
Abstract
While selective defunctionalizations are valuable in organic synthesis, hydrodeamination of primary amines poses challenges. Deuterodeamination, analogous to hydrodeamination, presents even greater difficulties due to its frequently slower deuteration rate, interference by hydrogenation and constraints in deuterated sources. This study introduces a reliable, robust, and scalable hydro- and deuterodeamination method capable of handling various primary amines. Defined by its mild reaction conditions, rapid completion, simplified purification facilitated by water-soluble byproducts, the method leverages deuterium oxide as a deuterium source and employs commercialized O-diphenylphosphinylhydroxylamine for deamination. Applied to a diverse range of biologically active molecules, it has consistently achieved high yields and efficient deuterium incorporation. By synergizing with site-selective C–H functionalization of primary aliphatic amines, our method reveals synthetic strategies utilizing nitrogen atom as a traceless directing group, encompassing deaminative alkylation, 1,1-deuteroalkylation, 1,1-dialkylation, 1,1,1-deuterodialkylation, C–H arylation, and 1,3-deuteroarylation. Emphasizing this innovation, the processes of deaminative degree-controlled deuteration have been developed.
Similar content being viewed by others
Introduction
Deuterium represents a naturally stable variant of hydrogen, distinguished merely by the inclusion of an extra neutron. The deuterium plays a significant role in advancing NMR spectroscopy techniques, mass spectrometry and drug developments1,2,3. Therefore, developing deuteration methods advances the discovery of functional molecules across diverse scientific fields. Given the widely presence of d-alkyl groups in current d-drugs3 and the prevalence of alkyl moieties in top-selling drugs4, the synthesis of d-labelled functionalized alkanes is particularly valuable. Despite the notable efficacy of hydrogen isotope exchange in the direct replacement of hydrogen with deuterium, the targeted incorporation of deuterium at inert aliphatic sites—specifically those devoid of acidity or proximity to radical-stabilizing moieties—presents a substantial obstacle in the absence of directing functionalities. Furthermore, the precise manipulation of the degree of deuteration, a controlled introduction of a defined quantity of deuterium at a particular site, remains unattainable5,6,7,8. d-Labelling approaches via functional-group transformations provide the ability to selectively incorporate deuterium atom at specific positions within a molecule9,10,11. While several functional-group transformations, such as reductive deuteration of double bonds12,13, dehalogenative14,15,16,17,18, decarboxylative19,20,21, and dehydroxylative22,23 deuterations, have been developed for this purpose, the diversity of organic compounds necessitates ongoing efforts to explore other functional-group transformations using abundant feedstock chemicals, aiming to fulfil the requirements of green and efficient synthesis.
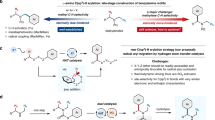
Amines are among the most prevalent functional groups found in natural products, pharmaceuticals, and synthetic intermediates. Its synthesis and derivatization can be accomplished through a diverse array of dependable methods24. Consequently, the establishment of an efficient deuterodeamination protocol holds the potential to substantially enhance the accessibility and chemical diversity of deuterated compounds. However, the development of deuterodeamination protocols is impeded by the low nucleofugality and high nucleophilicity of amines, alongside the robustness of the C–N bond. Furthermore, the inherent challenges of deuteration, including the potential for unintended hydrogenation reactions and the constraints and expenses linked to employing deuterated materials, further complicate the development of such protocols. The hydrodeamination process, like deuterodeamination in certain aspects, exhibits accelerated hydrogenation kinetics and leverages the abundant accessibility of hydrogen sources; nevertheless, its advancement remains constrained to a restricted scope of research and development25,26,27. The hydrodeamination method often necessitates the utilization of harsh conditions, which can have adverse effects on both the overall yield and the tolerance of functional groups28,29,30,31,32,33,34,35. Consequently, the two-step indirect deamination methods involving the pre-activation of the primary amines have been developed, in which the conversion of aliphatic primary amines to reactive isonitriles36,37 or Katritzky-type pyridinium salts38,39,40, represents widely employed strategies. One recent pioneering work of direct hydrodeamination from the Oestreich group uses B(C6F5)3 as catalyst and super stoichiometric PhSiH3 as the reductant (Fig. 1a)41. Despite enabling the hydrodeamination of benzyl amines and α-tertiary alkylamines, this method requires high temperature and anhydrous conditions, and suffers the intolerance with nucleophilic functional groups. Later, the Levin group developed a straightforward method for the direct hydrodeamination of primary amines via isodiazene intermediates, accomplished in a single step with high tolerance for diverse functional groups42. However, this technique requires strict exclusion of water and air, and often careful dropwise addition of the pre-prepared anomeric amide to prevent excessive temperature rise, and the substrate scope is limited to α-primary, α-secondary alkylamines and aryl amines. Therefore, it is crucial to pursue further research focused on developing a hydrodeamination methodology that is applicable to a broad spectrum of substrates and employs straightforward experimental procedures.

a. Development of direct hydrodeaminaton of primary amines. b. N-Deletion of alkylamines by DPPH. c. DPPH promoted hydro- and deuterodeamination of primary amines (This work).
Our previous research demonstrated that O-diphenylphosphinylhydroxylamine (DPPH), commonly referred to as Harger’s reagent43, promotes N-atom deletion44,45,46 of secondary amines, during which a rearrangement of dialkyltriazanium occurs (upper equation, Fig. 1b)47. We postulated that a similar intermediate, alkyltriazanium, could be generated and undergo a comparable rearrangement, resulting in the deamination of primary amines (down equation, Fig. 1b). Here, we introduce a reliable, robust, and scalable hydro- and deuteron-deamination reaction by DPPH, based on this hypothesis (Fig. 1c). This method effectively accommodates a diverse array of primary amines, spanning from α-primary to sterically hindered α-tertiary alkylamines and aryl amines, and presents numerous noteworthy benefits, including high efficiency and easy operation in a water-compatible environment. Moreover, it facilitates late-stage modifications for both pharmaceuticals and naturally occurring amines. Furthermore, a strategic approach for site-specific, degree-controlled deuteration48 has been developed, leveraging an amine as a traceless directing group.
Results
Initial optimization studies
The optimization of this reaction is detailed in Figures S1–S3 of the Supplementary Information (SI). We identified the standard conditions of hydrodeamination (S.C.H.) that require the addition of DPPH (2.2 equiv) and K2CO3 (2.2 equiv) to a solution of amine (0.4 mmol) in a 1:1 mixture of THF/H2O (Fig. 2). This setup facilitates the reaction to complete within 10 min at 50 °C. Under the standard conditions of deuterodeamination (S.C.D.) by substituting H2O with D2O and conducting the reaction under an argon atmosphere, we successfully synthesized the deuterated product.

a Standard conditions of hydro-(S.C.H.) or deuterodeamination (S.C.D.): amine a (0.4 mmol), DPPH (2.2 equiv), K2CO3 (2.2 equiv), H2O (2.0 mL, 3.3 mL of D2O for deuteration), THF (2.0 mL, 3.3 mL for deuteration), 10 min, 50 °C, under air (argon for deuteration). Isolated yield. b Using a•HCl (7a•maleate, 15a•H3PO3) instead of a, K2CO3 (3.2 equiv) was added. c K2CO3 (3.2 equiv), for 1 h. d DPPH (1.2 equiv), K2CO3 (1.2 equiv). e Under argon. f 20 min.g Racemized product. hdr = 1.3. idr = 1.1. jGC yield using dodecane as a standard. k DPPH (3.0 equiv), K2CO3 (3.0 equiv), 18-crown-6 (40 mol%), THF (4.0 mL). l Using D2O/THF (3.3 mL/0.67 mL) instead of D2O/THF (3.3 mL/3.3 mL). m Using d8-THF instead of THF.
Substrate scope
The versatility of both hydro- and deuteron-deamination was investigated in terms of functional-group compatibility and structural diversity. Amines with diverse electron properties, encompassing both electron-rich and electron-deficient alkylamines, serve as excellent substrates (Fig. 2). Amines with differing steric hindrance have been demonstrated as suitable substrates, arranging from α-primary (1a-7a, 23a), α-secondary and branched (9a-14a, 16a-17a, 21a, 22a), α-secondary and cyclic (8a, 15a, 18a-20a), α-tertiary and branched (24a-26a, 32a, 33a), to α-tertiary and cyclic (27a-30a, 31a) alkylamines. Remarkably, all these substrates demonstrated good to excellent yields for both hydro- and deuterodeamination, with high deuterium incorporation observed in the deuterodeamination reactions. These results are noteworthy given that the specific steric effects of the α-alkyl group have posed challenges in previous direct hydrodeamination reactions, as demonstrated in Levin’s Method42. For instance, to achieving reasonable yields for α-secondary alkylamines typically requires the careful dropwise addition of Levin’s reagent (anomeric amide), and the application of α-tertiary alkylamines has proven ineffective in their methodology42. In addition, the substrates contained various functional groups, including oxazolyl (1a), aldehyde (2a), ketone (13a), unprotected indolyl (3a-5a, 10a-12a, 21a-23a), purinyl (16a), triazolyl (17a), imidazolyl (18a), quinazolinyl (18a), electron-rich phenyl (24a), electron-deficient phenyl (19a), vinyl (15a, 19a, 20a), alkynyl (18a), ether (15a), oxime (7a), sulfonamido (8a), ester (9a, 10a, 15a, 21a-23a, 25a), primary amido (11a), secondary amido (15a, 21a-23a), tertiary amido (14a, 18a, 19a), carbamate (27a) and cyano (14a, 19a), all of which exhibited successful reactivity in our conditions. More interestingly, different nucleophilic functional groups, such as unprotected phenol group (5a, 6a, 9a, 33a), hydroxyl (6a, 14a, 26a, 28a, 32a) and carboxylic acid (12a, 33a) groups, were tolerant well. Various natural products and pharmaceutical molecules, including tryptamine (3a), 5-methoxytryptamine (4a), serotonin (5a), octopamine (6a), fluvoxamine (7a), tyrosinate (9a), tryptophanate (10a), tryptophanamide (11a), tryptophan (12a), saxagliptin (14a), oseltamivir (15a), valganciclovir (16a), sitagliptin (17a), linagliptin (18a), alogliptin (19a), memantine (31a), fingolimod (32a) and methyldopa (33a) were be used directly, providing ready access to value-added derivatives of feedstock compounds and their deuterated derivatives (>95% D). The valuable compounds 5-cholestene (20b) and its d-analogues (20c), and 18{19}-norabietatriene (30b) and its d-analogues (30c) were prepared from the derivatives of non-amino natural products cholesterol and dehydroabietic acid (see pages S6 and S14 in SI). The increasing recognition of the therapeutic potential of peptides has sparked numerous initiatives aimed at developing diverse late-stage functionalization of peptides49. Direct selective removal of the N-atom from peptides would be a useful and straightforward strategy to obtain peptide derivatives, however, it is shown to be not sufficient under the reported methods42. In our conditions, deamination of the terminal amino group in the dipeptide (21a) and tripeptide (22a), as well as the amino group in the side chain in the tetrapeptide (23a), were successfully achieved with good to excellent yields and high deuterium incorporation. The α-chiral centre is not retained during the transformation, as evidenced by the racemic product 29c obtained from the reaction of chiral metirosine 29a. Finally, aryl amines (34a-37a) were tested, yielding the corresponding hydrodeamination products (34b-37b) in good yields under modified reaction conditions (note k in Fig. 2, see details in Fig. S4 in SI). However, the deuterodeamination process was less efficient, resulting in a moderate yield and moderate deuterium incorporation for the desired d-aryl product 34c-37c.
Synthetic applications
The direct late-stage hydro- and deuterodeamination of readily available complex molecules may provide ready access to value-added feedstock compounds and their deuterated derivatives which are difficult to be achieved by known methods. We illustrate this advancement with three illustrative examples (Fig. 3a). Pseudosugar, unlike true sugar which exhibit an anomeric and a pyranose-furanose equilibrium in an aqueous solution, offers a stable and preferred conformation in solution50. This stability provides a clear understanding of the exact conformation of each hydroxyl group. The N-deletion of valiolamine (38a), without the need for protecting multiple hydroxyl groups, enables the generation of pseudo-α-D-sorbopyranose (38b) and its d-analogues (38c) directly. The simplified model system allows for a more focused exploration of the functional aspects and structural intricacies associated with D-sorbose without the complicating factors introduced by the dynamic equilibria observed in true sugars, potentially facilitating the bioactive studies of D-sorbose. Huperzine A (39a), an α-tertiary alkylamine, has received approval as a palliative drug for Alzheimer’s disease in China51. Mechanistic studies have revealed that the bridgehead amino group of huperzine A does not directly interact with the protein52, prompting interest in synthesizing deamino huperzine A (39b)53. The racemic form of deamino huperzine A (±39b) was successfully synthesized in ten steps, commencing from benzoquinone monoketal A, with an overall yield of 13%53. The synthetic route for chiral 39b from the N-atom deletion of chiral 39a has not been established, and it is conjectured that the steric hindrance of the amine group and the existence of various double bonds with distinct reactivities render this structure susceptible to disintegration, presenting challenges in removing the N-atom by conventional methods. Interestingly, under the standard conditions, chiral 39b and its d-analogues (39c) were successfully obtained in good yield and d-incorporation. Hydrodeamination of leelamine (40a), an α-primary alkylamine with low price, can afford abietatriene (40b) which is not commercially available and was used as a key starting material in several total syntheses54. Levin successfully devised the most efficient synthetic pathway, resulting in the isolation of 40b with a yield of 65%, achieved through the purification using silica-gel column chromatograph42. Under the standard conditions we established, abietatriene (40b) was obtained in an 85% isolated yield, and d-abietatriene (40c) was achieved with a remarkable 97% yield and 95% deuterium incorporation.

a. Representative examples for the convenient synthesis of valuable compounds and their deuterated derivatives. b. Gram-scale synthesis without column chromatograph. c. C–H functionalization and N-deletion synthetic sequence. d. Degree-controlled deuteration by using D2O. a Standard conditions of hydro- (S.C.H.) or deuterodeamination (S.C.D.): amine a (0.4 mmol), DPPH (2.2 equiv), K2CO3 (2.2 equiv), H2O (2 mL, 3.3 mL of D2O for deuteration), THF (2 mL, 3.3 mL for deuteration), 10 min, 50 °C, under air (argon for deuteration). b DPPH (4.2 equiv), K2CO3 (4.2 equiv). c Conditions for α-C–H alkylation: 4CzIPN or (1 mol%), Bu4N+N3 (10 mol%), tert-butyl acrylate (1.0 equiv), LED, MeCN, 20 h. d Using Ir[dF(CF3)ppy]2(dtbbpy)PF6 instead of 4CzIPN and tert-butyl acrylate (3.0 equiv). e Conditions for γ-C–H arylation: Pd(OAc)2 (10 mol%), HO2CCHO (20 mol%), AgTFA (1.5 equiv), 4-(BnO)C6H4I (1.5 equiv), HOAc, 100 oC, 15 h. f Yield for the two steps. g Conditions for α-H/D exchange: 4CzIPN (1 mol%), iPr3SiSH (30 mol%), EtOAc, D2O, LED, 48 h. h Using 3DPA2FBN (2 mol%) instead of 4CzIPN (1 mol%). i GC yield using dodecane as a standard.
Furthermore, the gram-scale hydrodeamination reactions for leelamine 40a and tryptamine 3a were successfully executed, yielding their respective products, 40b and 3b, with impressive efficiency (Fig. 3b). Specifically, 40b was obtained with a 97% yield and 94% purity, while 3b showcased a 99% yield and 95% purity. This was achieved simply through washing with acidic water, circumventing the need for traditional silica gel column chromatography, as detailed in SI. The results indicate that most byproducts from these reactions are water-soluble, which not only streamlines the purification process but also potentially enables the direct application of crude products in further synthesis steps. Such outcomes highlight the method’s practicality and its advantages in simplifying synthetic procedures.
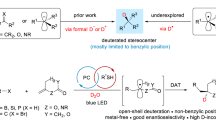
Through the seamless incorporation of the N-deletion methodology into synthetic transformations involving amines, innovative retrosynthetic approaches can be introduced. The amino group is a widely used directing group in C–H functionalization55. The combination of N-direct C–H functionalization with the N-deletion process, can establish a reliable gateway for traceless C–H functionalization (Fig. 3c). As an illustration, leveraging the recent advancements in the direct α C–H functionalization of unprotected primary amines by Cresswell et al. 56 we have developed various synthetic strategies, such as deaminative alkylation (41e) and 1,1-deteroalkylation (41f) of α-secondary amine 41d, 1,1-dialkylation (42e) and 1,1,1-deterodialkylation (42f) of α-primary amine (42d). Furthermore, selective γ-C–H functionalization of amines, developed by the Ge group57, was followed by N-deletion reactions, resulting in a C–H arylation process (43e) and a 1,3-deteroarylation process (43f) that employs nitrogen as a traceless directing group.
Inspired by recent breakthroughs in the direct substitution of α-C–H bonds in primary amines with deuterium via H/D exchange12,13,14,15,16,17,18, and the existing gap in the development of precision-controlled deuteration techniques48, we developed a methodology. This approach allows for precision-controlled deuteration, employing the nitrogen atom as a traceless directing group, as illustrated in Fig. 3d. Building upon the recent work of Wang et al. on the direct α-C–H deuteration of unprotected primary amines58, three distinct sets of reaction protocols were devised by utilize D2O as the deuterium source. As previous demonstrated, under the established standard conditions of deuterodeamination (S.C.D.), the protocol consistently yielded high monodeuteration across a variety of substrates (Figs. 2 and 3d). To achieve deaminative dideuteration of α-secondary alkylamines or trideuteration of α-primary alkylamines, the α-deuteration of amine was initially conducted via H/D exchange with D2O using an organophotocatalytic process58. After solvent removal, the resulting crude α-deuterated intermediates 41a, 44 a, 45a were directly subjected to S.C.D. With these conditions (route II), this method introduced 1.87D for the methylene in linear product 41c’, 1.89D for the methylene in cyclic product 44c’, and 2.89D for the methyl group in alkane 45c’. Employing the same synthetic procedure under the standard conditions of hydrodeamination (S.C.H.) instead of S.C.D. in the secondary step (route III) selectively installed 1.97 D in the methyl group of product 45b’. In summary, degree-controlled deuterations can be accomplished with satisfactory overall yields and high d-incorporation. Notably, these represent only a subset of the potential applications due to the richness of amine chemistry, and the extension to a broader range of examples promises to unveil additional functionalities and applications.
Mechanistic studies
To further understand the reaction mechanism, several experiments were performed (Fig. 4). The reaction of phenylethyl amine 42a was conducted at 0 °C with 1.2 equivalents of DPPH for 10 min (Fig. 4a). In the HRMS analysis of the reaction mixture, we identified two notable compounds with molecular weight of 137.1070 and 152.1189. These two compounds are likely phenylethyl hydrazine 42g (Mw + H+ = 137.1073) and a triazane derivative 42h (Mw + H+ = 152.1182). To further explore this, we subjected commercially available phenylethyl hydrazine 42g to our standard reaction conditions with CDCl3 as a co-solvent. Through this experiment, we were able to detect the formation of ethyl benzene 42b in 91% NMR yield. This result supports the hypothesis that alkyl hydrazine functions as one of the intermediates in this process. Previous research has shown that alkyl hydrazine can produce a defunctionalized product under mild oxidative conditions, involving an alkyl diazene intermediate59,60. Additionally, drawing upon Levin’s findings42,44 and our own research on N-deletion of secondary amines45,47,61,62, we consider the possibility of an alkyl isodiazene intermediate forming. To investigate these intermediates further, we directly compared various reactions (Fig. 4b). For example, when we treated benzyl amine 46a with DPPH, it failed to produce the expected deamination product, toluene, unlike the oxidation of benzyl hydrazine 46a’, which successfully yielded toluene at a 51% yield through a diazene intermediate60. Treating 1-naphthalenamine 47a did not produce any deamination product 47b, but led to a 76% NMR yield of the deaminative rearrangement product 47 d. This compares to a 57% NMR yield of 47b under Levin’s conditions42, suggesting the involvement of a [2,3]-sigmatropic rearrangement of an isodiazene intermediate63. Moreover, when propargylic amine 48a reacted with DPPH, it resulted in the production of alkyne 48b and allene 48 d at an approximate ratio of 1:17. This outcome closely aligns with the 1:8 ratio reported under the conditions described by Levin (indicative of an isodiazene intermediate) and contrasts with the 1:140 ratio observed under the conditions performed by Myers64, which initiates from propargylic alcohol 48a’ and includes a diazene intermediate in the process. These results suggest that an isodiazene, rather than a conventional diazene, may be involved in our reactions. To ascertain the rate-controlling step, we conducted an analysis of the kinetic isotope effect (KIE) by investigating the reaction of 28a in a solvent mixture of H2O and D2O, as well as the corresponding reactions of amine 49a in either H2O or D2O (as illustrated in Fig. 4c). This analysis showed a one-pot competition KIE with a ratio of kH/kD = 1.06 and a parallel KIE with a ratio of kH/kD = 1.13. Then, two radical trapping experiments were conducted, as depicted in Fig. 4d. In the first experiment, we used an equivalent amount of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) to trap radicals from the reaction of tryptamine 3a, successfully isolating the TEMPO capture product 3 d with a 42% yield. In the second experiment, Electron Paramagnetic Resonance spectra were recorded using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) for spin trapping in the reaction of phenylethyl amine 42a, and the DMPO-spin adduct 42i was observed. Furthermore, several radical clock experiments were conducted (Fig. 4e). While no direct deamination products 50b and 51b were observed, ring-opening products 50b’ and 51b’ were obtained with good yield in the reactions of cyclopropylmethyl radical clock 50a (kr ≈ 108 s−11) and its phenyl analogous 51a (kr ≈ 1011 s−11)65. In addition, the radical self-coupling products 51 d were detected obviously by GCMS in the reaction of 51a. Likewise, small quantities of the hydrodeamination product 52b, as well as the principal ring-closed products 52b’ and 52c’, were produced from the reactions involving the 5-vinylmethyl radical clock 52a (kr ≈ 107 s−11)65. These results indicate the rate of alkyl radical abstracting hydrogen is slower than the rearrangement rates of these radical clocks, suggesting a cage-escaping (k(cage escape) ≈ 1010 s−1)65,66, free alkyl radical is involved in the deamination process. According to these experimental results and previous studies67,68,69,70, a mechanism was proposed in Fig. 4f. Amine reacts with two equivalents of DPPH to form possible triazanium intermediate. Its rearrangement under basic conditions generates a primary isodiazene intermediate. Its decomposition produces an initial alkyl radical and a diazane radical (radical initiation). Then, alkyl radical abstract hydrogen from primary isodiazene intermediate to produce desired deamination product and alkyl diazenyl radical species. By releasing nitrogen gas, the rearrangement of the alkyl diazenyl radical species regenerates alkyl radical, finishing a chain, hydrogen-atom transfer process.

a. Detection of key intermediates. b. Comparison of diazene and isodiazene. c. Kinetic isotope effect (KIE) experiments. d. Radical trapping reactions. e. Radical clock experiments. f. Proposed mechanism of hydrodeamination. a Standard conditions of hydro- (S.C.H.) or deuterodeamination (S.C.D.): amine a (0.4 mmol), DPPH (2.2 equiv), K2CO3 (2.2 equiv), H2O (2 mL, 3.3 mL of D2O for deuteration), THF (2 mL, 3.3 mL for deuteration), 10 min, 50 °C, under air (argon for deuteration). b Using CDCl3 instead of THF. c NMR yield. d (4-CF3C6H4)CON(OCOtBu)(OBn) (1.2 equiv), THF, N2, 45 ° C. e PPh3 (1.5 equiv), DEAD (1.5 equiv), NBSH (1.5 equiv), THF, −15 °C. f Using a 1:1 mixture of D2O and H2O instead of pure H2O in S.C.H. g 33% yield of deamination product 2b. h Detected by GCMS.
Discussion
In this study, we present a groundbreaking direct hydro- and deuteron-deamination method capable of accommodating a wide range of primary amines. These include α-primary, α-secondary, sterically hindered α-tertiary alkylamines, and aryl amines. The reaction demonstrates exceptional tolerance to various functional groups, including nucleophilic phenols, hydroxyls, amides, and carboxylic acids. Impressively, the method has been successfully applied to versatile bio-relevant compounds, spanning pharmaceutical molecules, amino acids, amino sugars, peptides, and natural products. The reaction of aliphatic amines exhibits high yields for both hydrodeamination (up to 98%) and deuterodeamination (up to 99%), accompanied by an impressive 96% deuterium incorporation. This approach boasts several noteworthy features, including mild aqueous conditions, rapid completion within ten minutes, easy purification due to high yield and water-soluble major byproducts, the utilization of deuterium oxide as a deuterium source, and commercially available DPPH as the deamination reagent. Abundant synthetic methods exist for the synthesis and transformations of nitrogen-containing compounds, wherein N-atom plays a pivotal role. When combined with the deamination reaction, these methods open avenues for chemical conversions, offering a unique and versatile means to guide reactions toward desired outcomes, with nitrogen atom serving as a traceless directing group. Expanding on this concept, our approach, when combined with N-directed α- or γ-C–H functionalization, unlocks diverse strategic applications. These include deaminative alkylation, 1,1-deuteroalkylation, 1,1-dialkylation, 1,1,1-deuterodialkylation, C–H arylation, and 1,3-deuteroarylation. Combing with N-direct α-H/D exchange reaction, we have devised a one-pot process for selective deaminative mono-, di-, and tri-deuteration at the original amino site, enhancing the accessibility of degree-controlled deuterated compounds. These developments contribute to diverse applications in chemical and pharmaceutical sciences, emphasizing the versatility and impact of our methodology.
Methods
General procedures
In a 25 mL Schlenk tube equipped with a stirring bar, amine a (0.4 mmol, 1.0 equiv) was added, followed by THF (2.0 mL, or 3.3 mL for deuteration), H2O (2.0 mL, or 3.3 mL of D2O for deuteration), K2CO3 (121.6 mg, 2.2 equiv), and DPPH (205.2 mg, 2.2 equiv) in sequence. The tube was sealed (under argon for deuteration) and placed on a preheated heating module at 50 °C. The mixture was stirred vigorously at 800 rpm for 10 min. After cooling to room temperature, 5.0 mL of aqueous NaCl solution was added, and the mixture was extracted three times with 5.0 mL of ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4, and the product was purified by flash chromatography over silica gel to yield the desired compound.
Data availability
The data supporting the findings of this study are available within the paper and its Supplementary Information files. Raw data are available from the corresponding author on request.
References
Liuni, P., Olkhov-Mitsel, E., Orellana, A. & Wilson, D. J. Measuring kinetic isotope effects in enzyme reactions using time-resolved electrospray mass spectrometry. Anal. Chem. 85, 3758–3764 (2013).
Campobasso, N. & Huddler, D. Hydrogen deuterium mass spectrometry in drug discovery. Bioorg. Med. Chem. Lett. 25, 3771–3776 (2015).
Mullard, A. Deuterated drugs draw heavier backing. Nat. Rev. Drug Discov. 15, 219–221 (2016).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. Deuterium- and tritium-labelled compounds: applications in the life sciences. Angew. Chem. Int. Ed. 57, 1758–1784 (2018).
Junk, T. & Catallo, W. J. Hydrogen isotope exchange reactions involving C−H (D, T) bonds. Chem. Soc. Rev. 26, 401–406 (1997).
Atzrodt, J., Derdau, V., Fey, T. & Zimmermann, J. The renaissance of H/D exchange. Angew. Chem. Int. Ed. 46, 7744–7765 (2007).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. C−H Functionalisation for hydrogen isotope exchange. Angew. Chem. Int. Ed. 57, 3022–3047 (2018).
Prakash, G. et al. D. C–H deuteration of organic compounds and potential drug candidates. Chem. Soc. Rev. 51, 3123–3163 (2022).
Li, N., Li, Y., Wu, X., Zhu, C. & Xie, J. Radical deuteration. Chem. Soc. Rev. 51, 6291–6306 (2022).
Kopf, S. et al. Recent developments for the deuterium and tritium labeling of organic molecules. Chem. Rev. 122, 6634–6718 (2022).
Li, H., Shabbir, M., Li, W. & Lei, A. Recent advances in deuteration reactions. Chin. J. Chem. 42, 1145–1156 (2024).
Vang, Z. P., Hintzsche, S. J. & Clark, J. R. Catalytic transfer deuteration and hydrodeuteration: emerging techniques to selectively transform alkenes and alkynes to deuterated alkanes. Chem. Eur. J. 27, 9988–10000 (2021).
Luo, J., Lu, L., Montag, M., Liang, Y. & Milstein, D. Hydrogenative alkene perdeuteration aided by a transient cooperative ligand. Nat. Chem. 15, 1384–1390 (2023).
Soulard, V., Villa, G., Vollmar, D. P. & Renaud, P. Radical deuteration with D2O: catalysis and mechanistic insights. J. Am. Chem. Soc. 140, 155–158 (2018).
Constantin, T. et al. Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides. Science 367, 1021–1026 (2020).
Li, Y. et al. Organophotocatalytic selective deuterodehalogenation of aryl or alkyl chlorides. Nat. Commun. 12, 2894 (2021).
Li, P. et al. Facile and general electrochemical deuteration of unactivated alkyl halides. Nat. Commun. 13, 3774 (2022).
Wood, D. & Lin, S. Deuterodehalogenation under net reductive or redox-neutral conditions enabled by paired electrolysis. Angew. Chem. Int. Ed. 62, e202218858 (2023).
Li, N. et al. A highly selective decarboxylative deuteration of carboxylic acids. Chem. Sci. 12, 5505–5510 (2021).
Lu, Y.-C. & West, J. G. Chemoselective decarboxylative protonation enabled by cooperative earth-abundant element catalysis. Angew. Chem. Int. Ed. 62, e202213055 (2023).
Deng, C.-Q. et al. Chemoselective direct deuterodecarboxylation of free aliphatic carboxylic acids enabled by deuteron-coupled electron transfer. Chem. Catal. 4, 100899 (2024).
Spiegel, D. A., Wiberg, K. B., Schacherer, L. N., Medeiros, M. R. & Wood, J. L. Deoxygenation of alcohols employing water as the hydrogen atom source. J. Am. Chem. Soc. 127, 12513–12515 (2005).
He, B.-Q. & Wu, X. Deuterium- and electron-shuttling catalysis for deoxygenative deuteration of alcohols. Org. Lett. 25, 6571–6576 (2023).
Lawrence S. A. Amines: Synthesis, Properties and Applications. (Cambridge University Press, 2004).
Mo, F., Dong, G., Zhang, Y. & Wang, J. Recent applications of arene diazonium salts in organic synthesis. Org. Biomol. Chem. 11, 1582–1593 (2013).
Wang, Q., Su, Y., Li, L. & Huang, H. Transition-metal catalysed C–N bond activation. Chem. Soc. Rev. 45, 1257–1272 (2016).
Berger, K. J. & Levin, M. D. Reframing primary alkyl amines as aliphatic building blocks. Org. Biomol. Chem. 19, 11–36 (2021).
Nickon, A. & Sinz, A. Reductive deamination of aliphatic amines. J. Am. Chem. Soc. 82, 753–754 (1960).
Nickon, A. & Hill, A. S. A direct method for reductive deamination of aliphatic amines. J. Am. Chem. Soc. 86, 1152–1158 (1964).
Doldouras, G. A. & Kollonitsch, J. A direct, selective, and general method for reductive deamination of primary amines. J. Am. Chem. Soc. 100, 341–342 (1978).
Bumgardner, C. L., Martin, K. J. & Freeman, J. P. Deamination reactions of difluoroamine. J. Am. Chem. Soc. 85, 97–99 (1963).
Maier, W. F. et al. Direct removal of functional groups by catalytic hydrogenolysis. Angew. Chem. Int. Ed. 18, 939–940 (1979).
Wohlgemuth, R. Selective biocatalytic defunctionalization of raw materials. ChemSusChem 15, e202200402 (2022).
McFadden, T. P., Nwachukwu, C. I. & Roberts, A. G. An amine template strategy to construct successive C–C bonds: synthesis of benzo[h]quinolines by a deaminative ring contraction cascade. Org. Biomol. Chem. 20, 1379–1385 (2022).
Schwartz, Z., Valiton, C., Lovasz, M. & Roberts, A. G. Recent applications of ammonium ylide based [2,3]-sigmatropic and [1,2]-stevens rearrangements to transform amines into natural products. Synthesis 56, 87–106 (2023).
Quirós, I. et al. Isonitriles as alkyl radical precursors in visible light mediated hydro– and deuterodeamination reactions. Angew. Chem. Int. Ed. 136, e202317683 (2023).
Jiao, Z. et al. Unified approach to deamination and deoxygenation through isonitrile hydrodecyanation: a combined experimental and computational investigation. Angew. Chem. Int. Ed. 63, e202405779 (2024).
Rössler, S. L. et al. Pyridinium salts as redox-active functional group transfer reagents. Angew. Chem. Int. Ed. 59, 9264–9280 (2020).
Katritzky, A. R., Horvath, K. & Plau, B. Reductive deamination of primary amines. J. Chem. Soc. Perkin Trans. 1 1980, 2554–2560 (1980).
Wang, C. et al. Biomimetic dehydroamination of primary amines. ACS Catal. 13, 14205–14212 (2023).
Fang, H. & Oestreich, M. Reductive deamination with hydrosilanes catalyzed by B(C6F5)3. Angew. Chem. Int. Ed. 59, 11394–11398 (2020).
Berger, K. J. et al. Direct deamination of primary amines via isodiazene intermediates. J. Am. Chem. Soc. 143, 17366–17373 (2021).
Jinan, D., Mondal, P. P., Nair, A. V. & Sahoo, B. O-Protected NH-free hydroxylamines: emerging electrophilic aminating reagents for organic synthesis. Chem. Commun. 57, 13495–13505 (2021).
Kennedy, S. H., Dherange, B. D., Berger, K. J. & Levin, M. D. Skeletal editing through direct nitrogen deletion of secondary amines. Nature 593, 223–227 (2021).
Qin, H. T. et al. N-Atom deletion in nitrogen heterocycles. Angew. Chem. Int. Ed. 60, 20678–20683 (2021).
Hui, C. G., Brieger, L., Strohmann, C. & Antonchick, A. P. Stereoselective synthesis of cyclobutanes by contraction of pyrrolidines. J. Am. Chem. Soc. 143, 18864–18870 (2021).
Guo, T., Li, J., Cui, Z., Wang, Z. & Lu, H. C(sp3)–C(sp3) bond formation through nitrogen deletion of secondary amines using O-diphenylphosphinylhydroxylamine. Nat. Synth. 3, 913–921 (2024).
Zhou, X., Yu, T. & Dong, G. Site-specific and degree-controlled alkyl deuteration via cu-catalyzed redox-neutral deacylation. J. Am. Chem. Soc. 144, 9570–9575 (2022).
deGruyter, J. N., Malins, L. R. & Baran, P. S. Residue-specific peptide modification: a chemist’s guide. Biochemistry 56, 3863–3873 (2017).
Suami, T. Chemistry of pseudo-sugars. in Carbohydrate Chemistry. Topics in Current Chemistry, Vol. 154 (Springer, 1990)
Tang, X. C., He, X. C. & Bai, D. L. Huperzine A: a novel acetylcholinesterase inhibitor. Drugs Future 24, 647–663 (1999).
Raves, M. L. et al. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (–)-huperzine A. Nat. Struct. Biol. 4, 57–63 (1997).
Högenauer, K., Baumann, K. & Mulzer, J. Synthesis of (±)-desamino huperzine A. Tetrahedron Lett. 41, 9229–9232 (2000).
Mori, N., Kuzuya, K. & Watanabe, H. Synthesis of (−)-chamobtusin A from (+)-dehydroabietylamine. J. Org. Chem. 81, 11866–11870 (2016).
Yang, X. & Cao, X. Transition-metal-catalyzed remote C–H bond functionalization of cyclic amines. SynOpen 6, 286–305 (2022).
Ryder, A. S. H. et al. Photocatalytic α-tertiary amine synthesis via C−H alkylation of unmasked primary amines. Angew. Chem. Int. Ed. 59, 14986–14991 (2020).
Liu, Y. & Ge, H. Site-selective C–H arylation of primary aliphatic amines enabled by a catalytic transient directing group. Nat. Chem. 9, 26–32 (2017).
Meng, X., Dong, Y., Liu, Q. & Wang, W. Organophotocatalytic α-deuteration of unprotected primary amines via H/D exchange with D2O. Chem. Commun. 60, 296–299 (2024).
Corey, E. J., Wess, G., Xiang, Y. B. & Singh, A. K. Stereospecific total synthesis of (.+-.)-cafestol. J. Am. Chem. Soc. 109, 4717–4718 (1987).
Hoffman, R. V. & Kumar, A. Oxidation of hydrazine derivatives with arylsulfonyl peroxides. J. Org. Chem. 49, 4014–4017 (1984).
Zou, X., Zou, J., Yang, L., Li, G. & Lu, H. Thermal rearrangement of sulfamoyl azides: reactivity and mechanistic study. J. Org. Chem. 82, 4677–4688 (2017).
Qin, H., Guo, T., Lin, K., Li, G. & Lu, H. Synthesis of dienes from pyrrolidines using skeletal modification. Nat. Commun. 14, 7307 (2023).
Dherange, B. D. et al. Direct deaminative functionalization. J. Am. Chem. Soc. 145, 17–24 (2023).
Myers, A. G. & Zheng, B. New and stereospecific synthesis of allenes in a single step from propargylic alcohols. J. Am. Chem. Soc. 118, 4492–4493 (1996).
Newcomb, M. in Encyclopedia of Radicals in Chemistry, Biology and Materials (eds Chatgilialoglu, C. & Studer, A.) (Wiley, 2012).
Herk, L., Feld, M. & Szwarc, M. Studies of “Cage” reactions. J. Am. Chem. Soc. 83, 2998–3005 (1961).
Masson-Makdissi, J. et al. Evidence for dearomatizing spirocyclization and dynamic effects in the Quasi-stereospecific Nitrogen Deletion of Tetrahydroisoquinolines. J. Am. Chem. Soc. 146, 17719–17727 (2024).
Xue, J.-H., Li, Y., Liu, Y., Li, Q. & Wang, H. Site-specific deaminative trifluoromethylation of aliphatic primary amines. Angew. Chem., Int. Ed. 63, e202319030 (2024).
Steiniger, K. A., Lamb, M. C. & Lambert, T. H. Cross-coupling of amines via photocatalytic denitrogenation of in situ generated diazenes. J. Am. Chem. Soc. 145, 11524–11529 (2023).
Wright, B. A. et al. Skeletal editing approach to bridge-functionalized bicyclo[1.1.1]pentanes from azabicyclo[2.1.1]hexanes. J. Am. Chem. Soc. 145, 10960–10966 (2023).
Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (22071100 H.L., 22271148 H.L.).
Author information
Authors and Affiliations
Contributions
P.M., T.G., and H.L. designed the experiments. P.M. and T.G. performed the experiments and analysed the data. All authors participated in writing the manuscript. H.L. conceived and supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Osama El-Sepelgy and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, P., Guo, T. & Lu, H. Hydro- and deutero-deamination of primary amines using O-diphenylphosphinylhydroxylamine. Nat Commun 15, 10190 (2024). https://doi.org/10.1038/s41467-024-54599-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54599-y
This article is cited by
-
Photocatalyst-free photochemical deuteration via H/D exchange with D2O
Nature Communications (2025)
-
Deaminative Giese-type reaction
Nature Chemistry (2025)
