
Abstract
NK cells are endowed with tumor killing ability, nevertheless most cancers impair NK cell functionality, and cell-based therapies have limited efficacy in solid tumors. How cancers render NK cell dysfunctional is unclear, and overcoming resistance is an important immune-therapeutic aim. Here, we identify autophagy as a central regulator of NK cell anti-tumor function. Analysis of differentially expressed genes in tumor-infiltrating versus non-tumor NK cells from our previously published scRNA-seq data of advanced human prostate cancer shows deregulation of the autophagic pathway in tumor-infiltrating NK cells. We confirm this by flow cytometry in patients and in diverse cancer models in mice. We further demonstrate that exposure of NK cells to cancer deregulates the autophagic process, decreases mitochondrial polarization and impairs effector functions. Mechanistically, CCAAT enhancer binding protein beta (C/EBPβ), downstream of CXCL12-CXCR4 interaction, acts as regulator of NK cell metabolism. Accordingly, inhibition of CXCR4 and C/EBPβ restores NK cell fitness. Finally, genetic and pharmacological activation of autophagy improves NK cell effector and cytotoxic functions, which enables tumour control by NK and CAR-NK cells. In conclusion, our study identifies autophagy as an intracellular checkpoint in NK cells and introduces autophagy regulation as an approach to strengthen NK-cell-based immunotherapies.
Similar content being viewed by others

Introduction
Immunotherapies represent a groundbreaking discovery in the field of cancer and hold promise for revolutionizing tumor treatment. Among them, T-cell transfer therapies have been applied to treat a broad range of tumors with promising results1. However, the efficacy of T cell-based approaches relies on tumor antigen presentation and expression of major histocompatibility complex (MHC) molecules and it is affected by the engagement of checkpoints that hamper T cell activation. As a consequence, T cell-based immunotherapies showed limited clinical benefits, and innate cells, including natural killer (NK) cells, are now under investigation as candidates for novel strategies that harness innate immunity2,3,4,5,6. NK cells are specialized immune effector cells endowed with the ability to spontaneously identify and kill stressed, foreign, infected, and transformed cells7,8. NK cells are featured by a robust cytotoxic activity, based on the release of cytolytic granules and cytotoxic cytokines as well as on the antibody-dependent cellular cytotoxicity (ADCC) mechanism9,10. In addition, NK cells shape the anti-tumor adaptive immune response and engage with lymphoid and dendritic subsets in the tumor-microenvironment (TME)5,11. NK cell-mediated killing is independent from human leukocyte antigen (HLA)-restriction and it is orchestrated by a repertoire of activating and inhibitory receptors that recognize germline-encoded ligands expressed by transformed cells. Importantly, the absence of class I MHC molecules, often downregulated in cancer cells, hampers T-cell recognition but triggers NK elimination of tumor cells. As a consequence, NK cells may overcome resistance to T cell-based therapies, and they are attractive candidates for cancer therapy. Importantly, an increased abundance of NK cells in cancer patients is associated with improved overall survival in most solid tumors2. Adoptive transfer of allogenic NK cells and NK cell lines proved to be safe and showed encouraging efficacy in clinical trials in some hematological malignancies12,13,14,15,16. The promising initial successes obtained in treating hematological cancers further prompted the use of this therapeutic approach in solid tumors. However, despite the potent anti-tumor effect observed in pre-clinical models of different cancer types17,18,19,20, NK cell clinical efficacy is still insufficient in most settings. A major challenge is represented by the fact that, within the TME, NK cells have to deal with a highly immune-suppressive milieu responsible for their functional exhaustion. Within the tumor, NK cells are exposed to extracellular factors secreted by cells of the TME, resulting in a profound alteration of their transcriptional profile, differentiation status, and metabolic program, ultimately leading to the loss of their effector functions21,22. Therefore, strategies to increase the efficacy of NK-based immunotherapies are needed23. Knowledge of the pathways involved in NK cell activation and mechanisms exploited by tumor cells to hinder NK cell functionality will conduce to ad hoc strategies able to endow NK cells with superior tumor killing.
Autophagy is a highly evolutionary conserved process involved in the degradation of cellular components, including lipids, proteins, large aggregates, and organelles, sequestered in double-membrane autophagosomes that fuse with lysosome for degradation24. Autophagy allows the cell to survive in homeostatic conditions, the so-called “Goldilocks Zone”, and in response to stress by preventing cell death pathway activation. Consequently, a severe impairment of autophagy is detrimental to the cell25. At the same time, excessive activation of the autophagy machinery may result in cell death and, as a consequence, autophagy needs to be tightly regulated26. Several works demonstrated the key role of autophagy in regulating differentiation, survival, proliferation, and effector functions of innate and adaptive immune cell subsets, including neutrophils, macrophages, and T cells27. Nevertheless, up to date, few studies have explored the impact of autophagy on NK cell biology28,29,30 and the role of autophagy in modulating NK cell anti-tumor response still remains elusive.
To explore mechanisms behind NK cell impairment in solid tumors, we utilize prostate cancer (PCa) tissues, and we employ preclinical models of PCa. We unveil that autophagy is essential for NK cell-mediated killing. We show that cancer cells hinder the autophagic response by engaging the CXCR4-C/EBPβ pathway in tumor-infiltrating NK cells, thus hampering their anti-tumor function. We also demonstrate that the re-activation of the autophagic pathway by pharmacological exposure or genetic manipulation, rescues the functional defects of tumoral NK cells, thus improving their ability to kill cancer cells in vitro. Finally, we show that ex vivo priming of NK and CAR-NK cells with autophagy-activation strategies augments the control of tumor growth in vivo. Our study identifies autophagy as a previously unknown intracellular checkpoint in NK cells and proposes an off-the-shelf non-invasive approach to enhance NK-based immunotherapies in the clinic.
Results
Intra-tumoral NK cells are defective in PCa
Evidence exists that NK cells are defective in PCa, however the extent of such impairment and the mechanisms that cause NK cell dysfunction are still unclear31,32. To investigate the role and function of NK cells in prostate cancer, we employed the murine Pten null cancer model33 that closely resembles the disease progression seen in PCa patients. Pten prostate-specific deletion was obtained by crossing Ptenloxp/loxp mice to the Probasin-Cre transgenic line, in which the Cre recombinase is under the control of a prostate-specific Probasin promoter. The tumor develops through distinct stages from prostatic intraepithelial neoplasia (PIN) to invasive adenocarcinoma and, therefore, provides a valuable tool to unveil mechanisms linked to PCa initiation and progression as well as to develop new targeted therapies (Fig. 1A, B). To gain insights into tumor-induced alterations of the immune infiltrate, we analyzed by fluorescence-activated cell sorting (FACS) the composition of CD45+ cells in Ptenpc−/− prostates from 12 weeks old mice (Supplementary Fig. 1A), while Ptenpc+/+ wild type (WT) age-matched prostates were utilized as controls. Tumor development led to significant changes in the T cell compartment, including an increase in both conventional and regulatory CD4+ T cells, as well as CD8+ T cells, with enhanced proliferation and features of exhaustion (Supplementary Fig. 1B, C). Within the innate immune branch, there was an increase in macrophages and neutrophils, and a notable decrease in NK cells (Fig. 1C). Tumor-associated macrophages and neutrophils displayed an immunosuppressive phenotype (Supplementary Fig. 1D, E). Analysis of NK cells led to the identification of the four discrete stages of maturation, previously defined on the basis of CD11b and CD27 expression34 (Supplementary Fig. 1F). Tumor-infiltrating NK cells presented an enrichment of the more immature compartments (CD11b− CD27− and CD11b− CD27+), at the expense of the more mature subset (CD11b+ CD27−) (Fig. 1D). Additionally, NK cells were marked by lower levels of granzyme B and perforin compared to their healthy counterpart (Fig. 1E, Supplementary Fig. 1G). Tumor-infiltrating NK cells, isolated from Ptenpc−/− prostates (Fig. 1F), and splenic NK cells from Ptenpc−/− tumor-bearing mice (Supplementary Fig. 1H) presented a reduced ability to kill target cells when compared to splenic cells from WT mice, thus confirming their dysfunctional state, which could directly relate to their altered maturation stages. A deeper characterization of the phenotype of tumor-infiltrating NK cells revealed a reduced expression of the activating receptor NKG2D and of the amino acid transporter CD98, previously associated with NK cell effector functions35 (Supplementary Fig. 1I).

A Breeding scheme for the establishment of the Pten knockout (Ptenpc−/−) mouse model. B Hematoxylin and eosin staining of prostate tissues from Ptenpc+/+ and Ptenpc−/− mice. Original magnification, ×20. Scale bar: 100 μm. Representative of an experiment of n = 4 mice/group. C Bubble plot showing the average frequency, referred to CD45+ live cell gate, of immune cell subsets in Ptenpc+/+ and Ptenpc−/− prostate (n = 4 mice/group). D Flow cytometry analysis of NK cell maturation in Ptenpc+/+ and Ptenpc−/− prostate (n = 10 mice/group). E Flow cytometry analysis of effector molecule expression in NK cells in Ptenpc+/+ and Ptenpc−/− prostate, according to gating strategy in S1G (n = 10 Ptenpc+/+ mice/group, n = 6 Ptenpc−/− mice/group). F Cytotoxicity of sorted splenic NK cells from Ptenpc+/+ mice and tumor-infiltrating NK cells from Ptenpc−/− mice (three different mice/conditions). G Tumor volume. n = 5 mice/group. H Hematoxylin and eosin staining of prostate tissues from αNK1.1 or isotype-treated mice. Original magnification, ×20. Scale bar: 100 μm. I Bubble plot showing the average frequency, referred to CD45+ live cell gate, of immune cell subsets in Ptenpc−/− prostate of αNK1.1 or isotype-treated mice (n = 5 mice/group). J Experimental scheme for tumor-conditioned NK cell generation. K Flow cytometry analysis of NK cell maturation in tumor-conditioned and control NK cells (n = 5 biological replicates, independently collected batches of tumor-conditioned medium). L Flow cytometry analysis of effector molecule expression in tumor-conditioned and control NK cells (n = 8 biological replicates, data pooled from two independent experiments). M Quantification of IFNγ release (n = 4 biological replicates/group). N Experimental scheme for cytotoxicity assay. O, P Cytotoxicity of tumor-conditioned and control NK cells against YAC-1 (O) and Pten−/− (P) target cells (n = 6 biological replicates, independently collected batches of tumor-conditioned medium). Data in C and I are presented as bubble plot showing average values; two-tailed unpaired t test. Data in (D), (E), (G), (K), and (L) are presented as Min to Max box-and-whisker plot, the box extends from the 25th to 75th percentiles and the whiskers reach the sample maximum and minimum values, the median is indicated at center line and the mean value is indicated as “+”; two-tailed unpaired t test. Data in (F) are presented as scatter plot mean ± SEM; two-tailed unpaired t test. Data in (M) are presented as scatter plot with bar with mean ± SEM; two-tailed unpaired t test. Symbols in (O) and (P) represent mean and error bars indicate SEM; two-tailed unpaired t test for corresponding E:T ratio.
To further assess the contribution of NK cells within the prostate TME, we depleted NK cells by means of an anti-NK1.1 antibody in established tumors in the Ptenpc−/− model (Supplementary Fig. 1J) and we monitored tumor volume and immune cell infiltrate upon NK cell depletion. Surprisingly, neither tumor size (Fig. 1G) nor histopathological features (Fig. 1H) were affected by the treatment and minimal changes were observed within the TME (Fig. 1I). In particular, we noticed an enrichment in the CD8+ memory subsets (central and effector memory), marked by a reduction in granzyme B expression (Supplementary Fig. 1K, L), while no changes were appreciated in the myeloid compartment (data not shown).
Collectively, these data prompted us to hypothesize that tumor-infiltrating NK cells are functionally impaired, and therefore, their depletion produces only minimal changes in the TME. To further test our hypothesis, we took advantage of a syngeneic murine prostate cancer model, based on subcutaneous injection of Pten−/− Trp53−/− prostate cancer cells and we depleted NK cells at both early and late time point after tumor engraftment. Depletion of NK cells during tumor initiation determined a significant acceleration in tumor growth (Supplementary Fig. 2A–C) and a remodeling of both innate and adaptive compartments (Supplementary Fig. 2D). On the contrary, depletion of NK cells at a later time point had no effect on tumor growth and affected the TME at a lower extent (Supplementary Fig. 2E–H). Together, these findings are indicative of a functional impairment acquired by NK cells during tumor progression.
To gain molecular insights into the crosstalk between cancer and NK cells, we set up an in vitro model in which NK cells are exposed to the supernatant of tumor cells (Fig. 1J). The analysis of tumor-conditioned NK cell phenotype revealed a reshape in terms of subset abundance, with an enrichment of the more immature compartments (CD11b− CD27− and CD11b− CD27+) (Fig. 1K). In addition, the tumor secretome determined an impairment of NK cell effector functions, as underlined by decreased levels of granzyme B, perforin and IFNγ compared to the control condition (Fig. 1L, M) and a weakened cytolytic activity both against the YAC-1 and the Pten−/− cancer cell lines (Fig. 1N–P and Supplementary Fig. 2I).
Overall, these data highlight that prostate tumor determined a profound remodeling of the TME with NK cells showing a defective functional state.
Tumor-infiltrating NK cells show a dysfunctional autophagy
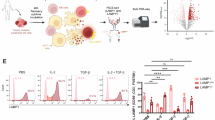
Results in the murine context encouraged us to further investigate the functionality of NK cells in human PCa. FACS analysis confirmed a decrease in the frequency of infiltrating NK cells in PCa tumor samples when compared to adjacent tissues (Fig. 2A). To deepen the mechanisms underlying NK cell defects within the prostate TME, we analyzed scRNA-seq data, that we previously generated, derived from patients affected by advanced PCa36 (Supplementary Fig. 3A). Gene set enrichment analysis (GSEA) performed on genes differentially expressed in tumor vs. non-tumor infiltrating NK cells showed an alteration of stress response mechanisms (autophagy), metabolic processes (metabolism of RNA), cytoskeleton dynamics (intracellular transport, signaling by GPCR) and effector functions (interferon gamma signaling) (Fig. 2B). Among the top three regulated pathways, we concentrated on autophagy, as several works have demonstrated that this process is a master regulator of differentiation and activation in immune cells28,29,37. Analysis of a gene signature related to autophagy activation38 suggested a selective shutdown of the autophagy pathway in tumor-infiltrating NK cells (Fig. 2C). FACS analysis, by means of CYTO-ID autophagy staining probe, highlighted an alteration of the autophagic process in tumoral NK cells compared to their normal counterpart (Fig. 2D). We then performed a re-clustering of scRNA sequencing data that allowed us to discriminate NK cell subclusters (Fig. 2E). According to differentially expressed genes (Fig. 2F) and taking advantage of previously published signatures39,40, we named each newly identified cluster: CD56dim CD16bright NK cells (C0 and C3), CD8+ T cells (C1 and C2), CD56bright CD16dim NK cells (C4), CD56bright CD16neg immature NK cells (C5) and CD56dim CD16dim NK cells (C6). The CD56dim NK subset was further classified according to CD57 expression, where CD57+ cells form a terminally mature population with a greater killing capacity41,42. Accordingly, we found an enrichment of classical effector molecules, including FGFBP2, GZMB, CST7, FCGR3A, GNLY and PRF1, in C0 compared to C3. Similarly, several NK cell activating receptors (KLRK1, CD160, CD244, NCR3 and NCR1) are highly expressed in C0 vs C3. Finally, CD56dim CD57− NK cells showed increased CD49a expression, accompanied by exhaustion-related checkpoint molecules such as PD-1, CD96, KLRG1, and LAG3, thus resembling CD49a+ NK cells, previously described in hepatocellular carcinoma43. Further characterization of NK cell subsets by flow cytometry revealed a remodeling of their maturation state, with an enrichment of the CD56bright CD16neg compartment at the expense of the more mature/cytotoxic CD56dim CD16bright subset (Fig. 2G, H). When looking at NK cell subsets, autophagy levels were significantly diminished in mature CD56dim CD16+ (Supplementary Fig. 3B), however, a lower autophagic level could be observed in most subpopulations. Data obtained in human PCa patients were further validated in Ptenpc−/− model, confirming the alteration of the autophagic pathway in tumor-infiltrating NK cells (Fig. 2I, J). Similarly to human NK cells, the mature CD11b+ CD27− subset showed a significant deregulation of the autophagic pathway (Supplementary Fig. 3C, D) that is paralleled by increased apoptosis (Supplementary Fig. 3E).

A Graph showing the frequency of NK cells, referred to CD45+ live cell gate, in tumor vs non-tumor samples (n = 10 samples/group) as determined by flow cytometry. B Dot plots of GSEA results illustrating the significant top 15 REACTOME pathways in tumor vs non-tumor NK cells (n = 3 samples/group). C Dot plot showing the proportion of normal and tumor NK cells expressing autophagy induction list genes, according to their average expression. D Graph showing the Median Fluorescence Intensity (MFI) of CYTO-ID on human NK cells in tumor vs non-tumor samples (n = 10 samples/group). E UMAP of NK cell populations in prostate (tumor and normal adjacent tissue), showing the formation of seven main clusters identified by marker genes. F Heat map showing scaled expression of the top 10 marker genes, for each NK cell cluster. G Contour plots of NK cell maturation status in tumor vs non-tumor samples, gated on CD3− CD56+ cells. H Flow cytometry analysis of NK cell main subsets in tumor vs non-tumor samples, according to the gates shown in G. I Graph showing the MFI of CYTO-ID on mouse NK cells in Ptenpc+/+ and Ptenpc−/− prostates (n = 11 mice/group). J Graph showing the frequency of LC3B+ cells in mouse NK cells in Ptenpc+/+ and Ptenpc−/− prostates (n = 9 Ptenpc+/+ mice/group, n = 6 Ptenpc−/− mice/group). Data in (A), (D), (H), and (I) are presented as before-after plot; two-tailed paired t test. Data in (B) are presented as balloon plot; permutation test. Data in (J) are presented as Min to Max box-and-whisker plot, the box extends from the 25th to 75th percentiles and the whiskers reach the sample maximum and minimum values, the median is indicated at center line and the mean value is indicated as “+”; two-tailed unpaired t test.
Overall, these data highlight that the transcriptional profile of NK cells is strongly affected by the tumor and suggest that autophagy is deregulated in tumor-infiltrating NK cells.
Autophagy is required for NK cell anti-tumor effector functions
We then explored in vitro the extent of autophagy deregulation on tumor NK cells by means of two different techniques: the autophagic vesicle-specific dye CYTO-ID and an autophagy LC3-antibody-based kit. Importantly, this kit allows to specifically quantify the lipidated and autophagosome-specific form of LC3 (LC3-II). We confirmed that exposure of murine NK cells to the supernatant of Pten−/− tumor cells negatively regulates the autophagic flux, as evaluated by both CYTO-ID (Fig. 3A) and lipidated LC3 staining (Fig. 3B, C). Importantly, these results were confirmed in diverse cancer models, as shown by exposure of NK cells to the supernatant of Pten−/− Trp53−/− cells (prostate), 4T1 cells (breast), ID8 cells (ovarian), B16-F10 cells (melanoma), MCA-203 cells (fibrosarcoma) and YAC-1 cells (lymphoma) (Supplementary Fig. 3F, G). To validate our findings in a human system, we used the human NK cell line NK-92, which shares several characteristics with primary NK cells44, and has been developed as an off-the-shelf product for clinical use in cancer cellular therapy16,45. Therefore, NK-92 cells represent a platform to validate murine data in a more clinical-like setting. Firstly, we evaluated whether the prostate tumor milieu was able to negatively affect NK-92 autophagic behavior, similarly to what we observed in murine NK cells. Notably, the exposure to tumor-derived secretome, derived from the human prostate cancer cell lines PC3 and LNCaP, determined a marked reduction of the autophagic flux, as evaluated by both FACS staining (Fig. 3D–F and Supplementary Fig. 3H) and western blot (WB) analysis (Fig. 3G, H). The decreased autophagic flux together with increased LC3-II basal levels (Fig. 3E) suggest a late-stage block in the autophagic machinery, possibly due to the inhibition of autophagosome-lysosome fusion and/or autophagosome degradation turnover46. Importantly, the alteration in the autophagic machinery accompanied a significant drop in NK cell killing ability (Fig. 3I, J and Supplementary Fig. 3I).

A Graph showing the ΔMFI of CYTO-ID (autophagic flux) on murine NK cells in tumor-conditioned and control condition (n = 7 biological replicates, data pooled from four independent experiments). B Histograms of lipidated LC3 staining in tumor-conditioned and control murine NK cells, with or without Reagent A (RA). C Graph showing the ΔMFI of LC3-II (autophagic flux) on murine NK cells in tumor-conditioned and control condition (n = 4 biological replicates, independently collected batches of tumor-conditioned medium). D Graph showing the autophagic flux on human NK-92 cells in tumor-conditioned and control condition (n = 4 replicates, results are representative of three independent experiments with similar results). E Histograms of LC3-II staining in tumor-conditioned and control NK-92 cells, with or without RA. F Graph showing the autophagic flux on NK-92 cells (n = 5 biological replicates, independently collected batches of tumor-conditioned medium). G, H Representative western blot image (G) and relative quantification (H) of LC3-II expression in NK-92 cells exposed to control or tumor-conditioned medium, with or without Chloroquine (CQ). β-actin was used as loading control (n = 6 biological replicates). I Cytotoxicity of control or tumor-conditioned NK cells, in presence or absence of Metformin, against YAC-1 target cells (n = 4 biological replicates, independently collected batches of tumor-conditioned medium). J Cytotoxicity of NK-92 cells, treated or not with tumor-conditioned medium, with or without Metformin, against PC3 target cells (n = 8 biological replicates, data pooled from two independent experiments). K Relative fold change of key autophagy-related genes in Beclin 1 overexpressing (BECN1 OE) NK-92 cells (n = 6 replicates, data pooled from two independent experiments). L, M Western blot analysis (L) and relative quantification (M) of BECN1 expression in scramble and BECN1 OE NK-92 cells. HSP90 was used as loading control (n = 4 biological replicates). N Graph showing the autophagic flux on scramble and BECN1 OE NK-92 cells, in tumor-conditioned and control condition (n = 4 biological replicates, independently collected batches of tumor-conditioned medium). O Cytotoxicity of scramble and BECN1 OE NK-92 cells, treated or not with tumor-conditioned medium, against PC3 target cells (n = 10 biological replicates, data pooled from two independent experiments). P Representative images of mitochondria in NK-92 cells treated or not with tumor-conditioned medium. Q Quantification of damaged mitochondria in NK-92 cells exposed or not to tumor-conditioned medium; more than 250 mitochondria in at least 25 different cells for each experimental group were analyzed. R Graph showing the levels of mitochondrial membrane potential (Δψm) in NK-92 cells, exposed or not to tumor-conditioned medium (n = 10 biological replicates, independently collected batches of tumor-conditioned medium). S Graph showing the proportion of MitoSox+ NK-92 cells exposed or not to tumor-conditioned medium (n = 5 biological replicates, independently collected batches of tumor-conditioned medium). T Graph showing TMRM incorporation in scramble and BECN1 OE NK-92 cells, exposed or not to tumor-conditioned medium (n = 8 biological replicates, independently collected batches of tumor-conditioned medium). U Graph showing the proportion of MitoSox+ scramble and BECN1 OE NK-92 cells exposed or not to tumor-conditioned medium (n = 6 biological replicates, independently collected batches of tumor-conditioned medium). Data in (A), (C), (D), (F), (H) and (Q–S) are presented as Min to Max box-and-whisker plot, the box extends from the 25th to 75th percentiles and the whiskers reach the sample maximum and minimum values, the median is indicated at center line and the mean value is indicated as “+”; two-tailed unpaired t test. Data in (I), (J), (N), (O), (T), and (U) are presented as Min to Max box-and-whisker plot, the box extends from the 25th to 75th percentiles and the whiskers reach the sample maximum and minimum values, the median is indicated at center line and the mean value is indicated as “+”; one-way ANOVA test with Holm–Šídák’s multiple-comparisons test. Data in (K) and (M) are presented as scatter plot with mean ± SEM; two-tailed unpaired t test.
To gain better insight into the link between autophagy and NK cell effector functions, we exposed murine NK cells to chloroquine (CQ), a pharmacological inhibitor of the autophagic flux. CQ administration induced a shift in NK cells phenotype, with an accumulation of the more immature subsets (CD11b− CD27− and CD11b− CD27+) (Supplementary Fig. 3J). Notably, the pharmacological abrogation of autophagy impaired NK cell effector functions, as demonstrated by the reduction of granzyme B and IFNγ expression (Supplementary Fig. 3K) as well as a significant suppression of their killing ability (Supplementary Fig. 3L). On the contrary, autophagy activation by means of Metformin, a well-known AMPK and autophagy activator47, strongly improved NK cell-mediated tumor killing (Fig. 3I, J), both in murine and human NK cells.
To validate our findings, we employed additional known autophagy activators. Firstly, we exposed NK cells to Rapamycin and Everolimus, two compounds that specifically inhibit mTORC1, thus determining autophagy activation. Unexpectedly, we observed a pronounced reduction of NK cell killing ability both in steady-state conditions and in presence of Pten−/−-conditioned medium (Supplementary Fig. 3M, N). However, these data were in accordance with previously published studies reporting that mTORC1 repression could inhibit NK cell effector functions48,49 and suggested that the effect of Metformin is independent from mTOR activation. We then tested two mTOR-independent drugs: Gemcitabine, an anti-metabolic nucleoside analog, that cause the early induction of autophagy through the activation of Vacuole Membrane Protein 1 (VMP1) expression50 and Carfilzomib, a second generation proteasome inhibitor, that activates autophagy via induction of the unfolded protein response and ATF451. Both molecules minimally affected NK cell killing ability in steady-state condition, however the co-treatment with tumor-conditioned medium restored their cytotoxic properties, similarly to what happened with Metformin treatment (Supplementary Fig. 3M, N).
As autophagy activators, including Metformin, could act on multiple pathways, we decided to specifically sustain autophagy in NK cells by genetic modification. We thus generated a NK-92 cell line overexpressing Beclin 1 (BECN1). BECN1 plays a pivotal role in the initiation and regulation of autophagy and its overexpression has been previously employed for autophagy hyperactivation52. Cells were modified in order to express the CRISPR/Cas9 synergistic activation mediator (SAM), consisting of an engineered protein complex for the transcriptional activation of endogenous genes. The system is based on three components: a nucleolytically inactive Cas9-VP64 fusion protein, a sgRNA incorporating two MS2 RNA aptamers and the MS2-P65-HSF1 activation helper protein53. We optimized the protocol to express all the components of the SAM system by means of a two-step lentiviral transduction (Supplementary Fig. 4A). Firstly, we validated BECN1 overexpression at both transcriptional and protein level (Fig. 3K–M). Then we confirmed the activation of the autophagic program, as evaluated by increased expression of key autophagy-related genes included in the “autophagy induction list”38 (Fig. 3K). Protein expression analysis confirmed increased levels of ATG7, ATG14 and p62 in BECN1 OE (Supplementary Fig. 4B–E). On the contrary, decreased levels of total LC3 were observed in BECN1 OE (Supplementary Fig. 4F, G). Notably, BECN1 overexpressing (BECN1 OE) NK-92 cells show a sustained autophagy and cytolytic activity, also when exposed to tumor-conditioned media (Fig. 3N, O).
Previous findings indicate that autophagy defects are associated with damaged mitochondria that in turn impairs NK cell functionality54. This evidence prompted us to investigate the status of mitochondrial fitness in tumoral NK cells. We thus performed transmission electron microscopy (TEM) analysis of control and tumor-exposed NK cells (Fig. 3P, Q and Supplementary Fig. 4H). The investigation of mitochondrial architecture revealed loss of integrity of the outer membrane, diffuse cristolysis and matrix disruption phenoma in NK cells exposed to tumor media. Collectively, these data indicate that NK cells exposed to tumor secretome accumulated damaged mitochondria, as confirmed by FACS staining that revealed a reduced percentage of cells containing functional mitochondria and a concomitant increase of cells accumulating dysfunctional mitochondria (Supplementary Fig. 4I–K). Accordingly, tumor exposure lead to the alteration of multiple parameters associated to mitochondrial fitness, including mitochondria membrane potential (Δψm), assessed by TMRM staining, mitochondrial reactive oxygen species production, evaluated by MitoSox staining, and intracellular lactate levels (Fig. 3R, S and Supplementary Fig. 4L). To evaluate whether autophagy impairment is associated to mitochondrial damage in tumoral NK, we performed FACS analysis of BECN1 OE NK92, in presence and absence of tumor media. In accordance with our hypothesis, BECN1 OE NK-92 cells showed higher TMRM levels if exposed to the tumor secretome, when compared to BECN1 WT cells (Fig. 3T). Additionally, genetic activation of autophagy by BECN1 overexpression prevented mitochondrial ROS accumulation in tumor-conditioned NK cells (Fig. 3U), suggesting that autophagy could regulate oxidative stress by removing damaged ROS-producing mitochondria.
Collectively these results demonstrate that autophagy regulates the anti-tumor functions of NK cells and that mitochondrial damage, associated to autophagy impairment, is implicated in the loss of function showed by tumoral NK cells.
Pharmacological and genetic activation of autophagy improves NK cell-mediated tumor clearance in vivo
Overall, our data led us to hypothesize that autophagy activation confers to NK cells a superior cytotoxic capability and may thus be exploited to improve NK cell tumor killing.
To substantiate our thesis by means of a more clinical approach, we adopted an experimental setting based on the subcutaneous injection of the Pten−/− Trp53−/− prostate cancer cell line in NSG mice, lacking mature T, B and NK cells. After tumor engraftment, mice received twice a week an infusion of splenic NK cells, treated or not with Metformin for 24 h before i.v. injection and washed before infusion (Fig. 4A). Of note, NK cells alone resulted to be completely ineffective in controlling tumor growth, while the administration of Metformin-treated NK cells determined a significant tumor control (Fig. 4B, C). The improved functionality of NK cells went hand-in-hand with the expansion of the cytotoxic compartment (Fig. 4D), the persistence of the autophagic activity (Fig. 4E) and the amelioration of key effector functions (Fig. 4F). The frequency of tumor-infiltrating NK cells was not affected, though the cells showed an activated phenotype, defined by increased NKG2D expression and a drop in KLRG1 expression (Supplementary Fig. 5A, B). Similar results were obtained in a different mouse model, based on the subcutaneous injection of B16-F10 melanoma cell line. Indeed, Metformin-treated NK cells were able to better control tumor growth, compared to their untreated condition (Supplementary Fig. 5C). Importantly, these cells have a superior ability to infiltrate the tumors as well as to maintain higher levels of effector molecules (Supplementary Fig. 5D, E). The relative abundance of NK cell subsets was not affected (Supplementary Fig. 5F). To determine the impact of Metformin on NK cells in an immunocompetent context, we administered Metformin by drinking water in WT tumor-bearing mice. Metformin treatment determined a slowdown in tumor growth and a reduction in tumor volume (Supplementary Fig. 5G, H). In accordance with our hypothesis, Metformin increased autophagy levels in NK cells (Supplementary Fig. 5I), and determined an enrichment of the CD11b+ CD27− cells (Supplementary Fig. 5J). Finally, autophagy activation by Metformin conferred a superior cytolytic ability to NK cells isolated from tumor-bearing mice (Supplementary Fig. 5K). To test the efficacy of autophagy activation in the human settings model, we subcutaneously injected NSG mice with PC3 cells. After engraftment, the mice received an infusion of NK-92 cells, treated or not with Metformin, twice a week (Fig. 4G). Untreated NK-92 cells were able to control tumor growth to a very limited extent, while Metformin-treated NK-92 cells caused a marked slowdown in tumor growth and a significant reduction in tumor volume (Fig. 4H,I). Metformin-treated NK-92 cells had a superior ability to infiltrate the tumor site (Fig. 4J and Supplementary Fig. 5L) and, despite unaffected levels of basal autophagy (Supplementary Fig. 5M), retained higher levels of some effector molecules, such as granzyme B (Fig. 4K). Finally, we employed BECN1 OE NK-92 cells in the above-described in vivo preclinical model (Fig. 4G). We found that BECN1 OE NK-92 cells presented a superior ability to control tumor growth, thus determining a marked reduction in tumor volume (Fig. 4L, M). Difference in abundance of tumor-infiltrating NK cells was not significant (Fig. 4N and Supplementary Fig. 5N), nevertheless cells showed higher levels of autophagy and effector molecules, including granzyme B, perforin and IFNγ (Fig. 4O, P). Strikingly, the analysis of peripheral blood at endpoint showed that BECN1 OE NK-92 cells are more abundant compared to their control counterpart (Fig. 4Q), suggesting a longer persistence and a protracted anti-tumor effect, also highlighted by enhanced autophagy levels and effector functions (Fig. 4R, S).

A Tumor model and treatment scheme of tumor-bearing mice with NK cells, treated or not with Metformin. B, C Tumor growth curves (B) and tumor volume at the end of the experiment (C) referred to mice treated as described in A (n = 5 control, n = 10 untreated NK cells, n = 12 Metformin-treated NK cells). D–F Graphs showing NK cell maturation status (D), frequency of LC3B+ cells (E), and effector molecule expression in tumor-infiltrating NK cells. n = 9 mice NK cells, n = 11 mice Metformin-treated NK cells. G Tumor model and treatment scheme of tumor-bearing mice with NK-92 cells, treated or not with Metformin. H, I Tumor growth curves (H) and tumor volume at the end of the experiment (I) referred to mice treated as described in G (n = 11 control, n = 11 NK-92 cells, n = 12 Metformin-treated NK-92 cells, data pooled from two independent experiments). J, K Graph showing the absolute number (J) and effector function expression (K) of tumor-infiltrating NK-92 cells. n = 11 control, n = 11 NK-92 cells, n = 12 Metformin-treated NK-92 cells. L, M Tumor growth curves (L) and tumor volume at the end of the experiment (M) referred to mice treated as described in G (n = 9 scramble NK-92 cells, n = 9 BECN1 overexpressing NK-92 cells). N Graph showing the absolute number of tumor-infiltrating NK-92 cells. n = 9 scramble NK-92 cells, n = 9 BECN1 overexpressing NK-92 cells. O, P Graphs showing the frequency of LC3B+ cells (O) and effector function expression (P) in tumor-infiltrating NK-92 cells. n = 9 scramble NK-92 cells, n = 9 BECN1 overexpressing NK-92 cells. Q Graph showing the frequency, referred to live cell gate, of circulating NK-92 cells in mice injected with control or BECN1 overexpressing NK-92 cells (n = 12 mice/group). R, S Graph showing the frequency of LC3B+ cells (R) and effector function expression (S) in peripheral blood circulating NK-92 cells. n = 12 sample/group. Symbols in (B), (H), and (L) represent mean and error bars indicate SEM; one-way ANOVA test with Holm-Šídák’s multiple-comparisons test (B, H) or two-tailed unpaired t test (L), comparing area under the curve (AUC). Data in (C) and (I) are presented as Min to Max box-and-whisker plot, the box extends from the 25th to 75th percentiles, and the whiskers reach the sample maximum and minimum values, the median indicated is at center line and the mean value is indicated as “+”; one-way ANOVA test with Holm-Šídák’s multiple-comparisons test. Data in (D–F), (J), (K), and (M–S) are presented as Min to Max box-and-whisker plot, the box extends from the 25th to 75th percentiles and the whiskers reach the sample maximum and minimum values, the median is indicated at center line and the mean value is indicated as “+”; two-tailed unpaired t test.
Activation of the CXCL12/CXCR4 axis inhibits autophagy in tumor-infiltrating NK cells
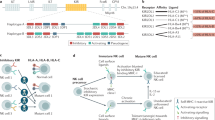
Seeking to identify specific tumor-driven alterations responsible for autophagy inhibition in tumor NK cells, we evaluated the mRNA expression levels of several autophagy‐associated genes in NK cells exposed to Pten−/−-conditioned medium (Fig. 5A). The qRT-PCR analysis revealed a broad shutdown of autophagy-related gene expression, involving different steps of the machinery, including main positive autophagy regulators, such as Irgm55, Mapk856 and Tp53, genes that promote autophagy initiation, including Ulk1, Ulk2, Ambra, Becn1, Prkaa1, Pik3ra and Pik3c357 and members of the ATG858 and ATG12 ubiquitin-like conjugation system59, engaged in autophagosome biogenesis. Finally, also the lysosomal degradation pathway was affected as evidenced by inhibition of Lamp1, Npc160, Dram261 and Ctsd62. Conversely, few molecules were upregulated by Pten−/−-derived medium and among them Fadd, Igf1 and Ins2, and the chemokine receptor Cxcr4 (Fig. 5A). Interestingly, CXCR4 has been recently described as a negative regulator of autophagy in immune and non-immune cells63,64. However, whether CXCR4 is involved in autophagy regulation in NK cells still remains unknown. Firstly, we found that the expression of CXCR4 was upregulated in tumor-conditioned murine NK cells both in vitro and ex vivo (Fig. 5B, C and Supplementary Fig. 6A, B) and in human tumor-exposed NK cells (Fig. 5D and Supplementary Fig. 6C–F). In murine NK cells CXCR4 upregulation was more evident within the mature CD11b+ CD27− subset (Supplementary Fig. 6G), thus supporting the hypothesis of a restricted rather than a widespread alteration. Notably, the exposure to the secretome of different tumor cell lines, determine, similarly to prostate tumors, the upregulation of CXCR4 (Supplementary Fig. 6H).

A Volcano plot showing the fold change expression of autophagy genes between tumor-conditioned and control NK cells (n = 2 biological replicates, NK cells from different mice). B–D Flow cytometry analysis of CXCR4+ cells in tumor-conditioned vs control NK cells (B, n = 7 biological replicates, independently collected batches of tumor-conditioned medium), Ptenpc+/+ vs Ptenpc−/− prostate-infiltrating NK cells (C, n = 6 Ptenpc+/+ mice and n = 5 Ptenpc−/− mice) and tumor vs non-tumor infiltrating NK cells (D, n = 5 samples/group). E UMAP representation of the 24 major cell types identified within CD45+ and CD45− infiltrating cells in Ptenpc−/− tumor tissues (n = 2 Ptenpc+/+ mice and n = 2 Ptenpc−/− mice). F CellPhoneDB intercellular communication analysis between NK cells and all the other cell clusters identified by scRNA-seq (n = 2 Ptenpc+/+ mice and n = 2 Ptenpc−/− mice). G Graph showing the autophagic flux on murine NK cells in tumor-conditioned NK cells, with or without Plerixafor (n = 5 biological replicates, independently collected batches of tumor-conditioned medium). H Cytotoxicity of tumor-conditioned NK cells, with or without Plerixafor, against YAC-1 target cells (n = 4 biological replicates, independently collected batches of tumor-conditioned medium). I Graph showing the autophagic flux on murine NK cells, in presence or absence of CXCL12 (n = 4 biological replciates, NK cells from different mice). J Cytotoxicity of control or CXCL12-treated NK cells against YAC-1 target cells (n = 5 biological replicates, NK cells from different mice). K Graph showing the autophagic flux on non-targeting (NT) and CXCR4 KO NK-92 cells, upon tumor-conditioned medium exposure (n = 5 biological replicates, independently collected batches of tumor-conditioned medium). L Cytotoxicity of NT and CXCR4 KO NK-92 cells, exposed to tumor-conditioned medium, against PC3 target cells (n = 12, biological replicates, data pooled from two independent experiments). M, N Tumor growth curves (M) and tumor volume at the end of the experiment (N) referred to mice treated as described in 4 G (n = 7 control, n = 7 NT NK-92 cells, n = 7 CXCR4 KO NK-92 cells). O Graph showing the absolute number of tumor-infiltrating NK-92 cells. n = 7 control, n = 7 NT NK-92 cells, n = 7 CXCR4 KO NK-92 cells. P, Q Flow cytometry analysis of effector functions in tumor-infiltrating (P) and circulating (Q) NK-92 cells. n = 7 control, n = 7 NT NK-92 cells, n = 7 CXCR4 KO NK-92 cells. Data in (A) are presented as volcano plot; two-tailed unpaired t test. Data in (B), (C), (G–L), and (O–Q) are presented as Min to Max box-and-whisker plot, the box extends from the 25th to 75th percentiles and the whiskers reach the sample maximum and minimum values, the median is indicated at center line and the mean value is indicated as “+”; two-tailed unpaired t test. Data in (D) are presented as as before-after plot; two-tailed paired t test. Symbols in (M) represent mean and error bars indicate SEM; one-way ANOVA test, comparing area under the curve (AUC). Data in (F) are presented as balloon plot; enriched ligand–receptor interactions were calculated based on permutation test. Data in (N) are presented as Min to Max box-and-whisker plot, the box extends from the 25th to 75th percentiles and the whiskers reach the sample maximum and minimum values, the median is indicated at center line and the mean value is indicated as “+”; one-way ANOVA test with Holm-Šídák’s multiple-comparisons test.
To gain more insights into the molecular features of tumor-infiltrating NK cells and their interactions with tumor cells, we performed scRNA-seq analysis of both CD45+ and CD45− cells isolated from Ptenpc−/− prostate tumors using Chromium-based scRNA-seq technology (10× Genomics) (Supplementary Fig. 6I). This allowed us to map the transcriptional landscape of Ptenpc−/− prostate, identifying 24 cell clusters, mainly immune cell subsets (Fig. 5E). Clusters were annotated based on canonical marker expression and differentially expressed genes (Supplementary Fig. 6J). Among CD45− cells, we recognized luminal and basal epithelial cells and fibroblasts. Among CD45+ cells, we identified various T cell subsets, macrophages, dendritic cells, monocytes and pDCs, as well as two subsets of neutrophils and B cells. We identified two NK cell clusters: C2 is composed by CD27+CD11b− cytokine-producing NK cells, in accordance with high expression of Xcl1 and Cd7 genes, while C13 is composed by CD27−CD11b+/ CD27+CD11b+ cytotoxic NK cells, according to the expression of Klra8, Klra4, Prf1, Cma1, Lgals1, Irf8 and Ly6c2. To then dissect the crosstalk between tumoral epithelial cells and the two NK cell clusters, we applied the CellPhoneDB algorithm65 that predicts cell-cell interactions. CellPhoneDB analysis unveiled a list of possible ligand-receptor pairs between epithelial and NK cells, that includes the CXCL12/CXCR4 axis (Fig. 5F). Of note, the CXCR4/CXCL12 interaction was exclusively present in the C13/C17 pair, possibly suggesting that this interaction has a prominent role in the cytotoxic compartment (Fig. 5F and Supplementary Fig. 6K). To test this mechanism in vitro, we treated NK cells with Plerixafor, a well-known CXCR4 inhibitor. Strikingly, stimulation of Pten−/−-conditioned NK cells with Plerixafor determined an increase in autophagic flux (Fig. 5G) as well as a rescue in NK cell killing ability (Fig. 5H). The inhibitory effect of CXCL12 on human NK cell behavior was confirmed by the rescue of both autophagic flux and cytolytic ability upon Plerixafor administration (Supplementary Fig. 7A, B). The blockade of CXCR4/CXCL12 axis determined a rescue in the mitochondrial potential levels (Supplementary Fig. 7C). Finally, administration of recombinant CXCL12 hinders both autophagic pathway (Fig. 5I and Supplementary Fig. 7D) and effector functions of NK cells (Fig. 5J and Supplementary Fig. 7E-G), which are restored upon Plerixafor administration, thus confirming the role of the CXCL12/CXCR4 axis in this context.
To validate our findings, we generated a CXCR4 knockout (CXCR4 KO) NK-92 cell line by means of the CRISPR/Cas9-mediated genome editing (Supplementary Fig. 7H). Importantly, we found that the lack of CXCR4 receptor induced in NK cells a marked resistance to tumor-mediated inhibitory effect on both autophagic flux and cytolytic functions (Fig. 5K, L). Abrogation of CXCL12 inhibitory effects in CXCR4 KO cells confirmed the role of the CXCR4 receptor in the regulation of both autophagy and lytic ability (Supplementary Fig. 7I, J).
To further corroborate our hypothesis, we employed CXCR4 KO NK-92 cells in vivo. We found that CXCR4 KO NK-92 cells were characterized by enhanced ability to control tumor growth, thus resulting in a marked reduction in tumor volume (Fig. 5M, N). Despite the lack of CXCR4 chemokine receptor, NK-92 cells similarly reach the tumor site (Fig. 5O and Supplementary Fig. 7K). Notably, both tumor-infiltrating and peripheral CXCR4 KO NK-92 cells were showed partially improved effector functions (Fig. 5P, Q).
Collectively, our results indicate that the CXCL12/CXCR4 axis is a key regulator of the autophagic behavior and the anti-tumor response in tumor-infiltrating NK cells.
C/EBP β activation impairs autophagy in tumor-conditioned NK cells
Seeking to explore the autophagy-related changes in tumor-infiltrating NK cells, we applied NicheNet analysis to predict ligand-target interactions that might drive gene expression changes in cells of interest66. We performed this analysis considering C13 and C17 NK cells as interacting cells and autophagy-related genes as pathway modulated by the interaction (Fig. 6A). NicheNet analysis identified tumor-derived factors involved in the transcriptional regulation of CXCR4, including TGFβ. Treatment with a selective TGFβ receptor inhibitor, LY2109761, abolished CXCR4 upregulation in both tumor-exposed and TGFβ-treated NK cells, thus confirming the role of TGFβ in this context (Fig. 6B). We also confirmed that TGFβ was released in conditioned media of both human and murine prostate cancer cell lines (Fig. 6C, D).

A Results of the NicheNet analysis performed on scRNA sequencing, described in 4E. B Graph showing the MFI of CXCR4 in NK-92 cells, treated with TGFβ or tumor-conditioned medium, in presence or absence of TGFβ receptor inhibitor LY2109761 (n = 6 replicates, independently collected batches of tumor-conditioned medium). C, D Quantification of TGFβ release from human (C) and murine (D) prostate cancer cell lines (n = 4 biological replicates). E, F Bar graph showing C/EBPβ activation in NK-92 cells upon administration of CXCL12 (E, n = 4 biological replicates) or PC3-derived conditioned media, with or without Plerixafor (F, n = 3 biological replicates). G, H Relative fold change of key autophagy-related genes in NK-92 cells treated or not with Helenalin Acetate (HA), in absence (G) or presence (H) of tumor-conditioned medium (n = 5 biological replicates). I Graph showing the autophagic flux on NK-92 cells, treated or not with HA, with or without tumor-conditioned medium (n = 5 biological replicates, independently collected batches of tumor-conditioned medium). J Cytotoxicity of control or tumor-conditioned NK-92 cells, in presence or absence of HA, against PC3 target cells (n = 5 biological replicates, independently collected batches of tumor-conditioned medium). K) Graph showing the autophagic flux on NK-92 cells, treated or not with HA, in presence or absence of CXCL12 (n = 6 biological replicates). L, M Tumor growth curves (L) and tumor volume at the end of the experiment (M) referred to mice treated with NK-92 cells, exposed or not to HA (n = 15 NK-92 cells, n = 15 HA-treated NK-92 cells). N Graph showing the absolute number of tumor-infiltrating NK-92 cells. n = 15 NK-92 cells, n = 15 HA-treated NK-92 cells. O, P Graph showing the frequency of LC3B+ cells (O) and effector function expression (P) in peripheral blood circulating NK-92 cells. n = 15 NK-92 cells, n = 15 HA-treated NK-92 cells. Data in (B) and (I–K) are presented as Min to Max box-and-whisker plot, the box extends from the 25th to 75th percentiles and the whiskers reach the sample maximum and minimum values, the median is indicated at center line and the mean value is indicated as “+”; one-way ANOVA test with Holm-Šídák’s multiple-comparisons test. Data in (C–F) are presented as scatter plot with mean ± SEM; one-way ANOVA test with Holm-Šídák’s multiple-comparisons test for (E) and (F). Data in (G) and (H) are presented as scatter plot with mean ± SEM; two-tailed unpaired t test. Symbols in (L) represent mean and error bars indicate SEM; two-tailed unpaired t test, comparing area under the curve (AUC). Data in (M–P) are presented as Min to Max box-and-whisker plot, the box extends from the 25th to 75th percentiles and the whiskers reach the sample maximum and minimum values, the median is indicated at center line and the mean value is indicated as “+”; two-tailed unpaired t test.
We then listed out the potential drivers of the altered autophagic behavior observed in NK cell subset, in accordance to Pearson correlation coefficient, indicative of the ability of each ligand to predict the expression of differentially expressed genes. CXCL12 emerged as one of the top ten ranked ligand (Fig. 6A) and its expression resulted to be limited to epithelial and fibroblast clusters (Supplementary Fig. 7L). CXCL12 was present in tumor secretome of both human and murine prostate cancer cell lines (Supplementary Fig. 7M, N). NicheNet analyses identified the CXCR4/CXCL12 pair as significant ligand-receptor interaction driving NK cell behavior (Fig. 6A). Mechanistically, NicheNet analyses performed on tumor-infiltrating NK cells uncovered C/EBPβ as a potential transcription factor downstream of CXCL12/CXCR4 axis (Fig. 6A). Accordingly, we found that C/EBPβ is activated upon exposure of NK-92 cells to CXCL12 (Fig. 6E). Strikingly, administration of tumor-conditioned media to NK-92 cells drives C/EBPβ activation that is hindered by Plerixafor treatment (Fig. 6F). Finally, to confirm the involvement of C/EBPβ in the tumor-mediated inhibition of both the autophagic pathway and the NK cell-mediated killing, we treated NK-92 cells with the sesquiterpene lactone helenalin acetate (HA), a natural compound known to be a potent C/EBPβ inhibitor67. HA has been previously reported to be specific towards C/EBPβ, however it may still exhibit off-target effects due to its ability to covalently interact with cysteine residues in other proteins68. We found that HA determined an activation of autophagic transcriptional program in both control and tumor-exposed NK cells (Fig. 6G, H). Importantly, the re-activation of autophagic flux by HA in tumor-conditioned and in CXCL12-treated NK cells (Fig. 6I, J) proved C/EBPβ participation in the fine-tuning of autophagy in this context. Of note, the inhibition of C/EBPβ resulted in the prevention of tumor-driven impairment of NK cell-mediated killing (Fig. 6K) and, similarly to CXCR4 blockade, in the partial rescue of mitochondrial membrane potential, suggesting a recovery of the pathway involved in the degradation of damaged mitochondria (Supplementary Fig. 7O). To test the involvement of C/EBPβ axis in vivo, we treated tumor-bearing mice, with NK-92 cells, pre-treated or not with HA. Importantly, HA-treated NK-92 cells showed a better ability to control tumor growth, compared to the untreated condition, thus determining a significant reduction in tumor volume (Fig. 6L, M). Exposure to HA conferred to NK-92 cells a better ability to infiltrate the tumor (Fig. 6N and Supplementary Fig. 7P). Finally, the analysis of peripheral blood at endpoint showed enhanced autophagy levels, despite unchanged levels of effectors molecules, in HA-treated NK-92 cells (Fig. 6O, P).
Overall, our results indicate that C/EBPβ is driver of the defective autophagy and functional behavior of tumor-conditioned NK cells.
Autophagy activation confers a superior anti-tumor ability to PD-L1.CAR NK cells
Recent works highlight the potential efficacy of PD-L1 targeting in the context of solid tumor treatment, thus providing the rationale to explore the safety profile of PD-L1 chimeric antigen receptor (CAR) NK cells in patients with solid tumors69,70. Importantly, a phase II trial is currently ongoing to test the efficacy of PD-L1.CAR NK cell immunotherapy in patients with recurrent/metastatic gastric or head and neck cancer (NCT04847466). Therefore, we further tested whether autophagy modulation can be synergistic with CAR-retargeting of NK cells. As a model we used NK-92 cells genetically modified with a PD-L1-specific CAR (PD-L1.CAR NK-92) (Fig. 7A). Importantly, we found that human PC3 cell line expresses PD-L1, thus representing a viable target for CAR-based therapy in prostate cancer (Fig. 7B, C). In vitro data confirmed that, similarly to parental cell line, the killing ability of PD-L1.CAR NK-92 cells was negatively affected by prostate cancer-conditioned medium and that Metformin was able to completely restored their functionality (Fig. 7D). In line with previous data, we proved that Plerixafor treatment was able to abrogate CXCL12-mediated inhibitory effect on both autophagic flux and lytic ability of PD-L1.CAR NK-92 cells (Supplementary Fig. 7Q, R). Finally, we validate our in vitro findings in the previously described tumor model (Fig. 4G). Indeed, in vivo, Metformin-treated PD-L1.CAR NK-92 cells better control tumor growth and infiltrated the tumor at higher levels compared to the untreated condition (Fig. 7E–G and Supplementary Fig. 7S). In addition, they were characterized by a significant increase in basal autophagy levels upon Metformin treatment (Fig. 7H) and showed a higher expression of effector molecules, including granzyme B and perforin (Fig. 7I).

A Scheme depicting the structure of PD-L1.CAR NK-92 used in the study. B Flow cytometry analysis of PD-L1.CAR expression on PD-L1.CAR NK-92 and parental NK-92 cells, by human recombinant PD-L1-Fc protein combined with anti-Fc secondary antibody. Filled gray areas indicate negative controls stained with secondary antibody only. Representative data from at least 3 independent experiments are shown. C Flow cytometry analysis of PD-L1 expression in PC3 cell line. Left plot represents Fluorescence Minus One (FMO) control, right plot represents cells stained with PD-L1 antibody. D Cytotoxicity of PD-L1.CAR NK-92 cells, treated or not with tumor-conditioned medium, with or without Metformin, against PC3 target cells (n = 4 biological replicates, independently collected batches of tumor-conditioned medium). E, F Tumor growth curves (E) and tumor volume at the end of the experiment (F) referred to mice treated as described in 4G (n = 7 control, n = 7 CAR NK-92 cells, n = 7 Metformin-treated CAR NK-92 cells). G Graph showing the absolute number of tumor-infiltrating NK-92 cells. n = 7 control, n = 7 CAR NK-92 cells, n = 7 Metformin-treated CAR NK-92 cells. H, I Graph showing the frequency of LC3B+ cells (H) and effector function expression (I) in tumor-infiltrating CAR NK-92 cells. n = 7 control, n = 7 CAR NK-92 cells, n = 7 Metformin-treated CAR NK-92 cells. Data in (D) and (F) are presented as Min to Max box-and-whisker plot, the box extends from the 25th to 75th percentiles and the whiskers reach the sample maximum and minimum values, the median is indicated at center line and the mean value is indicated as “+”; one-way ANOVA test with Holm-Šídák’s multiple-comparisons test. Symbols in (E) represent mean and error bars indicate SEM; one-way ANOVA test with Holm-Šídák’s multiple-comparisons test, comparing area under the curve (AUC). Data in (G), (H), and (I) are presented as Min to Max box-and-whisker plot, the box extends from the 25th to 75th percentiles and the whiskers reach the sample maximum and minimum values, the median is indicated at center line and the mean value is indicated as “+”; two-tailed unpaired t test.
Collectively, our results indicate that autophagy is an intracellular checkpoint in NK cells, and it can be exploited in vivo to improve NK-based cell therapy in cancer.
Discussion
NK cells anti-tumor capability is impaired in cancers, and mechanisms that drive NK cell dysfunction prejudice the efficacy of NK-cell-based therapies2. The mechanisms behind NK cell impairment are numerous, and a definitive therapeutic strategy to reinforce NK cell anti- tumor killing is still missing4. Here, we unveiled an uncovered role for autophagy in NK cell activation and killing ability in tumors. Tumor-infiltrating NK cells show a defective autophagy process in prostate cancer patients and mouse models. Autophagy impairment is induced by the engagement of the CXCL12/CXCR4/C/EBPβ pathway, and activation of the autophagy machinery by drugs or genetic alteration confers to NK and CAR-NK cells a superior anti-tumor function.
Previous studies have highlighted the dependency of lymphoid subsets, including T and B lymphocytes, on autophagy for differentiation, survival, and effectiveness, and more recently the impact of autophagy on NK cell development, survival, and anti-viral response in NK cells has been uncovered27,28,29,71. However, the role of autophagy in NK-cell anti-cancer response remains unknown. Our findings give insight in the extrinsic autophagy response in cancer and unveil that loss of autophagy in tumor-infiltrating NK cells significantly restricts their anti-tumor properties. Importantly, whether the modulation of autophagy may represent a therapeutic approach in cancer is still under debate. The breakdown products deriving from autophagy are recycled into metabolic pathways and cancer cells exploit this process to sustain their metabolism and proliferation. Accordingly, autophagy inhibition in many tumors induces cancer regression and even elicits a pro-inflammatory immune response72,73. However, systemic inhibition of autophagy may, in parallel, hinder the immune response against cancer, thus limiting the efficacy of this therapeutic approach. Here, we propose an off-the-shelf strategy that activates autophagy specifically in NK cells and may be considered as combinatorial approach to reinforce the efficacy of systemic autophagy modulators in cancer therapy. Here we employed a genetic overexpression of Beclin 1 to induce autophagy activation. Although Beclin 1 itself does not directly regulate the expression of ATG genes, its overexpression as well as the treatment with Tat-Beclin1 can induce autophagy74. This induction may lead to an increase in autophagic gene expression, probably through positive feedback mechanisms that enhance the overall autophagic process, as demonstrated for mTOR inhibition by Torin175.This suggests that Beclin 1 overexpression may indirectly support autophagy through feedback loops involving key transcription factors.
Importantly, a notable consequence of autophagy impairment is the dysfunction of mitochondria within tumor-conditioned NK cells. Dysfunctional mitochondria have been implicated in a variety of cellular processes, and the loss of mitochondrial function can severely compromise the capacity of NK cells to execute their anti-tumor activities. While our study demonstrates a strong association between autophagy impairment and mitochondrial dysfunction in tumor-infiltrating NK cells, it is important to acknowledge that the precise underlying causes of this relationship require further investigation. Several factors may contribute to the observed defects. For instance, alterations in the autophagic machinery, such as inadequate autophagosome formation or lysosomal dysfunction, could hinder the removal of damaged mitochondria. Furthermore, the TME itself may create conditions that disrupt autophagy in NK cells, thus indirectly affecting mitochondrial integrity. The specific molecular mechanisms and signaling pathways that link autophagy impairment to mitochondrial dysfunction remain areas of ongoing exploration.
Mechanistically, we identified CXCL12 as key factor that hinders autophagy in NK cells. Apart from its physiological role in organogenesis, angiogenesis and immune cell trafficking, the CXCL12/CXCR4 axis acts as a tumor-promoting pathway. Intrinsically, CXCR4 activation in cancer cells controls the transduction of downstream signalling pathways that sustain cell survival, proliferation, and metastatic spread76. In parallel, the expression of CXCL12 is prognostic of progression in multiple cancers, including prostatic adenocarcinoma77. The CXCL12/CXCR4 signalling pathway also supports the crosstalk between cancer and its microenvironment. On this line, CXCL12 released by stellate hepatocytes engages CXCR4 on NK cells and impairs cytotoxicity, thus favouring the metastatic outgrowth of breast cancer to the liver78. Accordingly, we found a specific tumor-driven induction of CXCR4 in NK cells as well as an inhibition of both autophagy and NK cell effector functions upon exposure to CXCL12. CXCR4 antagonists have emerged as potential therapeutic approaches, however, the pathway is pleiotropic and systemic toxicity so far precluded the application of inhibitors in the clinic. Nevertheless, specific modulation of CXCR4 on NK cells before cell infusion would overcome systemic toxicity, while sustaining the efficacy of cell therapy. Of note, here we unveiled a previously unknown role for C/EBPβ downstream CXCR4 in NK cells. C/EBPβ is a known transcription factors expressed by most tumor cell types and by myeloid subsets. Interestingly, C/EBPβ deleted models showed impaired tumor progression. Our findings indicate that C/EBPβ regulates NK cell activation and may thus be targeted to improve NK cell-based therapies in cancer.
In recent years, the concept of NK cells as candidate for therapy in cancer has gained interest. Promising results in blood cancers supported the development of strategies to augment NK cell efficacy and half-life once infused. Still challenges in non-hematopoietic cancers remain. Our results indicate that autophagy modulation can increase NK cell fitness and extend persistence in vivo in prostate cancer. Autophagy modulation in NK cells thus provides a translatable method to enhance NK and CAR-NK cell efficacy during manufacturing and may guide the design of NK cell therapies with improved efficacy in solid cancers.
Methods
Our research complies with all relevant ethical regulations
Experimental model and subject details
Animals and ethics statement
All mice used in this study were housed in individually ventilated cages in the specific pathogen-free animal facility at Humanitas Clinical and Research Center (Rozzano, Milan, Italy). Experimental/control animals were co-housed. Procedures involving animal handling and care conformed to protocols approved by the Humanitas Clinical and Research Center, in compliance with national and international law and policies. The study was approved by the Italian Ministry of Health. All efforts were made to minimize the number of animals used and their suffering, in accordance to the 3Rs principle79. Mice were euthanized by CO2 exposure. Ptenpc−/− mice were obtained by crossing Ptenloxp/loxp mice to the Probasin-Cre transgenic mice (B6.129S4-Ptentm1Hwu/J, JAX stock #006440, and Tg(Pbsn-cre)4Prb/J, JAX stock #026662), in order to achieve Pten prostate-specific deletion. Seven-week-old male C57BL/6 wild-type mice (JAX stock #000664) were obtained from Charles River Laboratories or were littermates of Ptenpc−/− mice. Seven-week-old male NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, JAX stock #005557) were obtained from Charles River Laboratories. Tumour size/burden did not exceed 1.5mm3. All mice received a standard chow diet.
Method details
Cell lines
Prostate cancer cell lines, including mouse cell line, PTEN-Cap8 (Pten−/−), and human cell lines, PC3 and LNCaP were obtained from ATCC. Non-prostatic cancer cell lines, including B16-F10, ID8, MCA-203, YAC-1 and 4T1, were obtained from ATCC.
Pten−/− Trp53−/− prostatic tumor cell line was provided by prof. Ronald De Pinho, MD Anderson. Cells were routinely maintained in RPMI medium supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin solution, 1% L-glutamine and 1% sodium pyruvate solution at 37 °C in 5% CO2. For Pten−/− prostatic tumor cells, 25 μg/ml bovine pituitary extract, 5 μg/ml human recombinant insulin and 6 ng/ml human recombinant epidermal growth factor were added to the complete medium. For Pten−/− Trp53−/− prostatic tumor cells, dihydrotestosterone was added to the complete medium.
NK-92 and PD-L1.CAR NK-92 cell lines were maintained in X-VIVO 20 medium supplemented with 12.5% heat-inactivated FBS, 12.5% heat-inactivated horse serum, 1% penicillin/streptomycin solution, 1% L-glutamine, 1% sodium pyruvate solution and 1% Non-Essential Amino Acid Solution, 500 U/mL IL-2). HEK293T cells were cultured in DMEM medium, supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin solution, and 1% L-glutamine.
Mouse procedures
Xenograft PCa model in immunodeficient background was obtained by subcutaneous injection of 3 × 106 Pten−/−Trp53−/− prostate cell line in male NSG mice. Xenograft melanoma cancer model in immunodeficient background was obtained by subcutaneous injection of 0.1 × 106 B16-F10 melanoma cell line in male NSG mice. Xenograft model of human PCa was achieved by injecting 3 × 106 PC3 cell line into male NSG mice. Tumor size was measured by caliper and quantified according to the formula: Volume = (D × d2)/2, where D = larger tumor diameter and d = smaller tumor diameter. At the end of the experiment, tumor size was measured by applying the following formula: Volume = (Width2 × Length)/2. For the prostatic tumors, the size of two anterior lobes was considered.
For adoptive NK cell transfer, enriched splenic NK cells were obtained by means of the NK Cell Isolation kit II, in accordance to the manufacturer’s instructions. Cells were stimulated in RPMI complete medium added of IL-12 (10 ng/mL), IL-15 (10 ng/mL) and IL-18 (50 ng/mL) in presence or absence of metformin (10 μM). After 24 h, cells were collected and cell viability was estimated by Trypan Blue exclusion. 106 NK cells (purity >95%) were intravenously injected in tumor-bearing mice according to the scheme (see Fig. 4A).
NK-92 and PD-L1.CAR NK-92 were stimulated in X-VIVO complete medium added of IL-2 (500 U/mL), in presence or absence of Metformin (10 μM). After 24 hours, cells were collected and cell viability was estimated by Trypan Blue exclusion. 3 × 106 cells were intravenously injected in tumor-bearing mice according to the scheme (see Fig. 4G).
For NK cell depletion experiment, tumor-bearing mice were intraperitoneally injected with αNK1.1 (clone PK136) antibody or recommended isotype control antibody according to the scheme (see Supplementary Figs. 1J, 2A, and E).
For metformin systemic treatment, male C57/BL6 were subcutaneous injected with 3 × 106 Pten−/−Trp53−/− prostate cancer cells. Starting from day 7 after tumor injection, mice were orally administered with metformin hydrochloride dissolved in drinking water (5 mg/mL).
Tumor infiltrate analysis by FACS
For analysis of tumor-infiltrating leukocytes, tumors were collected, cut into small pieces, and digested with Collagenase I (1 mg/mL for mouse tissue and 0.5 mg/mL for human tissue) for 45 mins at 37 °C on a rocking platform. After a quick digestion in 2.5% Trypsin and DNase I, single-cell suspension was obtained by mechanical dissociation through a syringe needle (18 G) and subsequent filtration on a 40-μm cell strainer. The composition of tumor infiltrate was determined by flow cytometry. All antibodies used are listed in Supplementary Table 1. Samples were analyzed with FACSymphony™ A5 Cell Analyzer.
Absolute cell counts of tumor-infiltrating NK-92 cells
To obtain an absolute count of tumor-infiltrating NK-92 cells CountBright™ Absolute Counting Beads were added to samples before FACS acquisition. We calculate the absolute number using the formula:
Absolute number was normalized on tumor weight.
Histopathology
Normal and tumor tissue samples were fixed in 4% neutral formalin for at least 24 h. Tissues were processed by ethanol dehydration and embedded in paraffin according to standard protocols. Sections (3 μm) were prepared for hematoxylin and eosin (H&E) staining. In all experiments, the histology was evaluated blindly.
Tumor-conditioned NK cells preparation
To obtain a tumor-conditioned medium, tumor cells were seeded in a complete RPMI medium, without any additional growth factors. After 72 h the confluence must not exceed 80%, so the initial number of seeded cells has been adjusted accordingly. In particular, we seeded:
-
13,300/cm2 Pten−/−
-
12,000/cm2 Pten−/− Trp53−/−
-
16,600/cm2 LNCaP
-
13,300/cm2 PC3
The tumor-conditioned medium was harvested and centrifuged to remove cellular debris at 800 × g for 10 mins at room temperature. Finally, the medium was collected without disturbing cellular debris on the bottom of the tube and stored at −80 °C till further use.
To obtain tumor-conditioned NK cells, both mouse and human NK cells were exposed for 24 h to tumor-conditioned medium mixed in a 1:1 ratio with fresh culture medium, added recombinant mouse IL-12, IL-15, and IL-18 or recombinant human IL-2, respectively.
For co-treatment of NK cells with autophagy-activating drugs, the following drugs were added to culture medium: Rapamycin (100 nM), Everolimus (20 nM), Gemcitabine (100 nM), Carfilzomib (10 nM) and Plerixafor (100 nM). CXCL12 was added to culture medium at a final concentration of 100 ng/mL. For C/EBPβ blockade experiments, cells were incubated with Helenalin Acetate (1 μM) or control vehicle (DMSO) for 4 h. Subsequently, they were exposed for 24 h to tumor-conditioned medium or left untreated. For TGFβ blockade experiments, cells were treated with LY2109761 (10 μM) or control vehicle (DMSO) for 1 h and then exposed to tumor-conditioned medium or TGFβ (10 ng/mL).
FACS-based autophagosome quantification using CYTO-ID
For ex vivo assessment of autophagy levels in NK cells using the CYTO-ID autophagy detection kit, cells were incubated with CYTO-ID (1:500) at 37 °C in a humidified atmosphere with 5% CO2 for 60 minutes before being washed with the CYTO-ID kit assay buffer diluted to 1× in distilled H2O. Cells were then stained with fixable viability dye, stained with surface antibodies. For in vitro autophagic flux measurement, NK cells were treated or not with tumor-conditioned medium (in presence or absence of specified drugs, including Plerixafor and Helenalin Acetate), as previously described, and then CQ was directly added to cells (10 μM). After 24 hours, cells were collected and stained with CYTO-ID, as previously described. Samples must be analyzed by flow cytometry as soon as possible to avoid drop in fluorescence intensity. Autophagic flux was calculated with the formula: Flux = MFI (Condition + CQ) – MFI (Condition–CQ).
FACS-based autophagosome quantification using LC3
For ex vivo analysis of autophagy levels in NK cells, cells were stained using an anti-LC3 antibody. Cells were stained with a fixable viability dye and then with surface antibodies. Cells were fixed and permeabilized with Fix/Perm buffer (Foxp3/Transcription Factor staining buffer kit) and then stained with anti-LC3 primary antibody (1:100), followed by an anti-rabbit secondary antibody. For in vitro analysis of autophagic flux, the Muse® Autophagy LC3-Antibody Based Kit was employed to measure the levels of autophagosome-associated LC3 (LC3-II). NK cells were treated or not with tumor-conditioned medium, as previously described, and then Autophagy reagent A was directly added to cells. After 4 hours, cells were treated with a specific permeabilization buffer (Autophagy reagent B) to selectively extract non-autophagosome associated cytosolic LC3-I and subsequently stained with anti-LC3 Alexa Fluor 555 conjugated antibody. Autophagic flux was calculated with the formula: Flux = MFI (Condition + reagent A) – MFI (Condition – reagent A).
NK cell in vitro killing assay
Murine NK cells used in in vitro killing assays were obtained, as previously described, from naïve or tumor-bearing mice. Tumor-conditioned NK cells were tested in a killing assay after an in vitro 24-hour stimulation, while NK cells from tumor-bearing mice were used immediately after isolation. For selected experiments, NK cells were sorted (CD3− F4/80− B220− Ly6G− NK1.1+) and immediately used for co-culture with target cells.
To distinguish effector from target cells, target cells were stained with CellTrace Violet (1 μM final concentration in PBS) for 20 mins at room temperature. 10,000 non-adherent cells (YAC-1) were seeded in a U bottom 96-well plate in complete RPMI media, while 20,000 adherent cells (Pten−/−, Pten−/− Trp53−/−, LNCaP, and PC3) were seeded in a flat bottom 96-well plate in complete RPMI media. For adherent cells, co-culture has been set following a 2-hour incubation for cell adhesion. NK cells were added to wells containing target cells, at the specified effector-to-target (E:T) ratio, and incubate for 4 h in 37 °C incubator. Cells were collected and then stained with a mix of Helix NPTM NIR and Annexin V FITC in Annexin V Binding Buffer for 15 mins at room temperature. FACS analysis was performed immediately with a BD FACSCanto II. We calculate the specific lysis using the formula:
Evaluation of NK cell subset viability in vitro
To evaluate NK cell subset viability upon tumor exposure, enriched splenic NK cells were exposed to tumor-conditioned medium or left untreated. After 24 h, cells were collected and then stained with surface antibodies (CD3, NK1.1, CD27 and CD11b). Finally, cells were stained with a mix of Helix NPTM NIR and Annexin V FITC in Annexin V Binding Buffer for 15 mins at room temperature. FACS analysis was performed immediately with a BD FACSymphony™ A5 Cell Analyzer.
Effector function measurement by FACS staining
To evaluate the expression of effector molecules in tumor-infiltrating NK cells, tumor single-cell suspensions were stained with fixable viability dye and then with surface antibodies. Then cells, were fixed and permeabilized with Fix/Perm buffer (Foxp3/Transcription Factor staining buffer kit) and finally stained with fluorescent-conjugated antibodies for Granzyme B, Perforin and IFNγ. For the analysis of in vitro-generated tumor-conditioned NK cells, Brefeldin A solution was added 4 h before the staining. All antibodies used are listed in Supplementary Table 1. Samples were analyzed with FACSymphony™ A5 Cell Analyzer.
Transmission Electron Microscopy and quantification of mitochondrial damage
NK-92 cells exposed to control or tumor-conditioned medium were centrifuged, washed with phosphate buffer (0.12 M) and fixed with 4% paraformaldehyde (PFA) and 2% glutaraldehyde (GA) for 6 hrs at room temperature. After fixation, cell pellets were fixed with 1% (wt/vol.) OsO4 in cacodylate buffer (0.12 M) for 2 hrs at RT. After dehydration in a graded series of ethanol preparations, pellets were cleared in propylene oxide, embedded in epoxy medium, and polymerised at 60 °C for 72 h. From each sample, 1 μm-section was cut with a Leica EM UC6 ultramicrotome, stained with Toluidine Blue and mounted on glass slides. Ultra-thin 60 nm-sections were obtained and counterstained with uranyl acetate and lead citrate, and images were obtained with an energy filter transmission electron microscope (Libra120) coupled with an yttrium aluminium garnet scintillator slow-scan CCD camera. To assess the percentage of damaged mitochondria more than 250 mitochondria in at least 25 different cells for each experimental group were analyzed on images acquired by the iTem software.
Mitochondrial functionality evaluation by FACS
Mitochondrial membrane potential and mitochondrial mass of NK-92 cells were assessed by staining cells with 20 nM of tetramethyl rhodamine methyl ester (TMRM) and 1 μM of MitoTracker Green at 37 °C for 30 mins. The gating strategy for the selection of cells containing functional and dysfunctional mitochondria is shown in Supplementary Fig. 4I. For quantification of mitochondrial reactive oxygen species, NK-92 cells were stained with MitoSox probe (5 μM) at 37 °C for 30 mins. Samples were analyzed with FACSymphony™ A5 Cell Analyzer.
Intracellular L-lactate quantification
To quantify intracellular L-lactate, we used the L-lactate assay kit, following manufacturer’s instructions. Briefly, control or tumor-exposed NK-92 cells were harvested, resuspended in Lactate Assay Buffer, and quickly homogenized. After centrifugation, supernatants were collected and subsequently treated with Deproteinizing Sample Preparation Kit, in order to quickly remove endogenous LDH that will degrade lactate. Samples were mixed with Lactate Assay Buffer, Lactate Substrate Mix, and Lactate Enzyme Mix in order to have a colorimetric reaction, measured at 450 nm OD.
The concentration of L-lactate in the test samples is calculated as:
Where:
La = amount of Lactic acid in the sample well calculated from a standard curve (nmol).
Sv = volume of sample added into the well (µL).
D = sample dilution factor.
Reclustering of human NK cells
Human PCa single-cell dataset was used as input for NK cells reclustering36. Clusters 3, 10, 13, already annotated as NK cell populations, were selected to generate a new clustering at a resolution level of 1.3. The new clustering generated a total of 7 NK cells subpopulations. Marker genes were calculated using the FindAllMarker function of Seurat pipeline (with default settings). The full list of marker genes is provided in Supplementary Data 1. Gene signatures enrichment analysis was performed by applying the AddModuleScore function of Seurat pipeline.
scRNA-seq processing (mouse)
Tumors were processed for FACS analysis as described above and stained with Fixable Viability Dye eFluor™ 780 and then with surface antibodies (CD45, CD31, and EpCAM). 50,000 CD45+ EpCAM− cells and 50,000 CD45+ EpCAM+ cells were sorted on a FACS Aria III. FACS-purified CD45+/CD45− cells were resuspended in 1 ml PBS plus 0.04% BSA and washed two times by centrifugation at 450 × g for 7 min. After the second wash, cells were resuspended in 30 μl and counted with an automatic cell counter (Countess II) to get a precise estimation of total number of cells recovered and of their concentration. Afterwards, CD45+/CD45− cells of each sample were loaded into one channel of the Single Cell Chip A using the Single Cell 3’ reagent kit v2 single cell reagent kit (10X Genomics) for Gel bead Emulsion generation into the Chromium system. Following capture and lysis, cDNA was synthesized and amplified for 14 cycles following the manufacturer’s protocol (10X Genomics). 50 ng of the amplified cDNA were then used for each sample to construct Illumina sequencing libraries. Sequencing was performed on the NextSeq550 Illumina sequencing platform following 10x Genomics instruction for reads generation. A sequencing depth of at least 30,000 reads/cell was obtained for each sample.
Single-cell RNAseq (mouse)
Raw sequencing data in the format of bcl files were converted in fastq files and aligned to the mouse reference genome mm10, taking advantage of the Cell Ranger Pipeline version 3.0.1 provided by 10X Genomics. After a quality check, we obtained a total of 13,843 cells from the two biological replicates. Filtered gene expression matrices from Cell Ranger were used as input for clustering analysis by Seurat R package (version 3.2.2; R version 4.0.3)80. We first processed each individual data set separately, considering the thresholds of 200, 50,000, and 0.25 for the number of genes, number of unique molecular identifiers (UMI), and mitochondrial content, respectively. For each data set, we selected the 2000 most variable genes. Subsequently, we used the FindIntegrationAnchors function to combine the datasets together, choosing 2000 anchor genes for integration. After integration, we ran principal component analysis (PCA) and used the first 72 principal components (PCs) to perform Louvain clustering and UMAP embedding. Finally, we obtained a total of 24 clusters (resolution level = 0.4). Marker genes analysis was performed using the FindAllMarkers function (setting default parameters). Full list of marker genes per cluster are provided in Supplementary Data 2.
NicheNet interaction analysis
We applied NicheNet method (NicheNet version 1.1.0)66 to predict ligand–receptors interactions existing between NK populations and epithelial cells. Seurat normalized data was imputed of missed counts with ALRA function, with a predicted rank-k approximation of 35. We restricted the analysis to the following gene sets: only ligands and receptors expressed in at least 30% of sender and receivers’ cells clusters were considered. Autophagy gene list, provided in Supplementary Data 3, was used as target gene set. Ligand activity scores were calculated, and only ligands with a positive Pearson correlation coefficient were considered. Heatmaps of ligand activity shows positive ligands ordered according to their z-scores. Full list of positive significant ligands is provided in Supplementary Data 4.
CellPhoneDB interaction analysis
In order to analyze cellular interactions among immune cell populations CellPhoneDB (version 2.0.0) was used with statistical_analysis method. Normalized gene expression matrices together with their cluster annotations were used as input (‘data’ slot from Seurat after ALRA imputation step). The enriched ligand–receptor interactions between two cell subsets were calculated based on permutation test. We selected as significant ligand–receptor pairs those interactions annotated as manually curated and with P value < 0.05. Full list of significant cell-cell interactions is provided in Supplementary Data 5. Circle plots of the total interaction count were performed using the mig_chord function implemented in the migest library (version 2.0.3).
Gene signature enrichment analysis
Enrichment analysis of autophagy gene signature was performed using the Seurat function AddModuleScore. Gene signature was used as input to compute a module score for each cell. Module scores along clusters were represented by violin plots.
Quantitative RT-PCR
Total RNA from NK cells or tumor cells was extracted using RLT lysis buffer or TRIzol Reagent, following the manufacturer’s recommendations. RNA was further purified using RNeasy Mini RNA isolation kit or TRIzol protocol for RNA isolation. cDNA was synthesized using 0.5–1 μg of total RNA by reverse transcription using High Capacity cDNA Reverse Transcription Kit. Quantitative real-time PCR was performed using SYBR Green PCR Master Mix using a QuantStudio 7 Flex Real-Time PCR System. Data were analyzed with the 2−ΔΔCT (Livak) Method. Arbitrary units (AU) were defined using the following transformation: 1000 × (2−ΔCT), where ΔCT = CT target – CT reference.
For screening of alterations in autophagy-related genes, tumor-conditioned NK cells were subjected to the predesigned 384-well panel Autophagy (SAB Target List) M384, according to the manufacturer’s protocol.
Immunoblotting
For evaluation of the protein level, cell pellets were washed in PBS, lysed in RIPA buffer, including protease/phosphatase inhibitors, and incubated for 10 minutes on ice. Cell lysates were then centrifuged at 21,000 × g for 15 minutes at 4 °C. The supernatants were collected, and proteins were quantified with a DC Protein Assay Kit. For immunoblot analyses, 50 μg of protein lysates per sample were denatured in 4× Loading dye+ β-Mercaptoethanol and boiled using a heating block at 95 °C for 5 minutes before loading to 12.5% SDS-PAGE gel. Following electrophoresis, proteins are transferred to 0.45-μm activated PVDF membrane using a wet transfer system. The membrane is then blocked for 1 h at room temperature in PBS + 5% milk and then probed with LC3, BECN1, p62, β-actin, and HSP90 antibodies. Following a 1-hour incubation with HRP-conjugated secondary antibodies, signal was detected using a Clarity Western ECL Substrate and Chemidoc system. Autophagic flux was calculated with the formula: Flux = LC3-II/β-actin (Condition + CQ)–LC3-II/β-actin (Condition–CQ).
The original uncropped WBs can be found in the Supplementary Information file.
FACS-based quantification of autophagic proteins
For quantification of autophagic proteins ATG7 and ATG14, scramble or BECN1 OE NK-92 cells were stained with fixable viability dye and, subsequently, fixed and permeabilized with Fix/Perm buffer (Foxp3/Transcription Factor staining buffer kit). Finally, cells were stained with anti-ATG7 PE-conjugated or anti-ATG14 APC-conjugated antibodies.
ELISA quantification of IFNγ, CXCL12, and TGFβ
The levels of IFNγ, CXCL12 and TGFβ were assessed in culture supernatants using commercially available ELISA kits, according to the manufacturer’s instructions. For quantification of IFNγ release, splenic NK cells were treated with tumor-conditioned medium or left untreated. After 24 h, medium was replaced with fresh complete RPMI medium for additional 24 h. Finally, the medium was collected and stored at −80 °C till further use.
Transcription factor analysis
For transcription factor analysis, NK-92 cells were starved for 4 h in fresh RPMI medium lacking serum and IL-2 to improve signal-to-background ratio. Subsequently, the cells were treated with CXCL12 or PC3-conditioned medium, in presence or absence of Plerixafor. Nuclear extracts from NK-92 cells were prepared using a commercially available Nuclear Extract Kit, according to the manufacturer’s instructions. These nuclear extracts were then subjected to TransAM C/EBPβ Transcription Factor ELISA Kit, according to the manufacturer’s instructions. Briefly, 10 μg nuclear protein was allowed to bind to an oligonucleotide-coated plate. C/EBPβ was detected by incubation with specific primary antibodies and HRP-conjugated secondary antibody. The colorimetric read-out was measured as optical density (OD, absorbance 450 nm), and it is proportional to transcription factor activity. Specific transcription factor activity was expressed as follow: OD450nm (Treated condition) – OD450nm (Untreated condition).
Lentiviral modification of NK-92 cells
To produce viral particles, HEK-293T cells were seeded in 150 mm dish and transfected using the Calcium Phosphate precipitation protocol to deliver a transfer plasmid with gene of interest, VSV-G envelope expressing plasmid pMD2.G and lentiviral packaging plasmid psPAX2. After 24 h the lentivirus-containing supernatant was harvested, filtered through a 0.45 μm pore size filter and concentrated at 50,000 × g for 2 h at 4 °C. After 48 h, the collection/concentration protocol was repeated.
The culture medium from the NK-92 cells was replaced with concentrated lentiviral supernatant diluted 1:2 with NK-92 medium for transduction81 added of polybrene (1000×, 10 μg/mL) and rhIL-2 (500 U/mL). After 90 mins of spinoculation (400 g at 32 °C), NK-92 cells were kept to final concentration of 0.5 × 106 cells/mL in a humidified atmosphere containing 5% CO2. The next day, the viral supernatant was replaced with the fresh NK-92 culture medium supplemented with 500 U/mL IL-2. After 72 h, antibiotic for selection was added to culture medium and kept until a 100% mortality was obtained in not-transduced cells (at least 4 d). To obtain BECN1 overexpressing cells, a two-step lentiviral transduction protocol was adopted. First, we generated a dCas9-VP64+ NK-92 cell line, upon transduction and subsequent selection with Blasticidin. Then we introduced both BECN1_sgRNA and MS2-P65-HSF1 vectors, in order to obtain the complete SAM system. The BECN1 overexpressing cell line was obtained by selecting with Zeocin and Hygromycin antibiotics, until a 100% mortality was obtained in dCas9-VP64+ NK-92 cells (at least 4 d).
To obtain CXCR4 KO cells, NK-92 cell line was transduced using the Edit-R All-in-one lentiviral system that allows to express both a gene-specific guide RNA and the Cas9 nuclease using a single vector. CXCR4 KO cells express EGFP selection marker that allows the selection by FACS sorting.
Generation of PD-L1.CAR NK-92 cells
PD-L1.CAR NK-92 was generated by lentiviral transduction with a second-generation CAR consisting of PD-L1-specific scFv, CD8 hinge, CD28 transmembrane/costimulatory, and CD3ζ intracellular signaling domains cloned into the pSIEW backbone, as described previously82. PD-L1.CAR NK-92 cells were further immunomagnetically enriched, resulting in >95% purity of PD-L1. CAR-expressing NK-92 cells at the time of analysis.
Statistical analysis
Statistical analyses were performed using two-tailed unpaired and paired Student’s t test, for comparison between two groups, or one-way ANOVA, for comparison between three or more groups, as specified. Data are presented as Min to Max box-and-whisker plot with the median indicated at the center line, and the mean value indicated as “+” or as a scatter plot with a bar with mean ± SEM. For tumor growth curve, symbols represent mean, and error bars indicate SEM. Exact p values are provided on the figures. Data were analyzed using GraphPad Prism 9 software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The scRNAseq data generated in this study have been deposited in the GEO database under accession GSE230311. Source data are provided with this paper.
References
June, C. H., O’Connor, R. S., Kawalekar, O. U., Ghassemi, S. & Milone, M. C. CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018).
Bald, T., Krummel, M. F., Smyth, M. J. & Barry, K. C. The NK cell–cancer cycle: advances and new challenges in NK cell-based immunotherapies. Nat. Immunol. 21, 835–847 (2020).
Guillerey, C., Huntington, N. D. & Smyth, M. J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 17, 1025–1036 (2016).
Myers, J. A. & Miller, J. S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 18, 85–100 (2021).
Bonavita, E. et al. Antagonistic inflammatory phenotypes dictate tumor fate and response to immune checkpoint blockade. Immunity 53, 1215–1229.e1218 (2020).
Molgora, M. et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 551, 110–114 (2017).
Morvan, M. G. & Lanier, L. L. NK cells and cancer: you can teach innate cells new tricks. Nat. Rev. Cancer 16, 7–19 (2016).
Cerwenka, A. & Lanier, L. L. Natural killer cell memory in infection, inflammation and cancer. Nat. Rev. Immunol. 16, 112–123 (2016).
Prager, I. & Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 105, 1319–1329 (2019).
Wang, W., Erbe, A. K., Hank, J. A., Morris, Z. S. & Sondel, P. M. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 6, 368 (2015).
Bottcher, J. P. et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 172, 1022–1037.e1014 (2018).
Liu, E. et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 382, 545–553 (2020).
Ruggeri, L. et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 295, 2097–2100 (2002).
Miller, J. S. et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 105, 3051–3057 (2005).
Tonn, T., Becker, S., Esser, R., Schwabe, D. & Seifried, E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J. Hematother. Stem Cell Res. 10, 535–544 (2001).
Tonn, T. et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 15, 1563–1570 (2013).
Gras Navarro, A. et al. NK cells with KIR2DS2 immunogenotype have a functional activation advantage to efficiently kill glioblastoma and prolong animal survival. J. Immunol. 193, 6192–6206 (2014).
Veluchamy, J. P. et al. In vivo efficacy of umbilical cord blood stem cell-derived NK cells in the treatment of metastatic colorectal cancer. Front. Immunol. 8, 87 (2017).
Geller, M. A. et al. Intraperitoneal delivery of human natural killer cells for treatment of ovarian cancer in a mouse xenograft model. Cytotherapy 15, 1297–1306 (2013).
Lee, S. J. et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer 15, 1011 (2015).
Cózar, B. et al. Tumor-infiltrating natural killer cells. Cancer Discov. 11, 34–44 (2021).
Wu, S.-Y., Fu, T., Jiang, Y.-Z. & Shao, Z.-M. Natural killer cells in cancer biology and therapy. Mol. Cancer 19, 120 (2020).
Childs, R. W. & Carlsten, M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat. Rev. Drug Discov. 14, 487–498 (2015).
Aman, Y. et al. Autophagy in healthy aging and disease. Nat. Aging 1, 634–650 (2021).
Chen, Y., Zhao, Y. & Mishra, P. K. Editorial: autophagy-mediated cell survival and death in disease progression and treatment. Front. Cell Dev. Biol. 10 https://doi.org/10.3389/fcell.2022.916347 (2022).
Galluzzi, L. et al. Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836 (2017).
Clarke, A. J. & Simon, A. K. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol. 19, 170–183 (2019).
López-Soto, A., Bravo-San Pedro, J. M., Kroemer, G., Galluzzi, L. & Gonzalez, S. Involvement of autophagy in NK cell development and function. Autophagy 13, 633–636 (2017).
Germic, N., Frangez, Z., Yousefi, S. & Simon, H. U. Regulation of the innate immune system by autophagy: monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 26, 715–727 (2019).
Gerada, C. & Ryan, K. M. Autophagy, the innate immune response and cancer. Mol. Oncol. 14, 1913–1929 (2020).
Pasero, C. et al. Inherent and tumor-driven immune tolerance in the prostate microenvironment impairs natural killer cell antitumor activity. Cancer Res. 76, 2153–2165 (2016).
Gallazzi, M. et al. Prostate cancer peripheral blood NK cells show enhanced CD9, CD49a, CXCR4, CXCL8, MMP-9 production and secrete monocyte-recruiting and polarizing factors. Front. Immunol. 11, 586126 (2020).
Wang, S. et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4, 209–221 (2003).
Chiossone, L. et al. Maturation of mouse NK cells is a 4-stage developmental program. Blood 113, 5488–5496 (2009).
Jensen, H., Potempa, M., Gotthardt, D. & Lanier, L. L. Cutting edge: IL-2-induced expression of the amino acid transporters SLC1A5 and CD98 is a prerequisite for NKG2D-mediated activation of human NK cells. J. Immunol. 199, 1967–1972 (2017).
Masetti, M. et al. Lipid-loaded tumor-associated macrophages sustain tumor growth and invasiveness in prostate cancer. J. Exp. Med. 219 https://doi.org/10.1084/jem.20210564 (2022).
Ma, Y., Galluzzi, L., Zitvogel, L. & Kroemer, G. Autophagy and cellular immune responses. Immunity 39, 211–227 (2013).
Bordi, M. et al. A gene toolbox for monitoring autophagy transcription. Cell Death Dis. 12, 1044 (2021).
Smith, S. L. et al. Diversity of peripheral blood human NK cells identified by single-cell RNA sequencing. Blood Adv. 4, 1388–1406 (2020).
Crinier, A. et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity 49, 971–986.e975 (2018).
Yang, C. et al. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat. Commun. 10, 3931 (2019).
Björkström, N. K. et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood 116, 3853–3864 (2010).
Sun, H. et al. Accumulation of tumor-infiltrating CD49a(+) NK cells correlates with poor prognosis for human hepatocellular carcinoma. Cancer Immunol. Res. 7, 1535–1546 (2019).
Klingemann, H. The NK-92 cell line-30 years later: its impact on natural killer cell research and treatment of cancer. Cytotherapy https://doi.org/10.1016/j.jcyt.2022.12.003 (2023).
Gong, J. H., Maki, G. & Klingemann, H. G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 8, 652–658 (1994).
Menzies, F. M., Moreau, K., Puri, C., Renna, M. & Rubinsztein, D. C. Measurement of autophagic activity in mammalian cells. Curr. Protoc. Cell Biol. Chapter 15, Unit 15.16 (2012).
Lu, G. et al. The effects of metformin on autophagy. Biomed. Pharmacother. 137, 111286 (2021).
Marçais, A. et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 15, 749–757 (2014).
Donnelly, R. P. et al. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 193, 4477–4484 (2014).
Pardo, R. et al. Gemcitabine induces the VMP1-mediated autophagy pathway to promote apoptotic death in human pancreatic cancer cells. Pancreatology 10, 19–26 (2010).
Zang, Y. et al. The next generation proteasome inhibitors carfilzomib and oprozomib activate prosurvival autophagy via induction of the unfolded protein response and ATF4. Autophagy 8, 1873–1874 (2012).
Levine, B., Liu, R., Dong, X. & Zhong, Q. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol. 25, 533–544 (2015).
Konermann, S. et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588 (2015).
Wang, S. et al. FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat. Commun. 7, 11023 (2016).
Chauhan, S., Mandell, M. A. & Deretic, V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 58, 507–521 (2015).
Wei, Y., Pattingre, S., Sinha, S., Bassik, M. & Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30, 678–688 (2008).
Dupont, N., Nascimbeni, A. C., Morel, E. & Codogno, P. In: International Review of Cell and Molecular Biology, Vol. 328 (ed Lorenzo Galluzzi) 1–23 (Academic Press, 2017).
Shpilka, T., Weidberg, H., Pietrokovski, S. & Elazar, Z. Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol. 12, 226 (2011).
Romanov, J. et al. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 31, 4304–4317 (2012).
Davis, O. B. et al. NPC1-mTORC1 signaling couples cholesterol sensing to organelle homeostasis and is a targetable pathway in niemann-pick type C. Dev. Cell 56, 260–276.e267 (2021).
Yoon, J. H., Her, S., Kim, M., Jang, I. S. & Park, J. The expression of damage-regulated autophagy modulator 2 (DRAM2) contributes to autophagy induction. Mol. Biol. Rep. 39, 1087–1093 (2012).
Sevlever, D., Jiang, P. & Yen, S. H. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 47, 9678–9687 (2008).
Coly, P.-M. et al. Chemotactic G protein-coupled receptors control cell migration by repressing autophagosome biogenesis. Autophagy 12, 2344–2362 (2016).
Ma, Q. et al. CXCR4 blockade in macrophage promotes angiogenesis in ischemic hindlimb by modulating autophagy. J. Mol. Cell. Cardiol. 169, 57–70 (2022).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 15, 1484–1506 (2020).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Jakobs, A. et al. An isoform-specific C/EBPβ inhibitor targets acute myeloid leukemia cells. Leukemia 30, 1612–1615 (2016).
Jakobs, A., Steinmann, S., Henrich, S. M., Schmidt, T. J. & Klempnauer, K. H. Helenalin acetate, a natural sesquiterpene lactone with anti-inflammatory and anti-cancer activity, disrupts the cooperation of CCAAT box/enhancer-binding protein β (C/EBPβ) and co-activator p300. J. Biol. Chem. 291, 26098–26108 (2016).
Bajor, M. et al. PD-L1 CAR effector cells induce self-amplifying cytotoxic effects against target cells. J. Immunother. Cancer 10, e002500 (2022).
Robbins, Y. et al. Tumor control via targeting PD-L1 with chimeric antigen receptor modified NK cells. Elife 9 https://doi.org/10.7554/eLife.54854 (2020).
Germic, N., Frangez, Z., Yousefi, S. & Simon, H.-U. Regulation of the innate immune system by autophagy: neutrophils, eosinophils, mast cells, NK cells. Cell Death Differ. 26, 703–714 (2019).
White, E., Mehnert, J. M. & Chan, C. S. Autophagy, metabolism, and cancer. Clin. Cancer Res. 21, 5037–5046 (2015).
Monkkonen, T. & Debnath, J. Inflammatory signaling cascades and autophagy in cancer. Autophagy 14, 190–198 (2018).
Shoji-Kawata, S. et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494, 201–206 (2013).
Settembre, C. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108 (2012).
Guo, F. et al. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 35, 816–826 (2016).
Parol-Kulczyk, M., Gzil, A., Ligmanowska, J. & Grzanka, D. Prognostic significance of SDF-1 chemokine and its receptors CXCR4 and CXCR7 involved in EMT of prostate cancer. Cytokine 150, 155778 (2022).
Correia, A. L. et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature 594, 566–571 (2021).
Hubrecht, R. C. & Carter, E. The 3Rs and humane experimental technique: implementing change. Animals (Basel) 9 https://doi.org/10.3390/ani9100754 (2019).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
Kumar, A. et al. Generation and validation of CRISPR-engineered human natural killer cell lines for research and therapeutic applications. STAR Protoc. 2, 100874 (2021).
Schönfeld, K. et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 23, 330–338 (2015).
Acknowledgements
We thank prof. Ronald De Pinho for kindly donating the Pten−/−; Trp53−/− cell line. The authors are grateful to Prof. Dr. Thomas J. Schmidt, Institute of Pharmaceutical Biology and Phytochemistry, University of Münster, Germany, for providing a sample of Helenalin Acetate. We thank Dr. Fabio Pasqualini and Dr. Fabio Grizzi for immunohistochemistry technical help, and Dr. Marco Erreni and Dr. Andrea Doni for imaging technical help. We thank Dr. Eduardo Bonavita for the revision of the manuscript. We also thank all the patients enrolled in the study. This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC Start up-ID 19141 to D. Di Mitri, Bridge grant-ID 27673 to D. Di Mitri) and Minsal (Gr-2016-02363531 to D. Di Mitri), FRBB Transcan3 (JTC2021, to D. Di Mitri) and FIS (Fondo Italiano per la Scienza to D. Di Mitri). D. Di Mitri also received funds from AIRC 5×1000 2019-ID 22757. Federica Portale is supported by a fellowship from Pezcoller Foundation. G. Marelli is supported by a fellowship from Associazione Italiana per la Ricerca sul Cancro (AIRC-ID 22588). J. Eitler receives funding from the German Research Foundation (DFG) through grant EI1223/2-1T. Tonn received funds from German Federal Ministry of Education and Research (Clusters4Future SaxoCell, 03ZU1111DA) and German Red Cross Blood Donation Service internal grants. E.L. is supported by the CRI Lloyd J. Old STAR (CRI award 3914) and by the Associazione Italiana per la Ricerca sul Cancro (AIRC IG 2022–ID 27391 and AIRC 5×1000 program UniCanVax 22757). Purchase of the BD FACSymphony A5 has been defrayed in part by Italian Ministry of Health (agreement 82/2015). Figures 1A, 4A, G, and Supplementary Figs. 1J, 2A, E were partly created with Biorender (Portale, F. (2024) BioRender.com/i53o287).
Author information
Authors and Affiliations
Contributions
F. Portale formulated the hypothesis, performed the experiments, designed the flow cytometry panels, and performed the related analysis, analysed data and interpreted the results, contributed to the writing. R. Carriero and P. Kunderfranco performed the sequencing analysis, interpreted the data, and revised the manuscript. S. Marchini supervised the RNA sequencing. G. Marelli, N. Morina, M. Pandini, M. Iovino, G. Meregalli, A. Garbin, M. Dambra and E. Magrini performed the in vitro and in vivo experiments. G. De Simone and C. Camisaschi gave technical support for the FACS sorting. G. Basso and J. Cibella performed the RNA-seq. T.J. Schmidt provided helenalin acetate. P. Colombo, M. Lazzeri and P. Casale provided the clinical samples from patients and revised the manuscript. T. Tonn, J. Eitler and W. Rackwitz provided PD-L1.CAR NK92 cell line and inputs for the project. M. Bordi and F. Cecconi provided support for the analyses of data and interpretation of results. E. Lugli designed the flow cytometry panels and revised the manuscript. A. Corbelli e F. Fiordaliso performed TEM experiments and performed analysis of imaging data. D. Di Mitri developed the concept, designed and supervised the experiments, interpreted the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
T. Tonn is named as an inventor on patents in the field of cancer immunotherapy. The remaining authors declare no competing interests.
Ethics statement
This study adheres to the principles outlined in the Declaration of Helsinki and the ethical standards set by our Institution. All human participants provided informed consent, and animal experiments were conducted with approval from the Institutional Animal Care. We promote diversity, equity, and inclusion in research, and we have disclosed any potential conflicts of interest.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Portale, F., Carriero, R., Iovino, M. et al. C/EBPβ-dependent autophagy inhibition hinders NK cell function in cancer. Nat Commun 15, 10343 (2024). https://doi.org/10.1038/s41467-024-54355-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54355-2
This article is cited by
-
Remodeling the tumor dormancy ecosystem to prevent recurrence and metastasis
Signal Transduction and Targeted Therapy (2026)
-
Inhibition of autophagy enhances the antitumor efficacy of T/CAR T cell against neuroblastoma
Journal of Experimental & Clinical Cancer Research (2025)
-
Therapeutic targeting of cell death-immune crosstalk in cancer to rewire the tumor immune microenvironment
Molecular Cancer (2025)
-
Bioinformatics identification and validation of pyroptosis-related gene for ischemic stroke
BMC Medical Genomics (2025)
-
Mitochondrial metabolism and cancer therapeutic innovation
Signal Transduction and Targeted Therapy (2025)
