenberg, M. F. et al. The structure of the multidrug resistance protein 1 (MRP1/ABCC1). crystallization and single-particle analysis. J. Biol. Chem. 276, 16076–16082 (2001).
Ramjeesingh, M., Kidd, J. F., Huan, L. J., Wang, Y. & Bear, C. E. Dimeric cystic fibrosis transmembrane conductance regulator exists in the plasma membrane. Biochem. J. 374, 793–797 (2003).
Denis, M. et al. Characterization of oligomeric human ATP binding cassette transporter A1 - Potential implications for determining the structure of nascent high density lipoprotein particles. J. Biol. Chem. 279, 41529–41536 (2004).
Gupta, S., Xie, J., Ma, J. & Davis, P. B. Intermolecular interaction between R domains of cystic fibrosis transmembrane conductance regulator. Am. J. Respir. Cell Mol. Biol. 30, 242–248 (2004).
Schillers, H., Shahin, V., Albermann, L., Schafer, C. & Oberleithner, H. Imaging CFTR: a tail to tail dimer with a central pore. Cell. Physiol. Biochem. 14, 1–10 (2004).
Xu, J. K., Liu, Y., Yang, Y. Y., Bates, S. & Zhang, J. T. Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J. Biol. Chem. 279, 19781–19789 (2004).
McDevitt, C. A. et al. Purification and 3D structural analysis of oligomeric human multidrug transporter ABCG2. Structure 14, 1623–1632 (2006).
Trompier, D. et al. Transition from dimers to higher oligomeric forms occurs during the ATPase cycle of the ABCA1 transporter. J. Biol. Chem. 281, 20283–20290 (2006).
Xu, J. et al. Oligomerization domain of the multidrug resistance-associated transporter ABCG2 and its dominant inhibitory activity. Cancer Res 67, 4373–4381 (2007).
Mo, W. & Zhang, J. T. Oligomerization of human ATP-binding cassette transporters and its potential significance in human disease. Expert Opin. Drug Metab. Toxicol. 5, 1049–1063 (2009).
Mo, W., Qi, J. & Zhang, J. T. Different roles of TM5, TM6, and ECL3 in the oligomerization and function of human ABCG2. Biochemistry 51, 3634–3641 (2012).
Nagata, K. O., Nakada, C., Kasai, R. S., Kusumi, A. & Ueda, K. ABCA1 dimer-monomer interconversion during HDL generation revealed by single-molecule imaging. Proc. Natl. Acad. Sci. USA. 110, 5034–5039 (2013).
Frixel, S. et al. Homooligomerization of ABCA3 and its functional significance. Int. J. Mol. Med. 38, 558–566 (2016).
Zhao, H., Lee, J. & Chen, J. The hemolysin A secretion system is a multi-engine pump containing three ABC transporters. Cell 185, 3329–3340.e13 (2022).
Hofmann, N. et al. Analysis of the proteolytic processing of ABCA3: identification of cleavage site and involved proteases. PLoS One 11, e0152594 (2016).
Xie, T., Zhang, Z., Yue, J., Fang, Q. & Gong, X. Cryo-EM structures of the human surfactant lipid transporter ABCA3. Sci. Adv. 8, eabn3727 (2022).
Mason, D. L., Mallampalli, M. P., Huyer, G. & Michaelis, S. A region within a lumenal loop of Saccharomyces cerevisiae Ycf1p directs proteolytic processing and substrate specificity. Eukaryot. Cell 2, 588–598 (2003).
Mason, D. L. & Michaelis, S. Requirement of the N-terminal extension for vacuolar trafficking and transport activity of yeast Ycf1p, an ATP-binding cassette transporter. Mol. Biol. Cell 13, 4443–4455 (2002).
Li, Z.-S., Szczypka, M., Lu, Y.-P., Thiele, D. J. & Rea, P. A. The yeast cadmium factor protein (YCF1) is a vacuolar glutathione S-conjugate pump. 271, 6509–6517 (1996).
Tommasini, R. et al. The human multidrug resistance-associated protein functionally complements the yeast cadmium resistance factor. Proc. Natl. Acad. Sci. USA. 93, 6743–6748 (1996).
Alvarez, C. P., Stagljar, M., Muhandiram, D. R. & Kanelis, V. Hyperinsulinism-causing mutations cause multiple molecular defects in SUR1 NBD1. Biochemistry 56, 2400–2416 (2017).
Hossack, J. A. & Spencer-Martins, I. Lipid composition of the yeast - lipomyces kononenkoae. European Journal of Applied Microbiology and Biotechnology 5, 273–278 (1978).
Lindberg, L., Santos, A. X. S., Riezman, H., Olsson, L. & Bettiga, M. Lipidomic profiling of saccharomyces cerevisiae and zygosaccharomyces bailii reveals critical changes in lipid composition in response to acetic acid stress. PLoS One 8, 1–12 (2013).
Morgan, B. et al. Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nat. Chem. Biol. 9, 119–125 (2013).
Cole, S. P. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J. Biol. Chem. 289, 30880–30888 (2014).
Lee, Y. M. et al. Identification and functional characterization of the natural variant MRP3-Arg1297His of human multidrug resistance protein 3 (MRP3/ABCC3). Pharmacogenetics 14, 213–223 (2004).
Martin, G. M., Kandasamy, B., DiMaio, F., Yoshioka, C. & Shyng, S. L. Anti-diabetic drug binding site in a mammalian KATP channel revealed by Cryo-EM. Elife 6 (2017).
Mazza, T. et al. Structural basis for the modulation of MRP2 activity by phosphorylation and drugs. Nat Commun 15, 1983 (2024).
Wang, J. et al. Placing steroid hormones within the human ABCC3 transporter reveals a compatible amphiphilic substrate-binding pocket. EMBO J 42, e113415 (2023).
Gupta, K. et al. The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 541, 421–424 (2017).
Khandelwal, N. K. & Tomasiak, T. M. Structural basis for autoinhibition by the dephosphorylated regulatory domain of Ycf1. Nat Commun 15, 2389 (2024).
Kanelis, V., Hudson, R. P., Thibodeau, P. H., Thomas, P. J. & Forman-Kay, J. D. NMR evidence for differential phosphorylation-dependent interactions in WT and DeltaF508 CFTR. EMBO J 29, 263–277 (2010).
Zhang, Z. & Chen, J. Atomic structure of the cystic fibrosis transmembrane conductance regulator. Cell 167, 1586–1597 (2016).
Liu, F., Zhang, Z., Csanády, L., Gadsby, D. C. & Chen, J. Molecular structure of the human CFTR ion channel. Cell 169, 85–95.e8 (2017).
Zhang, Z., Liu, F. & Chen, J. Conformational changes of CFTR upon phosphorylation and ATP binding. Cell 170, 483–491.e8 (2017).
Zhang, Z., Liu, F. & Chen, J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. USA. 115, 12757–12762 (2018).
Arbesu, M., Iruela, G., Fuentes, H., Teixeira, J. M. C. & Pons, M. Intramolecular fuzzy interactions involving intrinsically disordered domains. Front Mol Biosci 5, 39 (2018).
Mittag, T., Kay, L. E. & Forman-Kay, J. D. Protein dynamics and conformational disorder in molecular recognition. J Mol Recognit 23, 105–116 (2010).
Sasser, T. L., Lawrence, G., Karunakaran, S., Brown, C. & Fratti, R. A. The yeast ATP-binding cassette (ABC) transporter Ycf1p enhances the recruitment of the soluble SNARE Vam7p to vacuoles for efficient membrane fusion. J. Biol. Chem. 288, 18300–18310 (2013).
Hurst, L. R. & Fratti, R. A. Lipid rafts, sphingolipids, and ergosterol in yeast vacuole fusion and maturation. Front. Cell Dev. Biol. 8, 539 (2020).
Starr, M. L. & Fratti, R. A. The participation of regulatory lipids in vacuole homotypic fusion. Trends Biochem. Sci. 44, 546–554 (2019).
Starr, M. L. et al. Phosphatidic acid induces conformational changes in Sec18 protomers that prevent SNARE priming. J. Biol. Chem. 294, 3100–3116 (2019).
Paumi, C. M. et al. Mapping protein-protein interactions for the yeast ABC transporter Ycf1p by integrated split-ubiquitin membrane yeast two-hybrid analysis. Mol. Cell 26, 15–25 (2007).
Snider, J. et al. Mapping the functional yeast ABC transporter interactome. Nat. Chem. Biol. 9, 565–572 (2013).
Nji, E., Chatzikyriakidou, Y., Landreh, M. & Drew, D. An engineered thermal-shift screen reveals specific lipid preferences of eukaryotic and prokaryotic membrane proteins. Nat. Commun. 9, 4253 (2018).
Fay, J. F. et al. Cryo-EM visualization of an active high open probability CFTR anion channel. Biochemistry 57, 6234–6246 (2018).
Liu, F. et al. Structural identification of a hotspot on CFTR for potentiation. Science 364, 1184–1188 (2019).
Qian, H. et al. Structure of the human lipid exporter ABCA1. Cell 169, 1228–1239.e10 (2017).
Aller, S. G. & Segrest, J. P. The regulatory domains of the lipid exporter ABCA1 form domain swapped latches. PLoS One 17, e0262746 (2022).
Aller, S. G. et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 (2009).
Kim, Y. & Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 359, 915–919 (2018).
Jackson, S. M. et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 25, 333–340 (2018).
Spitz, O. et al. Type I Secretion Systems-One Mechanism for All? Microbiol. Spectrum 7, https://doi.org/10.1128/microbiolspec.PSIB-0003-2018 (2019).
Guo, H., Bueler, S. A. & Rubinstein, J. L. Atomic model for the dimeric Fregion of mitochondrial ATP synthase. Science 358, 936–940 (2017).
Di Trani, J. M. et al. Structure of the bc1-cbb3 respiratory supercomplex from Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA. 120, e2307093120 (2023).
Wang, H. L., Bueler, S. A. & Rubinstein, J. L. Structural basis of V-ATPase V region assembly by Vma12p, 21p, and 22p. Proc. Natl. Acad. Sci. USA 120, e2217181120 (2023).
Di Trani, J. M. et al. Structural basis of mammalian complex IV inhibition by steroids. Proc. Natl. Acad. Sci. USA. 119, e2205228119 (2022).
Keon, K. A., Benlekbir, S., Kirsch, S. H., Müller, R. & Rubinstein, J. L. Cryo-EM of the yeast V complex reveals distinct binding sites for macrolide V-ATPase inhibitors. Acs Chemical Biology 17, 619–628 (2022).
Yanofsky, D. J. et al. Structure of mycobacterial CIII2CIV2 respiratory supercomplex bound to the tuberculosis drug candidate telacebec (Q203). Elife 10, e71959 (2021).
Vasanthakumar, T. et al. Structural comparison of the vacuolar and Golgi V-ATPases from Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 116, 7272–7277 (2019).
Sydor, A. M. et al. Metal binding properties of Escherichia coli YjiA, a member of the metal homeostasis-associated COG0523 family of GTPases. Biochemistry 52, 1788–1801 (2013).
Marr, C. R., Benlekbir, S. & Rubinstein, J. L. Fabrication of carbon films with approximately 500nm holes for cryo-EM with a direct detector device. J. Struct. Biol. 185, 42–47 (2014).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Rubinstein, J. L. & Brubaker, M. A. Alignment of cryo-EM movies of individual particles by optimization of image translations. J. Struct. Biol. 192, 188–195 (2015).
Punjani, A. & Fleet, D. J. 3D variability analysis: resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 213, 107702 (2021).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol. 75, 861–877 (2019).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D. Struct. Biol. 74, 519–530 (2018).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248–254 (1976).
Lehrer, S. S. Solute perturbation of protein fluorescence. the quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry 10, 3254–3263 (1971).
Nikolaeva, O., Moiseyev, G., Rodgers, K. K. & Ma, J. X. Binding to lipid membrane induces conformational changes in RPE65: implications for its isomerohydrolase activity. Biochem. J. 436, 591–597 (2011).
Lakowicz, J. R. Principles Of Fluorescence Spectroscopy, 3rd ed., Springer, New York., (2006).
Liu, R. & Sharom, F. J. Fluorescence studies on the nucleotide binding domains of the P-glycoprotein multidrug transporter. Biochemistry 36, 2836–2843 (1997).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 50, D543–D552 (2022).
Deutsch, E. W. et al. The ProteomeXchange consortium at 10 years: 2023 update. Nucleic Acids Res 51, D1539–D1548 (2023).
Pickin, K. A. et al. Suppression of Ycf1p function by Cka1p-dependent phosphorylation is attenuated in response to salt stress. FEMS Yeast Res. 10, 839–857 (2010).
Acknowledgements
SCB was supported by a Queen Elizabeth II Graduate Scholarships in Science and Technology and an Alexander Graham Bell Canada Graduate Scholarship-Doctoral from the Natural Sciences and Engineering Research Council of Canada (NSERC). JLR was supported by the Canada Research Chairs program. This research was supported by NSERC Discovery Grants (RGPIN-2020-05835 to VK, and RGPIN-2023-04676 to JLR). CryoEM data were collected at the Toronto High-Resolution High-Throughput CryoEM Facility, supported by the Canada Foundation for Innovation and Ontario Research Fund. Mass spectrometry data was acquired and processed by Leanne Wybenga-Groot and Craig Simpson at the SPARC BioCentre, SickKids Hospital. The absorbances of the reactions for the Malachite Green assays were obtained using instrumentation in the Structural Biology Protein Core in the Peter Gilgan Centre for Research and Learning, SickKids Hospital. The authors thank Hanlin Wang for the yHW10 strain for a control for the yeast viability studies.
Author information
Authors and Affiliations
Contributions
V.K. conceived of the project and designed experiments with J.L.R. and S.C.B. S.C.B. created the yeast strain, purified the protein, performed ATPase assays, fabricated the cryo-EM specimen grids, processed cryo-EM images, constructed the Ycf1p atomic models, and conducted fluorescence quenching experiments. S.B. prepared cryo-EM specimens and collected images. Figures were prepared by S.C.B. and V.K., and the manuscript was written by S.C.B., V.K., and J.L.R.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Cédric Orelle for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bickers, S.C., Benlekbir, S., Rubinstein, J.L. et al. Structure of a dimeric full-length ABC transporter. Nat Commun 15, 9946 (2024). https://doi.org/10.1038/s41467-024-54147-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54147-8
This article is cited by
-
Structural insights into toxicant export mediated by ABCC2 in Arabidopsis thaliana
Nature Communications (2025)
