
Abstract
There remains a critical need for the precise control of CRISPR (clustered regularly interspaced short palindromic repeats)-based technologies. Here, we engineer a set of inducible CRISPR-based tools controllable by focused ultrasound (FUS), which can penetrate deep and induce localized hyperthermia for transgene activation. We demonstrate the capabilities of FUS-inducible CRISPR, CRISPR activation (CRISPRa), and CRISPR epigenetic editor (CRISPRee) in modulating the genome and epigenome. We show that FUS-CRISPR-mediated telomere disruption primes solid tumours for chimeric antigen receptor (CAR)-T cell therapy. We further deliver FUS-CRISPR in vivo using adeno-associated viruses (AAVs), followed by FUS-induced telomere disruption and the expression of a clinically validated antigen in a subpopulation of tumour cells, functioning as “training centers” to activate synthetic Notch (synNotch) CAR-T cells to produce CARs against a universal tumour antigen to exterminate neighboring tumour cells. The FUS-CRISPR(a/ee) toolbox hence allows the noninvasive and spatiotemporal control of genomic/epigenomic reprogramming for cancer treatment.
Similar content being viewed by others
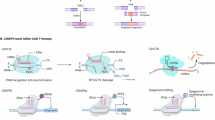


Introduction
The emergence of CRISPR technology has revolutionized numerous aspects of life science and medicine1,2,3,4,5. With a single guide RNA (sgRNA), the Cas9 nuclease can be targeted to, in principle, any accessible genomic locus next to a protospacer adjacent motif (PAM) to cause site-specific double-strand break (DSB), providing a powerful method for reprogramming endogenous genome and ultimately the phenotypes of organisms6,7. The subsequent development of CRISPRa and CRISPRi with nuclease-dead Cas9 (dCas9) further enabled transcriptional and epigenetic modifications of endogenous loci, demonstrating the power of CRISPR in regulating the genome at different levels8,9. As the CRISPR-based technologies advanced to translational applications and clinical trials, safety/controllability has become one of the major concerns, mainly due to the immunogenicity of Cas9-related proteins and their off-target effects accumulated during long-time expression in the cells10,11,12.
To address this, controllable CRISPR systems utilizing small molecules13,14,15, light16,17,18,19, or heat20,21 as external cues for induction have been developed. Small molecule-based systems can tightly control the time of action for CRISPR, but the diffusive characteristic of small molecules compromises the spatial precision. Light-based systems provide an elegant solution to control both the timing and location of CRISPR; however, they require light-sensitive proteins, which can be bulky and difficult to deliver or possibly immunogenic due to their non-human origins22,23. Also, the penetration depth of light with a maximum of millimeters limits its therapeutic applications, particularly in tissues tens of centimeters deep24. Heat-inducible CRISPR-dCas9 systems using near-infrared (NIR) and gold nanorods for heat generation have also been developed20,21, yet NIR can only penetrate a maximum of a few centimeters25, and the clinical usage of gold nanorods is restricted26,27.
Focused ultrasound (FUS) can penetrate tens of centimeters deep and directly induce localized hyperthermia in biological tissues28,29. It has been used for tissue ablation in patients at relatively high temperatures (> 60 °C)30,31,32,33 and for controlling transgene expression in vivo at mildly elevated temperatures, (42−43 °C) using heat-sensitive promoters such as the heat shock protein promoter (Hsp)34,35,36,37,38,39. We have previously developed FUS-inducible CAR (FUS-CAR)-T cells that can be ontogenetically activated by FUS for cancer therapy with reduced off-tumor toxicities40. Therefore, we hypothesize that FUS, with its penetration power and spatiotemporal precision, would allow the direct control of CRISPR without co-factors for genome editing and regulations at specific tissues and organs. Indeed, a recent study reported FUS-controllable genome editing of tumor cells via Cas9-containing nanocomplexes41. Yet, the short half-life and limited delivery efficiency (mainly in the liver) of the nano complexes restrict their broader clinical applications. Another recent proof-of-concept study also demonstrated FUS-controllable gene activation and base editing, but the direct control of the epigenome, as well as its translational potential for cancer treatment, has not been fully realized42.
Here, we present a set of sonogenetics-based CRISPR tools, including FUS-inducible CRISPRa (FUS-CRISPRa), FUS-inducible CRISPR epigenetic editor (FUS-CRISPRee), and FUS-inducible CRISPR (FUS-CRISPR). We show that this FUS-CRISPR(a/ee) toolbox can allow FUS-controllable genomic and epigenomic reprogramming in multiple cell types and in vivo with applications in synergistic cancer immunotherapy.
Results
Inducible upregulation of exogenous and endogenous genes via FUS-CRISPRa
To engineer a FUS-CRISPRa system with the heat-sensitive Hsp, we adopted the Ribozyme-gRNA-Ribozyme (RGR) strategy utilizing self-cleaving HH and HDV ribozymes that enables gRNA production from RNA polymerase II promoters like Hsp43,44. Upon FUS-induced heat stimulation (Supplementary Fig. 1, “Methods”), Hsp initiates production of the HHRibo-sgRNA-HDVRibo transcript, which undergoes self-cleavage to generate the sgRNA (Fig. 1a). The sgRNA then integrates with the constitutively expressed dCas9 and transcriptional factors (e.g., VP64, SAM45) to activate target gene expression (Fig. 1a).

a Schematic illustration of the FUS-CRISPRa system. b Normalized Fluc luminescence in cells engineered with P1-targeting FUS-CRISPRa and P1-driven Fluc quantified 24 h after different durations of HS. Readings were normalized to the CT group. P = 2.22 × 10−7, 6.92 × 10−10, 7.14 × 10−12, 5.90 × 10−14 from left to right. c Left, schematic illustration of FUS stimulation of cells in vitro; Right, normalized Fluc luminescence in cells engineered with P1-targeting FUS-CRISPRa and P1-driven Fluc quantified 24 h after FUS. Readings were normalized to the FUS- group. d Cells engineered with P1- and P2-targeting FUS-CRISPRa, P1-EYFP, and P2-ECFP were imaged 24 h after HS. Scale bar = 30 μm. e Relative IL1B mRNA expression in HEK 293 T cells engineered with hIL1B-targeting FUS-CRISPRa, normalized to IL1B mRNA level in wild type (WT) HEK 293 T cells. P = 5.78 × 10−9 at 6 h. f Pro-IL1B protein expression in wild-type (WT) cells or engineered cells in (e). MK: marker. g, h, Relative IL1B (g) or IFNβ (h) mRNA expression in RAW 264.7 cells engineered with FUS-CRISPRa targeting mouse IL1B (g) or IFNβ (h) gene, normalized to the corresponding mRNA levels in WT RAW 264.7 cells. In (g), P = 3.62 × 10−7 at 6 h, 1.74 × 10−6 at 12 h. In (h), 1.87 × 10−6 at 6 h, 6.20 × 10−9 at 12 h. In (b), CT, control, without HS; data are technical triplicates representative of three independent experiments. In (c), FUS +, with 20 min FUS stimulation at 43 °C; FUS-, without FUS stimulation; n = 3 biological replicates. In (d–h), HS, with 30 min HS; CT, without HS. In (e, g, and h), n = 3 technical replicates representative of two individual experiments. Bar heights represent means; error bars represent s.e.m. Two-tailed unpaired t test was used in (c), and two-way ANOVA followed by Sidak’s multiple comparisons test was used in (b, e, g, h). Source data are provided as a Source Data file. Created in BioRender. Liu, L. (2024) https://BioRender.com/u26w026.
We first tested the capability of FUS-CRISPRa in activating exogenous genes. In cells transfected with FUS-CRISPRa for the inducible expression of gRNA1 targeting a synthetic promoter P144 (Supplementary Fig. 2a), different durations of heat stimulation induced tunable expression of P1-driven firefly luciferase (Fluc, Fig. 1b). FUS stimulation (43 °C, 20 min) also induced a comparable level of Fluc activation in the engineered cells in vitro (Fig. 1c). We further applied FUS in vivo in mice and observed significant Fluc activation via FUS-CRISPRa as well (Supplementary Fig. 2b). In addition, we engineered cells with multiplexed FUS-CRISPRa containing Hsp-DsRed2-RG1R-RG2R, allowing simultaneous inducible production of multiplexed gRNA1 and gRNA2 targeting synthetic promoters P1 and P2 respectively44 (Supplementary Fig. 2c). Along with the Hsp-driven DsRed2 expression, the activations of P1-driven EYFP and P2-driven ECFP via FUS-CRISPRa were also observed in the cells with heat shock (HS), with minimal background signals in control (CT) cells without HS (Fig. 1d). Together, the above validated the design of FUS-CRISPRa with inducible gRNAs for multiplexed genome regulation.
We then applied FUS-CRISPRa to regulate endogenous gene expressions. We constructed an all-in-one piggyBac plasmid containing Hsp-RGR targeting the human IL1B (hIL1B) gene, which is a common target of CRISPRa46, together with the constitutive dCas9-SAM (Supplementary Fig. 2d) to generate cell lines accordingly (Methods). Quantification of hIL1B mRNA level and pro-IL1B protein expression in the engineered HEK 293 T cells at different time points after HS revealed a trend of heat-inducible upregulation of hIL1B through FUS-CRISPRa (Fig. 1e, f). No heat-inducibility of hIL1B was observed in wild-type (WT) cells (Supplementary Fig. 2e). To demonstrate the general applicability of FUS-CRISPRa, we also validated our design in mouse RAW 264.7 cells targeting mouse IL1B (mIL1B) and IFNβ (mIFNβ) genes (Fig. 1g, h). Heat itself did not significantly alter mIL1B and mIFNβ expression in WT RAW 264.7 cells (Supplementary Fig. 2f, g). As such, FUS-CRISPRa allows inducible activation of various exogenous and endogenous genes in different cell types.
FUS-CRISPRee-mediated epigenetic regulation for gene repression
We next sought to engineer FUS-CRISPRee for controllable gene repression for lasting periods through epigenetic reprogramming. CRISPRoff is an epigenetic memory writer composed of dCas9, DNA methyltransferase DNMT3A-3L domains, and KRAB domains reported to durably silence gene expression47 (Supplementary Fig. 3a). We co-transfected HEK 293 T cells with CRISPRoff and Hsp-RGR containing gRNA targeting ARPC2, a common target of dCas9-mediated gene repression48, to test heat-inducible gene repression. However, we did not observe significant ARPC2 downregulation after HS (Supplementary Fig. 3b). We also tested Hsp-RGR containing Zap70-targeting gRNA in Jurkat cells by electroporation, yet still did not observe Zap70 downregulation (Supplementary Fig. 3b). On the contrary, robust gene repression was observed when constitutive ARPC2 or Zap70 gRNA was co-transfected with CRISPRoff (Supplementary Fig. 3c). We hypothesized that the copy number of gRNA generated from Hsp-RGR after HS may not be sufficient to induce gene repression with CRISPRoff.
Therefore, we employed a different strategy to engineer FUS-CRISPRee by changing the inducible component from gRNA to dCas9 while incorporating the SunTag amplification system44. Since heat-inducible expression may result in a lower protein copy number than constitutive expression, we reasoned that having a heat-inducible dCas9-nxGCN4 and a constitutive scFv-regulator would allow a favorable stoichiometry to promote the recruitment of multiple copies of the regulators to a given dCas9 complex. We also replaced the Hsp with the previously developed synthetic heat-sensitive promoter 7H-YB with higher heat inducibility (Supplementary Fig. 4)49. As such, this FUS-CRISPRee system is composed of the 7H-YB promoter driving the dCas9 fused to eight repeats of GCN4, a constitutive EFS promoter driving a previously reported αGCN4-scFv-fused epigenetic regulator DNMT3A-3L, and the constitutive U6 promoter driving the gRNA50 (Fig. 2a and Supplementary Fig. 5a). FUS stimulation induces dCas9-8xGCN4 expression, allowing the recruitment of multiple copies of the epigenetic regulators through the scFv. As such, the complex is brought to the target locus by the gRNA to repress gene expression via DNA methylation (Fig. 2a).

a Schematic illustration of the FUS-CRISPRee system. b, c Representative flow cytometry data of CD81 (b) or CXCR4 (c) expression in FUS-CRISPRee-engineered Jurkat cells with gRNA targeting CD81 (b) or CXCR4 (c), or with non-targeting (NT) gRNA. The cells were stained with anti-CD81 (b) or anti-CXCR4 (c) antibodies four days after HS. d Relative CD81 mRNA expression 3 or 9 days after HS in cells in (b). P = 1.02 × 10−6 at 3 h in CD81 group. e, Relative CXCR4 mRNA expression in cells in (c). P = 2.99 × 10−7 at 3 h in CXCR4 group. f Percentage of CXCR4 + cells in Nalm6 cells engineered with CXCR4-targeting or NT FUS-CRISPRee with DNMT mutant with different treatments. g Kinetics of CXCR4 expression in cells engineered with CXCR4-targeting FUS-CRISPRee. h The migration ability (%) of the engineered FUS-CRISPRee Nalm6 cells in a transwell assay. In (b–h) HS, with 20 min HS; CT, without HS. In (f) FUS +; with 20 min FUS stimulation at 43 °C on cells in vitro. In d and (e), bar heights represent means of technical triplicates representative of two individual experiments. In (f and h), bar heights represent means of biological triplicates. Error bars represent s.e.m. Two-way ANOVA followed by Sidak’s multiple comparisons test was used in (d, e, and h). One-way ANOVA with multiple comparisons was used in (f). Source data are provided as a Source Data file. Created in BioRender. Liu, L. (2024) https://BioRender.com/h43m913.
We transduced Jurkat cells with the FUS-CRISPRee system containing gRNAs targeting surface markers CD81 or CXCR4, which can be quantified by staining. Cell surface staining of CD81 four days after HS showed a significant decrease in CD81 expression in the HS cells compared with non-heated control (CT) cells (53.8% vs. 91.7%, Fig. 2b). Similarly, CXCR4 expression was also repressed by HS (46.7% in HS vs. 90.8% in CT cells, Fig. 2c). HS itself did not affect CD81 or CXCR4 expression in the cells with non-targeting (NT) gRNA (Fig. 2b, c). The effect of FUS-CRISPRee-mediated gene repression was also confirmed by quantification of the corresponding mRNA levels (Fig. 2d, e). Similar gene repression effects were achieved in Nalm6 cells engineered with FUS-CRISPRee (Supplementary Fig. 5b–d).
CXCR4 is a chemokine receptor known to promote tumor growth and metastasis51,52,53. We, therefore, examined the effect of FUS-CRISPRee-mediated CXCR4 downregulation in Nalm6 tumor cells. We also replaced the WT DNMT in the original FUS-CRISPRee with a previously reported DNMT mutant of reduced off-target methylation (Supplementary Fig. 6a). A dramatic reduction of CXCR4 expression was seen in CXCR4 FUS-CRISPRee cells four days after HS compared with those without HS, and FUS stimulation was able to induce a comparable repression effect in the engineered cells (Fig. 2f). Dynamic tracking revealed that the CXCR4 expression in the cells with HS recovered to a level similar to that in the cells without HS in approximately 40 days, indicating a sustained but reversible effect of FUS-CRISPRee (Fig. 2g and Supplementary Fig. 6b). The FUS-CRISPRee-mediated gene repression was also confirmed by methylation analysis (Supplementary Fig. 6c). Transwell assays further demonstrated that the migration ability was compromised in cells with HS-induced CXCR4 downregulation (Fig. 2h). Taken together, our results suggest that FUS-CRISPRee allows inducible and reversible gene repression on different genes through epigenetic modulation in different cell types, allowing the control of cellular functions by ultrasound.
The SunTag-based FUS-CRISPRee platform is versatile in that it can be readily converted into a FUS-CRISPRa system by replacing the epigenetic regulators with transcription activators like VP64 (Supplementary Fig. 7a, b). We hence engineered such a system and tested its ability to activate P1-driven Fluc (Supplementary Fig. 7c). We observed robust Fluc activation with HS or FUS stimulation in multiple cell types and in vivo (Supplementary Fig. 7d, e), validating the design of SunTag-based FUS-CRISPRa. The induction level achieved by the SunTag-based FUS-CRISPRa system appeared lower than that of the ribozyme-based FUS-CRISPRa system when targeting the same P1-driven Fluc, possibly due to a higher basal level caused by the SunTag system.
FUS-CRISPR-mediated knockout of endogenous genes
One of the advantages of the FUS-inducible system is its ability to transiently activate regulators (e.g., Cas9) that may be immunogenic or toxic if expressed constitutively12. Following the development of FUS-CRISPRa and FUS-CRISPRee, we engineered FUS-CRISPR composed of inducible Cas9 and constitutive gRNAs (Fig. 3a and Supplementary Fig. 8a, b) and verified heat-inducible Cas9 expression in the engineered cells (Fig. 3b). In Jurkat T cells engineered with FUS-CRISPR targeting key signaling molecules CD3D or Zap70, HS induced CD3D knockout (KO) in 44.3% cells and Zap70 KO in 39.2% cells as quantified by genotyping PCR and sequencing (Fig. 3c). Basal levels of KO were observed in CT cells (13% for CD3D and 15.4% for Zap70), likely due to the leakage of the heat-sensitive promoters (Fig. 3c). To test whether HS-induced KO can affect cellular functions, we stimulated the Jurkat T cells with anti-T-cell receptor (TCR) antibody and quantified T-cell activation by CD69 staining. As expected, since CD3D is a subunit of the TCR complex and Zap70 is a critical mediator of the TCR signaling pathway, Jurkat cells with HS-induced KO of CD3D or Zap70 demonstrated significantly weakened TCR-dependent T-cell activation, reflected by CD69 expressions (Fig. 3d).

a Schematic illustration of the FUS-CRISPR system. b Heat-inducible Cas9 expression represented by eGFP signal under flow cytometry in engineered Jurkat cells. c Knockout efficiencies in Jurkat cells engineered with FUS-CRISPR targeting CD3D or Zap70 quantified four days after HS. N = 4 and 6 biological replicates for CD3D and Zap70, respectively. P = 8.10 × 10−5 for CD3D. d CD69 staining of WT or FUS-CRISPR-engineered Jurkat cells after TCR stimulation. eThe all-in-one FUS-CRISPR plasmid. f Percentage of CD81+ cells (left) and the representative flow cytometry profile (right) in U-87 MG cells engineered with CD81-targeting FUS-CRISPR quantified 8 days after HS. g Knockout efficiencies in Nalm6 cells engineered with FUS-CRISPR with different gRNAs targeting the PLK1 gene, quantified four days after HS. h Normalized cell number of the cells in (g) on Day 4 after HS. Cell number was normalized to Day 0. P = 9.18 × 10−8 for gRNA1, 2.27 × 10−9 for gRNA2. In (c, d, and f), HS, with 20 min HS; CT, without HS. In (g and h), HS, with 15 min HS; CT, without HS. Bar heights represent means; error bars represent s.e.m. In (f and g), n = 3 biological replicates. In (h), n = 3 technical replicates representative of two independent experiments. Two-tailed unpaired t test was used in (f), and two-way ANOVA followed by Sidak’s multiple comparisons test was used in (c, g, h). Source data are provided as a Source Data file. Created in BioRender. Liu, L. (2024) https://BioRender.com/c63k467.
To examine the feasibility of broad applications, we further engineered an all-in-one plasmid for FUS-CRISPR and tested it in multiple tumor cell lines (Fig. 3e). Surface staining of U-87 MG glioma tumor cells engineered with CD81-targeting FUS-CRISPR showed that HS induced significant CD81 KO (Fig. 3f). To explore the therapeutic applications of FUS-CRISPR, we generated Nalm6 tumor cells containing FUS-CRISPR targeting polo-like kinase 1 (PLK1, Supplementary Fig. 8c), a key regulator of cell cycle and an active target of cancer therapy18,54. HS induced PLK1 KO and significantly inhibited cell proliferation with different PLK1-targeting gRNAs (Fig. 3g, h and Supplementary Fig. 8d). In summary, FUS-CRISPR can be applied to control genome editing of endogenous genes and reprogramming of cellular functions.
Telomere disruption by FUS-CRISPR
In addition to genetic editing of single genes, we hypothesized that FUS-CRISPR can act with a higher editing efficiency on repetitive loci such as telomeres than on non-repetitive loci. It has been reported that telomere dysfunction can trigger catastrophic events leading to cell senescence and apoptosis55,56,57. We hence co-transfected HEK 293 T cells with FUS-CRISPR containing the gRNA targeting repetitive telomere sequences (Supplementary Fig. 8d) and HaloTag-fused 53BP1, a marker for DNA double-strand breakage (DSB) to report the genome editing sites. Fluorescence microscopy revealed that HS-induced DSB at multiple loci in the cells with telomere-targeting FUS-CRISPR, as evidenced by the dotted 53BP1 pattern, which was not observed in non-activated CT cells or cells with non-targeting NT FUS-CRISPR (Fig. 4a). We also co-transfected the cells with tagBFP-fused telomeric repeat binding factor 2 (TRF2) to mark the telomere loci58. Merged images of 53BP1 and TRF2 showed multiple colocalization puncta, confirming the presence and precision of FUS-CRISPR-induced DSB at telomeres (Fig. 4a).

a Nuclear distribution of tagBFP-TRF2 and HaloTag-53BP1 in FUS-CRISPR-engineered HEK 293 T cells with telomere-targeting gRNA or non-targeting (NT) gRNA. HS, with 30 min HS; CT, without HS. Right, enlarged image merging TRF2 and 53BP1 signals. Scale bar = 10 μm. b Normalized cell number of FUS-CRISPR-engineered Nalm6 cells with telomere-targeting gRNA or NT gRNA two (D2) or four (D4) days after HS. Cell number was normalized to Day 0. N = 4 biological replicates. c Heat-map of differential gene expression in Nalm6 cells engineered with telomere-targeting or NT FUS-CRISPR at 24, 48, or 96 h after HS. d The top three enriched GO terms in the HS group compared to the CT group in the telomere-targeting FUS-CRISPR cells in (c). e Volcano plot showing the downregulated (blue) and upregulated (red) genes between HS and CT groups in the telomere-targeting FUS-CRISPR cells in (c). f Schematic illustration of CAR-T cell attack on tumor cells. g Survival (%) of FUS-CRISPR-engineered Nalm6 tumor cells 72 h after culture with (w/T) or without (w/o T) αCD19CAR-T cells in the luciferase-based cytotoxicity assay. The survival (%) was normalized to CT, w/o T group. P = 7.62 × 10−8 for w/o T, 5.32 × 10−5 for w/ T. h Cytotoxicity (%) of CAR-T cells in the co-culture groups (w/ T) in (g). The cytotoxicity (%) was quantified as 100% – Tumor survival (%). In (g and h), n = 3 technical replicates. Data are representative of two independent experiments. In (b, c, g, and h) HS: with 10 min HS; CT, without HS. Bar heights represent means; error bars represent s.e.m. Two-way ANOVA followed by Sidak’s multiple comparisons test. Source data are provided as a Source Data file. Created in BioRender. Liu, L. (2024) https://BioRender.com/c41g432.
We then engineered Nalm6 tumor cells with telomere-targeting or NT FUS-CRISPR. Consistent with previous reports of telomere-dysfunction-related cell senescence and apoptosis, we observed that a relatively short duration of HS (10 min) significantly inhibited the proliferation of the cells engineered with telomere FUS-CRISPR, but not that of the cells with NT FUS-CRISPR, suggesting that telomere disruption rather than hyperthermia itself suppressed cell growth (Fig. 4b). Bulk RNA-seq further revealed that FUS-CRISPR-mediated telomere disruption led to the upregulation of multiple genes associated with the stress response p53 signaling pathway and apoptotic process (e.g., MDM2, FAS, BBC3) and the TNF family (e.g., CD70) in the engineered cells to trigger cell cycle arrest (Fig. 4c–e and Supplementary Fig. 9)59. This priming effect of FUS-CRISPR on tumor cells may hence not only cause the tumor cell cycle arrest and apoptosis but also induce T cell immune responses via TNF family60.
To test whether telomere disruption affects tumor killing by T cells, we employed anti-CD19 chimeric receptor antigen (CAR)-T cells specifically targeting CD19+ Nalm6 tumor cells (Fig. 4f and Supplementary Fig. 10). Fluc-expressing FUS-CRISPR Nalm6 cells with or without HS were co-cultured with CAR-T cells at a low effector-to-target (E:T) ratio of 1:20 for luciferase-based killing assay. The percentage of surviving tumor cells and the corresponding cytotoxicity of the CAR-T cells were quantified from Fluc luminescence 72 h after co-culture (Fig. 4g, h). CAR-T cells demonstrated significantly stronger cytotoxicity against Nalm6 cells with HS-induced telomere disruption than that against CT Nalm6 cells (84.6% vs. 54.3%), while similar cytotoxicities were observed against NT FUS-CRISPR Nalm6 cells with or without HS (59.2% and 61.2%, respectively, Fig. 4h). These results indicated that tumor cells with induced priming and telomeric DSB became more susceptible to CAR-T cell killing.
Tumor priming via FUS-CRISPR for enhanced CAR-T therapy
Encouraged by the effect of FUS-CRISPR-mediated telomere disruption in vitro, we investigated its therapeutic potential in vivo. We generated subcutaneous tumors in NSG mice using Fluc+ Nalm6 cells engineered with telomere FUS-CRISPR or NT FUS-CRISPR. The tumors were treated with (FUS +) or without (FUS-) 10 min FUS on Days 9 and 12 (Fig. 5a and Supplementary Fig. 11a). No significant difference in growth was observed between NT FUS-CRISPR tumors with or without FUS, indicating that FUS alone did not affect tumor growth (Supplementary Fig. 11b–e). In the mice bearing telomere FUS-CRISPR tumors, FUS + tumors exhibited mildly inhibited growth compared with the FUS- tumors from bioluminescence imaging (BLI), yet no statistically significant difference from caliper measurement (Fig. 5b–d). Both the FUS + and FUS- groups showed 0% survival at the end of observation (Fig. 5e). These results suggested that FUS-CRISPR-mediated telomere disruption alone was not sufficient for tumor treatment.

a Timeline of FUS-CRISPR-mediated telomere disruption experiment in NSG mice. b–d Tumor aggressiveness in the mice in (a) quantified by the total flux of the tumor from BLI measurement (b), the corresponding BLI images (c), and the tumor volume based on caliper measurement (d). e Survival curves of the tumor-bearing mice in (a). f Experimental timeline of FUS-CRISPR combined with CAR-T therapy in NSG mice. g–i Tumor aggressiveness in the mice in (f) quantified by the total flux of the tumor (g), the corresponding BLI images (h), and the caliper-measured tumor volume (i) In (g), P = 1.22 × 10−5. In (i), P = 8.82 × 10−6. j Survival curves of the tumor-bearing mice in (f). Data points represent means; error bands represent s.e.m.; n = 5 mice per group. Two-way ANOVA followed by Sidak’s multiple comparisons test was used in (b, d, g, and i). Log-rank (Mantel-Cox) test was used in (e and j). Source data are provided as a Source Data file. Created in BioRender. Liu, L. (2024) https://BioRender.com/r77l232.
Therefore, we hypothesized that a treatment strategy combining FUS-CRISPR-mediated telomere disruption for tumor priming and CAR-T therapy could synergistically lead to a more prominent therapeutic outcome. We accordingly generated subcutaneous tumors in mice using telomere FUS-CRISPR Nalm6 cells followed with (FUS + ) or without (FUS-) FUS stimulation (Fig. 5f). Ten days later, we injected a low dose of CAR-T cells intravenously in both FUS + and FUS- groups (Fig. 5f). We observed significantly suppressed growth of the tumors in the FUS + group compared to that of FUS- (Fig. 5g–i). The two groups of mice also showed different survival profiles: while all the mice in the FUS+ group survived, only 40% (two out of five) mice in the FUS- group responded to CAR-T therapy, and the rest 60% mice had reached euthanasia criteria due to tumor progression by the end of observation (Fig. 5j). In a control experiment using NT FUS-CRISPR tumors with CAR-T treatment in both FUS- and FUS+ groups (Supplementary Fig. 11f), only a mild inhibition of tumor growth in the FUS + group compared with the FUS- group but no significant difference in the survival rate was observed (Supplementary Fig. 11g–j). We further investigated the intratumoural frequencies of CAR-T cells in tumors with or without FUS-CRISPR-mediated telomere disruption. We generated a bilateral tumor model using telomere FUS-CRISPR Nalm6 cells followed by FUS stimulation on one tumor, systemic CAR-T cell administration, and collection of both tumors (Supplementary Fig. 12a). Flow cytometry analysis revealed an average of 18.3% CAR-T cells in the tumors with FUS stimulation, significantly higher than that of 9.0% in the tumors without (Supplementary Fig. 12b–d). These results demonstrated enhanced intratumoural frequencies of CAR-T cells associated with telomere-targeting FUS-CRISPR treatment. Taken together, telomere-targeting FUS-CRISPR can allow ultrasound-controllable genome editing and tumor priming for efficient CAR-T therapy to achieve synergistic therapeutic effects.
Clinically compatible FUS-CRISPR delivery for cancer cell reprogramming and immunotherapy
To further demonstrate the translational potential of our technology, we set off to use adeno-associated virus (AAV) to directly deliver FUS-CRISPR components into tumor cells in vivo. Meanwhile, in addition to tumor priming via FUS-CRISPR-mediated telomere disruption, we proposed to further prime the CAR-T cells by employing the synNotch design to overcome the less efficient gene delivery in vivo. In synNotch CAR-T cells, binding with a FUS-induced specific antigen A (“priming”) in the viral-infected subpopulation of cancer cells can induce the cleavage of the synNotch receptor and release of the fused transcription factor, activating the expression of a CAR against antigen B universally expressed on the whole population of cancer cells (“killing”)61,62,63. We hence engineered anti-CD19 synNotch CAR-T cells, where synNotch recognizes tCD19 (truncated CD19) and activates anti-PSMA (prostate-specific membrane antigen) CAR expression (Supplementary Fig. 13a). Meanwhile, we engineered a FUS-CRISPR circuit that allowed the disruption of telomeres and induction of tCD19 expression in PSMA + PC3 prostate cancer cells upon FUS stimulation (Supplementary Fig. 13b, c). As such, the FUS-induced tCD19 + PC3 cells can serve as “training centers” to trigger PSMACAR expression in synNotch CAR T cells, which in turn leads to the killing of all the PSMA + PC3 cells at the proximity of tumor site, both tCD19 + and tCD19-, via PSMACAR (Fig. 6a). This integration of FUS-CRISPR and synNotch CAR T can hence overcome two potential problems: (1) the lack of specific and clinically validated antigens for solid tumors; (2) the possibly less ideal efficiency of AAV gene delivery and FUS-induction in vivo.

a Schematic illustration of FUS-CRISPR-mediated synNotch CAR-T activation. Priming of synNotch CAR-T cells by FUS-CRISPR-induced tCD19 enables the killing of PSMA + PC3 cells. b Principle of FUS-CRISPR-mediated tCD19 expression. The tCD19 gene is split by tandem repeated sequences flanking a Cas9 cutting site, which can be recombined into functional tCD19 after Cas9 cutting and single-strand annealing (SSA). c FUS-CRISPR-mediated tCD19 expression in PC3 cells quantified by anti-CD19 antibody staining. N = 3 biological repeats. d Cell death (%) of PC3 cells in (c) without (w/o T) or with (w/ T) co-culture with αCD19-synNotch PSMACAR-T cells. P = 2.85 × 10−5 for w/ T. N = 3 technical replicates representative of two independent experiments. e Timeline of in vivo experiment in NSG mice. f BLI images showing tumor aggressiveness. g Tumor aggressiveness quantified by the total flux of the tumor from BLI measurement. P = 1.49 × 10−6 at 29, 1.14 × 10−11 at 32. N = 5 mice. h Survival curves of the tumor-bearing mice. N = 5 mice. i Immunofluorescence images of the tumor sections. GFP: PC3 cells; mCherry: synNotch CAR-T cells. j Quantification of CAR-T cell percentage in the tumors on Day 28 via flow cytometry. N = 4 mice. In (c, d), CT: without HS; HS: with 15 min HS. FUS-: no FUS treatment. FUS + 10 min FUS stimulation. Error bars and error bands represent s.e.m. Two-tailed unpaired t test was used in (c). Two-way ANOVA followed by Sidak’s multiple comparisons test was used in (d, g). Log-rank (Mantel-Cox) test was used in (h). Two-tailed paired t test was used in (j). Source data are provided as a Source Data file. Created in BioRender. Liu, L. (2024) https://BioRender.com/o33v949.
We first tested this design in vitro by infecting the PC3 cancer cells with two AAVs. One AAV contained the inducible Cas9 driven by the heat-inducible 7H-YB promoter, and the other AAV contained U6-driven gRNAs targeting a truncated CD19 (tCD19) reporter and the telomere, respectively, followed by the tCD19 reporter (Supplementary Fig. 13b). The tCD19 reporter was composed of the tCD19 gene split by tandem repeated sequences flanking the gRNA targeting site, which could be recombined into functional tCD19 after FUS-CRISPR-mediated double-strand break (DSB) followed by single-strand annealing (SSA)-mediated repair64,65 (Fig. 6b). We observed that HS induced tCD19 expression in 12.2% of the engineered PC3 cells as compared to a basal expression of 1.2% (Fig. 6c). Co-culture of HS-treated PC3 cells with anti-CD19 synNotch PSMACAR-T cells led to the death of 71.8% of the PC3 cells, while minimal cell death was observed without the presence of synNotch CAR-T cells, or when the PC3 cells without HS stimulation were co-cultured with synNotch CAR-T cells (Fig. 6d).
To test this in vivo, we generated subcutaneous PC3 tumors (PSMA +, Fluc + ) in NSG mice. When the tumors were approximately 50 mm3 (Day 17), FUS-CRISPR AAVs were delivered into PC3 cells via intratumoural injection (Supplementary Fig. 14, “Methods”). The PC3 tumors were treated with or without FUS stimulation on Day 20 and Day 25. Anti-CD19 synNotch PSMACAR-T cells were injected intravenously on Day 23. On Day 28, tumors were harvested and subjected to immunofluorescence imaging or flow cytometry analysis. Tumor aggressiveness in the remaining mice was continuously monitored (Fig. 6e). Significant inhibition of tumor growth was observed in the tumors with FUS treatment compared to those without (Fig. 6f, g). A significantly higher survival rate was also achieved in the FUS+ group compared to the FUS- group (100% vs. 0% by Day 38, Fig. 6h). Immunofluorescence imaging and flow cytometry revealed dramatically increased intratumoural frequencies of CAR-T cells in the FUS-treated tumors compared to the nontreated ones (17.8% vs. 5.7%, Fig. 6i, j). Increased tCD19 expression was also observed with FUS treatment, indicating FUS-CRISPR-mediated induction of tCD19 expression, which served as the priming antigen for synNotch CAR-T cells (Supplementary Fig. 15). Our results hence indicate that FUS-CRISPR-mediated in vivo reprogramming of cancer cells enables synNotch CAR-T cells to achieve anti-tumor effects.
We further tested this FUS-CRISPR-mediated “training center” strategy using LNCaP tumor cells that endogenously express high levels of PSMA. In NSG mice bearing bilateral Fluc + LNCaP tumors, we introduced FUS-CRISPR AAVs locally at one tumor site, applied FUS to both tumors and administrated synNotch CAR-T cells intravenously (Supplementary Fig. 16a). We observed significantly inhibited tumor growth on the side with FUS-CRISPR AAV injection (Supplementary Fig. 16b. c), providing further evidence of the spatiotemporal control of the developed technology.
Discussion
We developed a FUS-CRISPR(a/ee) toolbox including FUS-controllable CRISPRa, CRISPRee, and CRISPR systems that allowed inducible control of genetic and epigenetic reprogramming by FUS. We demonstrated inducible upregulation, downregulation, and knockout of exogenous and/or endogenous genes in multiple cell types in vitro and in vivo using FUS. We induced multiple DSBs at telomere sites in tumor cells via telomere-targeting FUS-CRISPR, which primed tumors for efficient killing by cytotoxic CAR-T cells in vitro and in vivo. We further delivered FUS-CRISPR in vivo using AAV to reprogram tumor cells and prime the synNotch CAR-T cells via the “training center” strategy to attack the entire population of cancer cells. These synergistic strategies enhanced the efficacy of CAR-T therapy against relatively resistant tumors.
CRISPR-Cas9 proteins have been a powerful tool for genome editing, but can evoke adaptive immune responses and tissue damages in vivo, and are therefore potentially pathogenic if used to correct inherited genetic defects to treat diseases66. Protein engineering to remove immunogenic epitopes and humanize these synthetic proteins to circumvent this issue can be difficult owing to the high diversity of the human leukocyte antigen (HLA) loci67. Using our sonogenetics approach, the transiently induced genomic and epigenomic regulators can be cleared in a timely manner to mitigate or evade the adaptive immune response, offering an option for genome editing and gene therapy at specific tissues/organs.
Ultrasound and its integration with genetic engineering and synthetic biology have revolutionized the control of genetics and cellular functions in live animals with unprecedented penetration depth at tens of centimeters40,68,69. Despite its high temporal resolution (e.g., hundreds of frames per second), the spatial resolution of traditional ultrasound is, however, limited at submillimeter levels70. With recent developments in acoustic reporter genes (ARGs) and functional ultrasound localization microscopy, ultrasound imaging can achieve spatial resolutions in micrometers and at single-cell levels71,72,73. Similarly, it is expected that the ultrasound control of genetics and cellular functions can reach the level of single cells and subcellular compartments. The FUS-CRISPR(a/ee) toolbox developed in this work can further allow the ultrasound-guided regulation in the dimensions of genome and epigenome at single-base precision74. Moreover, FUS-CRISPR(a/ee) can be integrated with different CRISPR regulators and gRNAs, and such a modular design should enable the targeting of, in principle, any accessible genomic locus for various reprogramming purposes. As such, our technology should provide a versatile platform to allow the remote and noninvasive control of genome and epigenome in specific tissues/organs of genetically engineered animals with high spatiotemporal resolution.
The current study has several limitations despite the encouraging results. For instance, the therapeutic application of FUS-CRISPRee at the current stage is limited by the possibly immunogenic yeast-origin SunTag module as well as the system’s bulky size, which is challenging for AAV packaging. Future development of synthetic biology should provide further opportunities for therapeutic applications. Also, leakage was still observed in several cases. Although these leakage levels are considerably lower than constitutive Cas9/dCas9 expression levels and may thus have weaker immunogenicity, additional efforts are needed to identify heat-sensitive promoters with lower leakage and higher activation potential, possibly via directed evolution. In short, future studies may focus on engineering more ideal FUS-CRISPR(a/ee) components and/or extending this technology for disease treatment beyond CAR-T therapy.
Methods
Ethics statement
Animal studies were approved in Protocols S15285, 21479 and FT-WuYQ-1 by the Institutional Animal Care and Use Committee (IACUC) at University of California, San Diego, University of Southern California, and Peking University, respectively. All researchers complied with animal-use guidelines and ethical regulations during research.
General cloning
Plasmids were constructed by Gibson Assembly (NEB, E2611L), T4 ligation (NEB, M0202L), or Golden Gate Assembly. PCR was performed using synthesized primers (Integrated DNA Technologies) and Q5 DNA polymerase (NEB, M0491). The sequences of the constructed plasmids were verified by Sanger sequencing (Azenta). Plasmids used in this study and their corresponding templates are listed in Supplementary Table 1. The sequences of the gRNAs were obtained from literature and listed in Supplementary Table 245,46,47,48,56,75,76,77.
General cell culture
Cell lines HEK 293 T (CRL-3216), Jurkat (TIB-152), RAW 264.7 (TIB-71), PC3 (CRL-1435), U-87 MG (HTB-14) were from American Tissue Culture Collection (ATCC, Manassas, VA). The Nalm6 cell line was a gift from Michel Sadelain Lab. The LNCaP cell line was a gift from Keyue Shen Lab. HEK 293 T and RAW 264.7 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, 10569010) supplemented with 10% fetal bovine serum (FBS) (Gibco, 10438026) and 1% penicillin-streptomycin (P/S) (Gibco, 15140122). Jurkat and Nalm6 cells were cultured in Roswell Park Memorial Institute Medium (RPMI 1640) (Gibco, 22400105) with 10% FBS and 1% P/S. Primary human T cells were cultured in complete RPMI 1640 supplemented with 100 U/ml recombinant human IL-2 (PeproTech, 200-02). All mammalian cells were cultured at 37 °C in a humidified 5% CO2 incubator.
Gene delivery methods
General plasmid transfection in HEK 293 T cells was performed using Lipofectamine 3000 transfection reagent (Invitrogen, L3000001) according to the manufacturer’s protocol.
For piggyBac-based cell line generation (Fig. 1e–h), the piggyBac transposon vector (Supplementary Fig. 2d) and the piggyBac transposase plasmid (SBI, PB210PA-1) were delivered into cells at a ratio of 2.5:1 by Lipofectamine transfection in HEK 293 T cells or by electroporation in Raw 264.7 cells using the Lonza 4D-Nucleofector and the SF kit (Lonza, V4XC-2032). Puromycin selection (5 μg/ml) was applied for 10 days.
Electroporation in Jurkat cells was performed as previously described78. Briefly, ten million Jurkat cells were resuspended in 500 μl of OptiMEM containing 20 μg Hsp-RGR or U6-gRNA plasmid and 20 μg CRISPRoff plasmid (Supplementary Fig. 3b, c) in a 4 mm cuvette and electroporated at 270 V, 950 μF (exponential wave, infinite resistance) using the Bio-Rad Gene Pulser Xcell Electroporation System. Cells were transferred to prewarmed culture media immediately after electroporation.
For lentiviral transduction, the lentivirus was produced by transfecting HEK 293 T cells with the transfer plasmid, packaging plasmid, and envelope plasmid using calcium phosphate-mediated transfection method (Promega, E1200) and harvesting the supernatant 48–72 h after transfection. For transduction of cell lines, 100–500 μl of unconcentrated lentivirus was added to 1 × 105 cells. For transduction of primary human T cells, the lentivirus was concentrated using Lenti-X™ Concentrator (Takara, 631232), followed by transduction as detailed in the Isolation, culture, and lentiviral transduction of primary human T cells section. FACS was performed to enrich the engineered cell populations when transduction efficiency was lower than 90% for cell lines or lower than 60% for primary T human cells.
In vitro heat shock
Cells were resuspended in regular culture media in 8-strip PCR tubes with 50 μl per tube and received heat shock (HS) in a thermal cycler (Bio-Rad, 1851148) for various durations before returning to normal culture condition. Samples heated with the thermal cycler were labeled as “HS” in the figures (as opposed to heating via FUS, which was labeled as “FUS +”). All in vitro HS experiments were performed at 43 °C.
Activation of exogenous genes via FUS-CRISPRa
For Fig. 1b, HEK 293 T cells were co-transfected with three FUS-CRISPRa plasmids (Supplementary Fig. 2a) at 1:1:1 ratio using Lipofectamine in a 12-well plate with 900 ng total DNA per well. Approximately 18 h after transfection, cells were resuspended in a culture medium, equally aliquoted into PCR tubes, and subjected to different HS treatments. The content of each individual PCR tube was added to individual wells containing 150 μl prewarmed medium in a 96-well plate (Corning, 3904) and returned to normal cell culture condition. The luminescence of each well was measured 24 h later using the Bright-Glo substrate (Promega, E2610) and a Tecan Infinite M200 Pro plate reader.
For Fig. 1d, HEK 293 T cells were co-transfected with four FUS-CRISPRa plasmids (Supplementary Fig. 2c) at 1:1:1:1 ratio using Lipofectamine in a 12-well plate with 1 μg total DNA per well. HS was performed 18 - 24 hours after transfection. Imaging was performed 24 h after HS, as described in the Fluorescence microscopy section.
Quantitative PCR
Total RNA was extracted from cells using Quick-RNA Microprep Kit (Zymo Research, R1050) and reverse transcribed to obtain cDNA using SuperScript™ IV Reverse Transcriptase (Invitrogen, 18090010). Quantitative PCR (qPCR) was performed using iTaq Universal SYBRRTM Green Supermix (Bio-Rad, 1725121) with primers listed in Supplementary Table 4.
Western blot analysis
Cells/tumors were harvested and homogenized with RIPA buffer (Cell signaling Technology, 9806S) containing protease and phosphatase inhibitor cocktail (Merck, 04693116001 and 4906837001). The same amount of protein lysate was loaded into a pre-cast polyacrylamide SDS-PAGE gel (Bio-Rad, 3450123) and ran at 30 mA for 90 min. The separated proteins were transferred onto 0.45 μm PVDF membrane (Bio-Rad, 1620184) at 230 mA for 100 min. After blocking with TBS-T (Tris-buffer saline containing 0.1% Tween 20) containing 5% powdered milk for 60 min, membrane was incubated with primary antibodies against IL1B (Abcam, Ab2105) and β-actin (Santa Cruz, sc-69879) overnight at 4 °C subsequently and the corresponding HRP-conjugated secondary antibodies, followed by chemiluminescence detection using a Bio-Rad ChemiDoc XRS + gel imager. The antibodies used for Western blot are listed in Supplementary Table 3.
Fluorescence microscopy
Microscopic images were taken with a Nikon Eclipse Ti inverted microscope with a cooled charge-coupled device (CCD) camera. For Fig. 1d and Supplementary Fig. 10b, HEK 293 T or primary human T cells were dropped onto uncoated glass-bottom dishes (Cell E&G, GBD00002-200), followed immediately by imaging. For Fig. 4a, HEK 293 T cells were resuspended in staining media (regular media containing Janelia Fluor® HaloTag® Ligands at 1:2000 dilution) and seeded onto fibronectin (Sigma Aldrich, F1141)-coated glass-bottom dishes. Three hours later, the staining media were washed out three times and replaced with regular media. Images were taken 6 hours after seeding.
Staining and flow cytometry
Staining was performed using fluorophore-conjugated antibodies according to manufacturers’ protocols. The antibodies used for flow cytometry are listed in Supplementary Table 3. Flow cytometry analysis was performed using BD Accuri C6 or SONY SH800. Gating was based on non-engineered cells with the same staining (if any) as the gating strategy illustrated in Supplementary Fig. 4a. Flow cytometry data were analyzed using FlowJo software (FlowJo).
Methylation detection
CXCR4-targeting FUS-CRISPRee Nalm6 cells without HS (CT) or 10 days after HS were used. Genomic DNA was extracted from cells using Quick-DNA Miniprep Plus Kit (Zymo Research, D4068). Bisulfite conversion was performed using EpiJET Bisulfite Conversion Kit (Thermo Scientific, K1461). PCR was performed using primer pairs
5’-GAGGTGGGTAGTTGGAAGTTTTTAG-3’, 5’-ATAATTTAACCTCCCCTTTAACACC-3’ (for region 1), 5’-GGGATTTAAGGGGGAGATATATGTAG-3’, 5’-AAAACCTAAATACTCCAATAACCAC-3’ (for region 2), 5’-GTTTTTTGTTTATTGTGTTGGGAGA-3’, 5’-TACATATATCTCCCCCTTAAATCC-3’ (for region 3) followed by Sanger sequencing (Supplementary Fig. 6c). The results were analyzed using QUMA, a quantification tool for methylation analysis79.
Transwell migration assay
7.5×104 Fluc+ cells in 100 μl culture medium were seeded onto Polycarbonate Membrane Transwell inserts (Corning, 3422). 600 μl culture media containing 10 ng/ml CXCR4 ligand CXCL12 (Peprotech, 300-28 A) were added to the transwell lower chambers as the chemoattractant. The cells in the inserts and the lower chambers were collected separately 3 h later, followed by quantification of luminescence as described above.
TCR stimulation in Jurkat cells
Jurkat cells were cultured in cell culture medium containing 1.7 μg/ml anti-TCR antibody (Sigma-Aldrich, 05-919) overnight followed by anti-CD69 antibody staining (Biolegend, 310910).
Isolation, culture, and lentiviral transduction of primary human T cells
Human peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats (Excellos) using lymphocyte separation medium (Corning, 25-072-CV), sorted with Pan T Cell Isolation Kit (Miltenyi, 130-096-535) to obtain primary human T cells, and activated by adding Dynabeads (Gibco, 11141D) at 1:1 bead-to-cell ratio. Two to three days later, T cells were mixed with lentivirus at the multiplicity of infection (MOI) equal to 5 in Retronectin (Takara, T100B)-coated culture plates and centrifuged at 1800 × g for 1 h at 32 °C for lentiviral transduction before returning to normal culture condition. Approximately one week later, T cells (with Dynabeads removed) were used for downstream applications or cryopreserved for future usage.
Quantification of knockout (KO) efficiency
Genomic DNA was extracted from cells using Quick-DNA Miniprep Plus Kit (Zymo Research, D4068). An approximately 500 bp fragment flanking the gRNA target site in the genome of engineered or WT cells was amplified by PCR with primers designed through NCBI Genome Data Viewer and Primer-BLAST (Supplementary Table 5). Sanger sequencing of the PCR products was performed to obtain trace files, which were uploaded to TIDE (TIDE created by Bas van Steensel lab, http://shinyapps.datacurators.nl/tide/) to quantify the KO efficiency.
T7E1 assay
T7E1 assay was performed to verify genome editing in Nalm6 cells engineered with PLK1-targeting FUS-CRISPR with or without HS. T7E1 assay was performed using primers 5’-TGCGAATGGTTGTGGACAGTGTTAAG-3’,
5’-AGTCTGTGAAGAATAGGGAGGAGTAGAG-3’ and the Alt-R® Genome Editing Detection Kit following the manufacturer’s protocol (IDT, 1075931).
Quantification of cell proliferation in vitro
Cells were stained with a live/dead dye AOPI (Nexcelom, CS2-0106) and counted using an automated cell counter (Nexcelom, Cellometer K2) to determine the cell number before seeding (Day 0). The same number of cells were then seeded in a 24-well plate for different groups. Cell culture media were refreshed every two days. At the time points specified in the corresponding figure legends (Figs. 3h, 4b), cells were collected and counted again as described above to determine the number of live cells, which was then normalized to the seeding cell number on Day 0 to obtain the normalized cell number.
Bulk RNA-seq
Nalm6 cells engineered with telomere-targeting or NT FUS-CRISPR were subjected to 10 min HS or no treatment (CT). Total RNA was collected at 24, 48, and 96 h after HS using the RNA microprep kit (Zymo Research, R1050) and sent for bulk RNA-seq (Novogene). RNA-seq data analysis was performed as previously described80. Briefly, raw RNA-seq reads were first preprocessed using Ktrim software (v1.4.1)81 to remove sequencing adapters and low-quality cycles; PCR duplicates (i.e., reads with identical sequences) and ribosomal RNAs were then removed using in-house programs, and the remaining reads were aligned to the human genome (build GRCh38/hg38) using STAR software (v2.7.9a)82; expression quantification were performed using featureCounts software (v2.0.3)83 against RefSeq gene annotation84; differential expression analysis were performed using DESeq2 software (v1.26.0)85; genes with an expression change larger than 1.5-fold and adjusted p-value smaller than 0.05 were considered as differentially expressed genes (DEGs). Functional annotation of the DEGs was performed using DAVID webserver86. RNA-seq results from the three-time points (24, 48, and 96 h) in the same treatment group were considered as three repeats for data analysis in Fig. 4d, e, and Supplementary Fig. 9.
Luciferase-based in vitro cytotoxicity assay
For Fig. 4g, h, 2 × 104 Fluc+ FUS-CRISPR-engineered Nalm6 cells with 10 min HS (HS) or without (CT) were cultured alone (w/o T), or mixed with αCD19CAR-T cells at an E:T ratio of 1:20 and co-cultured (w/ T) in 96-well plates. Culture media were renewed at 48 h by replacing one-third volume of the supernatant with fresh media. Fluc luminescence was measured 72 h after co-culture using the Bright-Glo Luciferase Assay System (Promega, E2610) and a Tecan Infinite M200 Pro plate reader. Fluc luminescence represents the amount of surviving Nalm6 tumor cells.
For Fig. 6d, 2 × 104 Fluc+ PC3 cells infected with FUS-CRISPR AAVs (Supplementary Fig. 13a) with 15 min HS (HS) or without (CT) were either cultured alone (w/o T) or co-cultured with αCD19-synNotch PSMACAR-T cells 24 h after HS at E:T = 1:1 (w/ T) in 96-well plates. Fluc luminescence was measured 24 h after co-culture as described above.
Animals
Six-to-eight weeks old male NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from Jackson Laboratory, UCSD Animal Care Program, or Shanghai Model Organisms Center, Inc. Animals were housed under a 12 light/12 dark cycle, 18–23 °C, and 40–60% humidity. Sex was not considered in the study design. Findings should be independent of sex. Only male mice were used in this study for consistency.
In vivo bioluminescence imaging
In vivo bioluminescence imaging (BLI) of firefly luciferase signals was performed using Lumina LT Series III (PerkinElmer) or Ami HTX (Spectral Instruments Imaging). Firefly luciferase substrate D-luciferin (GoldBio, LUCK-1G) was administered intraperitoneally, followed by BLI approximately 10 min later until the capture of the peak signal. Images were analyzed with Living Image software (PerkinElmer) or Aura Imaging Software (Spectral Instruments Imaging). The integrated luminescence reading within a fixed region of interest (ROI) over the tumor was used to represent the tumor size.
FUS system
We developed a FUS system with a real-time Proportional-Integral-Derivative (PID) temperature control feedback loop for generating localized hyperthermia in vitro and in vivo (Supplementary Fig. 1a). A focused 1.15-MHz single-element transducer was fabricated in-house using a pre-focused modified PZT (diameter: 70 mm, radius of curvature: 65 mm, DL-47, Del Piezo Specialties) with a 20 mm hole in the center. A coupling cone (length: 65 mm) with an opening (diameter: 4 mm) at the tip was 3D-printed and glued to the transducer to hold degassed water through the acoustic path and to guide the ultrasound focus. The opening at the tip of the cone was sealed with an acoustically transparent thin film (Chemplex, 100). Deionized water was degassed with a vacuum pump (Vevor). A function generator (Sanford Research System, SG386) and a 50 dB power amplifier (E&I, 325LA) were used to feed pulsed sine waves to the transducer.
For FUS stimulation on cells in vitro (Supplementary Fig. 1b, c), cells were resuspended in a 50 μl medium in a PCR tube. The cell-containing PCR tube was fixed on the acoustic absorber (Precision Acoustics, F28-SMALL) below the transducer. A needle-type thermocouple (Physitemp Instruments, MT-29/2HT) was inserted into the tube to measure the temperature of the cell medium with a thermometer (Omega, HH806AU). Acoustic gel (Aquasonic, 26354) was applied between the transducer and the tube.
For in vivo FUS stimulation (Supplementary Fig. 1d, e), the anesthetized mouse was placed on its side on the animal bed with an embedded acoustic absorber. The animal bed is placed on a heating plate (Auber Instruments, WSD-30B) set to 37 °C to maintain the body temperature of the anesthetized mouse. The needle-type thermocouple was inserted into the tumor region subcutaneously to measure the temperature. Acoustic gel was generously applied. The FUS transducer was placed above the mouse to focus on the tumor. Stable heat generation and induction of heat-sensitive transgene expression in vitro and in vivo using this FUS system were validated (Supplementary Fig. 1f–h).
The temperature readings were fed to a PID controller in real-time to adjust the output power of the function generator to maintain the focal temperature at the target value. All in vivo FUS stimulation was targeted at 43 °C for 10 min or less with 90–95% duty cycle and 500 ms PRT. The code repository for the PID controller and the device interfaces can be found at https://github.com/phuongho43/ultrasound_pid.
In vivo tumor model
For the Nalm6 tumor model, 2 × 105 Fluc+ Nalm6 cells were injected subcutaneously into NSG mice on Day 0. FUS stimulation (43 °C, 10 min) targeted at the tumor region was performed on Day 9 and Day 12 in the FUS + groups. 2 × 106 CD19CAR-T cells were administered intravenously on Day 10 in the indicated groups. Tumor aggressiveness was monitored by BLI and caliper measurement (volume = length × width2/2). Tumor sizes did not exceed the approved maximum of 1.5 cm in diameter.
AAVs were purchased from the GT3 Core Facility of the Salk Institute or Vigene Biosciences. Regarding the delivery efficiency of FUS-CRISPR AAVs in vivo, since neither of the FUS-CRISPR AAVs contains fluorescent protein markers, we co-infected the two FUS-CRISPR AAVs with a third AAV expressing constitutive GFP (pAAV-CMV-GFP, Supplementary Fig. 14). A mixture of the three AAVs (of similar titers) at 1:1:1 ratio (50 μl each) was injected intratumourally when the subcutaneous tumors reached approximately 50 mm3. Five days later, the tumors were harvested, snap-frozen in OCT (SAKURA, 4583), and subsequently sectioned. The sections were fixed and stained with DAPI and imaged using a Leica Stellaris SP8 microscope.
For the PC3 tumor model, 1 × 106 PSMA+ Fluc+ PC3 cells were injected subcutaneously into NSG mice on Day 0. The two FUS-CRISPR AAVs of similar titers (Supplementary Fig. 13b) were mixed at a 1:1 ratio (50 μl each) and injected intratumourally on Day 17 when the PC3 tumors were approximately 50 mm3. Tumors were treated with or without FUS stimulation (43 °C, 10 min) on Day 20 and Day 25. On Day 23, 8 × 106 αCD19-synNotch PSMACAR-T cells were injected intravenously. On Day 28, some mice were sacrificed, and their tumors were harvested and sectioned as described above. The sections were fixed and stained with DAPI. Some tumor sections were further stained with anti-CD19 primary antibody (ABclonal, A19013) and AF647-conjugated secondary antibody (Invitrogen, A32795). Confocal images were acquired using a Leica Stellaris SP8 microscope. Tumor aggressiveness in the remaining mice was monitored by BLI.
For the LNCaP tumor model, 1 × 106 Fluc+ LNCaP cells were injected subcutaneously into both flanks of NSG mice on Day 0. On Day 15, the two FUS-CRISPR AAVs were mixed at a 1:1 ratio (50 μl each) and injected locally at the tumor site. Tumors were treated with or without FUS stimulation (43 °C, 10 min) on Day 18 and Day 25. On Day 23, 5 × 106 αCD19-synNotch PSMACAR-T cells were injected intravenously. Since LNCaP has been reported to be low tumorigenic even in immunodeficient mice87, and the tumors were barely palpable throughout the experimental period, we, therefore, used BLI of Fluc luminescence instead of caliper measurement to quantify tumor aggressiveness.
Software
Data were graphed and the corresponding statistical analysis was performed in GraphPad Prism 9.0.0. Microscopy images were analyzed in Fiji ImageJ2 2.3.0.
Statistics & reproducibility
The detailed statistical analysis methods were described in the corresponding figure legends. No statistical method was used to predetermine sample size. No data were excluded from the analyses. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the results of this study are available within the paper and its Supplementary Information. The RNA-seq data generated in this study have been deposited in the NCBI GEO database under accession code GSE279445. Source data are provided in this paper.
Code availability
The code repository for the PID controller and the device interfaces for the in-house built FUS system can be found at https://github.com/phuongho43/ultrasound_pid88.
References
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Pickar-Oliver, A. & Gersbach, C. A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 20, 490–507 (2019).
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020).
Xu, J. et al. Gene editing in rabbits: Unique opportunities for translational biomedical research. Front. Genet. 12, 642444 (2021).
Niu, D. et al. Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9. Science 357, 1303–1307 (2017).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Gilbert, L. A. et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 (2013).
Tay, L. S., Palmer, N., Panwala, R., Chew, W. L. & Mali, P. Translating CRISPR-Cas therapeutics: Approaches and challenges. CRISPR J. 3, 253–275 (2020).
Kellogg, E. H. et al. What are the current bottlenecks in developing and applying CRISPR technologies? Cell Syst. 13, 589–593 (2022).
Mehta, A. & Merkel, O. M. Immunogenicity of Cas9 Protein. J. Pharm. Sci. 109, 62–67 (2020).
Li, R. et al. Generation and validation of versatile inducible CRISPRi embryonic stem cell and mouse model. PLoS Biol. 18, e3000749 (2020).
Danziger, O., Patel, R. S., DeGrace, E. J., Rosen, M. R. & Rosenberg, B. R. Inducible CRISPR activation screen for interferon-stimulated genes identifies OAS1 as a SARS-CoV-2 restriction factor. PLoS Pathog. 18, e1010464 (2022).
Lundin, A. et al. Development of an ObLiGaRe doxycycline inducible Cas9 system for pre-clinical cancer drug discovery. Nat. Commun. 11, 4903 (2020).
Nihongaki, Y., Kawano, F., Nakajima, T. & Sato, M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 33, 755–760 (2015).
Wu, X., Huang, H., Yu, B. & Zhang, J. A blue light-inducible CRISPR-Cas9 system for inhibiting progression of melanoma cells. Front. Mol. Biosci. 7, 606593 (2020).
Yu, Y. et al. Engineering a far-red light–activated split-Cas9 system for remote-controlled genome editing of internal organs and tumors. Sci. Adv. 6, eabb1777 (2020).
Wang, X. et al. A far-red light-inducible CRISPR-Cas12a platform for remote-controlled genome editing and gene activation. Sci. Adv. 7, eabh2358 (2021).
Gamboa, L. et al. Heat-triggered remote control of CRISPR-dCas9 for tunable transcriptional modulation. ACS Chem. Biol. 15, 533–542 (2020).
Chen, X., Chen, Y., Xin, H., Wan, T. & Ping, Y. Near-infrared optogenetic engineering of photothermal nanoCRISPR for programmable genome editing. Proc. Natl. Acad. Sci. USA 117, 2395–2405 (2020).
Lan, T.-H., He, L., Huang, Y. & Zhou, Y. Optogenetics for transcriptional programming and genetic engineering. Trends Genet. S0168-9525, 00140–00148 (2022).
Wang, T., Liu, S., Huang, Y. & Zhou, Y. Red‐shifted optogenetics comes to the spotlight. Clin. Trans. Med. 12, https://doi.org/10.1002/ctm2.807 (2022).
Polesskaya, O. et al. Optogenetic regulation of transcription. BMC Neurosci. 19, 12 (2018).
Pan, Y. et al. Near-infrared upconversion-activated CRISPR-Cas9 system: A remote-controlled gene editing platform. Sci. Adv. 5, eaav7199 (2019).
Jahangiri-Manesh, A. et al. Gold nanorods for drug and gene delivery: An overview of recent advancements. Pharmaceutics 14, 664 (2022).
Gerosa, C. et al. Gold nanoparticles: A new golden Era in oncology? Pharmaceuticals 13, 192 (2020).
Hill, C. R. & ter Haar, G. R. High intensity focused ultrasound—potential for cancer treatment. BJR 68, 1296–1303 (1995).
Elhelf, I. A. S. et al. High intensity focused ultrasound: The fundamentals, clinical applications and research trends. Diagn. Interv. Imaging 99, 349–359 (2018).
Fite, B. Z. et al. Immune modulation resulting from MR-guided high intensity focused ultrasound in a model of murine breast cancer. Sci. Rep. 11, 927 (2021).
Sofuni, A., Asai, Y., Mukai, S., Yamamoto, K. & Itoi, T. High-intensity focused ultrasound therapy for pancreatic cancer. J. Med. Ultrason. https://doi.org/10.1007/s10396-022-01208-4 (2022).
Panzone, J., Byler, T., Bratslavsky, G. & Goldberg, H. Applications of focused ultrasound in the treatment of genitourinary cancers. Cancers 14, 1536 (2022).
Shoji, S. et al. Development and future prospective of treatment for localized prostate cancer with high-intensity focused ultrasound. J Med Ultrason. https://doi.org/10.1007/s10396-021-01183-2 (2022).
Madio, D. P. et al. Invited. On the feasibility of MRI-guided focused ultrasound for local induction of gene expression. J. Magn. Reson. Imaging 8, 101–104 (1998).
Smith, R. C., Machluf, M., Bromley, P., Atala, A. & Walsh, K. Spatial and temporal control of transgene expression through ultrasound-mediated induction of the heat shock protein 70B promoter in vivo. Human Gene Therapy 13, 697–706 (2002).
Guilhon, E. et al. Image-guided control of transgene expression based on local hyperthermia. Mol. Imag. 2, 11–17 (2003).
Deckers, R. et al. Image-guided, noninvasive, spatiotemporal control of gene expression. Proc. Natl. Acad. Sci. USA 106, 1175–1180 (2009).
Wang, S., Zderic, V. & Frenkel, V. Extracorporeal, low-energy focused ultrasound for noninvasive and nondestructive targeted hyperthermia. Future Oncol. 6, 1497–1511 (2010).
Abedi, M. H. et al. Ultrasound-controllable engineered bacteria for cancer immunotherapy. Nat. Commun. 13, 1585 (2022).
Wu, Y. et al. Control of the activity of CAR-T cells within tumours via focused ultrasound. Nat Biomed Eng 5, 1336–1347 (2021).
Chen, X. et al. Non-invasive activation of intratumoural gene editing for improved adoptive T-cell therapy in solid tumours. Nat. Nanotechnol. 18, 933–944 (2023).
Liu, P. et al. Sonogenetic control of multiplexed genome regulation and base editing. Nat. Commun. 14, 6575 (2023).
Gao, Y. & Zhao, Y. Self‐processing of ribozyme‐flanked RNAs into guide RNAs in vitro and in vivo for CRISPR‐mediated genome editing. J. Integr. Plant Biol. 56, 343–349 (2014).
Nissim, L., Perli, S. D., Fridkin, A., Perez-Pinera, P. & Lu, T. K. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol. Cell 54, 698–710 (2014).
Joung, J. et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 12, 828–863 (2017).
Konermann, S. et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588 (2015).
Nuñez, J. K. et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 184, 2503–2519.e17 (2021).
Yeo, N. C. et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 15, 611–616 (2018).
Miller, I. C. et al. Enhanced intratumoural activity of CAR T cells engineered to produce immunomodulators under photothermal control. Nat. Biomed. Eng. 5, 1348–1359 (2021).
Hofacker, D. et al. Engineering of effector domains for targeted DNA methylation with reduced off-target effects. IJMS. 21, 502 (2020).
Bianchi, M. E. & Mezzapelle, R. The chemokine receptor CXCR4 in cell proliferation and tissue regeneration. Front. Immunol. 11, 2109 (2020).
Burger, J. A. & Kipps, T. J. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood 107, 1761–1767 (2006).
Shen, W., Bendall, L. J., Gottlieb, D. J. & Bradstock, K. F. The chemokine receptor CXCR4 enhances integrin-mediated in vitro adhesion and facilitates engraftment of leukemic precursor-B cells in the bone marrow. Exp. Hematol. 29, 1439–1447 (2001).
Chiappa, M. et al. Present and future perspective on PLK1 inhibition in cancer treatment. Front. Oncol. 12, 903016 (2022).
Abdisalaam, S. et al. Dysfunctional telomeres trigger cellular senescence mediated by cyclic GMP-AMP synthase. J. Biol.Chem. 295, 11144–11160 (2020).
Kim, H. et al. CRISPR-Cas9 Mediated telomere removal leads to mitochondrial stress and protein aggregation. IJMS. 18, 2093 (2017).
Sahin, E. et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470, 359–365 (2011).
Rademacher, A., Erdel, F., Trojanowski, J., Schumacher, S. & Rippe, K. Real-time observation of light-controlled transcription in living cells. J. Cell Sci. 130, 4213–4224 (2017).
Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 6, a026104 (2016).
Croft, M. The role of TNF superfamily members in T-cell function and diseases. Nat. Rev. Immunol. 9, 271–285 (2009).
Morsut, L. et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell 164, 780–791 (2016).
Roybal, K. T. et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164, 770–779 (2016).
Choe, J. H. et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 13, eabe7378 (2021).
Ren, C., Xu, K., Segal, D. J. & Zhang, Z. Strategies for the enrichment and selection of genetically modified cells. Trends Biotechnol. 37, 56–71 (2019).
Ren, C. et al. Dual-reporter surrogate systems for efficient enrichment of genetically modified cells. Cell Mol. Life Sci. 72, 2763–2772 (2015).
Chew, W. L. et al. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods 13, 868–874 (2016).
Moreno, A. M. et al. Immune-orthogonal orthologues of AAV capsids and of Cas9 circumvent the immune response to the administration of gene therapy. Nat. Biomed. Eng. 3, 806–816 (2019).
Maresca, D. et al. Biomolecular ultrasound and sonogenetics. Annu. Rev. Chem. Biomol. Eng. 9, 229–252 (2018).
Ibsen, S., Tong, A., Schutt, C., Esener, S. & Chalasani, S. H. Sonogenetics is a non-invasive approach to activating neurons in Caenorhabditis elegans. Nat. Commun. 6, 8264 (2015).
Couture, O., Hingot, V., Heiles, B., Muleki-Seya, P. & Tanter, M. Ultrasound localization microscopy and super-resolution: A state of the art. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 65, 1304–1320 (2018).
Renaudin, N. et al. Functional ultrasound localization microscopy reveals brain-wide neurovascular activity on a microscopic scale. Nat. Methods 19, 1004–1012 (2022).
Sawyer, D. P. et al. Ultrasensitive ultrasound imaging of gene expression with signal unmixing. Nat. Methods 18, 945–952 (2021).
Macé, E. et al. Functional ultrasound imaging of the brain. Nat. Methods 8, 662–664 (2011).
Rabinowitz, R. & Offen, D. Single-base resolution: Increasing the specificity of the CRISPR-Cas system in gene editing. Mol. Ther. 29, 937–948 (2021).
Sanson, K. R. et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 9, 5416 (2018).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014).
Shifrut, E. et al. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 175, 1958–1971 (2018).
Huang, Z. et al. Engineering light-controllable CAR T cells for cancer immunotherapy. Sci. Adv. 6, eaay9209 (2020).
Kumaki, Y., Oda, M. & Okano, M. QUMA: quantification tool for methylation analysis. Nucleic Acids Res. 36, W170–W175 (2008).
Peng, Q. et al. Engineering inducible biomolecular assemblies for genome imaging and manipulation in living cells. Nat. Commun. 13, 7933 (2022).
Sun, K. Ktrim: an extra-fast and accurate adapter- and quality-trimmer for sequencing data. Bioinformatics 36, 3561–3562 (2020).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Kawada, M. et al. Establishment of a highly tumorigenic LNCaP cell line having inflammatory cytokine resistance. Cancer Lett. 242, 46–52 (2006).
Phuong Ho. Ultrasound Control of Genomic Regulatory Toolboxes for Cancer Immunotherapy. Github https://doi.org/10.5281/zenodo.13909749 (2024).
Acknowledgements
This work was supported in part by grants from NIH EB029122 (Y. Wang), GM140929 (Y. Wang), HL121365 (Y. Wang), HD107206 (Y. Wang), and CA262815 (Y. Wang) to Y. Wang, NIH K01EB035649 (L.L.) to L.L. and NIH EB032822 (G. Kwong) to G. Kwong. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was supported by the GT3 Core Facility of the Salk Institute with funding from NIH-NCI CCSG: P30 014195, an NINDS R24 Core Grant, and funding from NEI.
Author information
Authors and Affiliations
Contributions
Y. Wu, Z.H., Y.L., and Y. Wang conceived and designed the experiments; Y. Wu, Z.H., Y.L., P. He., Yu. Wang., L.Y., X.W., S.G., X.Z., C.Y., Y.S., and Z. Yuan performed the experiments; Y. Wu, Y.L., Z.H., K.S., and L.L. analyzed the data; C.Y., Y. Zeng, P. Ho., L.Z., Q.Z., Y. Zhao, and G.K. contributed materials; Y. Wu, Z.H., Y.L., T.L., S.C., L.L., and Y. Wang wrote and revised the paper. All authors approved the final version.
Corresponding authors
Ethics declarations
Competing interests
Y. Wang is a scientific co-founder and consultant of Cell E&G Inc. and Acoustic Cell Therapy Inc. These financial interests do not affect the design, conduct, or reporting of this research. G.K. is a co-founder of Glympse Bio and Port Therapeutics. This study could affect his personal financial status. The terms of this arrangement have been reviewed and approved by Georgia Tech in accordance with its conflict-of-interest policies. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks John Lee, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, Y., Huang, Z., Liu, Y. et al. Ultrasound Control of Genomic Regulatory Toolboxes for Cancer Immunotherapy. Nat Commun 15, 10444 (2024). https://doi.org/10.1038/s41467-024-54477-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54477-7
This article is cited by
-
Modular engineering of thermoresponsive allosteric proteins
Nature Chemical Biology (2026)
-
A compact and inducible dCas12f-based CRISPRa platform for programmable in vivo gene activation
Nature Communications (2026)
-
Frontiers in mechanobiology and mechanomedicine
Med-X (2025)
