
Abstract
The development of efficient and robust catalysts for hydrogen evolution reaction is crucial for advancing the hydrogen economy. In this study, we demonstrate that ultra-low coordinated hollow PtRuNi-Ox nanocages exhibit superior catalytic activity and stability across varied conditions, notably surpassing commercial Pt/C catalysts. Notably, the PtRuNi-Ox catalysts achieve current densities of 10 mA cm−2 at only 19.6 ± 0.1, 20.9 ± 0.1, and 21.0 ± 0.1 mV in alkaline freshwater, chemical wastewater, and seawater, respectively, while maintaining satisfied stability with minimal activity loss after 40,000 cycles. In situ experiments and theoretical calculations reveal that the ultra-low coordination of Pt, Ru, and Ni atoms creates numerous dangling bonds, which lower the water dissociation barrier and optimizing hydrogen adsorption. This research marks a notable advancement in the precise engineering of atomically dispersed multi-metallic centers in catalysts for energy-related applications.
Similar content being viewed by others


Introduction
Hydrogen, acclaimed as a pristine energy vector, has the potential to radically transform global energy paradigms by providing a zero-emission alternative to conventional fossil fuels1,2,3. Concurrently, the utilization of renewable energy sources to power electrochemical water splitting offers a sustainable and environmentally friendly method for effective hydrogen production4,5. Given the scarcity of pure water resources, utilizing chemically contaminated wastewater presents a viable alternative3,4. When deploying these catalysts in real wastewater environments, the complex composition of wastewater introduces additional challenges. Variations in electrolytes and the presence of harmful substances can expressively impact the stability and activity of the catalysts6. Consequently, the development of catalyst materials that can operate stably across diverse electrolytic environments, particularly those capable of resisting various interfering factors present in wastewater, has become a pivotal area of research7,8. Furthermore, ruthenium (Ru), one of the most affordable noble metals, and its compounds exhibit a remarkable capability in facilitating the water dissociation step9,10. This makes them excellent supports for platinum (Pt) atoms, enabling the full exploitation of their catalytic potential for the HER11,12. Nonetheless, a major limitation of Ru is its susceptibility to oxidation under actual water splitting conditions, resulting in a swift decline in catalyst activity due to rapid deactivation during the electrochemical process13,14. Additionally, the passivation behavior of Pt(Ru) under operational conditions remains unclear, thereby hindering the systematic development of stable Pt(Ru)-based materials optimized for HER performance in real-world applications.
Cavity nanocages featuring twinned, distortion-formed multi-metal-oxide interfaces present unique lattice and surface characteristics—such as steps, defects, and dangling bonds—that enhance catalytic reactions by providing numerous active sites and elevated surface energy15,16,17,18,19. Meanwhile, these polymetallic nanocages structures with twin-distortion-modulated interfaces and ultra-low coordination enhance surface free energy, which substantially improves the adsorption and activation of critical intermediate on the nanocage matrix16,20. However, the rational synthesis of Pt(Ru)-based cavity nanocages is difficult due to its lattice mismatch with other metals, such as nickel (Ni)21. To this end, it has been demonstrated that encapsulating metallic alloys within metal oxides can stabilize ultra-low coordination interfaces for the HER, thereby maintaining a stable current density during real-world operational tests22. Such catalysts should not only enhance interactions with substrate oxides and improve hydrolytic dissociation but also boost catalytic stability and activity23. Despite the formidable challenges involved, advancing Ru-based catalysts with accurately defined local structures and environments is essential for improving hydrogen production under actual working conditions.
Aiming at this issue, we develop PtRuNi alloy-oxide cavity nanocages with twin boundary distortion structures (referred to as PtRuNi-Ox) through a straightforward solid-liquid synthesis method. Employing transmission electron microscopy (TEM) and synchrotron X-ray absorption spectroscopy (XAS), we observe that the polymetallic-metal oxide interfaces form metal atoms with ultra-low coordination numbers, resulting in numerous active sites due to dangling bonds. The unique structure of the alloy-oxide nanocages regulated by mixed oxidation states also provides an efficient channel for electron transport. Twin boundary distortion induces stress and strain effects, notably increasing defects at the interface and modulating the electronic configuration of the catalyst surface, thereby enhancing catalytic efficiency. The PtRuNi-Ox catalyst exhibits outstanding HER activity in alkaline freshwater, delivering the current density of 10 mA cm−2 at a low overpotential of 19.6 mV, and maintains excellent stability after 40,000 cycles of cyclic voltammetry (CV), surpassing commercial Pt/C. Additionally, to reach 10 mA cm−2 in alkaline chemical wastewater and seawater electrolytes, PtRuNi-Ox requires only 20.9 and 21.0 mV overpotentials, respectively, demonstrating its potential in real wastewater treatment and environmental protection.
Results
Materials synthesis and characterization
A straightforward atmospheric solvothermal method was employed to regulate the reducing agent at different rates, promoting metal ion reduction and facilitating alloy formation, as illustrated in Fig. 1a. The resulting PtRuNi-Ox hollow nanocages possess a high-surface-area open framework that provides an abundance of active sites for catalytic reactions24. Comparative catalysts (PtNi-Ox, RuNi-Ox, and PtRu-Ox) prepared using similar synthetic methods also possess similar structures (Supplementary Figs. 1–3), except the morphology of Pt-Ru binary catalysts obtained was distinctly different from the small nanoparticles typical of other hollow structure catalysts investigated in this system, indicating that the atomic size and chemical properties of Ni likely contribute to its role in supporting and stabilizing these nanocages. The hollow cavity structure of these nanocages is evident in high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images, shown in Fig. 1b and Supplementary Fig. 4. A more distinct visualization of the hexagonal facet structure of the nanocages is provided by transmission electron microscopy (TEM) in Fig. 1c and S4, and further detailed through aberration-corrected scanning TEM (AC-STEM) insets of Fig. 1c. Meanwhile, numerous twin boundaries within the nanocage framework are identified in Fig. 1d and Supplementary Fig. 5, likely resulting from synthesis variations or lattice mismatches24. These twin boundaries likely result from synthesis variations and lattice mismatches during crystal growth25. Higher synthesis temperatures (e.g., 300 °C) lead to more stable twin boundaries by increasing atomic mobility, allowing atoms to rearrange into stable structures. In contrast, lower temperatures (e.g., 200 °C) cause irregular and unstable boundaries due to limited atomic movement (Supplementary Fig. 6). Reaction time also affects boundary stability, with shorter times leading to more defects and unstable boundaries due to inadequate atomic rearrangement (Supplementary Fig. 7). Twin boundary analysis, presented in Fig. 1e, reveals an ordered arrangement of Pt, Ru, and Ni atoms within the nanocage framework, suggesting a synergistic interaction among these elements, that contributes to a stable structure and positive catalytic performance. During the synthesis of the heteroepitaxial structure composed of different metal atoms, lattice mismatch leads to an accumulation of strain energy, this mismatch-induced localized stress concentration facilitates the formation of twin boundaries, thereby enhancing the stability and performance of the nanocage framework26,27.

a Schematic diagram of the solvothermal synthesis method of PtRuNi-Ox catalyst. b HAADF-STEM, c TEM images of PtRuNi-Ox, and magnified HAADF-STEM images of a typical nanocage in the inset. d, e Atomic phase images of PtRuNi-Ox, FFT patterns generated from e in the inset. f HRTEM images of PtRuNi-Ox shows the lattice fringes with interface sign. g Magnified HAADF-STEM images of the interface framework and the corresponding atomic strength profile analysis, and the red dashed circles represent atomic defects at the boundary. h EDS mapping images of PtRuNi-Ox.
High-resolution TEM images (HRTEM) of PtRuNi-Ox (Fig. 1f) reveal lattice spacings of 0.17, 0.21, 0.22, and 0.25 nm, corresponding to the RuO2 (2 2 0), RuO2 (2 1 0), RuO2 (0 1 1), and Pt(Ru) (1 1 1) facets, respectively28,29. These observations further confirm the formation of the PtRuNi alloy–oxide interface in PtRuNi-Ox. Further investigations into atomic vacancies, illustrated in Fig. 1g, reveal obvious vacancies within the twin nanocage framework and surface structure, primarily attributed to stress and strain effects induced by twin boundary deformation30. This observation supports the existence of a low-coordination state for the metal atoms (Pt, Ru, and Ni), wherein the reduction in coordination number exposes more metal atoms at surface positions, thereby enhancing catalyst activity and stability30. Elemental distribution analysis, as demonstrated in Fig. 1h and Supplementary Fig. 8, confirms that the nanocage framework predominantly comprises Pt, Ru, and Ni metals, while the surface is largely made up of their oxides, further validated by Inductively Coupled Plasma-Atomic Emission Spectrometry (ICP-OES) tests (Supplementary Table 1). All these findings confirm the formation of alloy–oxide interface in PtRuNi-Ox, which provides additional active sites for producing hydrogen.
A range of spectroscopic analyses was carried out to examine the structural and electronic properties of PtRuNi-Ox, emphasizing the beneficial twin cavity structure. X-ray diffraction (XRD) confirms the complex structure arising from the combination of PtRuNi alloy and oxides (Supplementary Fig. 9). The peaks at 40.3°, 46.8°, and 68.4° correspond to the (111), (200), and (220) planes of Pt (#87-647), respectively. These peaks highlight the ordered structure facilitated by the twin cavities, which enhance catalytic activity due to optimal atomic arrangement and exposure2. In addition, small peaks related to RuO2 (#65-2824) and PtNi (#65-2797) can be observed20. The XPS spectrum of Pt in the 4f region (Supplementary Fig. 10) displays two groups of peaks, indicating the presence of Pt(0) (71.8 and 75.1 eV) and Pt(II) (72.5 and 75.7 eV). The 4f7/2 and 4f5/2 peaks represent different spin–orbit splitting states of Pt atoms, with 4f7/2 corresponding to a spin state of 1/2 and 4f5/2 to 3/2. Variations in the intensity ratio (I4f7/2/I4f5/2) can indicate changes in the oxidation state of Pt or its interactions with the environment31. Notably, the Pt 4f binding energy in PtRuNi-Ox shifts negatively by 0.2 eV compared to that of PtNi, suggesting complex electronic interactions within the twin cavities that promote effective electron transfer among Pt, Ni, and Ru, thereby enhancing the catalytic efficiency26. In the Ni 2p spectrum (Supplementary Fig. 11), the peaks at 851.4, 855.4, and 856.6 eV indicate the presence of Ni(0), Ni(II), and Ni(III), respectively32. This multivalent nature allows Ni to flexibly participate in different steps of the HER, thereby facilitating the adsorption and dissociation of reaction intermediates33. Supplementary Fig. 12 shows peaks at 461.6 and 483.9 eV in the Ru 3p region attributed to Ru(0), while peaks at 463.7 and 486.0 eV are attributed to Ru(IV)20. The Pt 4f, Ni 2p, and Ru 3p spectra further detail the mixed valence states contributing to the alloy-oxide environment within PtRuNi-Ox, which can provide more electronic states and energy levels, potentially increasing electron mobility. This enhanced electron mobility can markedly improve the overall catalytic efficiency, as more effective electron transfer accelerates the formation and conversion of reaction intermediates, thereby increasing the reaction rate34.
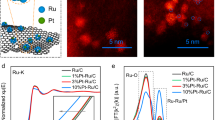
Subsequent investigations into the chemical states and local atomic configurations were performed using X-ray absorption spectroscopy (XAS) powered by synchrotron radiation. The Ru K-edge XANES data (Fig. 2a) indicate that the absorption edge of PtRuNi-Ox is positioned between those of Ru foil and RuO2, confirming the oxidation state (Ruδ+) of Ru in PtRuNi-Ox. The Fourier-transformed k2-weighted EXAFS spectra, depicted in Fig. 2b, identify two distinct chemical bonds within PtRuNi-Ox, with major peaks at approximately 2.0 and 2.8 Å illustrating the formation of Ru–O and Ru–Ru bonds, respectively. Furthermore, model-based EXAFS fitting was applied to PtRuNi-Ox. As shown in Fig. 2c, Ru exhibits an oxidation peak between 1 and 2 Å and a metal bond between 2 and 3 Å, indicating that isolated Ru atoms in PtRuNi-Ox are coordinated by adjacent O atoms from oxide domains and neighboring Ru(Ni) atoms from alloy areas. Consequently, detailed AC-STEM and EXAFS analyses reveal that Ru entities are uniformly distributed on the PtRuNi-Ox interfaces. In the case of Pt, the Pt L3-edge in PtRuNi-Ox closely resembles that of Pt foil (Fig. 2d). The Pt L3-edge FT-EXAFS spectrum shows a primary radial distance of around 2.7 Å, corresponding to the Pt–M (M = Pt/Ru/Ni) bond, while a peak at ~2.0 Å is attributed to Pt–O bonding29. Meanwhile, Fig. 2e and f indicate that Pt–O has about one coordination, with Pt–Ni and Pt–Ru forming the main coordination, consistent with the near-edge structure35. Similarly, the Ni K-edge in PtRuNi-Ox lies between those of Ni foil and NiO, verifying the valence state of Ni ranging from 0 to +2 (Fig. 2g). The Ni K-edge FT-EXAFS spectra of PtRuNi-Ox (Fig. 2h) corroborate this conclusion with fitting results (Fig. 2i), indicating that Ni not only bonds with oxygen (Ni–O) but also forms stable alloy structures with Pt or Ru (Ni–Pt or Ni–Ru). This alloying enhances the electron transfer capability of the catalyst, creating synergistic active sites that reduce the overpotential of the HER32.

Normalized XANES spectra for (a) Ru K-edge, d Pt L3-edge, and g Ni K-edge of PtRuNi-Ox. FT-EXAFS spectra for (b) Ru, e Pt, and h Ni of PtRuNi-Ox, and EXAFS R-space fittings for (c) Ru, f Pt, and i Ni of PtRuNi-Ox, R in x-axis represents radial distance.
To better distinguish the bonding information and atomic distribution of PtRuNi-Ox, wavelet transform (WT) analysis of k3-weighted EXAFS spectra was conducted (Supplementary Fig. 13). PtRuNi-Ox clearly shows Pt–M (M = Pt/Ru/Ni) and M (M = Pt/Ru/Ni)–O bonds. Quantitative FT-EXAFS fitting (Supplementary Figs. 14, 15 and Table 2) reveals that the average coordination numbers (CNs) for Pt–Ni and Pt–Ru are approximately 4.5 and 1.7, respectively, lower than those observed in Pt foil (12). Similarly, the EXAFS parameter at the Ru K-edge and Ni K-edge presents lower average CNs for Ru–Ni (~1) and Ru–Pt (~1.6), and Ni–Pt (~3.9) compared to those in Ru foil (12) and Ni foil (12). These findings suggest that the introduction of multiple twin interfaces effectively reduces the average CN36. Specifically, the ultra-low unsaturated coordination of Pt, Ru, and Ni atoms generates extensive dangling bonds within the hollow nanocage structure, thereby increasing adsorption and reaction capabilities, markedly impacting the catalytic reactivity.
High HER performance of PtRuNi-Ox
The HER performance of PtRuNi-Ox was evaluated in an N2-saturated 1.0 M KOH solution. For comparison, under the same experimental conditions, PtNi-Ox, RuNi-Ox, and commercial Pt/C were used as references. As shown in Fig. 3a, PtRuNi-Ox exhibits an overpotential of only 19.6 ± 0.1 mV to reach 10 mA cm−2, which is notably lower than that of commercial Pt/C (23.0 ± 0.1 mV), PtNi-Ox (28.3 ± 0.4 mV), and RuNi-Ox (31.8 ± 0.1 mV). Remarkably, PtRuNi-Ox maintains a relatively low overpotential even at comparatively high current densities (η100 = 40.1 ± 0.3 mV, Supplementary Fig. 16). To analyze the catalytic kinetics of HER, Tafel plots were generated based on HER polarization curves (Fig. 3b)20, detailed data are listed in Supplementary Table 3. The results reveal that PtRuNi-Ox has a much lower Tafel slope (15.0 ± 1.0 mV dec−1) than others (Supplementary Figs. 17 and 18), which indicates the fastest HER kinetics among the catalysts tested. Moreover, the HER performance metrics of PtRuNi-Ox, including η10, exchange current density (j0), mass activity, and turnover frequency (TOF) at an overpotential of 50 mV, were systematically compared with those of other catalysts (Supplementary Fig. 19, the equations refer to Supplementary Note 3). PtRuNi-Ox demonstrates a mass activity of 4.91 ± 0.5 A mg−1 at η = 50 mV, which is remarkably higher than PtNi-Ox (0.82 ± 0.2 A mg−1), RuNi-Ox (0.28 A ± 0.1 mg−1), and even commercial Pt/C (0.48 ± 0.1 A mg−1). Additionally, the TOF of PtRuNi-Ox at η = 50 mV is 2.59 ± 0.1 s−1, far exceeding that of PtNi-Ox (0.52 ± 0.1 s−1) and RuNi-Ox (0.25 s−1). These results confirm that PtRuNi-Ox possesses superior intrinsic activity compared to the other catalysts.

a Electrochemical polarization curves with iR compensation (The electrode working area is π × 0.15 × 0.15 cm2, and the solution resistances used for compensation are shown in Supplementary Fig. 21), b performance parameter comparisons (the standard deviations were obtained through three reduplicative measurements in Supplementary Figs. 16, 17 and 19), c the cyclic polarization curves for PtRuNi-Ox and Pt/C in alkaline freshwater. d Nyquist plots of the PtRuNi-Ox, PtNi-Ox, RuNi-Ox, and Ni catalysts in alkaline freshwater, respectively. e Electrochemical polarization curves in alkaline chemical wastewater with iR compensation (The solution resistance used for compensation is shown in Supplementary Fig. 21). f Comparison of η10 and mass activity of as-prepared catalysts in alkaline chemical wastewater. g The cyclic polarization curves for PtRuNi-Ox and Pt/C in alkaline chemical wastewater. h The performance polarization curves of PtRuNi-Ox in various alkaline electrolytes. i Polarization curves were measured with two electrode systems.
Besides outstanding HER activity, PtRuNi-Ox also exhibits higher stability than Pt/C and excellent long-term stability. The continuous chronoamperometry test results (Fig. 3c) show that PtRuNi-Ox does not exhibit any obvious degradation while operating at 10 mA cm−2 for 90 h (Supplementary Fig. 20). Moreover, a positive shift of only 2 mV in η10 can be observed after 40,000 sweeping cycles. As illustrated in Supplementary Table 4, PtRuNi-Ox outperforms the other catalysts across all performance metrics. This underscores the critical role of low coordination in enhancing HER performance, positioning PtRuNi-Ox as one of the most exceptional Ru-based electrocatalysts for alkaline HER reported to date. Nyquist plots (Fig. 3d) and the corresponding fitted equivalent circuit diagram (Supplementary Fig. 22), recorded at an overpotential of 50 mV, reveal that the semicircle associated with the PtRuNi-Ox system is obviously smaller compared to those of PtNi-Ox and RuNi-Ox. This indicates that PtRuNi-Ox owns the lowest resistance. Moreover, the high-frequency region is indicative of the electronic conductivity within the inner layer or at the interface of the catalyst, while the low-frequency region corresponds to charge transfer processes occurring at the interface between the electrolyte and the catalyst14. The double-layer capacitance (Cdl) measurements confirm that PtRuNi-Ox possesses a larger active surface area (Supplementary Fig. 23). Additionally, to more accurately describe the electrochemical active surface area (ECSA) involved in catalysis by noble metals (the equation refers to Supplementary Note 4), hydrogen underpotential deposition (Hupd) peaks were captured37. As shown in Supplementary Fig. 24, the shaded regions highlight the adsorption and desorption processes of H. By comparing the areas of shaded regions, it can be confirmed that PtRuNi-Ox possesses a larger ECSA, consistent with Cdl measurement results, confirming that Ni with Pt or Ru may also provide additional surface active sites, which lower the adsorption energy barrier, further enhancing the reaction efficiency.
The substantial amount of water required for large-scale hydrogen production poses a notable challenge due to freshwater scarcity. Direct hydrogen production from real chemical wastewater presents a promising strategy to mitigate this issue4,6. To gain further insight into hydrogen production in real wastewater, we analyzed the HER performance of the prepared catalysts under chemical wastewater conditions. It typically exhibits a slightly alkaline pH for wastewater. Consequently, HER data were measured under alkaline conditions to better simulate the actual conditions of wastewater treatment. As illustrated in Fig. 3e, PtRuNi-Ox demonstrates an overpotential of 20.9 ± 0.1 mV at 10 mA cm−2 in 1.0 M KOH chemical wastewater. This value is markedly lower than those of commercial Pt/C (26.2 ± 0.1 mV), PtNi-Ox (32.3 ± 0.2 mV), and RuNi-Ox (33.4 ± 0.3 mV) (Supplementary Fig. 25). A similar trend is maintained even at a current density of 100 mA cm−2 (ηPtRuNi-Ox < ηPtNi-Ox < ηPt/C). Additionally, PtRuNi-Ox exhibits the lowest Tafel slope indicating the fastest HER kinetics.
More importantly, PtRuNi-Ox shows a mass activity of 1.85 ± 0.2 A mg−1 at an overpotential of 50 mV in chemical wastewater, as depicted in Fig. 3f. Supplementary Figs. 26 and 27 further demonstrates that PtRuNi-Ox exhibits the lower charge transfer resistance and larger active area in the wastewater environment, confirming its superior electrochemical performance. Durability tests were conducted using chronopotentiometry in 1.0 M KOH chemical wastewater at a fixed current density of 10 mA cm−2 without iR compensation. As shown in Fig. 3g and Supplementary Fig. 28, PtRuNi-Ox maintains a stable current density with only a 4 mV positive shift in η10 after 15,000 sweeping cycles, highlighting its excellent long-term stability.
To evaluate the compatibility of PtRuNi-Ox in various electrolytes, we expanded the HER performance testing to include different water qualities. Considering the organic composition of the chemical wastewater and prioritizing safety and controllability in the experimental process, methanol and ethanol were chosen as potential electrolytes. Figure 3h shows polarization curves measured in alkaline electrolytes of freshwater, seawater, and wastewater containing various alcohols. The activity sequence of PtRuNi-Ox at 10 mA cm−2 is as follows: ηfreshwater (19.6 mV) < ηwastewater (20.9 mV) < ηseawater (21.0 mV) < ηMeOH (23.0 mV) < ηEtOH (26.0 mV). Overall, the performance of chemical wastewater in electrolysis is inferior to that of freshwater. This disparity is primarily attributable to the presence of impurities in chemical wastewater, such as organic compounds and heavy metals, which are adsorbed on the catalyst surface. This adsorption obstructs active sites and diminishes catalytic activity, thereby increasing the electron transfer resistance and reducing the overall efficiency of the electrolysis process37. However, comparative studies have demonstrated that the PtRuNi-Ox catalyst owns remarkable resilience against these contaminants, retaining its catalytic activity even in polluted environments. The unique composition of this catalyst facilitates the selective HER activity within the complex matrices of wastewater, thereby ensuring effective hydrogen production. To further demonstrate the suitability of PtRuNi-Ox for simultaneous hydrogen evolution in alkaline chemical wastewater, we conducted a test using a two-electrode system with PtRuNi-Ox and RuO2 as cathodes and anodes, respectively, immersed in an alkaline chemical wastewater electrolyte solution. As shown in Fig. 3i, PtRuNi-Ox exhibits a notably lower potential of 1.54 V at 10 mA cm−2 compared to 1.55 V for Pt/C‖RuO2 in 1.0 M KOH solution, indicating its superior electrocatalytic activity.
Analysis of structure–activity relationships
In the process of catalyst design, the d-band center is a critical parameter that influences the binding strength of intermediates on the catalyst surface. A more negative d-band center generally indicates weaker binding with intermediates (*OH)38,39. Therefore, theoretical density functional theory (DFT) calculations were employed to analyze the d-band centers and the binding energies of *OH across various catalysts (Supplementary Table 5). As shown in the table, PtRuNi-Ox exhibits the most negative d-band center, indicating the weakest binding with *OH. Experimental results validated our computational predictions, confirming that PtRuNi-Ox demonstrates superior catalytic activity due to its optimal d-band center. The weaker OH binding on PtRuNi-Ox ensures efficient OH desorption following H2O adsorption, thereby promoting a continuous and efficient HER process. Achieving a balance between the adsorption and desorption of intermediates is crucial for optimizing catalytic performance under alkaline conditions.
In the alkaline HER process, the adsorption behavior of OH is closely related to hydrogen spillover40. An increase in *OH adsorption on the catalyst surface can enhance water dissociation, thereby promoting hydrogen generation41. However, this adsorption behavior maintains a dynamic balance, where hydrogen spillover regulates the OH adsorption distribution, preventing excessive accumulation at specific sites and ensuring uniform surface reactions. To demonstrate potential atomic-scale hydrogen spillover in PtRuNi-Ox during alkaline HER electrocatalysis, in situ Raman experiments and electrochemical assessments were conducted, as illustrated in Fig. 4a–d. The peak at ~700–800 cm−1 in the test results originates from the surface-adsorbed OH intermediate (Fig. 4b)42. In the alkaline HER process, direct spectral evidence is provided by the acquisition of *OH adsorbed on the surface of the catalyst. Combined with XPS analysis, it is evident that the high-valence metal states in the PtRuNi-Ox structure have milder adsorption energies towards H2O, H, and OH during the interface reaction process43.

a Schematic diagram of in-situ Raman device. b The in-situ Raman spectra of PtRuNi-Ox during alkaline HER. c Bode plots and d the fitting Rct values of PtRuNi-Ox during HER in alkaline freshwater, respectively. e Bode plots and f the fitting Rct values of PtRuNi-Ox during HER in alkaline chemical wastewater, respectively. g Charge density variation in the PtRuNi alloy and oxide heterostructure model, where blue and yellow areas represent regions of electron accumulation and depletion, respectively. Calculated Gibbs free energy of (h) H* adsorption and i OH* adsorption at PtRuNi-Ox surface and interface site, respectively. j Schematic diagram of HER at PtRuNi-Ox interfaces.
According to principles derived from Faradaic capacitance, the oxidation reaction predominantly generates capacitance through *OH adsorption44. This is corroborated by data showing that the PtRuNi-Ox catalyst displays higher capacitance values, as illustrated in Supplementary Fig. 29, which correlates with enhanced *OH adsorption during the reaction. The presence of adsorbed OH appears to facilitate water dissociation at the catalyst interface, thus boosting catalytic activity45. Moreover, the gradual increase in the weak δPt-OH peak at ~1150 cm−1 with voltage variations provides additional evidence of increased *OH adsorption during the HER process46. The distinct structure of the twin multi-interface in PtRuNi-Ox optimizes the adsorption of active metal species onto *H and *OH intermediates, promoting both the Volmer step and efficient OH desorption, which are essential for catalysis in alkaline media. Additionally, the abundance of low-coordination sites within the PtRuNi-Ox structure, characterized by their unsaturated chemical bonds and unique electronic structures, effectively facilitates the adsorption and dissociation of H2O. These low-coordination sites enhance the adsorption and binding of hydrogen atoms, thereby substantially improving the efficiency of the catalytic reactions47.
Ion aggregation and electron transfer dynamics at the electrode–electrolyte interface during the reaction were analyzed using in-situ electrochemical impedance spectroscopy (EIS)48. Bode plots (Supplementary Fig. 29) show a decrease in phase angles at high frequencies under identical voltage conditions in both freshwater and chemical wastewater, suggesting enhanced electron transfer rates on PtRuNi-Ox surfaces, as a key factor governing reaction resistance during HER49. The shift towards higher frequencies and a decreasing phase angle with increasing voltage, as depicted in Fig. 4c and e, further signify accelerated charge transfer. To fully reflect the impedance characteristics of the samples, frequency variation curves of the impedance modulus (|Z|) and phase angle are in all Bode plots, providing an intuitive display of impedance and phase characteristics at different frequencies50. Supplementary Figs. 30 and 31 exhibit the behavior of the RuNi-Ox, PtNi-Ox, and PtRuNi-Ox catalysts, that in alkaline freshwater, both the impedance modulus and phase angle possess a consistent trend frequency, indicating stable electrochemical behavior. In contrast, the complexity and impurities in chemical wastewater obviously affect the impedance of the catalyst in the high-frequency region, leading to pronounced variations in impedance modulus and phase angle. Furthermore, the fitted Rct results in different electrolytes are given in Fig. 4d and f, which show that PtRuNi-Ox possesses a faster charge transfer rate than RuNi-Ox and PtNi-Ox. To further explore this phenomenon, the HER performance of the PtRuNi alloy and PtRuNi-Ox (Supplementary Fig. 32) was compared, and the results indicated that the HER performance of the PtRuNi alloy was inferior to that of PtRuNi-Ox. This inferiority is due to the electronic interactions among Pt, Ru, and Ni atoms in the PtRuNi alloy through alloying effects, leading to a redistribution of electron density, and thus affecting catalytic activity51. This comparison underscores the significance of the distinct electronic interactions within the twin cavities of PtRuNi-Ox for optimizing electron transfer, consequently enhancing the catalytic performance.
Finally, the outstanding alkaline HER activity of PtRuNi-Ox was elucidated through DFT calculations19,49. Atomic models of PtRuNi-Ox, shown in Fig. 4g and Supplementary Fig. 33 (calculate details in Supplementary Note 1 and Specific atomic coordinates refer to Supporting data 1), demonstrate the notable electron redistribution due to varying electronegativities, primarily from Ru to central Pt atoms, confirming insights from XPS measurements. Bader charge analysis reveals higher electron concentrations at interfacial metal sites, promoting an advantageous redistribution at the d-band centers of the metals and refining reactant adsorption energies52,53. The calculated adsorption-free energies for the intermediate H species indicate thermodynamically favorable conditions for H adsorption at the interface edge (Fig. 4h and i). This site specificity enhances the adsorption of OH, thus facilitating the water dissociation in alkaline media and positively influencing catalytic activity54. The detailed reaction mechanism, illustrated in Fig. 4j, underscores the pivotal role of the alloy-oxide interface in tuning *H and *OH adsorption behaviors, thereby elevating the HER performance of the PtRuNi-Ox catalyst.
Discussion
In summary, PtRuNi-Ox was designed as a nanocage catalyst with an ultra-low coordination cavity structure. This intricate alloy framework featured a metal oxide cage surface, specifically engineered to include abundant twin interfaces and atomic vacancies. Experimental investigations revealed that PtRuNi-Ox had superior HER activity and durability in 1.0 M KOH electrolytes, outperforming Pt/C and most previously reported catalysts in alkaline electrolytes, including those derived from freshwater, seawater, and chemical wastewater containing various alcohols. Further insights were gained through in-situ Raman spectroscopy and electrochemical impedance spectroscopy (EIS) measurements, which confirmed that the ultra-low coordination cavity structure of PtRuNi-Ox facilitated the effective electron transfer within the inner-layer/interface. This enhanced electron mobility notably improved the HER efficiency by optimizing the adsorption of H and OH intermediates and accelerating the reaction kinetics. Overall, the findings from this study are not only pivotal for advancing the understanding of catalytic mechanisms in alkaline environments but also emphasize the importance of structural innovation in enhancing the functionality and application scope of catalytic materials.
Methods
Chemicals
Commercial Pt/C catalyst (20 wt%) was purchased from Johnson Matthey (UK). Nickel (II) acetylacetonate (Ni(acac)2, AR) was obtained from Aladdin (China). Ruthenium (III) chloride hydrate (RuCl3·3H2O, AR), Potassium chloroplatinate (K2PtCl6, AR), and KOH (85 wt%) were obtained from Sinopharm Chemical Reagent Co. Ltd. (China). Dodecylamine (DDA, AR), and octadecene (ODE, AR) were purchased from Alfa Aesar. Nafion solution (5 wt%) was purchased from Adamas-beta Chemical Co. (Switzerland). Milli-Q deionized water (DI water, 18.25 MΩ cm) was employed in all experiments. All reagents were used as received without further purification. The working glass carbon electrode, reference electrode, and graphite electrode used in electrochemical tests are all from Tianjin Ada Hengsheng Technology Co., Ltd.
Synthesis of PtRuNi-Ox
Ni(acac)2 (0.5 mM), RuCl3·3H2O (0.25 mM), and potassium chloroplatinate (K2PtCl6) (0.25 mM) as the metal precursors, then 6 mL DDA was used as a reducing agent and 10 mL ODE as a solvent and surfactant. The mixture was added into the three-necked flask (250 mL), then heated from room temperature to 300 °C at 7 °C/min and maintained at 300 °C for 20 min. After the reaction was cooled to room temperature, the resulting black product was separated by centrifugation and washed several times with heptane. The same synthetic method for PtNi-Ox except without RuCl3·3H2O, RuNi-Ox except without K2PtCl6, and Ni except without RuCl3·3H2O and K2PtCl6.
Materials characterization
The atomic structures of the PtRuNi-Ox were examined using a JEOL ARM 200CF transmission electron microscope at 200 kV, equipped with double spherical aberration correctors. HAADF images were taken at an acceptance angle of 90–370 mrad. The STEM used a DCOR+ spherical aberration corrector for the electron probe, which was aligned with a standard gold sample before observations. The mass fraction of the metal was determined using ICP-OES technology on a Thermo Fisher iCAP PRO instrument. Powder X-ray diffraction (XRD) patterns were recorded using graphite monochromatized Cu K radiation (λ = 0.154060 nm) and a D/max 2500VL/PC diffractometer (Japan) over a scan range of 5–90° in 2θ. X-ray photoelectron spectra (XPS) were obtained using an Al K radiation-powered scanning X-ray microprobe (PHI 5000 Versa, ULACPHI, Inc.), with binding energies of other elements calibrated to the C1s peak at 284.6 eV. The Athena and Artemis programs of the Demeter data analysis packages, using FEFF6 software, were utilized for EXAFS data reduction, analysis, and fitting. Performing chronoamperometry tests using the CHI 760E workstation, with in situ data collected in conjunction with Raman (Thermo Fisher DXR2). Operation E1S data were measured by Metrohm Autolab PGSTAT302N. All electrochemical measurements were performed by an electrochemical workstation (CHl 760E, CH Instruments Ins, Shanghai, China).
Electrochemical measurements
Conducted at room temperature (~25 °C) on a CHI 760E electrochemical workstation. A 3 mm diameter (0.07 cm2 area) L-type glassy carbon electrode (GCE) coated with catalysts, a Hg/HgO reference electrode, and a graphite rod counter electrode were used. The alkaline freshwater electrolyte was freshly prepared within a week by dissolving 66 g of KOH in 1000 mL of deionized water. The preparation of the chemical wastewater electrolyte involves allowing the wastewater directly sourced from the plant to settle for 24 h. After settling, the clear liquid is filtered using filter paper. The filtered liquid is then diluted fifty times. Subsequently, 66 g of KOH is dissolved into 1000 mL diluted wastewater. The prepared electrolyte is stored in a sealed volumetric flask and kept in a cabinet to protect it from light. The pH of alkaline freshwater and wastewater electrolytes is shown in Supplementary Fig. 34.
The working electrode was prepared by dispersing 5 mg of catalysts and 10 μL of 5 wt% Nafion solution in 1 mL of a mixed solvent (600 μL ethanol and 390 μL water), followed by 30 min of ultrasonication to form a dispersion. A specified volume of catalyst ink was dropped onto clean carbon paper and air-dried. Prior to electrochemical testing, the GCE was polished with Al2O3 slurry and rinsed with Milli-Q water to ensure a clean surface. 8.4 μL of the catalyst solution was applied to the clean GCE. The load capacity is calculated by the following formula (1):
Here c is the concentration of the sample solution, V is the volume of the sample solution dropped on the electrode, and SGCE is the area of the glassy carbon electrode.
N2-saturated 1.0 M KOH was used as the electrolyte for HER. The electrochemical tests were conducted in a 100 mL electrolytic cell, with the electrolyte volume ranging from 50 to 60 mL. The distance between the three electrodes was equal (Supplementary Fig. 35). The chemical wastewater used in this study was sourced from an actual chemical manufacturing facility, Jiangsu Zhongshan New Material Co., LTD. The sampling location was the outlet of the wastewater collection pool (Supplementary Fig. 36 and Table 6).
Polarization curves were recorded at a scan rate of 5 mV s−1 with 100% iR-correction. The stability measurements in this study were conducted using CV methods. For the CV tests, the scan rate was set at 0.1 V/s, with a potential range from 0 to −0.1 V (vs. RHE).
The Hg/HgO electrodes used in the experiment were calibrated, and the specific calibration method is shown in Supplementary Note 2. Test the LSV curve in the range of −1 to 1 V. The voltage value is read at a current of 0 and compared with the standard potential of Hg/HgO (0.1989 V). An error within 2 mV is considered acceptable.
All the electrochemical measurements were converted to the reversible hydrogen electrode (RHE) by the formula (2):
The solution resistance is corrected using the following formula (3):
where i is the measured current, and the uncompensated resistance (R) was determined using EIS, and the R value for freshwater electrolyte is 7.5 Ω and the R value for chemical wastewater electrolyte is 7.9 ± 0.1 Ω. The HER polarization curves without iR compensation are presented in Supplementary Fig. 37.
In situ Raman
In situ Raman spectra were obtained using a Raman spectrometer (Thermo Fisher DXR2) with excitation from a 532 nm argon ion laser. PtRuNi-Ox served as the working electrode, Hg/HgO as the reference electrode, and a platinum wire as the counter electrode, which was separated by an anion exchange membrane (Fumasep FAA-3-50). During HER measurements in 1.0 M KOH, Raman spectra were recorded at various potentials via chronoamperometry on a Chenhua electrochemical workstation. Detailed test conditions are presented in Supplementary Fig. 38.
Data availability
The data that support the plots are available within this paper and its Supplementary Information. All other relevant data that support the findings of this study are available from the corresponding authors on reasonable request. Source data are provided with this paper.
Change history
09 January 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-55867-1
References
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294–303 (2012).
Gao, X. et al. Ultra-efficient and cost-effective platinum nanomembrane electrocatalyst for sustainable hydrogen production. Nano-Micro Lett. 16, 108 (2024).
Xiao, X., Li, Z., Xiong, Y. & Yang, Y.-W. IrMo nanocluster-doped porous carbon electrocatalysts derived from cucurbit[6]uril boost efficient alkaline hydrogen evolution. J. Am. Chem. Soc. 145, 16548–16556 (2023).
Huang, Y. et al. Nitrogen-doped porous molybdenum carbide and phosphide hybrids on a carbon matrix as highly effective electrocatalysts for the hydrogen evolution reaction. Adv. Energy Mater. 8, 1701601 (2018).
Yang, J., Zhu, C., Li, W.-H., Zheng, X. & Wang, D. Organocatalyst supported by a single-atom support accelerates both electrodes used in the chlor-alkali industry via modification of non-covalent interactions. Angew. Chem. Int. Ed. 63, e202314382 (2024).
Luo, Y. et al. High-loading as single-atom catalysts harvested from wastewater towards efficient and sustainable oxygen reduction. Energy Environ. Sci. 17, 123–133 (2024).
Liu, J.-J. et al. Achieving high-efficient photoelectrocatalytic degradation of 4-chlorophenol via functional reformation of titanium-oxo clusters. J. Am. Chem. Soc. 145, 6112–6122 (2023).
Xu, X. et al. Corrosion-resistant cobalt phosphide electrocatalysts for salinity tolerance hydrogen evolution. Nat. Commun. 14, 7708 (2023).
Yao, R. et al. Stable hydrogen evolution reaction at high current densities via designing the Ni single atoms and Ru nanoparticles linked by carbon bridges. Nat. Commun. 15, 2218 (2024).
Mu, X. Q. et al. Symmetry-broken Ru nanoparticles with parasitic Ru–Co dual-single atoms overcome the volmer step of alkaline hydrogen oxidation. Angew. Chem. Int. Ed. 63, e202319618 (2024).
Liu, R. et al. Enhanced metal-support interactions boost the electrocatalytic water splitting of supported ruthenium nanoparticles on a Ni3N/NiO heterojunction at industrial current density. Angew. Chem. Int. Ed. 62, e202312644 (2023).
Zhu, S. et al. The role of ruthenium in improving the kinetics of hydrogen oxidation and evolution reactions of platinum. Nat. Catal. 4, 711–718 (2021).
Zhang, D. et al. Construction of Zn-doped RuO2 nanowires for efficient and stable water oxidation in acidic media. Nat. Commun. 14, 2517 (2023).
Huang, Z. et al. Implanting oxophilic metal in PtRu nanowires for hydrogen oxidation catalysis. Nat. Commun. 15, 1097 (2024).
Ding, H. et al. Highly crystalline iridium-nickel nanocages with subnanopores for acidic bifunctional water splitting electrolysis. J. Am. Chem. Soc. 146, 7858–7867 (2024).
Zhu, J. et al. Janus nanocages of platinum-group metals and their use as effective dual-electrocatalysts. Angew. Chem. Int. Ed. 60, 10384–10392 (2021).
Zheng, Y. et al. Scalable and controllable synthesis of Pt–Ni bunched-nanocages aerogels as efficient electrocatalysts for oxygen reduction reaction. Adv. Energy Mater. 13, 2204257 (2023).
Mu, X. et al. Constructing symmetry-mismatched RuxFe3−xO4 heterointerface-supported Ru clusters for efficient hydrogen evolution and oxidation reactions. Nano Lett. 24, 1015–1023 (2024).
Gan, T. & Wang, D. Atomically dispersed materials: ideal catalysts in atomic era. Nano Res. 17, 18–38 (2024).
Zhao, M. et al. Hollow metal nanocrystals with ultrathin, porous walls and well-controlled surface structures. Adv. Mater. 30, 1801956 (2018).
Li, K. et al. Enhanced electrocatalytic performance for the hydrogen evolution reaction through surface enrichment of platinum nanoclusters alloying with ruthenium in situ embedded in carbon. Energy Environ. Sci. 11, 1232–1239 (2018).
Han, J. et al. Strong metal-support interaction facilitated multicomponent alloy formation on metal oxide support. J. Am. Chem. Soc. 145, 22671–22684 (2023).
Li, Y. et al. Ruthenium nanoclusters and single atoms on α-MoC/n-doped carbon achieves low-input/input-free hydrogen evolution via decoupled/coupled hydrazine oxidation. Angew. Chem. Int. Ed. 63, e202316755 (2024).
Zeng, L. et al. Single-atom Cr-N4 sites with high oxophilicity interfaced with Pt atomic clusters for practical alkaline hydrogen evolution catalysis. J. Am. Chem. Soc. 145, 21432–21441 (2023).
She, X. et al. Pure-water-fed, electrocatalytic CO2 reduction to ethylene beyond 1,000 h stability at 10 A. Nat. Energy 9, 81–91 (2024).
Gu, J. et al. Twinning engineering of platinum/iridium nanonets as Turing-type catalysts for efficient water splitting. J. Am. Chem. Soc. 146, 5355–5365 (2024).
Li, X. et al. Phase transformation induced transitional twin boundary in body-centered cubic metals. Acta Mater. 249, 118815 (2023).
Hong, Y. et al. Double-walled tubular Heusler-type platinum–ruthenium phosphide as all-pH hydrogen evolution reaction catalyst outperforming platinum and ruthenium. Adv. Energy Mater. 14, 2304269 (2024).
Song, K. et al. Interfacial engineering of cobalt thiophosphate with strain effect and modulated electron structure for boosting electrocatalytic hydrogen evolution reaction. Adv. Funct. Mater. 34, 2312672 (2024).
Jin, H., Ha, M., Kim, M. G., Lee, J. H. & Kim, K. S. Engineering Pt coordination environment with atomically dispersed transition metal sites toward superior hydrogen evolution. Adv. Energy Mater. 13, 2204213 (2023).
Zhu, Q. et al. Hierarchical twinning governed by defective twin boundary in metallic materials. Sci. Adv. 8, eabn8299 (2022).
Hou, M. et al. Microenvironment reconstitution of highly active Ni single atoms on oxygen-incorporated Mo2C for water splitting. Nat. Commun. 15, 1342 (2024).
Huang, H. et al. Manipulation of oxidation states on phase boundary via surface layer modification for enhanced alkaline hydrogen electrocatalysis. Adv. Mater. 2405128 https://doi.org/10.1002/adma.202405128 (2024).
Yang, J., Li, W., Wang, D. & Li, Y. Electronic metal–support interaction of single-atom catalysts and applications in electrocatalysis. Adv. Mater. 32, 2003300 (2020).
Chen, X. et al. Rational design of dynamic interface water evolution on turing electrocatalyst toward the industrial hydrogen production. Adv. Mater. 36, 2401110 (2024).
Chao, T. et al. Reversible hydrogen spillover at the atomic interface for efficient alkaline hydrogen evolution. Energy Environ. Sci. 17, 1397–1406 (2024).
Shah, A. H. et al. Platinum surface water orientation dictates hydrogen evolution reaction kinetics in alkaline media. J. Am. Chem. Soc. 146, 9623–9630 (2024).
Cassol, G. S. et al. Ultra-fast green hydrogen production from municipal wastewater by an integrated forward osmosis-alkaline water electrolysis system. Nat. Commun. 15, 2617 (2024).
Li, J. et al. Unveiling the pivotal role of d2−2 electronic states in nickel-based hydroxide electrocatalysts for methanol oxidation. Angew. Chem. Int. Ed. 63, e202404730 (2024).
Zhang, H. et al. Tailoring oxygen reduction reaction kinetics of Fe−N−C catalyst via spin manipulation for efficient zinc–air batteries. Adv. Mater. 36, 2400523 (2024).
Li, Y. et al. Hydrogen spillover mechanism at the metal–metal interface in electrocatalytic hydrogenation. Angew. Chem. Int. Ed. 63, e202407810 (2024).
Zhong, M. et al. Modulating the electronic structure of Ni(OH)2 by coupling with low-content Pt for boosting the urea oxidation reaction enables significantly promoted energy-saving hydrogen production. Energy Environ. Sci. 17, 1984–1996 (2024).
Fan, X. et al. Surface-enriched single-Bi-atoms tailoring of Pt nanorings for direct methanol fuel cells with ultralow-Pt-loading. Adv. Mater. 36, 2313179 (2024).
Chen, X. et al. Revealing the role of interfacial water and key intermediates at ruthenium surfaces in the alkaline hydrogen evolution reaction. Nat. Commun. 14, 5289 (2023).
Xia, W. et al. Fabricating highly active Pt atomically dispersed catalysts with the co-existence of Pt-O1Ni1 single atoms and Pt sub-nanoclusters for improved hydrogen evolution. Appl. Catal. B: Environ. Energy 354, 124074 (2024).
Qian, S. et al. Tailoring coordination environments of single-atom electrocatalysts for hydrogen evolution by topological heteroatom transfer. Nat. Commun. 15, 2774 (2024).
Ji, S. et al. Effect of interfacial interaction on electrocatalytic activity and durability of Pt-based core–shell nanocatalysts. ACS Catal. 14, 11721–11732 (2024).
Li, X., Yan, Y., Yao, Y. & Liu, Y. Three-in-one tandem catalysis for alkaline hydrogen evolution reaction on Pt/CoV-LDHs. Chem. Eng. J. 489, 151237 (2024).
Hao, Y. et al. Methanol upgrading coupled with hydrogen product at large current density promoted by strong interfacial interactions. Energy Environ. Sci. 16, 1100–1110 (2023).
Zhang, B. et al. A strongly coupled Ru–CrOx cluster–cluster heterostructure for efficient alkaline hydrogen electrocatalysis. Nat. Catal. 7, 441–451 (2024).
Li, J. et al. Highly active and stable metal single-atom catalysts achieved by strong electronic metal–support interactions. J. Am. Chem. Soc. 141, 14515–14519 (2019).
Zhu, Y. et al. Facilitating alkaline hydrogen evolution reaction on the hetero-interfaced Ru/RuO2 through Pt single atoms doping. Nat. Commun. 15, 1447 (2024).
Zhou, K. L. et al. Platinum single-atom catalyst coupled with transition metal/metal oxide heterostructure for accelerating alkaline hydrogen evolution reaction. Nat. Commun. 12, 3783 (2021).
Zheng, Y., Jiao, Y., Vasileff, A. & Qiao, S.-Z. The hydrogen evolution reaction in alkaline solution: from theory, single crystal models, to practical electrocatalysts. Angew. Chem. Int. Ed. 57, 7568–7579 (2018).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22234005 and 21974070), the Natural Science Foundation of Jiangsu Province (BK20222015). The authors thank BL14W1 station at the Shanghai Synchrotron Radiation Facility and the High Performance Computing Center of Nanjing Tech University for supporting the computational resources.
Author information
Authors and Affiliations
Contributions
Y.Z.(Yue Zhang) and X.M. conceived the project and designed the experiments, Y.Z. and X.M. synthesized and characterized the materials, Y.Z.(Yue Zhang), X.M., Z.L., H.Z., and S.L. performed data analysis, Y.Z.(Yifan Zhang) performed the Raman experiment. Z.L. and H.Z. performed DFT calculations, Z.Z., S.M., S.L., D.W., and Z.D. reviewed and revised the manuscript draft, and Z.D. supported this project. The manuscript was written through the contributions of all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Mu, X., Liu, Z. et al. Twin-distortion modulated ultra-low coordination PtRuNi-Ox catalyst for enhanced hydrogen production from chemical wastewater. Nat Commun 15, 10149 (2024). https://doi.org/10.1038/s41467-024-54513-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54513-6
