
Abstract
Zika virus (ZIKV) infection can result in a birth defect of the brain called microcephaly and other severe fetal brain defects. ZIKV enters the susceptible host cells by endocytosis, which is mediated by the interaction of the envelope (E) glycoprotein with cellular surface receptor molecules. However, the cellular factors that used by the ZIKV to gain access to host cells remains elusive. Here, we report that the extracellular domain of integrin beta 4 (ITGB4) is an entry factor of ZIKV. ITGB4 mediates ZIKV infection by directly interacting with the E glycoprotein of ZIKV, and ITGB4 knockout hampers the binding and replication of ZIKV to host cells. A functional monoclonal antibody against ITGB4 or the soluble forms of ITGB4 could decrease the binding and infection of ZIKV to permissive cell lines. Importantly, the ITGB4 antibody blocks the infection of ZIKV to mouse placenta, thus protecting the fetuses from ZIKV infection. Together, our study has demonstrated that ZIKV infection involves ITGB4 dependent binding.
Similar content being viewed by others

Introduction
Zika virus (ZIKV) is a mosquito-transmitted flavivirus which was first isolated from a febrile rhesus macaque in Uganda in 1947. In 2007, a large epidemic of Asian genotype ZIKV was reported in Yap Island and Guam, Micronesia, and then a further outbreak was reported in France between 2013 and 20141, resulting in over 1.5 million infections. In November 2015, Brazil experienced a sharp rise in the number of cases of neonatal microcephaly, which corresponds temporally and geographically to the ZIKV outbreak. Subsequently, mounting evidence suggests that ZIKV infection in pregnant women can lead to congenital abnormalities and fetal demise2,3,4. ZIKV infection is observed in both the amniotic fluid of pregnant women with microcephalic fetuses and microcephalic fetal brain tissues5. The following experiments further prove that ZIKV infection in impregnate mice can cause placental damage, fetal microcephaly and even demise6,7. These results indicates that ZIKV can cross blood placenta barrier (BPB) and cause the damage of fetal brain. In addition, ZIKV infections in adults have been linked to Guillain-Barré syndrome8,9. Available treatments for ZIKV infection are still limited, as there is currently no effective chemoprophylaxis or vaccination against infection.
Similar to dengue virus and Japanese encephalitis virus (JEV), ZIKV enters into susceptible cells by endocytosis which is mediated by the interaction of the E glycoprotein with cellular surface receptor molecules. Several cell surface proteins, such as AXL, TYRO3, TIM1/4 and DC-SIGN, are reported to be involved in ZIKV entrance and infection. Earlier studies shows that AXL functions as an attachment or entry factor for ZIKV in keratinocytes and endothelial cells10,11. Moreover, the expression of AXL in human neural progenitor cells (hNPCs) promotes the efficient infection of ZIKV, which further highlights AXL as a candidate entry receptor of ZIKV in hNPCs12. However, mounting recent studies demonstrate that AXL may does not function as a host cell receptor of ZIKV since the genetic ablation of AXL does not protect hNPCs or mice from ZIKV infection13,14. Hastingset also reports that AXL is not required for ZIKV infection in mice15. The reason for AXL promoting ZIKV infection is due to its antagonism of type I interferon signaling14,16,17. Besides, TYRO3 is not necessary factors for ZIKV infection in mice15. Other candidate proteins for ZIKV entry, such as DC-SIGN, TIM1, and TIM4 showed little to no expression in hNPCs by gene expression analysis14. Studies above called into the reasonable question about the role of AXL, TYRO3, DC-SIGN, TIM1 and TIM4 in ZIKV entry. Recently, integrin ITGAV/ITGB5 was identified as an internalization factor but not a binding factor for ZIKV during neural stem cells infection18. Despite those important research progresses, the factors or determinants required for ZIKV binding and entry are still largely unknown. There may be yet unidentified cell surface molecules that functions in ZIKV entry.
In this work, we find that ITGB4 mediates the ZIKV infection. ITGB4 homodimer or ITGB4/ITGA6 heterodimer can interact with E glycoprotein of ZIKV to mediate ZIKV infection. The lack of ITGB4 or the soluble forms of ITGB4 decreases the binding and infection of ZIKV to permissive cell lines. Importantly, the functional monoclonal antibody against ITGB4 could protect mouse from ZIKV infection. Therefore, this work uncovers the role of ITGB4 in ZIKV infection and supply a potential treatment against ZIKV infection.
Results
ITGB4 interacts with ZIKV E protein
To identify cell surface proteins that interact with ZIKV E envelope protein, we conducted a tandem affinity purification (TAP) experiment and mass spectrometry. A specific 200 kDa band was observed in SDS-PAGE analysis (Fig. 1a). Mass spectrometry analysis of the tryptic peptides identified a single pass type I membrane protein ITGB4 which was expressed on the cell surface (Fig. 1b). The results from immunoprecipitation further determined the interaction of ZIKV E with ITGB4 ectodomain (Figs. 1c and S1f). Considering that ITGB4 usually forms heterodimer with ITGA6 on cell surface and functions as a laminin receptor, we indeed analyzed the interaction between ZIKV E and endogenous ITGB4/ITGA6. The results from western blotting showed that ZIKV E could also interact with endogenous ITGB4/ITGA6 heterodimer (Figs. 1d and S1g). To further verify ITGB4 or ITGB4/ITGA6 could bind with ZIKV E, we expressed and purified the recombinant ZIKV E, RuV E1, JEV E, and the ectodomain of ITGB4 or ITGB4/ITGA6 bearing with His tag (Figs. S1a–S1e), and then incubated ZIKV E in ITGB4 or ITGB4/ITGA6 coated plate and measured bond ZIKV E by HRP-conjugated anti-E antibody. As a control, the purified Ruv E1 and JEV E could not directly interact with ITGB4 or ITGB4/ITGA6. However, ITGB4 or ITGB4/ITGA6 indeed interacted with ZIKV E directly (Figs. 1e and S1h). Besides, ITGB4 or ITGB4/ITGA6 could also interact with live ZIKV virions (Fig. 1f), which further demonstrated the ITGB4 could directly interact with ZIKV E.

a Hun-7 cell lysates were incubated with ZIKV-E or RuV-E1 proteins and the affinity-isolated proteins were subjected to SDS-PAGE. The red arrowhead indicates the specifically isolated protein using the ZIKV-E protein (n = 3 of independent experiments). b ITGB4-derived tryptic fragments as determined by mass spectrometry. The tryptic fragments corresponding to ITGB4 protein sequence (Gene bank: NM_000213) is indicated in red. c Immunoprecipitation assay showing specific binding between ZIKV-E and the ectodomain of ITGB4. Lysates of 293 cells were incubated with ZIKV-E or RuV-E1 immobilized on anti-Flag M2 monoclonal antibody-conjugated agarose beads. The precipitates were washed by DDM-PBS and eluted with Flag peptide. The eluents were then separated by SDS-PAGE and immunoblotted with the related antibodies (n = 3 of independent experiments). d Immunoprecipitation assay showing specific binding between ZIKV-E and ITGB4/ITGA6 heterodimer in Hun-7 cells. Lysates of Hun-7 cells were incubated with ZIKV-E or RuV-E1 immobilized on anti-FLAG M2 monoclonal antibody-conjugated agarose beads. The precipitates were treated as in (c) (n = 3 of independent experiments). CL: Hun-7 cell Lysates. e ZIKV-E protein interacted with ITGB4. Purified ITGB4, ITGB4/ITGA6 or BSA were coated on 96-well plate overnight and incubated with flag tagged ZIKV-E or RuV-E1 for 3 h at 4°C. ZIKV-E or RuV-E1 was detected by HRP-conjugated anti-Flag antibody. The data were expressed as Mean ± SD of three independent experiments. **p = 0.0057, ****p < 0.0001, two-sided, Student’s t test. f ZIKV virions interacted with the ectodomain of ITGB4. Purified ITGB4, ITGB4/ITGA6 or BSA was coated on 96-well plate overnight and incubated with MR766 or Mock (PBS) for 4 h at 4°C. The bound virus was detected by HRP-conjugated anti-E antibody. The data were expressed as Mean ± SD of three independent experiments. ***p = 0.0002, **p = 0.032, two-sided, Student’s t test. g, h Susceptibility of Vero, Hun-7, A549, LLC-MK2, hNPCs and HEK-293T cells to ZIKV. Cells were infected with ZIKV MR766 at an MOI = 0.05. 48 h post infection and then visualized using a human antibody that recognizes the structural protein E (green). Cell nuclear were stained with DAPI (blue). n = 3 of independent experiments. All scale bars are 50 μm.
ITGB4 mediates ZIKV infection
The cell surface expression and transcription of ITGB4 could be detected in ZIKV permissive cell lines, such as Vero, Hun7, LLC-MK2, A549 and hNPCs, but except for 293 T cell lines (Figs. S1i and S1j). ZIKV E protein binding and ZIKV infection was observed in all detected ZIKV permissive cell lines, while 293 T cells were not susceptible to ZIKV E binding (Figs. 1g, h). To probe the specificity of ITGB4 in ZIKV entry, the ZIKV genome RNA was transfected into the 293 T cells. Results showed ITGB4 is not essential for viral replication once the viral genome has entered the cell (Figs. 2a, b). The findings elucidate that 293 T cells are not naturally susceptible to ZIKV infection due to the deficient expression of ITGB4, highlighting ITGB4’s role in viral entry.

a Western blot analysis of the ZIKV in 293 T cells transfected with ZIKV genome RNA. 2 ug purified ZIKV genome RNA was transfected into 293 T cells for 24 h. Then, the supernatant of 293 T cells was transferred into Vero cells (n = 3 of independent experiments). Control: Vero cells infected with ZIKV for 24 h. Mock: Vero-E6 cells. b Viral titers in cell supernatants were determined by TCID50 experiment in Vero-E6 cells. The data were expressed as Mean ± SD of five independent experiments. *P = 0.0204, two-sided, by multiple comparisons by One-way ANOVA test. Control: Vero cells infected with ZIKV for 24 h. 293T-ZIKV: Vero cells incubated with the supernatant of 293 T cells transfected with ZIKV genome RNA. 293 T: Vero cells were incubated with the supernatant of 293 T cells. c Cells were subsequently inoculated for 1 h with the ZIKV MR766-protein mixes on ice followed washing three times with PBS. The virus titer was determined by plaque assay on Vero cells at 48 h after infection. The data were expressed as Mean ± SD of three independent experiments. ****p < 0.0001 (Vero), ****p < 0.0001 (A549), **P = 0.0045 (Hun7), ***P = 0.0002 (Hun7), ****p < 0.0001 (MK2), **P = 0.0013 (hNPCs), ****p < 0.0001 (hNPCs), two-sided, multiple comparisons by One-way ANOVA test. d Cells were infected with ZIKV as described above. After incubated with virus-protein mixes for 1 h on ice, cells were washed with cold PBS. Total RNA was then extracted and ZIKV RNA levels that represent viral particles bind to cell surface were quantified by RT-PCR assay. The GAPDH was used as internal control gene. The data were expressed as Mean ± SD of three independent experiments. Control: PBS buffer. **P = 0.0038 (Hun7, top), ***P < 0.0003 (Hun7, middle), *P = 0.0241 (Hun7, bottom), **P = 0.0022 (Vero, top), *P = 0.0475 (Vero, bottom), *P = 0.0117 (MK2), **P = 0.0072 (A549, top), *P = 0.0006 (A549, middle), *P = 0.0072 (A549, bottom), *P = 0.0102 (hNPCs, top), ***P = 0.0002 (hNPCs, middle), **P = 0.0032 (hNPCs, bottom), two-sided, multiple comparisons by One-way ANOVA test. e ZIKV MR766 was preincubated with the indicated concentrations of soluble ITGB4/ITGA6 or BSA. Cells were subsequently inoculated for 1 h with the virus-protein mixes on ice. Cells were washed three times with PBS and the virus titers were determined by plaque assay on Vero cells at 48 h after infection. The data were expressed as Mean ± SD of three independent experiments. ***P = 0.003 (Vero), ****p < 0.0001 (Vero), ***P = 0.0001 (A549), ****p < 0.0001 (A549), **P = 0.0022 (Hun7), ***P = 0.0002 (Hun7), **P = 0.0034 (MK2), ***p = 0.0007 (MK2), *P = 0.0257 (hNPCs), **P = 0.0016 (hNPCs), two-sided, multiple comparisons by One-way ANOVA test. f Cells were infected with ZIKV that preincubated with the indicated concentrations of soluble ITGB4/ITGA6 or BSA. Incubated with virus-protein mixes on ice for 1 h, cells were washed three times with cold PBS. Total RNA was then extracted and ZIKV RNA levels were determined by RT-PCR assay. The data were expressed as Mean ± SD of three independent experiments. Control: PBS buffer. **P = 0.0055 (Hun7, top), ***P = 0.0002 (Hun7, middle), **P = 0.0099 (Hun7, bottom), *P = 0.0197 (Vero, top), **P = 0.0017 (Vero, bottom), *P = 0.0227 (MK2, top), **P = 0.0011 (MK2, middle), *P = 0.0403 (MK2, bottom), **P = 0.0061 (A549, top), ***P = 0.0001 (A549, middle), **P = 0.0033 (A549, bottom), **P = 0.0064 (hNPCs, top), ***P = 0.0004 (hNPCs, middle), *P = 0.0232 (hNPCs, bottom), two-sided, multiple comparisons by One-way ANOVA test.
To ascertain that the binding of ITGB4 to ZIKV E mediates ZIKV infection, we performed an infection inhibition assay with soluble ITGB4 homodimer or ITGB4/ITGA6 heterodimer. The preincubation of ITGB4 homodimer reduced the binding of ZIKV to host cells (Figs. 2c and S2a). The decreasing of viral particles was observed in ITGB4-pretreated cells, which showed ITGB4 dose-dependent (Figs. 2d and S2b). In contrast, BSA treatment showed no obvious effect on ZIKV binding at the detected concentrations (Figs. 2c, S2a, and S2b). Since the heterodimer of ITGB4/ITGA6 could directly interact with ZIKV E protein, we indeed analyzed whether the heterodimer could affect ZIKV infection. Results showed that the preincubation of ITGB4/ITGA6 heterodimer also significantly reduced ZIKV binding and infection in host cells (Figs. 2e, 2f, and S2a).
ITGB4 antibody (13H10) inhibits ZIKV infection
To provide further evidence of ITGB4 acting as a receptor for ZIKV infection, an antibody (13H10) that specific binds to the ITGB4 homodimer or ITGB4/ITGA6 heterodimer with a high affinity was generated (Figs. 3a, S3a, S3b and S3c). Comparing with isotype-control group, pre-incubation with 13H10 decreased ZIKV infections in different ZIKV permissive cell lines (Figs. 3b, 3c and S3d). In the presence of the 13H10, the binding of infectious virus to ITGB4 or ITGB4/ITGA6 was striking decreased (Fig. S3e). In addition, pre-incubation with 13H10 had no obvious effects on binding and replication of another flavivirus JEV in A549 cells (Figs. S4c, S4d and S4e), indicating that ITGB4 may be a specific factor that mediates ZIKV infection.

a A surface plasmon resonance assay (SPR) characterizing the binding between ITGB4/ITGA6 and 13H10 antibody. The 13H10 antibody were immobilized on the chip at about 300 response units. Concentrations of ITGB4/ITGA6 was used to flow over the chip surface. 13H10 was ITGB4 specific antibody. b Cells were treated with various concentrations of 13H10 or isotype antibody for 1 h and infected with ZIKV with MOI = 0.05. Virus titers were determined by plaque assay on Vero cells at 48 h after infection. The values in the graph represent the mean ± SD of n = 3 independent experiments. ***P = 0.0082 (Vero), ****p < 0.0001 (Vero), **P = 0.0052 (A549), ***p = 0.0005 (A549), ***P = 0.0002 (Hun7), ****p < 0.0001 (Hun7), ****p < 0.0001 (MK2), **P = 0.0046 (hNPCs), ***P = 0.0002 (hNPCs), two-sided, multiple comparisons by One-way ANOVA test. c Cells were preincubated with isotype antibody or indicated concentrations of 13H10 antibody for 1 h and then challenged with ZIKV with MOI = 10 at 4 °C for 1 h. Cells were washed, and total RNA was extracted. ZIKV RNA was quantified by RT-PCR. The results were expressed as ZIKV RNA levels relative to the expression of the GAPDH internal control gene. The data were expressed as Mean ± SD of three independent experiments. ****P < 0.0001 (Hun7, top), ***P = 0.0001 (Hun7, middle), **P = 0.0042 (Hun7, bottom), ***P = 0.0002 (Vero, top), **P = 0.0019 (Vero, middle), *P = 0.0192 (Vero, bottom), ****P < 0.0001 (A549, top), ****P < 0.0001 (A549, middle), **P = 0.0029 (A549, bottom), ***P = 0.0002 (MK2, top), **P = 0.0025 (MK2, middle), *P = 0.0216 (MK2, bottom), ***P = 0.0003 (hNPCs, top), **P = 0.0060 (hNPCs, middle), *P = 0.0169 (hNPCs, bottom), two-sided, multiple comparisons by One-way ANOVA test.
The results above showed that ITGB4 facilitated ZIKV binding and infection. Since 293 T is a ZIKV un-permissive cell line19, we then over-expressed ITGB4 or ITGA6 in 293 T cells to detect ZIKV infection (Fig. S4a). Results showed that the virus binding on cell surface or virus titers in supernatant were elevated in ITGB4-overexpressed 293 T cells (Fig. 4a). However, ITGA6-overexpression did not affect ZIKV binding and infection (Fig. 4a). The soluble ITGB4, ITGB4/ITGA6 protein or 13H10 mono-antibody could weaken ZIKV infection in ITGB4-overexpressed 293 T cells (Figs. 4b, 4c and S4b). Therefore, these results proved that it was ITGB4, not ITGA6, which acted as the binding factor for ZIKV.

a HEK-293T cells were transfected with plasmids encoding human ITGB4 or ITGA6 for 48 h and infected with ZIKV as described above. Cells were subsequently inoculated for 1 h with the virus-protein mixes on ice. Cells were washed three times with PBS and the virus titers were determined by plaque assay on Vero cells at 48 h after infection. The values in the graph represent the mean ± SD of three independent experiments. ****P < 0.0001 (SZ_SMGC-1, top), **P = 0.0012 (MR766, top), ***P = 0.0007 (MR766, bottom), two-sided, multiple comparisons by One-way ANOVA test. b HEK-293T cells were transfected with plasmids encoding human ITGB4 for 48 h, and then infected with ZIKV that preincubated with the indicated concentrations of soluble ITGB4, ITGB4/ITGA6 or BSA. After incubated with 1 h with virus-protein mixes on ice, cells were washed three times with cold PBS. ZIKV RNA levels were determined by RT-PCR assay. The data were expressed as Mean ± SD of three independent experiments. **P = 0.0032 (left, top), ****P < 0.0001 (left, bottom), **P = 0.0014 (right, top), ****P < 0.0001 (right, bottom), two-sided, multiple comparisons by One-way ANOVA test. c Effects of ITGB4 antibody on ZIKV SZ_SMGC-1 binding. HEK-293Tcells were transfected with plasmids encoding human ITGB4 were treated with the indicated amount of 13H10 antibody or an isotype antibody before incubation with ZIKV. 1 h after incubation on ice, cells were washed, and total RNA was extracted. ZIKV RNA was quantified by RT-PCR. The data were expressed as Mean ± SD of three independent experiments. ***p = 0.0003, *p = 0.0129, two-sided, multiple comparisons by One-way ANOVA test.
ITGB4 stays out of ZIKV particle assembly
To further determine the roles of ITGB4 in ZIKV infection, an ITGB4 knock-out A549 cell line (A549ITGB4−/−) was generated by deleting exons 3-12 of the ITGB4 gene with Cas9 system, which was determined by flow cytometry (Fig. 5a). ITGB4 deficiency strikingly weakened ZIKV binding with cells (Fig. 5b). Correspondingly, virus titers in the A549ITGB4−/− cell lines were also obviously decreased (Fig. 5c). Consequently, the number of ZIKV positive cells was evidently decreased in A549ITGB4−/− cell lines (Fig. 5d), whereas the re-expression of ITGB4 in A549ITGB4−/− cell lines rescued ZIKV infection (Fig. 5e). To elucidate the specificity of ITGB4 in ZIKV entry, the ZIKV genome RNA was directly introduced into A549ITGB4−/− cells via transfection. Notably, both the parental A549 and the A549ITGB4−/− cell lines exhibited comparable capacities for ZIKV particle production upon successful delivery of the viral genome, suggesting ITGB4 is not an essential host factor for ZIKV particle assembly once the viral genome is inside the host cell (Fig. 5f).

a The expression of ITGB4 on cell surface of A549 (orange line) and A549ITGB4−/− (red line) was monitored by flow cytometry. Cyan line represents A549ITGB4−/− cells stained with normal mouse IgG. b RT-PCR analysis showing the binding of ZIKV to A549 and A549ITGB4−/−cells. Cells were incubated with ZIKV SZ_SMGC-1 or ZIKV MR766 with MOI = 10 at a multiplicity of infection at 4°C for 45 min. Cells were washed, and total RNA was extracted. ZIKV RNA was quantified by RT-PCR. The data were expressed as Mean ± SD of five independent experiments. ****p < 0.0001, two-sided, Student’s t test. c Cells were infected with ZIKV with MOI = 0.05. Virus titers were determined by plaque assay on Vero cells at 48 h after infection. The values in the graph represent the mean ± SD of three independent experiments. ***P = 0.002(SZ_SMGC-1), ***P = 0.001(MR766), two-sided, Student’s t test. d Cells were infected with ZIKV as described above. The percentages of infected cells were measured by flow cytometry 48 h post infection. Mock represents cells with no ZIKV infection. e The complementation expression of ITGB4 in A549ITGB4−/− cells restored ZIKV infectivity. A549ITGB4−/− cells were transfected with plasmids expressing ITGB4, ITGA6 or empty vector. 24 h post infection, cells were infected with ZIKV MR766 at MOI of 0.05 for 48 h. ZIKV positive cells were counted by immunofluorescence assay. The data were expressed as Mean ± SD of three independent experiments. **p = 0.0024, two-sided, Student’s t test. f Viral titers in A549 and A549ITGB4−/− cells transfected with ZIKV genome RNA determined by TCID50 experiment in Vero-E6 cells. 2 ug purified ZIKV genome RNA was transfected into A549 and A549ITGB4−/− cells by using Lipofectamine 3000 for 24 h. And then, the supernatant of A549 cells was transferred into Vero-E6 cells. The data were expressed as Mean ± SD of six independent experiments and multiple comparisons by One-way ANOVA test. ND: Vero cells incubated with the supernatant of A549ITGB4−/− cells.
13H10 protects from ZIKV infection
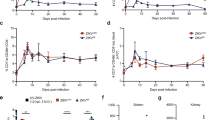
To evaluate the protective roles of 13H10 in host defense against viral infection in vivo, the WT male C57BL/6 mice were infected with ZIKV SZ_SMGC-1 after intraperitoneal injection with anti-Ifnar1 and 13H10 mAb. As control, the mice from Isotype-control mAb group started to lose weight on day 3 post-infection and started to regain weight on day 7 post-infection. However, the mice from 13H10 mAb treated group showed a delayed body weight loss and quickly body weight recovery (Fig. 6a). Importantly, 13H10 treatment strongly decreased ZIKV induced mice death (Fig. 6b). RT-PCR results demonstrated that 13H10 obviously reduced viremia at the indicated times (Fig. 6c). To further determine the protective role of 13H10 in ZIKV infection, WT male mice treated with anti-Ifnar1 mAb were injected with 13H10 mAb at 1 or 3 days after subcutaneous inoculation with ZIKV. The results from RT-PCR showed that viral RNA level was significantly reduced in serum and brain from 13H10 mAb treated mice at 1 day post-infection. However, 13H10 showed no protective role at 3 days post-infection (Figs. 6d and 6e).

a, b Four to five weeks old WT male mice (n = 12 per group from three independent experiments) were intraperitoneal treated with anti-Ifnar1 mAb (2.5 mg each mouse). 18 h later, mice were treated with 1 mg of isotype-control mAb or 13H10 followed by subcutaneous infection with 104 PFU of ZIKV-SZ_SMGC-1. Mouse weight loss (a) and survival (b) was observed and recorded daily until 16 d.p.i. Data were expressed as Mean ± SD, two-sided, **P = 0.0016, log-rank test). c Viremia measurements at days 1 through 4 after infection with ZIKV as determined by plaque assay. The data were expressed as Mean ± SD of four independent experiments. **P(1) = 0.0047, ***P(2) = 0.0002, **P(3) = 0.0012, ***P(4) = 0.0003, two-sided, Student’s t test. d and e Four to five weeks old WT male mice were treated with anti-Ifnar1 mAb. 24 h later, mice were treated with 1 mg of isotype-control mAb or 13H10 followed by subcutaneous inoculation with 104 PFU of ZIKV-SZ_SMGC−1. At day +3, viral RNA in serum (d) brain (e) was measured by RT-PCR. n = 7 mice per group (d) and n = 9 mice per group (e). The data were expressed as Mean ± SD of three independent experiments. ***P = 0.0003 (d) ***P = 0.0001 (e) two-sided, Student’s t test. Dashed lines indicate the limit of detection of the assay.
Placental damage and fetal injury occur in pregnant mice after ZIKV infection6,20,21. To assess the protective ability of 13H10, we firstly measured the distributions of 13H10 in organs of pregnant mouse and investigated the penetration of 13H10 to fetus. The in vivo fluorescent images showed that the fluorescent signals that represent 13H10-Cy5 were shown at the uteri, liver, spleen, kidney, heart and small intestine (Figs. S6a and S6c). Besides, the signal of 13H10-Cy5 could also observed in the uteruses (Figs. S6b and S6d). These results suggested that 13H10 was able to enter into blood circulation and organs of pregnant mice and penetrated into fetuses.
Next, we sequentially administered WT pregnant C57BL/6 mice with anti-Ifnar1 mAb and 13H10 mAb prior to inoculation with ZIKV (Fig. 7a). Analysis of peripheral blood obtained at E13.5 post-infections from ZIKV-infected dams showed lower viremia in mice receiving 13H10 antibodies, and striking differences in tissue viral loads in the maternal brain, placenta and fatal heads were observed in the pregnant dams (Fig. 7b). Histological analysis showed reductions in the area and thickness of the labyrinth and junctional layer of the placenta in isotype-treated dams, which was obviously reversed with 13H10 pre-treatment after ZIKV infection (Figs. 7c, 7h and S7e). RNA FISH analysis determined the presence of ZIKV RNA in the maternal placenta in the isotype-control mAb treated mice. In contrast, no obvious RNA FISH signals that represents viral RNA was observed in placentas of mice treated with 13H10 (Figs. 8a and S7d). Fetuses harvested at E13.5 from pregnant dams treated with 13H10 have a bigger size compared to those from the isotype group (Fig. 8b). ZIKV infection resulted in about 70% rate of fetal resorption rate in isotype-control mAb treated group compared to a 30% rate in the dams treated with 13H10 (Fig. 8c). Gross morphological analysis of fetuses obtained at E13.5 from ZIKV-infected mice treated with isotype-control mAb showed fetal growth restriction and demise compared to those from mice treated with 13H10 mAb (Fig. 8d). Considering that Ifnar1−/− mice are an accepted model for studying ZIKV pathogenesis22, we then used this mouse model to repeat the above experiment. The results showed that 13H10 mAb could also protect Ifnar1−/− mice from ZIKV infection (Figs. S7a, S7b and S7c).

a Scheme of antibody injection and ZIKV challenge of pregnant dams. Eight to nine weeks-old pregnant WT dams (n = 12 per group) were treated with anti-Ifnar1 (2.5 mg each mouse) at E4.5 or E5.5. 18 h later, mice were treated with a single 1 mg dose of isotype control antibody or 13H10 by intraperitoneal injection followed by subcutaneous inoculation with 104 PFU of ZIKV-SZ_SMGC−1. Mice were euthanized on E13.5. b ZIKV RNA levels in maternal serum (n = 10), maternal brain (n = 12), fetal heads (n = 14), and placenta (n = 11). The dashed line indicates the limit of detection of the qRT-PCR assay. The data were expressed as Mean ± SD of three biological replicates. **P = 0.0031 (serum), **P = 0.0091 (brain), **P = 0.0025 (head), ****P < 0.0001 (placenta), two-sided, Student’s t test. c Measurement of thickness at indicated areas of placentas from ZIKV infected dams after isotype control or 13H10 antibody treatment. The data were expressed as Mean ± SD of three independent experiments, n = 8 in all the measurement but n = 7 in placenta junctional thickness measurement. **P = 0.0060 (labyrinth area), ****P < 0.0001 (labyrinth thickness), **P = 0.0025 (placenta area), **P = 0.0093 (junctional thickness), **P < 0.0001 (junctional area), two-sided, multiple comparisons by One-way ANOVA test.

a RNA in situ hybridization staining of placentas at E13.5. The images are representative of several independent placentas from multiple dams. Black arrows indicate cells positive for ZIKV RNA. (n = 3 of independent experiments) b Measurements of fetus body size at E13.5. Each dot represents data from eight individual placenta. The data were expressed as Mean ± SD of three biological replicates. ***P = 0.0005, two-sided, Multiple comparisons by One-way ANOVA test. c The percentage of fetuses survival on E13.5 (n = 17 for 13H10, n = 14 for isotype control antibody). Data were representative of 3 independent experiments. ****p < 0.0001, two-sided, Student’s t test. d Representative images of E13.5 uterus recovered from ZIKV-infected dams after isotype control or 13H10 antibody treatment. Most fetuses carried by isotype control antibody treated dams died in utero and had undergone resorption, leaving only the residual abnormal placenta.
Considering that ZIKV preferential infect mononuclear cytotrophoblasts (CTBs) and multi-nucleated syncytiotrophoblasts (STBs) from the organotypic culture system of chorionic villus explants from first and second trimester human placentas, we next analyzed the expression of ITGB4 in mouse. The results showed that ITGB4 could be detected in mouse, which further determined the vital role of ITGB4 in ZIKV infection (Fig. S7f).
The results showed that ITGB4 interacted with the ZIKV-E protein and functions as a surface receptor to mediate ZIKV infection. The addition of soluble ITGB4 or an anti-ITGB4 antibody could block viral binding and replication. Furthermore, the ITGB4 knockout and rescue experiments have demonstrated its essential role in ZIKV infection. Importantly, an anti-ITGB4 antibody was able to decrease the death of mice infected with ZIKV and to protect mouse fetuses from ZIKV infection. These data demonstrated that ITGB4 played a pivotal role in ZIKV infection.
Discussion
Integrins have been demonstrated to mediate the entry of various viruses23,24,25,26,27. For flaviviruses, such as JEV and WNV, ITGB3 may participate in virus binding and internalization28. ITGAV/ITGB5 was shown to mediate ZIKV internalization but not binding during neural stem cells infection18. Here we showed that ITGB4 mediated the binding between ZIKV and various cells lines including hNPCs, indicating that integrins may play different roles in ZIKV entry. Unlike other beta integrins that could interact with multiple alpha subunits, ITGB4 has been shown to interact with only ITGA629. Beyond its adhesive functions in epithelial cells, ITGB4/ITGA6 heterodimers functions in their signal transduction pathways within the central nervous system (CNS)30,31,32,33. ZIKV infects neural stem/progenitor cells, astrocytes and immature neurons in the developing brain and leads to microcephaly and other brain abnormalities in infants20,34,35. It has been reported that ITGB4 is expressed in several kinds of neural cells including astrocytes, schwann cells and neural stem cells36,37. ITGB4 anchors astrocytes to the vascular basal lamina to increase the resistance of the blood brain barrier and promotes differentiation of neural stem cells. Thus, there was a strong correlation between ITGB4 expression and ZIKV susceptibility in various CNS cells. ZIKV may first infect astrocytes by using ITGB4 and indeed destroy the blood brain barrier. The infection of astrocytes contributes to subsequent viral dissemination. A detailed investigation of correlation between ITGB4 distribution and ZIKV infection in types of cells of blood brain barrier would help for the explanation of ZIKV penetration.
Fetus is at a relatively high risk of microcephaly if the mother is infected with ZIKV during the first trimester of pregnancy35,38. Serious birth defects were observed in about 1 in 12 fetuses or infants of pregnant women with ZIKV infection in the first trimester. Studies show that trophoblast from first trimester expresses the ITGA6 and ITGB439. Thus, the ITGB4 mediated ZIKV infection in trophoblasts may contribute to the penetration of ZIKV to fetus. Taking the reasons that high level of ITGB4 expression in human and mouse placentas, a monoclonal antibody to ITGB4 (13H10) could prevent ZIKV infection in placenta and keeps blood placental barrier integrity. With these new insights regarding to the entrance of ZIKV, a number of small compounds, antibodies and peptides could be developed for the treatment or prevention of ZIKV infection by blocking the ZIKV-ITGB4 interaction site, or by inducing a conformation in ITGB4 that is unfavorable to binding. Further in-depth characterization of the binding interface of ZIKV E and ITGB4 by resolving the structure of ITGB4/ZIKV-E or virions/ITGB4 complex may provide important insights into this possibility. ZIKV remains to be a threat to human health. The present study is expected to help in ZIKV-related disease control and treatment.
Methods
Ethics statement
All animals used in the experiments were handled according to the guidelines of the Institutional Animal Care and Use Committee of the Institute of Microbiology, Chinese Academy of Sciences (IMBS2D(3)-SC04-2014). The C57BL/6 mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. IFNα/β receptor-deficient C57BL/6 mice (Ifnar1−/−) were purchased from Jackson Laboratories and bred in a specific-pathogen-free facility at Institute of Microbiology, CAS. Mice were housed under controlled environmental conditions with ambient temperature maintained at 25 ± 2 °C and relative humidity at 50−55%. Animals were kept on a standard 12: 12 light: dark cycle, with environmental parameters monitored throughout the experimental period.
Cells and virus
African green monkey kidney epithelial cells (Vero, CCTCC, GDC0029), Human embryonic kidney fibroblast cells (293 T, CCTCC, GDC0187) and Rhesus monkey kidney epithelial cells (LLC-MK2, CCTCC, GDC0156) were obtained from China Center for Type Culture Collection (CCTCC). Hun7 (ATCC number HB-8065, human hepatoma cell line derived from a liver tumor) and A549 cells (CCTCC, GDC0063) were maintained by our laboratory. Cells were propagated and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with antibiotics (penicillin and streptomycin) and 10% fetal bovine serum at 37 °C in the presence of 5% CO2. hNPCs were generated from pluripotent stem cells (iPSCs) as previously described40.
Asian ZIKV strain, SZ_SMGC-1, isolated from a patient infected in Samoa (GenBank accession number: KU866423), was kindly provided by Yuhai Bi (Institute of Microbiology, Chinese Academy of Sciences). ZIKV strain MR766 (GenBank accession number: MK105975.1) was provided by Shihua Li (Institute of Microbiology, Chinese Academy of Sciences). JEV-SA14 was provided by Bo Zhang (Wuhan Institute of Virology, Chinese Academy of Sciences). Virus stocks were propagated in Vero cells and titrated by the Reed-Muench method.
Antibodies, primers and plasmids
The following antibodies were used in this study: anti-ITGB4 (Abcam, ab29042); anti-Flag (Sigma, F1804); anti-ITGA6 (Abcam, ab20142); anti-His (Proteintech, 66005-1-Ig); anti-GAPDH (Proteintech, 60004-1-Ig); anti-ZIKV Envelope antibodies labeled with FITC (Z6-FITC antibody) were generated from activated PBMC of hospitalized patients41. Anti-ZIKV Envelope antibodies labeled with HRP by horseradish peroxidase mark kit (LinKine).
ITGB4 monoclonal antibody was produced in mouse by using the ectodomain of ITGB4 by GenScript (Nanjing, China). Briefly, Balb/c mice were immunized and boosted with ITGB4. Mice spleen cells were harvested for hybridoma fusion 60 days after the first immunization. Indirect ELISA was then performed for screening with immunogen and positive hybridomas are expanded. 30 hybridomas with the best screening result are selected for isotyping and antibody expression. An antibody (13H10) with the best virus blocking capability and high ITGB4 or ITGB4/ITGA6 binding ability were purified and selected out for further experiments.
Plasmids that used for full length ITGB4 (#16039) and ITGA6 (#53352) expression were obtained from Addgene. Primers that used in relative quantitative PCR are ZIKV-Q-F, ZIKV-Q-R, GAPDH-F, and GAPDH-R (Supplementary Table S1). Viral RNA copies were calculated by Real-Time PCR.
Protein expression and purification
The codon-optimized coding sequences for the ectodomain of the RuV-E1 (Uniprot number P07055) from strain RA27/3 and ITGB4 (NCBI: txid9606) were synthesized and incorporated into the pFastBacTM1 (Invitrogen) plasmid for protein production using the Bac-to-Bac baculovirus expression system as previous described42. The proteins were fused with an N-terminal gp67 secretive signal peptide and a 6×His affinity tag to facilitate protein expression and purification. Both the RuV-E1 and ITGB4 proteins were expressed using Hi5 cells (Invitrogen) as soluble proteins and were captured from the cell culture supernatant by immobilized metal affinity chromatography (IMAC) with a HisTrap HP 5 mL column (GE Healthcare). The eluted fractions were pooled and further purified by size-exclusion chromatography (SEC) with a Superdex 200 column (GE Healthcare) equilibrated with a buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl for final purification. The resulting products reached a purity of more than 95% and were concentrated to 10 mg ml−1 for further experiments.
The coding sequence for residues 1-409 of ZIKV-E was cloned into pET21a vector with C-terminal Flag and 6×His affinity tag. The coding sequence for residues 1–406 of JEV-E was cloned into pET19b vector with a C-terminal Flag affinity tag. Both the ZIKV-E and JEV-E proteins were expressed in Escherichia coli strain BL21(DE3) as inclusion bodies and then refolded in vitro using the method as previously described43,44. Subsequently, the refolded protein was concentrated and then further purified by size-exclusion chromatography (SEC) with a Superdex 200 column (GE Healthcare) equilibrated with a buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 5% glycerol.
Immunoprecipitation and identification of ITGB4
Hun-7 cells were lysed with 0.3% DDM lysis buffer (pH 7.6, PBS supplemented with 1 mM Mg2+, 2% glycerol, 0.3% DDM) containing protease-inhibitor cocktail. The lysate was clarified by centrifugation and pre-immunoprecipitated with anti-Flag monoclonal antibody-conjugated agarose beads (to eliminate background binding of anti-Flag antibodies that may be present in the envelope protein tag preparations). The lysates were then incubated with proteins previously bound to anti-Flag M2 monoclonal antibody-conjugated agarose beads. The precipitates were washed twice in 0.3% DDM lysis buffer (pH 8.5, 2% glycerol, 1 mM Mg2+) and twice in 0.5% DDM lysis buffer (pH 8.0, 1% glycerol) and then eluted with Flag peptide (0.2 mg/ml). Eluted proteins were heat denatured and separated by 12% SDS-PAGE (Bio-Rad). A portion of the gel was stained with Coomassie G-250 stainer (Bio-Rad). Gels containing the specific protein band captured by the E-Flag were excised, digested in the gel with sequencing-grade trypsin (Promega), and subjected to peptide sequencing by tandem mass spectrometry.
Flow cytometry
Cells were harvested at the indicated times and fixed in 4% PFA for 10 min at room temperature. After three washes in phosphate buffered saline (PBS). Cells were blocked with 5% bovine serum albumin (BSA) in PBS and incubated for 1 h at room temperature. After being washed, the cells were resuspended in PBS with 1% BSA and incubated for 2.5 h at 4 °C with the primary antibody at a final concentration of about 1 μg/ml. Cells were then incubated for 1 h with the fluorescent secondary antibody. The cells were then subjected to flow cytometry analysis (FlOWJO Version 7.6.4) after they had been washed with PBS.
Surface plasmon resonance technology (SPR) analysis
The purified ZIKV E, ITGB4 and ITGB4/ITGA6 proteins were analyzed by an analytical gel-filtration assay with a calibrated Superdex®75 10/300 GL column (GE Healthcare) as previously described45. The samples were also analyzed by SDS-PAGE. The SPR analysis of affinity was performed using a BIAcore T100 with CM5 chips (GE Healthcare) linked with anti-his antibody at 25 °C as previously described45. Briefly, the buffers for all proteins used were exchanged to a BIAcore buffer (10 mM pH 7.5 HEPES, 150 mM NaCl, and 0.005% (v/v) Tween-20). After protein was immobilized on the chip at about 100 response units, gradient concentrations of 13H10 or isotype-control antibody were used to flow over the chip surface with the response units measured at single cycle. The binding kinetics were analyzed with the software BIA evaluation Version 4.1 using a 1:1 Langmuir binding model. The affinity between 13H10 and ITGB4/ITGA6 heterodimer or ITGB4 were analyzed by SPR.
BioLayer Interferometry
The affinity between 13H10 mAb and ITGB4 was measured by BLI on an OctetRed 96 (ForteBio). The ITGB4 protein was randomly biotinylated on surface-exposed lysine side chains with succinimidyl-6-(biotinamido) hexanoate, and then purified using a ZebaSpin desalting column (Fisher, catalog number 89882) and diluted to 50 µg/ml. The sensors (super streptavidin) were pre-wet in dialysis buffer for 15 min before use and then loaded with biotinylatedITGB4 proteins for 15 min. 13H10 mAb or isotype-control was serially diluted (800, 400, 200, 100, 50, and 25 nM) in a 96-well plate. The measurements were carried out automatically at room temperature. Experiments were performed in three biological replicates with curve fitting using a 2:1 (Heterogeneous Ligand) model.
In vivo fluorescence Iimaging
Pregnant C57/B6 mice (8–12 weeks old, E12.5) were assigned randomly to two groups and injected intravenously 50 μg 13H10-Cy5 in 100 μl PBS or 100 μl PBS without antibody in caudal vein. 1 h later, the distribution of 13H10-Cy5 in mice were imaged by the IVIS lumina K series III in vivo imaging system (PerkinElmer, Waltham, MA) and the radiant efficiency (ps−1 cm−2 sr−1) (mW−1 cm2) was calculated using the Living Image 4.4 software. To determine the distribution of 13H10-Cy5 in organs, pregnant mice were killed by intraperitoneal injection of sodium pentobarbital. The dissected uteri, livers, spleens, kidneys hearts, small intestine and fetuses were imaged and the average radiant efficiency was calculated.
Immunofluorescence Assays (IFA)
Cells were transfected plasmids encoding ITGA4 or ITGB6 bearing Flag tag with PEI (1 mg/ml). After infected with ZIKV virus at indicated time, cells were washed with PBS three times and then fixed with 4% paraformaldehyde (PFA) in PBS for 10 min at room temperature. The fixed cells were permeabilized with 0.5% Triton X-100 in PBS for 10 min and then rinsed three times in PBS for 5 min each. Then permeabilized cells were blocked with 5% BSA in PBS for 30 min at 37 °C, incubated overnight with the corresponding primary antibody against Flag at 4 °C overnight, followed by incubation with Fluor 488- or Alexa Fluor 546-conjugated secondary antibodies for 1 h at room temperature. ZIKV infection was analyzed by staining the permeabilized cells with anti-ZIKV envelope protein antibodies labeled with FITC.
ZIKV binding assays
Cells were treated with 13H10 or isotype antibody for 1 h and challenged with ZIKV at 4 °C for 1 h. Cells were washed with chilled PBS for three times, and lysed using TRIzol regent. And then the binding viral RNA was quantified by qPCR. The results were expressed as ZIKV RNA levels relative to the expression of the GAPDH internal control gene.
In some cases, ZIKV was preincubated with concentrations of soluble ITGB4 or BSA. Cells were subsequently inoculated with the virus-protein mixes on ice for 1 h. Cells were then washed three times with cold PBS to remove the unbound virions. Total RNA was then extracted from cells with TRIzol regents. ZIKV RNA levels that represent viral particles bind to cell surface were quantified using a quantitive RT-PCR assay.
Elisa binding assay
To detect the direct interaction between ZIKV and ITGB4 or ITGB4/ITGA6, Purified ITGB4, ITGB4/ITGA6 or BSA were coated (550 ng/well) in 100 μl TBS (adding 10 mM MgCl2) overnight at 4 °C. Plates were then blocked with 5% BSA in TBS for 1 h at 37 °C. ZIKV virions (106 PFU), ZIKV-E or RuV-E1 were added to each well and incubated at 4 °C for 2 h. Unbound virions in supernatants were removed by washing the plates with TBS five times. Then, 0.5 mg/ml HRP labeled antibody were added and incubated at 37 °C for 1 h. TMB substrate was added and the intensity at 450 nm that representing the binding virions was measured.
Immunohistochemistry
Mouse fetuses were removed by Caesarian-section at E13.5, fixed in 4% paraformaldehyde and then sectioned at 5 μm with Leica CM3050 S. For immunohistochemistry, sections were immersed in blocking solution and then incubated in primary antibodies against ITGB4 overnight. Subsequently, sections were rinsed and reacted with fluorescent secondary antibodies.
Hematoxylin-eosin (HE) staining
Harvested placentas were fixed in 10% neutral buffered formalin (Fisher) at room temperature and embedded in paraffin. Tissues were sectioned at a thickness of 5 μm on a Leica RM2255 rotary microtome and then were de-paraffinized in xylene and rehydrated with a gradient (100-50%) of ethanol, followed by a wash in distilled water. Lately, Sections were stained with hematoxylin and eosin (H&E) and then examined with Leica APERIO VERSA 8.
RNA fluorescence in situ hybridization
RNA FISH was performed using RNAscope®2.5HD Regent Kit-RED (#313911, Advanced Cell Diagnostics) according to the manufacturer’s instructions. In short, Formalin-fixed paraffin-embedded tissue sections were deparaffinized at 60 °C for 1 h, and then washed by dimethylbenzene and ethyl alcohol at room temperature. Endogenous peroxidases were quenched with H2O2 for 10 min at room temperature. Target Retrieval was conducted at 100-104 °C for 15 min. The multiple hybridization and signal amplification were performed at 40 °C in a hybridization oven. The probe targeting ZIKV RNA was designed and synthesized by Advanced Cell Diagnostics (#467871). Positive and negative (targeting bacterial gene dapB) control probes also were obtained from Advanced Cell Diagnostics (#313911 and #310043, respectively). Tissues were counterstained with Gill’s hematoxylin and then visualized using Leica APERIO VERSA 8.
ITGB4 Knock out cell line
CRISPR-Cas9 mediated ablation of the ITGB4 gene was achieved with transposon plasmid (provided by Haixing Bioscience) containing expression cassettes for hSpCas9, Puro and chimeric guide RNA. To target exon 3-12 bp of the ITGB4 gene, two guide RNA gRNA-F and gRNA-R (Supplementary Table S1) were selected through the http://crispr.mit.edu website. Plasmid containing the guide RNA sequence was electro-transfected into cells using Neon™ transfection system according to the manufacturer’s instructions (ThermoFisher Scientific). 24 h post-transfection, cells were selected by puromycin (2 µg/ml). After three days, single colonies were transferred into 96-well plates. To determine the presence of insertions or deletions in ITGB4 targeted clones, genomic DNA was isolated using a Blood/Cultured cells DNA Kit (Simgen, 3002050) and the exon 3-12 bp of ITGB4 was amplified using 2×Taq Master Mix (Dye Plus; Vazyme, P112) with primers flanking exon 3-12 bp. PCR product was sequenced by Sanger sequencing (GENEWIZ, China). Clones with mutations in both alleles were selected for downstream studies. Depletion of ITGB4 expression was screened by western blot and flow cytometry. All clones were maintained under the same conditions as parental cells.
RT-PCR
Total RNA was extracted from cells using TRIzol. RNA concentration was quantified by using a Nanodrop spectrophotometer at 260 nm. Single-stranded cDNA synthesis was carried out by reverse transcription kit (Promega). PCR was then performed to analyze cDNA levels of integrin α subunits and β subunits. The levels of GAPDH were also analyzed as an internal control. The primers used for cDNA analysis and chimeric protein construction are shown in Supplementary Table S1.
Animal experiments
To evaluate the protective role of 13H10 in the host’s defense against viral infection in vivo, the 4 to 5-weeks-old WT male C57BL/6 mice were intraperitoneal injected with 2.5 mg anti-Ifnar1 mAb (MAR1-5A3). 24 h later, mice were treated with 1 mg of isotype-control or 13H10 mAb followed by subcutaneous infection with 104 FFU of ZIKV. Mice were monitored for 16 days after ZIKV infection. In some experiments, WT mice were treated with indicated doses of 13H10 by intraperitoneal injection at day 1 or 3 after ZIKV infection.
The 8 to 9-week-old pregnant C57BL/6 mice (E4.5-5.5) were sequential intraperitoneal injected with 2.5 mg anti-Ifnar1 mAb (MAR1-5A3, Leinco Technologies). 24 h later, the mice were treated with a single 1 mg dose of isotype-control or 13H10 antibody by intraperitoneal injection followed by subcutaneous inoculation with 104 PFU of ZIKV. The mice were sacrificed at E13.5 and placentas, fetuses, maternal tissues and serum were harvested from the infected mice.
The 8 to 9-week-old Ifnar1−/− female mice were mated with wild type C57/B6 male mice. At E5.5, dams were treated with 400 μg of isotype-control or 13H10 mAb. After 2 h, dams were infected with 104 FFU of ZIKV through the subcutaneous inoculation. Dams were harvested at E13.5. Viral burden in the maternal brain, placenta and serum were measured by RT-PCR. Fetus size was measured as the crown-rump length × occipital-frontal diameter of the head.
Measurement of viral burden
ZIKV-infected tissues were weighed and homogenized in 600 μl or 200 μl PBS. Samples were centrifuged at 2000 × g for 10 min and then stored at −80 °C. For some samples, viral burden was determined by plaque assay. ZIKV RNA was extracted from the tissue and serum samples, then ZIKV RNA levels were determined by Taqman one-step qRT-PCR. Viral burden was expressed on a log10 scale as viral RNA equivalents per g or ml after calculated with a standard curve produced using serial tenfold dilutions of ZIKV RNA. The ZIKV RNA was detected using the published primer (ZIKV-R-F, ZIKV-R-R and ZIKV-RNA-Probe, Supplementary Table S1)46.
Statistics
All statistics were done using GraphPad Prism 9.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available within the article and its supplementary materials. Source data are provided with this paper.
Change history
07 April 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-58610-y
References
Cao-Lormeau, V. M. & Musso, D. Emerging arboviruses in the Pacific. Lancet 384, 1571–2 (2014).
Brasil, P. et al. Zika virus infection in pregnant women in Rio de Janeiro. N. Engl. J. Med 375, 2321–2334 (2016).
Sarno, M. et al. Zika virus infection and stillbirths: a case of hydrops fetalis, hydranencephaly and fetal demise. PLoS Negl. Trop. Dis. 10, e0004517 (2016).
Ventura, C. V., Maia, M., Bravo-Filho, V., Góis, A. L. & Belfort, R. Jr. Zika virus in Brazil and macular atrophy in a child with microcephaly. Lancet 387, 228 (2016).
Mercado, M. et al. Zika virus detection in amniotic fluid and Zika-associated birth defects. Am. J. Obstet. Gynecol. 222, 610.e1–610.e13 (2020).
Miner, J. J. et al. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 165, 1081–1091 (2016).
Yockey, L. J. et al. Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol. 3, eaao1680 (2018).
Araujo, L. M., Ferreira, M. L. & Nascimento, O. J. Guillain-Barré syndrome associated with the Zika virus outbreak in Brazil. Arq. Neuropsiquiatr. 74, 253–5 (2016).
Cao-Lormeau, V. M. et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet 387, 1531–1539 (2016).
Retallack, H. et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl Acad. Sci. USA 113, 14408–14413 (2016).
Savidis, G. et al. Identification of zika virus and dengue virus dependency factors using functional genomics. Cell Rep. 16, 232–246 (2016).
Nowakowski, T. J. et al. Expression analysis highlights AXL as a candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell 18, 591–6 (2016).
Wang, Z. Y. et al. Axl is not an indispensable factor for Zika virus infection in mice. J. Gen. Virol. 98, 2061–2068 (2017).
Wells, M. F. et al. Genetic ablation of AXL does not protect human neural progenitor cells and cerebral organoids from Zika virus infection. Cell Stem Cell 19, 703–708 (2016).
Hastings, A. K. et al. TAM receptors are not required for Zika virus infection in mice. Cell Rep. 19, 558–568 (2017).
Chen, J. et al. AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol 3, 302–309 (2018).
Meertens, L. et al. Axl mediates ZIKA virus entry in human glial cells and modulates innate immune responses. Cell Rep. 18, 324–333 (2017).
Wang, S. et al. Integrin αvβ5 internalizes Zika virus during neural stem cells infection and provides a promising target for antiviral therapy. Cell Rep. 30, 969–983.e4 (2020).
Hamel, R. et al. Biology of Zika virus infection in human skin cells. J. Virol. 89, 8880–96 (2015).
Cugola, F. R. et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534, 267–71 (2016).
Yockey, L. J. et al. Vaginal exposure to Zika virus during pregnancy leads to fetal brain infection. Cell 166, 1247–1256.e4 (2016).
Lazear, H. M. et al. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19, 720–30 (2016).
Jackson, T., Mould, A. P., Sheppard, D. & King, A. M. Integrin alphavbeta1 is a receptor for foot-and-mouth disease virus. J. Virol. 76, 935–41 (2002).
Jokinen, J. et al. Molecular mechanism of alpha2beta1 integrin interaction with human echovirus 1. Embo j. 29, 196–208 (2010).
Schornberg, K. L. et al. Alpha5beta1-integrin controls ebolavirus entry by regulating endosomal cathepsins. Proc. Natl Acad. Sci. USA 106, 8003–8 (2009).
Summerford, C., Bartlett, J. S. & Samulski, R. J. AlphaVbeta5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat. Med 5, 78–82 (1999).
Wang, X., Huang, D. Y., Huong, S. M. & Huang, E. S. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat. Med 11, 515–21 (2005).
Chu, J. J. & Ng, M. L. Interaction of West Nile virus with alpha v beta 3 integrin mediates virus entry into cells. J. Biol. Chem. 279, 54533–41 (2004).
Feltri, M. L. et al. Beta 4 integrin expression in myelinating Schwann cells is polarized, developmentally regulated and axonally dependent. Development 120, 1287–301 (1994).
Dang, J. et al. Zika virus depletes neural progenitors in human cerebral organoids through activation of the Innate Immune Receptor TLR3. Cell Stem Cell 19, 258–265 (2016).
Hogervorst, F., Kuikman, I., von dem Borne, A. E. & Sonnenberg, A. Cloning and sequence analysis of beta-4 cDNA: an integrin subunit that contains a unique 118 kd cytoplasmic domain. Embo j. 9, 765–70 (1990).
Li, C. et al. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 19, 672 (2016).
Noronha, L., Zanluca, C., Azevedo, M. L., Luz, K. G. & Santos, C. N. Zika virus damages the human placental barrier and presents marked fetal neurotropism. Mem. Inst. Oswaldo Cruz 111, 287–93 (2016).
Cauchemez, S. et al. Association between Zika virus and microcephaly in French Polynesia, 2013-15: a retrospective study. Lancet 387, 2125–2132 (2016).
El Costa, H. et al. ZIKA virus reveals broad tissue and cell tropism during the first trimester of pregnancy. Sci. Rep. 6, 35296 (2016).
Schouest, B. et al. Transcriptional signatures of Zika virus infection in astrocytes. J. Neurovirol 27, 116–125 (2021).
Su, L., Lv, X. & Miao, J. Integrin beta 4 in neural cells. Neuromolecular Med 10, 316–21 (2008).
Molnár, Z. & Kennedy, S. Neurodevelopmental disorders: risks of Zika virus during the first trimester of pregnancy. Nat. Rev. Neurol. 12, 315–6 (2016).
Aplin, J. D. Expression of integrin alpha 6 beta 4 in human trophoblast and its loss from extravillous cells. Placenta 14, 203–15 (1993).
Tang, Q. Y. et al. UTX regulates human neural differentiation and dendritic morphology by resolving bivalent promoters. Stem Cell Rep. 15, 439–453 (2020).
Ma, W. et al. Zika virus causes testis damage and leads to male infertility in mice. Cell 168, 542 (2017).
Li, Q. et al. Structural and functional characterization of neuraminidase-like molecule N10 derived from bat influenza A virus. Proc. Natl Acad. Sci. USA 109, 18897–902 (2012).
Dai, L. et al. Structures of the zika virus envelope protein and its complex with a flavivirus broadly protective antibody. Cell Host Microbe 19, 696–704 (2016).
Liu, X. et al. The structure differences of Japanese encephalitis virus SA14 and SA14-14-2 E proteins elucidate the virulence attenuation mechanism. Protein Cell 10, 149–153 (2019).
Song, H. et al. Molecular basis of arthritogenic alphavirus receptor MXRA8 binding to chikungunya virus envelope protein. Cell 177, 1714–1724.e12 (2019).
Lanciotti, R. S. et al. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 14, 1232–9 (2008).
Acknowledgements
This work was supported by National Natural Science Foundation of China (32060039 of X.D.H, 82030033 of T.S.T, 92254301 of T.S.T, 82271428 of C.M.L, and 81921006 of T.S.T), the Program of Inner Mongolia Natural Science Foundation of China (2022SHZR1769 of X.D.H), and National Key R&D Program of China (2023YFA1801900 of T.S.T). We thank Xiaolan Zhang and Tong Zhao for technical help with confocal microscopy and flow cytometry in this study. We also thank Prof. Paul Chu for his kind help during the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
S.Z. and X.H. conceived the project, designed the experiments, and modified the manuscript. T.T. and C.L. analyzed the data and wrote the manuscript. N.D. performed the expression and purification of the recombinant proteins, and the SPR analysis. H.C. and J.W. conducted the animal experiments. H.C., L.S., and X.H. performed the ZIKV binding and neutralization assays. R.L. performed Flow Cytometry and RT-PCR assay. R.W. and Y.Y. conducted the Immunoprecipitation and Immunofluorescence assays. H.C., J.W., and X.H. performed the RNA fluorescence in situ hybridization, HE staining, and immunohistochemistry. All authors commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cong, H., Wang, J., Du, N. et al. ITGB4/CD104 mediates zika virus attachment and infection. Nat Commun 15, 10729 (2024). https://doi.org/10.1038/s41467-024-54479-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54479-5
