
Abstract
Next-generation T-cell-directed vaccines for COVID-19 focus on establishing lasting T-cell immunity against current and emerging SARS-CoV-2 variants. Precise identification of conserved T-cell epitopes is critical for designing effective vaccines. Here we introduce a comprehensive computational framework incorporating a machine learning algorithm—MHCvalidator—to enhance mass spectrometry-based immunopeptidomics sensitivity. MHCvalidator identifies unique T-cell epitopes presented by the B7 supertype, including an epitope from a + 1-frameshift in a truncated Spike antigen, supported by ribosome profiling. Analysis of 100,512 COVID-19 patient proteomes shows Spike antigen truncation in 0.85% of cases, revealing frameshifted viral antigens at the population level. Our EpiTrack pipeline tracks global mutations of MHCvalidator-identified CD8 + T-cell epitopes from the BNT162b4 vaccine. While most vaccine epitopes remain globally conserved, an immunodominant A*01-associated epitope mutates in Delta and Omicron variants. This work highlights SARS-CoV-2 antigenic features and emphasizes the importance of continuous adaptation in T-cell vaccine development.
Similar content being viewed by others

Introduction
The emergence of the COVID-19 pandemic, which is attributed to the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), spurred the rapid development of many vaccines1. The effective deployment of vaccines that integrate the SARS-CoV-2 spike (S) protein has played a pivotal role in safeguarding millions of individuals from severe illness through stimulation of both antibody and cell-mediated immune responses2. However, the S protein has been experiencing significant mutations during the pandemic, leading to the emergence of antibody escape mechanisms by SARS-CoV-2 variants3,4,5. This underscores the importance of promoting T-cell immunity directed towards conserved SARS-CoV-2 antigens, which could provide more robust protection against severe disease caused by current and forthcoming hypermutated variants6,7,8. To address this objective, at least two T-cell-directed vaccines, CoVac-1 and BNT162b4, have recently been developed and are undergoing clinical trials9,10,11,12. CoVac-1, a multi-peptide-based T-cell activator, aims to induce broad and enduring SARS-CoV-2 T-cell immunity and is currently advancing to phase III clinical trials (NCT04954469)9,10,11. BNT162b4, a T-cell-directed mRNA-based vaccine, encodes conserved non-S antigens and is undergoing clinical evaluation in conjunction with the Omicron-updated bivalent BNT162b2 (NCT05541861)12.
To enhance the design of next-generation T-cell vaccines targeting future SARS-CoV-2 variants, the creation of comprehensive digital maps that encompass the entire spectrum of SARS-CoV-2 peptides presented by HLA molecules, along with a detailed understanding of their mutational dynamics is of paramount importance13,14,15. To achieve this goal, the development of unbiased and scalable hardware and software is needed16. In this regard, Mass Spectrometry (MS)-based immunopeptidomics stands out as a powerful scalable method for the unbiased identification of HLA-associated peptides17,18,19. In fact, MS has been increasingly applied in recent years to characterize the SARS-CoV-2 immunopeptidome20,21,22,23,24,25,26,27,28,29,30. These efforts have not only revealed conventional SARS-CoV-2 T-cell epitopes but also uncovered unconventional epitopes, including those arising from noncanonical translation, often escaping detection by conventional epitope mapping approaches22,23.
From a technical standpoint, MS-based immunopeptidomics involves the sequential processes of isolating HLA-associated peptides through immunoaffinity capture, peptide elution, and acquisition by Liquid Chromatography coupled to tandem Mass Spectrometry (LC-MS/MS)17,31,32. The subsequent matching of peptide sequences to the tandem mass spectra obtained from MS/MS (i.e. peptide-spectrum matches (PSMs)) is executed using proteomics database search engines such as SEQUEST33, Comet34, MS-GF + 35, MSFragger36, SpectroMine37, PEAKS38, or Andromeda, which is included in the MaxQuant environment39,40. These computational tools compare each experimental MS/MS spectrum against a set of theoretical MS/MS spectra derived for every candidate peptide based on a provided protein or peptide sequence database, assigning a score to each PSM and reporting those with the top scores. Since each experimental MS/MS spectrum is matched to at least one peptide sequence, many of those matches are false positives. To control the downstream false discovery rate (FDR), decoy peptides, representing shuffled or reversed versions of peptide sequences from the “target” protein sequence database41, are also used to match experimental MS/MS spectra by database search engines. Subsequently, target and decoy PSMs serve as input for computational post-processing tools like PeptideProphet42,43,44 and Percolator45,46. Such tools combine database search engine scores, as well as sequence and spectrum properties that are useful for discrimination between target and decoy PSMs. Since the search space of possible HLA-bound peptides is larger than that of peptides typically obtained in standard proteomics experiments, the number of false positives tend to be higher in immunopeptidomics experiments47. To attempt to address this issue, there have been additional machine learning (ML) and deep learning (DL) tools described (e.g. DeepRescore, Prosit, MS2Rescore, MSBooster) that add features (i.e. peptide fragmentation patterns and retention times) to Percolator input files to further enhance validation of peptides in immunopeptidomics48,49,50,51. Another strategy identifying peptides from MS/MS spectra without the use of protein sequence database was recently proposed to yield de novo identification of HLA-peptides52.
The rules of antigen processing and presentation (APP) have been studied, assessed, and incorporated into prediction algorithms to predict T-cell epitopes53,54. For instance, MHCflurry55,56 and NetMHCpan57,58,59 are two widely used algorithms that merge scores derived from APP properties. MHCflurry encompasses two essential predictors: an “antigen processing” predictor, which models MHC allele-independent effects like proteosomal cleavage, and a “presentation” predictor that combines processing predictions with HLA-peptide binding affinity (BA) predictions to yield a composite “presentation score (PS)”. On the other hand, NetMHCpan generates both HLA-peptide binding affinity (BA) and eluted ligand (EL) prediction scores. Previous studies have applied those scores as a target-decoy discriminative factor for validating PSMs in immunopeptidomics but face challenges in maintaining consistency in the treatment of PSMs60,61,62. In principle, a more comprehensive incorporation of those scores into the modeling process for PSM confidence assessment could significantly enhance the sensitivity and accuracy of HLA-peptide identification at a fixed FDR, which is particularly important in the context of developing a robust platform for optimal vaccine design. Such an approach allows to directly control FDR in contrast with the popular practice in the field of immunopeptidomics, where score filtering is applied after the FDR estimation process63. The inclusion of APP prediction scores in discriminating target from decoy PSMs relies, however, on the performance of the prediction algorithms in addition to require prior knowledge of HLA alleles expressed in the samples. To overcome this limitation, a recent development involves the creation and implementation of a sequence encoder strategy, which aims to learn primary sequence elements that distinctly characterize HLA-peptides64. This approach has a greater discovery potential since it considers both well-established and potentially less-characterized sequence motifs65. However, unlike APP prediction scores, encoded peptide sequences cannot be easily incorporated into a PSM confidence assessment software package such as Percolator. This limitation arises from Percolator’s underlying support vector machine (SVM) model, which lacks inherent flexibility in accommodating variable-length sequence inputs. Therefore, more versatile PSM confidence assessment methods are needed to replace Percolator and leverage standard PSM quality features, APP prediction scores and peptide amino acid sequences in order to enhance the validation of HLA-I-specific PSMs in immunopeptidomics.
While MS-based immunopeptidomics demonstrates significant capabilities, its accessibility remains somewhat restricted, impeding researchers’ capacity to conduct direct measurements of SARS-CoV-2 immunopeptidomes and their associated mutational dynamics on a large population scale16,66. In contrast, the widespread availability of genome sequencing technologies has facilitated the extensive sequencing of over 14 million SARS-CoV-2 sequences and 3,423 lineages, including B.1.1.7 (alpha), B.1.617.2 (delta), B.1.1.529 (omicron) and its hypermutated variant BA.2.86 (Pirola)67. Such large-scale genomic data have been used to track the mutational dynamics of T-cell epitopes to study T-cell escape mechanisms by SARS-CoV-2 variants14. Large-scale genome sequencing of SARS-CoV-2 has also been useful to study the intra-host variation and evolutionary dynamics of SARS-CoV-2 populations in COVID-19 patients68. Genomic heterogeneity of the virus within a host can indeed be analysed by capturing intra-host Single Nucleotide Variations (iSNVs)68,69. Those intra-host variations are generated by mutations initiated randomly in a small fraction of viruses during infection, providing a mutational pool shaping the rapid global evolution of the virus68,69,70. Intra-host genetic diversity of SARS-CoV-2 lineages have been studied from relatively large cohorts of COVID-19 patients70,71, both in unvaccinated and vaccinated individuals72, but the integration of such large-scale genomic data with ML-enhanced immunopeptidomics for informing vaccine design against SARS-CoV-2 variants remains largely unexplored.
In this work, we show a unique computational framework and analysis platform that provide valuable insights for T-cell vaccine design against SARS-CoV-2 variants. This platform comprises six modules (Fig. 1): (1) MS-based immunopeptidomics of new and publicly available datasets, (2) ML-based MHCvalidator, (3) intra-host genomic variations of SARS-CoV-2 populations from a large cohort of 100,512 infected patients, (4) T-cell epitope immunogenicity, (5) EpiTrack for monitoring the geo-temporal mutational dynamics of vaccine-relevant T-cell epitopes across 14.6 million SARS-CoV-2 sequences and 3,423 lineages, and (6) selection of immunogenic and stable T-cell epitopes to inform T-cell vaccine design (BNT162b4 is shown as an example tested in this study). The analysis platform is applicable to any viruses. Below, we describe the development of MHCvalidator and show its utility to boost the unbiased discovery of conserved T-cell epitope vaccine candidates through population-scale multi-omic data integration and improved immunopeptidomics sensitivity.

(1) MS-based immunopeptidomics for data acquisition, (2) MHCvalidator for HLA-I-specific PSMs confidence assessment and optimal identification of both canonical and non-canonical HLA-I viral peptides, (3) population-scale analysis of SARS-CoV-2 proteome diversity using intra-host databases, (4) T-cell epitope immunogenicity assessment, (5) EpiTrack for geo-temporal analysis of epitope conservation across variants, (6) selection of immunogenic and stable epitopes to inform optimal T-cell vaccine design. T-cell epitopes encoded by the BNT162b4 mRNA-based vaccine were analyzed in this study. Created in BioRender. Hamelin, D. (2024) BioRender.com/l76m979.
Results
MHCvalidator replaces Percolator for PSM confidence assessment in immunopeptidomics
MHCvalidator enables validation of PSMs from MS-based immunopeptidomics experiments, integrating both database search metrics and MHC interaction/presentation predictors into the discriminant function (for details, see https://github.com/CaronLab/mhc-validator and the Methods section ‘MHCvalidator design’). Briefly, MHCvalidator features a multi-layer perceptron (MLP) neural network validator (NN-validator) as its core component and was designed to be run in three different configurations that can be combined to obtain maximum gain in confidence of PSMs (Fig. 2a and Supplementary Fig. 1). Search results from a database search engine (e.g. Comet) are used as input files (PIN or csv).
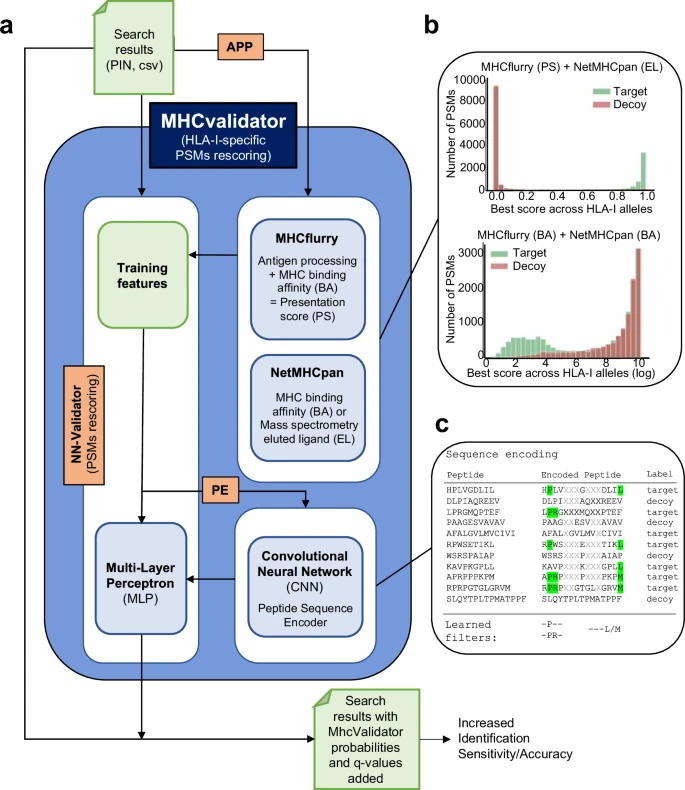
a Schematic illustrating the main components, workflow and possible configurations of MHCvalidator. The components governing the configurations of MHCvalidator are NN-validator, APP and PE (orange). NN-validator represents the core component for PSMs confidence assessment. It accepts input files (PIN, csv/tsv) and processes training features via a multi-layer perceptron (MLP); APP provides antigen processing and presentation prediction scores via MHCflurry and NetMHCpan; PE provides encoded peptide sequences via a convolutional neural network (CNN). b Example distributions of target-decoy PSMs after integration of various prediction scores generated by MHCflurry and NetMHCpan. c Cartoon illustrating the sequence encoding process and the learned filters for PSM rescoring based on sequence composition.
Using an immunopeptidomic dataset generated from JY cells (HLA-A*02:01; -B*07:02; -C*07:02), we showed superior performance of NN-validator over Percolator at all FDR below 5% (Fig. 3a), particularly in low-input samples (Supplementary Fig. 1c). In addition to NN-validator, MHCvalidator integrates two other key components: APP prediction algorithms (NetMHCpan and MHCflurry) and a convolutional neural network (CNN) based peptide sequence encoder (PE) (Fig. 2a). APP integrates prediction scores, which clearly show discriminating power in distinguishing between target and decoy PSMs from database search (Fig. 2b and Supplementary Fig. 2). PE encodes the peptide sequences and feeds them directly into the MLP of NN-validator as additional numerical features (Fig. 2c) (see Methods for details). Thus, MHCvalidator is de-facto inherently designed to generate a list of high-confidence HLA class I-specific PSMs.

The analyses were performed using immunopeptidomic MS data generated from JY cells. a Number of HLA-I-specific PSMs identified below a given FDRs by Percolator (dotted line) and four configurations of MHCvalidator (NN-validator only: blue, NN-validator and PE: green, NN-validator and APP: orange, NN-validator, PE and APP: red). b Venn diagram showing the number of high-confidence HLA-I peptides validated by MHCvalidator and Percolator. c Representative motifs extracted using the MixMHCp 2.1 tool from high-confidence peptides that were identified by both Percolator and MHCvalidator (upper motifs), and uniquely by MHCvalidator (lower motifs) in (b). d Mirror images of a representative MS/MS spectra showing alignments of fragment ions generated from Prosit prediction (bottom) vs native/endogenous peptide uniquely identified by MHCvalidator (top). Different values were generated for each peptide tested (right). Distribution of delta retention time (e), spectral angle (f), Person correlation (g) and Spearman correlation (h) for peptides uniquely identified with MHCvalidator versus those identified by both Percolator and MHCvalidator. Source data are provided as a Source Data file.
MHCvalidator outperforms percolator to identify HLA-I self-peptides in cell lines
Next, we tested the configurations of MHCvalidator in different immunopeptidomics experiments. On the dataset generated from JY cells, our results show that all MHCvalidator configurations outperformed Percolator in validating HLA-I-specific PSMs (Fig. 3a, b). Notably, the “NN-validator+PE + APP” configuration consistently delivered the most favorable results across all PSM-level FDRs below 5%. At peptide-level FDR 1%, a total of 4775 high-confidence HLA-I-specific peptides were validated by both MHCvalidator (NN-validator+PE + APP) and Percolator while 1,537 high-confidence HLA-I-specific peptides were uniquely identified by MHCvalidator (Fig. 3b and Supplementary Data 1). Percolator identified 3,238 high-confidence HLA-I-specific peptides with only 64 that were not detected by MHCvalidator. MHCvalidator (NN-validator+PE + APP) therefore yielded a ~ 150% increase in identified peptides. PSMs that were uniquely determined as high-confidence by MHCvalidator showed typical HLA binding motifs associated to A*02:01, B*07:02 and C*07:02 from JY cells (Fig. 3c). To rigorously validate these PSMs independent of motif filtering, we incorporated supplementary validation techniques such as MS/MS spectrum prediction similarity and retention time prediction (see “Methods”). A comparison was made between two groups: (1) PSMs uniquely identified by MHCvalidator, and (2) PSMs identified by both Percolator and MHCvalidator. Specifically, we computed delta retention time, spectral angle, Pearson correlation, and Spearman correlation for each PSM (Fig. 3d). Our findings indicate that PSMs validated by MHCvalidator exhibited similar value distributions to those validated by both Percolator and MHCvalidator (Fig. 3e–h). These validation steps confirm the reliability of the PSMs identified uniquely by MHCvalidator, ensuring that these peptides are not false positives and adding robustness to our findings beyond motif-based filtering. Moreover, our analysis showed that MHCvalidator outperformed DeepRescore, a Percolator-dependent complementary tool for PSM rescoring49, across all PSM-level FDRs below 5% (Supplementary Fig. 3a, b). Thus, MHCvalidator significantly boosts the sensitivity of peptide identification at a fixed FDR compared with filtering the data using individual HLA binding prediction scores post-peptide validation.
To compare the performance between MHCvalidator and Percolator for a large panel of HLA-I alleles, we used 310 publicly available MS raw files generated from 71 HLA class I mono-allelic cell lines73. For a total of 157,973 high-confidence HLA-I-specific peptides identified by both Percolator and MHCvalidator (1% FDR), 99% of those identified by Percolator were also deemed as high-confidence by MHCvalidator while 45,388 additional peptides (28%) were uniquely identified by MHCvalidator, thereby representing a 1.4-fold improvement in overall performance over Percolator (Supplementary Fig. 4a). On average, MHCvalidator yielded ~1.34, ~1.59, and ~1.97 times more high-confidence HLA-A, -B, and -C-specific PSMs compared to Percolator, respectively (Supplementary Fig. 4b). MHCvalidator under the NN-validator+PE + APP configuration achieved increased identification sensitivity relative to Percolator for 70 out of 71 alleles represented in the experiments (Supplementary Fig. 4c). We also observed that the increased PSM validation rate was HLA allele-dependent, showing a median increase of ~1.47 and reaching up to a ~5.3-fold increase for HLA-C*07:01 (Supplementary Fig. 4c). Interestingly, we observed an increase in peptide identification capabilities of MHCvalidator compared to Percolator when the ratio of target PSMs vs decoy PSMs decreases (Supplementary Fig. 4c). This hints that MHCvalidator’s benefits over Percolator (i.e. increased peptide identification sensitivity) are greater when the quality of experiments and datasets appear to decrease. In experiments where we observed a high-confidence PSM fold-increase (MHCvalidator/Percolator) greater than 4.0, 99% of the peptides identified by Percolator overlapped with those identified by MHCvalidator, while the latter enabled a 2.9-fold increase in peptide identification (3,095 vs 1,076 peptides), most of them being HLA-C peptides and matching their corresponding binding motifs (Supplementary Fig. 4c). These results suggest that MHCvalidator performs more efficiently in low-input samples (e.g. less peptides or peptides of lesser abundances) containing a lower fraction of target PSMs.
To gain a deeper understanding of the performance of MHCvalidator with low-input samples, we performed an evaluation using MS data generated from twofold serial dilutions (undiluted, 2x, 4x, 8x and 16x) of HLA-I peptides isolated from JY cells (Fig. 4a). Immunopeptidomics MS data were searched using Comet, then PSM confidence was assessed by MHCvalidator (NN-validator+PE + APP at 1% PSM-level FDR) or Percolator (1% PSM-level FDR and NetMHCpan4.1 EL %rank<2). To establish a benchmark, we employed the set of peptides deemed of high-confidence by Percolator in the undiluted sample as the benchmarking reference (Fig. 4a). We then evaluated the performance of MHCvalidator and Percolator at each dilution point relative to the benchmarking reference. Notably, MHCvalidator achieved a higher sensitivity than Percolator for peptide identifications versus the benchmarking reference at all dilution points, with the least improvement in the undiluted sample (~1.5-fold increase) and the greatest improvement in the most diluted sample (~3.6-fold increase) (Fig. 4a). We also observed that MHCvalidator consistently validated more peptides than Percolator did in the previous dilution point. For instance, MHCvalidator yielded ~3,250 high-confidence peptides in the 4x dilution, while Percolator yielded ~2500 high-confidence peptides in the 2x dilution. Furthermore, the majority of the peptides deemed high-confidence by MHCvalidator overlapped with the benchmarking reference, even in the most diluted sample in which MHCvalidator performs best. Thus, our results indicate that MHCvalidator excels at assessing the confidence of PSMs from low-input samples.

a Histogram illustrating the number of HLA-I-specific peptides that were deemed of high-confidence by MHCvalidator and Percolator (y-axis) following twofold serial dilutions of HLA-I peptides isolated from JY cells (x-axis). Fold-increase of peptides identified by MHCvalidator over that of Percolator is indicated for each dilution. The benchmarking reference used for comparisons corresponds to the peptides that were identified by Percolator in the undiluted sample (–). Legend: Peptides identified by Percolator (blue) and MHCvalidator (red) found in the benchmarking reference; high-confidence peptides not found in the benchmarking reference by Percolator (pale blue) and MHCvalidator (pale red). Distribution of XCorr values (b) and peptide length (c) for PSMs found uniquely with MHCvalidator versus those found with Percolator from the most diluted JY sample (16x). We performed a standard independent 2-sample t-test that assumes equal population variances for these instances. Box plot showing the number of HLA-I-specific PSMs “deemed high-confidence” that were found in a yeast proteome (d) or human proteome digested with Lys-C (e) using Percolator and the four configurations of MHCvalidator (NN-validator only, NN-validator and PE, NN-validator and APP, as well as NN-validator with PE and APP). Boxplots/error bars are based on 1550 samples derived from the monoallelic dataset (d). The LysC digestion analysis is based on a subset of these data, 145 samples in total that were randomly selected from the complete monoallelic dataset (e). Boxplots are given in Inter Quartile Ranges (IQRs) where the box extends from the first quartile (Q1) to the third quartile (Q3) of the data, with a line at the median. The whiskers extend from the box to the farthest data point lying within 1.5x the inter-quartile range (IQR) from the box. Flier points are those past the end of the whiskers. Source data are provided as a Source Data file.
To determine the possible features that could explain this difference in sensitivity in low-input samples, we compared quality of PSM (Xcorr values) and peptide length between PSMs identified uniquely by MHCvalidator and those identified by Percolator, specifically in the 16x-diluted sample. Our analysis shows that, on average, the Xcorr values for peptides uniquely identified by MHCvalidator were significantly lower compared to those identified by Percolator (p = 1.2'7 × 10−46) (Fig. 4b). However, we found no significant difference in peptide length between the peptides uniquely identified by MHCvalidator and those identified by Percolator (p = 0.072) (Fig. 4b). These results indicate that one of the differences lies in the quality of PSMs, suggesting that MHCvalidator has a higher sensitivity for detecting peptides with lower values for this identification confidence score compared to Percolator.
To rigorously evaluate the specificity of MHCvalidator compared to Percolator, we designed challenging test scenarios in which a human immunopeptidomic dataset was searched against (1) a S. cerevisiae/yeast proteome (no enzyme search), and (2) a human proteome (LysC-digest search). The rationale behind this choice stems from the distinct peptide composition of yeast compared to humans, as well as the presence of larger peptides in a LysC-digested human proteome in contrast to tryptic peptides.
This test involved 29 MS files generated from the HLA-I monoallelic cell lines. This MS dataset was searched against the yeast proteome and the LysC-digested human proteome. Subsequently, the results were subjected to confidence assessment using (1) the four different configurations of MHCvalidator (NN-Validator, PE, APP, PE + APP), and (2) Percolator. To ensure specificity, peptide sequences that were identical in the yeast and human digest databases were removed from the yeast peptide database. Upon examination of the distributions of PSMs deemed of high confidence across the various MHCvalidator configurations and Percolator, we observed that <25 PSMs in average were from the yeast proteome search and therefore constitute likely false positives. This was observed for all configurations, which also displayed visual similarities with Percolator’s observed distribution, and with no statistically significant differences (p value = 0.7369) in terms of the number of yeast PSMs deemed of high-confidence (Fig. 4d and Supplementary Fig. 3c). Similar results were generated from the LysC-digested human proteome (Fig. 4e and Supplementary Fig. 3d). These results suggest that (1) the addition of the APP and PE training features did not allow MHCvalidator to memorize specific PSMs to boost the number of IDs, and that the identification of false-positive PSMs remained a random process; and (2) that MHCvalidator’s false-positive identification rate is similar to Percolator’s.
Taken together, our analyses demonstrate that MHCvalidator is highly sensitive and specific, outperforming Percolator for robust confidence assessment of HLA-I-specific PSMs in immunopeptidomics experiments.
MHCvalidator identifies known and novel MS-detectable SARS-CoV-2 HLA-I peptides with canonical and non-canonical properties
To demonstrate the potential of MHCvalidator for the unbiased discovery of potential CD8+ epitope vaccine candidates, we sought to reanalyze immunopeptidomics data generated from SARS-CoV-2 infected cells23. In the original study, three cell lines expressing different combinations of HLA-I alleles were infected: Calu-3 (HLA-A*24:02, -A*68:01, -B*07:02, -B*51:01, -C*15:02), IHW01070 (HLA-A*01:01, -A*02:01, -B*08:01, -B*40:01, -C*04:04, -C*07:01) and HEK293T cells (HLA-A*02:01, -A*03:01, -B*07:02, -C*07:02). Here, the same raw MS data were searched with Comet against the human proteome and the same SARS-CoV-2 proteome used in the original publication, and the resulting PSMs were then rescored using MHCvalidator (NN-validator+PE + APP) or Percolator, both at PSM-level FDR 5%. Notably, Comet results that were validated with MHCvalidator achieved a ~ 2.2-fold increase in the number of confidently identified HLA-I SARS-CoV-2 peptides (24 peptides) in comparison with the original method (11 peptides) (Fig. 5a, b). In contrast, Comet results that are deemed high-confidence by Percolator (PSM-level FDR 5% and NetMHCpan %rank<2) yielded only 6 high-confidence HLA-I SARS-CoV-2 peptides, all of which were also deemed high-confidence by MHCvalidator (Supplementary Fig. 4d). None of the identified SARS-CoV-2 peptides were detected in the non-infected cells. The identified SARS-CoV-2 peptides were then assigned to their respective HLA-I allele using prediction scores generated by NetMHCpan57 and HLAthena73 (Fig. 5b and Supplementary Fig. 5). Using an in vitro HLA binding assay, most peptides confidently assigned to a specific HLA allele expressed in the corresponding cell line were confirmed to bind their respective HLA allele (Fig. 5c, d). Furthermore, we gained confidence in the amino acid sequences of the MS-detectable SARS-CoV-2 peptides by comparing the tandem mass spectra of synthetic peptides with the experimental spectra and observed high correlation between fragment ions (average Pearson r for all peptides = 0.9) (Fig. 5e and Supplementary Fig. 6).

a Venn diagram showing the number of high-confidence SARS-CoV-2 HLA-I peptides identified by the original method described by Nagler et al., and by MHCvalidator’s optimal configuration (NN-validator+PE + APP). Overlapping peptides are shown. Peptides selected for immunogenicity experiments are also indicated. b Table showing the list of SARS-CoV-2-derived peptides identified by MHCvalidator. Source protein, NetMHCpan/HLAthena prediction score and HLA allele assignment are indicated in the table. A reference number is shown for peptides that have already been detected by MS in previous studies; if not detected before by MS, ‘New’ is indicated. ND: not determined. c Histogram showing the proportion of confirmed assigned peptides (y-axis) for their respective HLA-A or -B allele (x-axis). HLA assignment was predicted in (b), and confirmed by in vitro HLA binding assay. Number of peptides (assigned/total) per allele is shown on top of each bar. d Heatmap illustrating the measured binding affinity (IC50 nM) across different HLA-A and -B alleles for all assigned peptides in (b). e Mirror spectral image showing alignments of fragment ions in MS/MS spectra of synthetic vs native MHCvalidated-peptides. Two representative peptides tested for immunogenicity are shown along with the Pearson correlation coefficient between the two MS/MS spectra. f Peptides were classified into five categories. Source data are provided as a Source Data file.
Out of eleven SARS-CoV-2 peptides that were identified in Nagler et al., seven were confirmed using our method (Fig. 5a). Those peptides include STTTNIVTR (A*68:01, nsp3), HSSGVTREL (C*15:02, nsp1) and TGSNVFQTR (A*68:01, S), as well as three Nucleocapsid (N)-derived peptides with overlapping amino acids, i.e. RITFGGPSD (unassigned), NAPRITFGGP (B*07:02) and APRITFGGP (B*07:02) (Fig. 5b). Notably, we also confirmed the identification of the non-canonical out-of-frame peptide GPMVLRGLIT (B*07:02), which originate from S.iORF1/2 (ORF9a), as evidenced with translations by ribo-seq74, and detected by MS in another independent study22 (Fig. 5b).
In addition to the above previously reported peptides, a set of 17 SARS-CoV-2 peptides were physically detected for the first time by MS. These MS-detectable SARS-CoV-2 peptides were classified into five different categories: (1) non-canonical out-of-frame, (2) non-canonical junction-driven, (3) canonical in-frame, (4) overlapping, and (5) > 11-mers (Fig. 5f). Notably, we discovered one novel non-canonical out-of-frame peptide RLGSPLSL (C*15:02) originating from N.iORF1/2, eight novel canonical in-frame peptides originating from both structural and non-structural SARS-CoV-2 proteins (S, nsp3, nsp6, nsp13, nsp16), a subset of five unassigned additional N-derived peptides sharing the same overlapping amino acid characteristics as mentioned above, and two unassigned 12- and 13-mers originating from N and ORF7a, respectively (Fig. 5b, f).
In-depth analysis of all MHCvalidator-confirmed peptides using a ribo-seq-derived database revealed an unexpected category of non-canonical HLA-I SARS-CoV-2 peptide: the non-canonical junction-driven peptides LPYPQILLL. This peptide originates from a shorter/truncated version of the S antigen (Fig. 6a) resulting from a short deletion→fusion event (or junction), which involved the removal of 31 nucleic acids at position5’23594-236243’ (Fig. 6b). Interestingly, this deletion occurs at a furin-like cleavage site and was recently discovered and referred to as a “leader-independent junction“74. Most importantly, the junction event creates an altered reading frame ( + 1-frameshift), and consequently, translation of the non-canonical peptide LPYPQILLL followed by a premature stop codon (Fig. 6b). In vitro HLA binding assay showed that the LPYPQILLL peptide strongly binds two common HLA-B7 supertype alleles (B*07:02 and B*51:01), and predicted to bind additional common alleles of the same supertype (Figs. 5d, 6b). To our knowledge, this is the first time that a junction-driven HLA-I peptide has been reported. Together, MHCvalidator identified MS-detectable HLA-I SARS-CoV-2 peptides spanning both canonical and non-canonical properties.

a Amino acid sequence in the Wuhan-1 (wild-type) and the truncated (deletion) Spike proteins. The uniquely generated peptide sequence due to the deletion is highlighted in brown. The LPYPQILLL peptide is emphasized by being bolded and circled. Created in BioRender. Hamelin, D. (2024) BioRender.com/k07e042. b The deletion (or leader-independent junction) from position 5’−23594 to 23624-3’ at the mRNA level, and the resulting +1 frameshift at the amino acid level is illustrated. Measured (non-italic) or predicted (italic) HLA binding affinity of the junction-dependent peptide LPYPQILLL (orange) is indicated for several HLA-B alleles, which all belong to the B7 supertype family. c Histogram illustrating the number of patients from the intra-host database showing a deletion/junction-driven +1 or +2 frameshift, or no frameshift (in-frame), in more than 100 reads. Deletions were analyzed between position 5’−23,623 and 23,693-3’. d Table and violin plot indicating the lengths of the deleted nucleic acid sequences (average, max and min) leading to in-frame, +1 or +2 frameshift. Source data are provided as a Source Data file.
Intra-host analysis of the non-canonical junction-driven B7 epitope encoded by the truncated S antigen
Numerous transcripts originating from non-canonical junctions have been documented for SARS-CoV-2 in vitro75,76. Hence, we hypothesized that multiple non-canonical junction/truncation events might occur within the S antigen to generate +1-frameshifts during SARS-CoV-2 infections in vivo, possibly leading to production and presentation of the non-canonical junction-driven peptide in B7+ individuals. To test this, we built and interrogated a unique intra-host dataset comprising 100,512 high-quality RNA libraries sequenced from 100,512 infected COVID-19 patients (see Methods for details). This dataset provides a comprehensive representation of intra-host variations in SARS-CoV-2, including mutations and non-canonical junctions that may have arisen during the course of infection in a large and diverse cohort of patients. Using this unique intra-host dataset, we searched for deletions (junctions/truncations) of any lengths in the vicinity of the transcriptomic region described above (between the genomic positions 23,623 and 23,693 of the S antigen), that could induce the necessary frameshift to produce the LPYPQILLL peptide. Our analysis revealed that out of 100,512 COVID-19 patients, ~1100 of them (~1%) had a deletion in the region of interest, each deletion supported by more than 100 reads (Fig. 6c and Supplementary Fig. 7a). Notably, ~850 patients (~0.8%) showed a predominant +1-frameshift, resulting in the coding of the non-canonical junction-driven LPYPQILLL peptide (Fig. 6c and Supplementary Fig. 7a). Deletion lengths were highly variable, ranging from 1 to 100 nucleic acids, with an average deletion length of 1.3 nucleotides (Fig. 6d and Supplementary Fig. 7b). Moreover, we noted that ∼25% of the observed +1 frameshift events can be attributed to two specific deletions affecting a single nucleotide at positions 5’-23649 (T) and 5’-23657 (T) (Supplementary Fig. 7c). Assuming that our intra-host dataset is representative of the SARS-CoV-2 infected human population, our results suggest that ∼0.8% of B7+ individuals may exhibit presentation of the LPYPQILLL peptide during infection. Given that 35% of the human population are B7+77, our analysis hints at the possibility that ∼0.3% of the human population could present the non-canonical junction-driven S epitope to CD8 + T cells. However, further experiments are essential to rigorously test and validate this observation in future studies. If validated, frameshifted viral antigens generated within the host during infections could constitute a currently untapped reservoir of T-cell epitopes, with the potential to play a role in infection control, disease severity, and vaccine design.
SARS-CoV-2 HLA-I peptides uncovered by MHCvalidator elicit CD8 + T-cell responses in individuals with COVID-19
To evaluate the immunogenicity of the HLA-I SARS-CoV-2 peptides detected by MS and MHCvalidator, including the junction-driven B7 peptide described above, we next performed ELISpot assays with peripheral blood mononuclear cells (PBMCs) from HLA-matched COVID-19 convalescent individuals [(A*68:01; n = 10), (B*07:02; n = 14), (A*68:02; n = 10), (A*02:01; n = 25), (B*51:01; n = 12)] and monitored IFNγ secretion in response to each peptide validated by MS and MHCvalidator (Fig. 7a, b). As positive controls, we compared the T-cell responses with S MegaPool and PepPool CEF, as described78. We also used the peptide YLQPRTFLL (A*02:01) as positive control since it was observed as the most reactive/immunodominant SARS-CoV-2 CD8+ epitope in several independent studies79,80,81,82. Interestingly, the non-canonical out-of-frame peptide GPMVLRGLIT (B*07:02), previously documented as non-immunogenic22, inducted a relatively potent CD8+ response in one particular B*07:02-matched individual (~500 SFU/106 PBMCs) (Fig. 7a). Furthermore, we show that the non-canonical junction-driven peptide LPYPQILLL induced a CD8+ response in ~7% and ~42% of B*07:02 and B*51:01 individuals, respectively (Fig. 7a, c). As expected, the immunodominant peptide YLQPRTFLL elicited a CD8+ response in 11 out of 25 HLA-A*02:01 individuals (~44%). Overall, ~85% (11 out of 13) of all peptides binding their respective HLA-I allele(s), and tested for immunogenicity, elicited CD8 + T cell responses in HLA-matched individuals (Fig. 7c). Consistently, the MHCvalidator-identified peptides induced a positive CD8+ response with an average frequency of ~25% ± ~16% (Fig. 7d). Response frequency was peptide-dependent and showed a correlation value (r) of 0.568 with predicted HLA binding affinity (Fig. 7d).

a Graph showing IFNγ secreting cells per million (y-axis) in response to the peptides identified by MS and MHCvalidator (x-axis). Data were generated by ELISpot for the indicated HLA types. N: number of HLA-matched PBMCs/individuals tested. The immunodominant peptide YLQPRTFLL is indicated as positive control (+Ctrl); ratio of individuals responding to it is indicated (red). b Representative well image of ELISpot assay. c Pie chart showing the fraction of MHCvalidator-discovered peptides tested for immunogenicity by ELISpot. Tables showing peptide sequences, rate of HLA-matched individuals responding to the corresponding peptide, and immune epitope database (IEDB) identification number (ID). Novel immunogenic peptides (orange) and previously reported immunogenic peptides (blue). d Graph showing correlation between predicted HLA binding affinity (y-axis) and response frequency by ELISpot (x-axis). The A*02:01- and A*68:01-associated peptide RTIKVFTTV, shown to be immunogenic by ELISpot and DNA-barcoded pMHC multimers is indicated. e Peptide-specific T-cell responses identified using DNA-barcoded pMHC multimers in four patients in the acute phase of SARS-CoV-2 infection. Confirmed response are colored and the size of the colored dots is according to the estimated frequency. Two patients with RTIKVFTTV and YLQPRTFLL are indicated. Source data are provided as a Source Data file.
To further strengthen our immunogenicity data, we compared our list of MHCvalidator-identified peptides to all experimentally validated reactive SARS-CoV-2 peptides found in the Immune Epitope Database (IEDB). Notably, 10 out of 13 MHCvalidator-identified SARS-CoV-2 HLA-I peptides were annotated with an IEDB ID (2023/10/10) (Fig. 7c). For instance, the immunogenicity of the peptide RTIKVFTTV (A*02:01), measured by MS and ELISpot in our study, was previously validated by a DNA-barcoded peptide-MHC multimer assay (Fig. 7e)83. Moreover, three MHCvalidator-identified peptides, shown as immunogenic in our study, have never been reported before (Fig. 7c). Together, this study provides a proof-of-concept that MHCvalidator enables unbiased discovery of immunogenic viral T-cell epitopes from infected cells.
MHCvalidator confirms presentation of non-spike epitopes encoded by the T-cell-directed vaccine BNT162b4
To showcase the effectiveness of MHCvalidator in the context of T-cell-directed vaccines, we conducted a reanalysis of immunopeptidomic data generated by DDA MS. The data originated from HLA-I monoallelic cell lines transfected with the BNT162b4 mRNA vaccine currently being clinically evaluated (NCT05541861)12. Applying Comet+MHCvalidator (NN-validator+PE + APP) for immunopeptidomic MS data analysis, we successfully identified six high-confidence SARS-CoV-2 HLA-I peptides encoded by BNT162b4 (Fig. 8a and Supplementary Fig. 8). All high-confidence peptides were predicted to be localized in non-membrane regions according its Protter topology (Supplementary Fig. 8)84. None of these peptides were detected from the corresponding non-transfected cells. This finding is consistent with the outcome of the original study, where identification of the exact same peptides was achieved using the Spectrum Mil MS Proteomics Software v6.012, hence providing robust validation. For instance, MHCvalidator confirmed presentation of the peptide TTDPSFLGRYM (nsp3; A*01:01) and its variant form TTDPSFLGRY (nsp3; A*01:01), the latter reported as highly immunodominant, particularly in hospitalized patients83. In addition, MHCvalidator confirmed presentation of another variant of this peptide from infected cells [HTTDPSFLGR (nsp3; A*68:01)] (Fig. 5). Notably, among the 16 MHCvalidator-identified CD8+ epitopes presented by SARS-CoV-2-infected cells or BNT162b4-transduced cells, 9 of them (56%) are encoded by nsp3. This observation suggests that nsp3 could potentially serve as a dominant source of protective epitopes for the development of next-generation T-cell-directed vaccines. Thus, MHCvalidator offers evidence of epitope presentation in immunopeptidomics experiments associated with T-cell-directed vaccines designed to protect against hypermutated SARS-CoV-2 variants.

a Schematic of the BNT162b4 mRNA vaccine. b Comprehensive (GISAID, 2020-2023) mutation rate of CD8+ epitopes identified from SARS-CoV-2-infected cells (Orange); BNT162b4 mRNA vaccine (Green); and a control consisting of 9-mers spanning the complete SARS-CoV-2 proteome (White). For all epitopes shown, the rate of mutation was expressed as the number of alternative epitopes found across the GISAID database (with a minimum of 10 GISAID sequences per alternative epitope) divided by the total number of GISAID sequences for which the epitope had sequencing coverage, presented in log10. c (Bottom) Proportion of GISAID sequences over time (2020-2023) for which the TTDPSFLGRY epitope (BNT162b4 mRNA vaccine, MHC-Validator-identified) was unmutated (Cyan) or mutated (purple, dark blue, light blue and green, in order of descending prevalence). Only top alternative epitopes (found in >1000 GISAID sequences) shown here. (Top) cumulative count of GISAID sequences over time. d Variant of Concern (VOC) associated with top alternative epitopes. The color scale corresponds to the number of GISAID sequences for which an alternative epitope is associated with a VOC. e Geographic map of the prevalence of top TTDPSFLGRY alternative epitopes (top: TTDP/LSFLGRY, Delta; bottom: TTDP/SSFLGRY, Omicron), with a focus on European countries. The color scale represents the proportion of GISAID sequences generated by each country featuring the alternative epitope in question, thus normalizing for country-specific sequencing bias. Source data are provided as a Source Data file.
EpiTrack enables geo-temporal conservation analysis of vaccine-relevant, MHCvalidator-identified CD8+ epitopes
There is currently a lack of information concerning the mutational profile of SARS-CoV-2 epitopes recognized by BNT162b4-induced CD8 + T cells. To gain knowledge in this regard, we developed EpiTrack and analyzed the mutational landscape of 16 MHCvalidator-identified CD8+ epitopes, including 6 encoded by the T-cell directed BNT162b4 vaccine. Global and temporal analysis of these epitopes was possible thanks to the extensive genome sequencing initiatives for SARS-CoV-2. Briefly, we compiled an exhaustive list of pandemic-wide alternative epitopes by extracting and translating the relevant nucleotide sequences taken from GISAID from the final dataset (see “Methods”). To gain insight into the evolutionary trends of each peptide, the prevalence and geo-temporal dynamics of all respective alternative peptides were tracked both worldwide and regionally using EpiTrack. All analyses were repeated on a set of end-to-end 9-mers spanning the entire SARS-CoV-2 canonical proteome to assess mutational dynamics in the context of the viral proteome. Overall, 11 of 16 MHCvalidator-identified CD8+ epitopes, including 4 of 6 BNT162b4 vaccine epitopes, show negligible diversification across the pandemic, with at most 1% of sequences worldwide carrying alternative epitopes (Fig. 8b). The remaining 5 peptides, including two BNT162b4 vaccine epitopes, carry mutations with frequencies ranging between 4.4% and 53.7% of sequences. Amongst these, BNT162b4 vaccine epitopes TTDPSFLGRY and TTDPSFLGRYM are of particular clinical interest due to their significant immunodominance83. Specifically, two mutations occurred within both epitopes, namely ORF1a/nsp3 P1640L and P1640S (Fig. 8c–e and Supplementary Fig. 9f). The former, identified in 404,743 GISAID sequences, was found amongst Delta sub-lineages, while the latter was identified within 200,770 GISAID sequences and found amongst Omicron sub-lineages (Fig. 8d). Neither mutation is predicted to abrogate the presentation of either peptide by its respective HLA allele, A*01:01, likely due to their occurrence on a non-anchor residue. Predictions using the IEDB immunogenicity predictor85 suggest that replacing proline might negatively impact the immunogenicity of both epitopes; however, these predictions should be approached with caution and validated with experiments. Other highly mutated MHCvalidator-identified epitopes included B*07:02 epitopes NAPRITFGGP and RANNTKGSL, mutated in 53.7% and 7.5% of sequences, respectively (Supplementary Fig. 9a and Fig. 8b). These findings put in evidence the non-trivial mutational dynamics of immunodominant, vaccine-relevant epitopes, thus promoting the need for continued monitoring of evolutionary trends within T-cell vaccine candidates.
Discussion
MS-based immunopeptidomics has emerged as a valuable strategy for identifying naturally presented HLA-associated peptides, offering insights for the design of T-cell vaccines against a spectrum of diseases, including cancer, viruses and other pathogens86,87,88,89. However, the continued advancement of this technique necessitates hardware and software solutions to enhance its robustness, sensitivity and specificity, ultimately expanding its deployment and impact in vaccinology and immunology16. Developing accurate computational frameworks and analysis platforms is important for assessing the efficacy of next-generation T-cell vaccines, especially in the context of rapidly mutating viruses such as SARS-CoV-2. To address this need, we have developed an approach aimed at unbiased identification of viral T-cell epitopes. Central to our approach is MHCvalidator, a ML method tailored for the confidence assessment of PSMs obtained from immunopeptidomics experiments. Its design allows all high-confidence PSMs to be considered as likely interesting antigenic peptides, eliminating the need for a post-validation filtering step. In the current version of MHCvalidator, two configuration modules are available: APP and PE. The latter option aims to address scenarios where MHC binding motifs are not well characterized, such as with non-classical MHC molecules like HLA-E90 or in species with unique MHC systems, like bats91. In these cases, the neural network cannot exclusively rely on known binding motifs (APP module) for accurate predictions. The PE option, when combined with APP also provides the most sensitive PSM identifications with high confidence. The PE option may therefore offer value by identifying other significant features or signals within the data, potentially enhancing the accuracy of the model’s predictions. Furthermore, MHCvalidator is highly versatile, capable of integrating various data property, probability, or scores as features to discriminate false from true PSMs. In principle, this flexibility enables the incorporation of additional numeric peptide feature predictions, such as retention time92, fragment intensities in MS2 spectra93,94,95, ion mobility coefficient/collisional cross sections64,96, HLA ligand presentation55,73,97, and immunogenicity54,98 into MHCvalidator’s input. Such additional features could improve the performances of MHCvalidator in the future. In addition, while the current version of MHCvalidator is designed for HLA-I immunopeptidomics experiments, its adaptable framework theoretically allows for a similar approach for HLA-II experiments. Moreover, other processes of APP acting at proximal regions of epitopes could also be integrated into the modeling process of MHCvalidator when subjected to scoring. This has relevance in the context of SARS-COV-2 variants since the SARS-CoV-2 Omicron BA.1 spike G446S mutation, located just outside the N-terminus of a cognate CD8 + T-cell epitope, was recently shown to improve antigen processing/presentation and antiviral T-cell recognition through tripeptidyl peptidase II (TPPII), a post-proteasomal protease that mediates antigen processing99. Furthermore, MHCvalidator, by learning and incorporating rules and predictions of cryptic peptides, including polypeptides created by posttranslational peptide splicing100,101,102,103,104, could become a valuable tool for discriminating between true and false spliced peptides without the need for additional experiments to validate their identifications. If further developed and tested, MHCvalidator could therefore be applied to continue the development of databases dedicated to immunopeptidomics, such as SysteMHC Atlas105,106,107. Thus, the first version of MHCvalidator represents a foundational database search-based PSM confidence assessment tool in immunopeptidomics, similar to what DTASelect108 and PeptideProphet42 were upon their creation in proteomics and were later followed by Percolator45.
In our study, MHCvalidator enabled the validation of non-canonical SARS-CoV-2 T cell epitopes through three distinct approaches: validation of peptide sequences using synthetic peptides, in vitro HLA peptide binding assays, and T cell immunogenicity assays. T cell immunogenicity was assessed using ELISpot assays on HLA-typed PBMCs sourced from a local cohort of convalescent COVID-19 patients109. We recognize the limitations associated with our sample size, encompassing 10 to 25 data points per peptide-HLA combination. This sample size was selected based on our prior experiences in COVID-19 research, where peptide pools successfully stimulated PBMCs, leading to robust T-cell responses detectable by ELISpot assays, and permitted statistical analyses78,110. In this study, we employed a Binary Response Presentation strategy, resonating with methodologies in existing literature, particularly in scenarios where minimal responses are evoked by single peptide stimulation111,112. Using the immunodominant A*02:01 peptide YLQPRTFLL, nearly half of the patients (11 out of 25) exhibited positive responses to stimulation, aligning with established expectations. In addition, the immunogenicity of identified peptides was confirmed when defining a response as positive if there was more than a twofold increase in specific spot number relative to the negative control, a threshold for positivity used in some studies113. Thus, despite the potential concerns about sample size and the binary nature of our data interpretation, we argue that our methodology is well-supported by experimental context and established precedents in the field. Our results provide a crucial cornerstone for subsequent studies, which would ideally include larger cohorts.
An interesting observation in our study was the detection of a B7-associated SARS-CoV-2 non-canonical epitope generated by a truncated version of the S antigen. This truncated version occurs at a junction-dependent region, first reported by Finkel et al.74. This junction was initially observed in a cell line and was unique to their dataset. In our study, we first wanted to verify if the exact same deletion was present in vivo within infected patients to validate the observation made by Finkel, and to estimate the prevalence of this non-canonical T-cell epitope in humans. To achieve this, we built and interrogated our intra-host database composed of thousands of SARS-CoV-2 sequences that were isolated and sequenced directly from infected individuals. Such databases have indeed proven to be increasingly powerful to track intra-host variation and evolutionary dynamics of SARS-CoV-2 populations in COVID-19 patients68,69,70,114. Interestingly, we did not find the exact same deletion as observed in vitro, but we did observe a large number of deletions of various lengths (1 to 100 nucleic acid) in several regions of S leading to altered reading frames. Notably, among ∼100,000 COVID-19 patients, ∼1100 had a deletion, and ∼850 exhibited a + 1-frameshift resulting in the coding of the non-canonical B7 epitope for reads detected at >100 copies. Whether the expression levels are sufficient to present the non-canonical B7 epitope requires further exploration. Nevertheless, it is tempting to speculate that producing of a truncated version of S, likely nonfunctional, could represent a form of defective ribosomal product (DRiP) that would be rapidly targeted to the proteasome for quick degradation and subsequent epitope presentation115. Given the presence of such frameshifted antigens, non-canonical polypeptides could be processed similarly during infection, possibly representing an untapped source of frameshifted T-cell epitopes to combat viral infection. Whether this phenomenon is specific to S, other SARS-CoV-2 antigens, or any viruses, remains an open question, and would require larger intra-host databases from diverse viral species. Nevertheless, understanding the significance of those unexplored non-canonical epitopes in controlling infection is paramount and necessitates further investigation. Additionally, exploring whether specific truncation events are associated with distinct clinical phenotypes (e.g., long COVID) could offer valuable insights. Such knowledge might pave the way for designing phenotype-specific vaccines to address the unique needs of affected individuals.
As new SARS-CoV-2 variants continue to emerge, recent reports have provided compelling evidence of mutations within immunodominant T-cell epitopes presented by prevalent HLA molecules116,117,118. This holds significant implications for T-cell evasion, including intra-host T cell evasion observed within immunocompromised patients119, and the emergence of hypermutated variants like BA.2.86, which was speculated to possess higher potential for evading T-cell immunity in larger populations120,121. The extensive diversity of HLA alleles at the population level somewhat mitigates concerns regarding mutations affecting T-cell epitopes. Nevertheless, our study underscores that maintaining a vigilant tracking of the mutational dynamics of T-cell vaccine targets is an important process to ensure the sustained efficacy of T-cell-directed vaccines over the next decades. This holds particular significance for vaccines that incorporate only a limited number of epitopes, as exemplified by the CoVac-1 T-cell vaccine, which comprises only six synthetic peptides9. In our study, we investigated the mutational dynamics of MHCvalidator-identified peptides encoded by the T-cell vaccine BNT162b4, detecting six peptides despite the mRNA coding for 2,257 unique predicted peptide-HLA-I pairs across 105 HLA-I alleles. Potential explanations for this discrepancy include the sensitivity of the detection method not being sufficiently high or the destruction of most peptides by intracellular proteases. Notably, we observed that the vaccine protein’s topology includes membrane regions and a potential localization within the endoplasmic reticulum (ER) membrane. If accurate, the ER-associated degradation (ERAD) pathway could play a role in displacing such proteins from the ER membrane, facilitating access to the proteasome for protein degradation and subsequent peptide generation and presentation122. However, it is conceivable that this process may not be optimal for efficient peptide presentation and for inducing a robust T-cell response against the vaccine targets. In this context, immunopeptidomics emerges as a valuable tool to identify and quantify the absolute abundance of peptides presented, employing various protein engineering designs123,124. Future research endeavors will be essential to address these questions and further refine our understanding of the intricate interplay between vaccine design, antigen processing pathways, and the subsequent T-cell immune response against hyperconserved epitopes.
Recently, modeling methods have emerged to predict mutations in future variants of concern125. With further refinement in the context of T-cell epitopes, these modeling approaches could significantly enhance our ability to predict the probability of mutations in T-cell vaccine targets. This, in turn, would allow us to foresee the durability of T-cell vaccines right from the outset of a pandemic. The integration of the machine learning-enhanced immunopeptidomics method and the global epitope conservation analysis presented in this study represent a significant step in this direction. As this framework undergoes further development, we envision its potential to build and track an evolving digital model of the actionable SARS-CoV-2 immunopeptidome. Such a model would be instrumental in informing the formulations of next-generation vaccines not only against SARS-CoV-2 variants but also against other rapidly evolving pathogens86,87,88. The continuous refinement of this approach holds promises for enhancing our ability to adapt and respond effectively to the dynamic landscape of viral evolution in the development of protective T-cell-directed vaccines.
Methods
Ethics
Our research complies with all relevant ethical regulations. RECOVER protocols were approved by the Research Ethics Board (REB) at the Sainte-Justine University Hospital and Research Center under study MP-21-2021-3035 and in each of the five participating centers in the Province of Québec. Written informed consent was obtained from all participants during the recruitment period, and ongoing consent was reviewed at each subsequent visit. The sex of participants was determined by self-reporting and considered in the study design, ensuring that symptomatic and asymptomatic groups were matched for sex, age, ethnicity, and other factors. No specific gender-based analysis was conducted as the primary focus was on immune responses to SARS-CoV-2 injection. Detailed sex-disaggregated data is provided in the supplementary materials and source data files in Nantel et al.78.
Cell line and reagents
JY cell line (human lymphoblastoid B-cells) was purchased from ATCC and cultured in RPMI 1640 supplemented with 10% FBS and 1% pen/strep. Anti-human HLA-A, -B, -C (W6/32, #BE0079) was purchased from BioXcell, Polyprep chromatography column (#7311553) and Combined inhibitor EDTA-free (#A32961) from Bio-Rad and Solid phase extraction disk ultramicrospin column C18 (#SEMSS18V, 5–200 µl) from The Nest Group. 1.5 ml and 2.0 ml microcentrifuge tubes (Protein LoBind Eppendorf #022431081 and #02243100), Low retention tips Eppendorf (10 µl #2717349, 20 µl #2717351, 200 µl #2717352), acetonitrile (#A9964), trifluoroacetic acid (TFA, #AA446305Y), formic acid (#AC147930010), chaps (#22020110GM), PBS (Buph, phosphate buffer saline packs, #28372), CNBr activated sepharose 4B (#45000066) and ammonium bicarbonate (#A643-500) were purchased from Fisher.
Cell culture and immunopurification of HLA-class I peptides from JY cells
JY cells were seeded at 0.5 × 106 cells/ml, incubated at 37 C° with 5% CO2 and expanded to obtain 100 million cells. Cells were harvested and centrifuged at 180 × g for a period of 5 min at room temperature. The culture medium was removed by aspiration and the cell pellets were washed gently by pipetting up and down with 5 ml of PBS and centrifuge again. After removing the PBS by aspiration, the cell pellets were stored at –80 degrees Celsius until used.
Immunopurification of HLA-class I peptides32,63. To isolate MHC class I peptides, a frozen pellet of 1 × 108 cells was resuspended in 500 µL of PBS by pipetting up and down until homogenization. The volume of the cell pellet suspension was measured and transferred into a new tube 2 mL microcentrifuge tube. Equivalent volume of cell lysis buffer (1% chaps in PBS containing protease inhibitors, 1 pellet/10 mL) was added to the cell suspension (final concentration of the lysis buffer of 0.5% Chaps), followed by an incubation for 60 min at 4 °C using a rotator device and centrifugation at 18,000 × g for 20 min at 4 °C. The cell lysis supernatant containing the MHC-peptides complexes was transferred in a new 2.0 mL microcentrifuge tube and kept on ice until used for the immunopurification. Next, 80 mg of sepharose CNBr activated beads were coupled with 2 mg of antibody. Sepharose antibody-coupled beads were incubated with the cell lysate supernatant in a 2.0 ml Low binding microcentrifuge tube overnight at 4 °C with rotation. The next day, a Bio-Rad column was installed onto a rack and pre-rinsed with 10 ml of buffer A (150 mM NaCl and 20 mM Tris–HCl pH 8). The beads-lysate mixture was transferred into the Bio-Rad column and the bottom cap was removed to discard unbound cell lysate. Beads retained in the Bio-Rad column were washed sequentially with 10 ml of buffer A (150 mM NaCl and 20 mM Tris–HCl pH 8), 10 ml of buffer B (400 mM NaCl and 20 mM Tris–HCl pH 8), 10 ml of buffer A and 10 ml of buffer C (20 mM Tris–HCl pH 8.). MHC-peptides complexes were eluted from the beads by adding 300 µl of 1% TFA, pipetting up and down 4–5 times and collecting the flowthrough. This step was repeated once and the flowthroughs were collected and combined in a new 2.0 ml tube. MHC class I peptides were desalted and eluted using a C18 column. First, the C18 column was pre-conditioned with 200 µl of (1) methanol, (2) 80% acetonitrile/0.1%TFA and (3) 0.1%TFA and spun at 1545 × g in a fixed rotor to collect and discard the flowthroughs. Then, the MHC-peptides complexes previously collected in 600 µl of 1% TFA were loaded (3 × 200 µl) into the pre-conditioned C18 column, spun and flowthroughs were discarded. A final wash was performed with 200 µl of 0.1% TFA and spun again. Finally, the C18 column was transferred onto a 2.0 ml Eppendorf tube and MHC class I peptides were eluted with 3 × 200 µl of 28%ACN 0.1%TFA. The flowthrough containing the eluted peptides was stored at –20 degrees Celsius for MS analysis. Prior to LC-MS/MS analysis, the purified MHC class I peptides were evaporated to dryness using a vacuum concentrator with presets of temperature 45 °C, for 2 h, vacuum level: 100 mTorr and vacuum ramp: 5.
MS/MS analysis and peptide identification from JY cells for the serial dilution experiment
Vacuumed sample 1 (undiluted) was resuspended in 50 µl of 4% formic acid (FA). Twofold dilution was performed by mixing 25 µl of undiluted sample with 25 µl of 4%, and so on. Two technical replicates of 10 µl per sample were loaded and separated on a home-made reversed-phase column (150-μm i.d. by 250 mm length, Jupiter 3 µm C18 300 Å) with a gradient from 5.6 to 30% ACN-0.1% FA and a 600-nl/min flow rate on an Easy nLC-1000 connected to an Orbitrap Eclipse (Thermo Fisher Scientific). Each full MS spectrum was acquired at a resolution of 240000, an AGC of 4E5 and an injection time of 50 ms, followed by tandem-MS (MS-MS) spectra acquisition on the most abundant (Top 10) multiply charged precursor ions for a maximum of 3 s. Tandem-MS experiments were performed using higher energy collisional dissociation (HCD) at a collision energy of 34%, a resolution of 30000, an AGC of 1.5E5 and an injection time of 300 ms.
Mass spectrometry database search
Raw mass spectrometry files were converted to mzML format using ThermoRawFileParser v 1.3.4126. All data was searched using the Comet search engine (v. 2021 rev 0) with the following settings: precursor mass tolerance: 10 ppm; fragment bin size: 0.02 Da; peptide length range: 8–15 amino acids; digest enzyme: non-specific; charge state: 1–4; output format: PIN. For JY cell lines and SARS-CoV-2 infected cell lines, no fixed modifications were used and variable modifications were set to deamidation of asparagine and glutamine and oxidation of methionine. For the mono-allelic cell lines, carbamidomethylation of cystein was set as a fixed modification and variable modifications were deamidation of asparagine and glutamine and oxidation of methionine with a maximum of 3 variable modifications per peptide. The JY and mono-allelic cell line data were searched against a reference human proteome downloaded from Uniprot (downloaded 2021-05-28). The SARS-CoV-2 infection data was searched against the combined human and SARS-CoV-2 FASTA file provided in the original publication (PXD025499). For each searches, a reversed protein decoy database was appended to each FASTA file.
Validation using Percolator
Where indicated, database search results were validated using Percolator v3.05.045. Test and train FDRs were set to 0.01 and the Cpos and Cneg arguments were undefined, allowing Percolator to determine them using cross-validation. The parameters were set to output PSM results for both targets and decoys.
Validation using DeepRescore
Where indicated, samples were validated with DeepRescore, a deep learning-based algorithm for peptide identification confidence rescoring that considers predictions of peptide retention time and MS2 spectra on top of the Percolator peptide validation algorithm49. Comet database search results were used as input for DeepRescore validation. Comet searches were performed as described above with the exception that the output format was set to.pepxml instead of.pin. For DeepRescore analysis, raw files were converted to.MGF files using msConvert by ProteoWizard127 (http://www.proteowizard.org/download.html). DeepRescore was then run using the default parameters. Resulting peptides were filtered using 1% FDR cutoff and subsequently with a NetMHCpan4.1 cutoff <=2.0 to directly compare peptide quantities with Percolator and MHCvalidator.
MHCvalidator design
MHC validator can be run in three different configurations that can be combined to obtain maximum gain in confidence of PSMs: NN-validator: NN-validator represents the core component for PSMs confidence assessment. PE: PE provides encoded peptide sequences via a convolutional neural network (CNN). APP: APP provides multiple antigen processing and presentation prediction scores via MHCflurry and NetMHCpan.
Data input. The preferred input data format accepted by MHCvalidator is tab-delimited text files (TSVs). Specifically, we developed MHCvalidator using a standard Percolator input (PIN) file as the input data because of the rich feature set already present in this format. However, MHCvalidator can process any TSV-format search results. While numerical features (e.g. database search scores) are expected, the absolute minimum features required for MHCvalidator to function are peptide sequences and target-decoy labels (either as a separate feature or encoded in protein IDs with a decoy tag). MHCvalidator also provides a parser for PEPXML format search results, but PIN format is preferred as validation and testing has only been carried out using this input format.
Feature engineering. All peptide sequences present in the input data are processed using NetMHCpan and/or MHCflurry. Binding affinity and eluted ligand scores from NetMHCpan, and affinity, presentation, and processing scores from MHCflurry are added to the features present in the input data. Binding affinity predictions are transformed to a log scale before being added, with values first being clipped to a minimum value of 1e-7. NetMHCpan is run from a user-indicated installation path. Because NetMHCpan does not support the use of multiple CPUs, in order to facilitate its practical use on the typically large list of peptides, the list is split into smaller chunks which are processed concurrently. MHCflurry is automatically installed as a dependency of MHCvalidator and runs natively from within Python. For PE training, the peptide sequences are first transformed into numerical representations. In brief, similar to the sequence encoding used by MHCflurry 1.3.0, the peptides are first middle-padded with an “X” amino acid to a length of 15. They are then encoded with a BLOSUM62 matrix to which an “X” amino acid has been added (with a substitution frequency of 1 for itself and 0 for every other amino acid). These encoded sequences are the input for the PE convolutional neural network.
Artificial neural network architecture. MHCvalidator makes use of two different feed-forward artificial neural networks. The first is a multilayer-perceptron (MLP) with defaults of two hidden layers and a width of 5-times the number of input features. The second, optional network, couples the architecture of the first to a convolutional neural network that encodes peptide sequences into a numerical vector of length 6. The convolutional neural network consists of a single 1D convolutional layer with 12 filters of size 4 and stride 3, followed by a 1D max pooling layer of size 2. The output is flattened and fully connected to the output layer, which is fed to the MLP as additional training features. This model is trained simultaneously with the MLP. All hidden layers in both architectures are connected with a default dropout out of 0.5. Most model hyperparameters are exposed in the Python API and can be tuned for the dataset of interest (e.g. number of layers, layer width, dropout, batch size, number of epochs, convolutional filter size, etc.). Much like Percolator, MHCvalidator is designed with flexibility in mind. Any additional features (e.g. probabilities or scores) can be used as features and the use of the artificial neural networks is optional.
Training and predicting. When training and predicting, a K-fold cross-validation is used. The MS data is split into a variable number of sets, as defined by the user with a default of 3. Predictions made on the validation splits during the cross-validation are reported and used for calculating q-values and FDR thresholds. Because MHCvalidator can use peptide sequences or values derived from peptide sequences as training features, the splits are constructed such that duplicate peptide sequences will not be present between any training and validation sets.
Comparison of Percolator and MHCvalidator for HLA allele-specific PSMs identifications
In order to benchmark Percolator with MHCvalidator, Percolator target PSMs (1% or 5% FDR cut-off) were annotated with NetMHCpan4.1 binding predictions. Data were then filtered to keep only PSMs with an eluted ligand (EL) %Rank cut-off <=2.0, as generally performed to gain confidence in HLA-specificity15,23,63,128,129,130. Resulting PSMs were used for comparison with MHCvalidator. Different combinations of the above-described configurations (NN-validator, NN-validator+PE, NN-validator+APP, NN-validator+PE + APP) were applied to Comet outputs and compared to percolator as specified. MHCvalidator results were not filtered because the method already incorporates presentation and HLA binding affinity prediction scores into the modeling process. Peptide identifications were obtained by keeping only unique PSMs based on best score for both percolator and MHCvalidator. It is noteworthy that MHCvalidator and percolator were applied to each PIN (Percolator INput) file separately as opposed to applying each software to batches of files or replicates. Only unique peptides were reported across replicates, where applicable.
Statistics & reproducibility
All available data from immunopeptidomics experiments were utilized in this study. Statistical analyses were performed using two-sample t-tests where specified, and data visualizations, including boxplots, were created using the default settings of the Matplotlib library. All available data from immunopeptidomics experiments were utilized in this study. Statistical analyses were performed using two-sample t-tests where specified, and data visualizations, including boxplots, were created using the default settings of the Matplotlib library. No statistical method was used to predetermine sample size. No data were excluded from the analyses; the experiments were not randomized; the Investigators were not blinded to allocation during experiments and outcome assessment.
Peptide clustering
Several deconvolution methods are available for analyzing binding motifs of HLA-associated peptides131,132,133. We applied MixMHCp 2.1132,134 to analyze 9-mer HLA-I peptides with the default settings and the number of maximum motifs set to 5. Upon completion of deconvolution, motifs were manually analyzed and assigned to JY HLA allotypes.
Validation of PSMs found uniquely with MHCvalidator
Peptide-spectrum matches found uniquely with the MHCValidator software were evaluated using MS2 spectrum prediction similarity and retention time prediction. MS2 spectrum predictions were made with Prosit (https://www.nature.com/articles/s41592-019-0426-7) using the Non-tryptic 2020 HCD model. MAPDP (https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00859) was used to execute Prosit and compute spectrum similarity using three metrics: normalized spectral contrast angle, Pearson correlation, and Spearman correlation. Retention times were predicted using the Prosit 2019 iRT prediction model and calibrated to the experimental ones using the calibration algorithm of DeepLC (https://www.nature.com/articles/s41592-021-01301-5). The mirror plot was drawn using spectrum-utils (0.4.2) (https://pubs.acs.org/doi/10.1021/acs.analchem.9b04884) and distribution histograms using seaborn (0.13.1) (https://joss.theoj.org/papers/10.21105/joss.03021).
SARS-CoV-2 peptide identification
To identify SARS-CoV-2 peptides, a similar strategy to that in Nagler et al. was used23. Because of possible ambiguity in the identification of leucine and isoleucine residues, all isoleucine residues were substituted with leucine in the following steps. Peptides that were assigned to both human and SARS-CoV-2 proteins were considered to be human in origin. The remaining peptides that were uniquely assigned to SARS-CoV-2 proteins were then compared with all six reading frames of the human non-coding regions (https://www.gencodegenes.org/human/release_19.html) and pseudogenes (http://www.pseudogene.org/Human/Human90.txt) used in Nagler et al. Peptides that could be attributed to human non-coding or pseudogene regions were considered to be human in origin.
In vitro HLA-peptide binding assays
Peptides binding to class I HLA molecules were quantitatively measured using classical competition assays based on the inhibition of binding of a high affinity radiolabeled peptide to purified HLA molecules, as detailed elsewhere135. Briefly, HLA molecules were purified from lysates of EBV transformed homozygous cell lines by affinity chromatography by repeated passage over Protein A Sepharose beads conjugated with the W6/32 (anti-HLA-A, -B, -C) antibody, following separation from HLA-B and -C molecules by pre-passage over a B1.23.2 (antiHLA B, C) column. Protein purity, concentration, and the effectiveness of depletion steps was monitored by SDS-PAGE and BCA assay. Peptide affinity for respective class I molecules was determined by incubating 0.1–+1 nM of radiolabeled peptide at room temperature with 1 µM to 1 nM of purified HLA in the presence of a cocktail of protease inhibitors and 1 µM B2microglobulin. Following a two-day incubation, HLA bound radioactivity was determined by capturing MHC/peptide complexes on W6/32 antibody coated Lumitrac 600 plates (Greiner Bioone, Frickenhausen, Germany). Bound cpm was measured using the TopCount (Packard Instrument Co., Meriden, CT) microscintillation counter. The concentration of peptide yielding 50% inhibition of the binding of the radiolabeled peptide was calculated. Under the conditions utilized, where [label]<[MHC] and IC50 ≥ [MHC], the measured IC50 values are reasonable approximations of the true Kd values. Each competitor peptide was tested at six different concentrations covering a 100,000-fold dose range, and in three or more independent experiments. As a positive control for inhibition, the unlabeled version of the radiolabeled probe was also tested in each experiment.
T cell immunogenicity
Subjects and samples collection: The study subjects were composed of 5 groups of previously infected health care workers (HCWs) who were recruited following a PCR-confirmed SARS-CoV-2 infection as part of the RECOVER study (n = 48). Participants were selected based on their HLA types at enrollment: (1) A68:01 (n = 10), (2) B07:02 (n = 14), (3) A68:02 (n = 10), (4) A02:01 (n = 25) and (5) B51:01 (n = 12). Some patients may share two or three different HLA types. Blood samples were collected at enrollment around 6.1 ± 2.4 months after infection into acid–citrate–dextrose tubes (ACD, BD) in each of the five participating centers in the Province of Québec, shipped to the Mother-Child Biobank at the CHU Sainte-Justine where peripheral blood mononuclear cells (PBMCs) were isolated according to standard operation procedures (SOPs) using SepMate™ tubes (Stemcell Technologies, Canada). PBMCs were cryopreserved in complete RPMI (Gibco) with 10% DMSO and stored in liquid nitrogen until used. Participants were recruited from August 17, 2020, to April 8, 2021.
IFN-γ ELISpot assay. Cell-mediated immune (CMI) response was estimated by ELISpot assay. Frozen PBMCs were rapidly thawed at 37 °C and rested overnight. PBMCs were plated at 400,000 cells per well (200,000 cells for positive controls) into MultiScreenHTS-IP Filter 96-well plates (Millipore, Massachusetts, US) pre-coated with an anti-IFN-γ antibody. PBMCs were then stimulated with 10 µg/mL of single peptides identified by Mass spectrometry (MS) immunopeptidomic (STTTNIVTR, TGSNVFQTR, HTTDPSFLGR, RTIKVFTTV, APRITFGGP, NAPRITFGGP, GPMVLRGLIT, RANNTKGSL, LPYPQILLL, AADLDDFSKQLQ, HSSGVTREL, YSGVVTTV, KLPDDFTGC, YLQPRTFLL, TLNDLNETL, VPYNMRVI). PBMCs were then incubated for 48 h at 37 °C, 5% CO2. Spots were revealed using BIO-RAD Alkaline Phosphatase Conjugate Substrate Kit. The resulting ELISpots were analyzed using CTL ImmunoSpot. S5 UV Analyzer (Cellular Technology Ltd, OH). Unstimulated cells and cells stimulated with 10 μg/mL of single peptide HIV pol HLA-A*0201 (ILKEPVHGV) (10 μg/mL, JPT Peptide Technologies, Berlin, Germany) were used as negative controls and cytostim (5 uL/ml, Miltenyi, Gaithersburg, MD), spike megapool of peptides (1 μg/mL, JPT Peptide Technologies, Berlin, Germany), PepPool CEF (CD8) (1 μg/mL, JPT Peptide Technologies, Berlin, Germany) were used as positive controls. In this assay, a response was defined as positive if it was greater than or equal to the mean of negative control (response to HIV single peptide) + 3.
Building a SARS-CoV-2 intra-host dataset
We compiled a comprehensive intra-host dataset, which included Illumina amplicon paired-end sequencing libraries of SARS-CoV-2 obtained during the initial two years of the COVID-19 pandemic, collected between 2020 and 2021. We ensured a representative sampling strategy across both time and geographical locations, while taking advantage of large amount of data from countries particularly involved in genomic surveillance efforts, with the United Kingdom (UK) and the United States of America (USA) contributing significantly (51% and 14% of the dataset, respectively). For each month, we randomly selected libraries based on their availability in the National Center for Biotechnology Information (NCBI): up to 5000 from the UK, up to 1000 from the USA, and up to 2000 from other global regions, resulting in a potential monthly total of 8,000 libraries, leading to the acquisition of a total of 100,512 libraries. Subsequently, each library underwent preprocessing, where Illumina sequencing adapters and low-quality reads (Phred score <20) were removed using TrimGalore! V.0.6.0. The trimmed libraries were then mapped to the SARS-CoV-2 reference genome (NC045512.2) using BWA meme v.0.7.17-r1188, resulting in BAM files. To further refine the data, we employed the iVar pipeline for primer trimming, using the ARTIC Network V3, V4, and V4.1 amplicon designs as these three kits were predominant in the sequencing centers within our dataset during the sampling period. The samtools mpileup tool, with specific parameters (-Q 20 -q 0 -B -A -d 600000), was used to generate pileup files containing read information for each BAM file. Finally, the tool pileup2base was used to convert each pileup files to the more comprehensive base file format. This process provided details such as the depth of coverage per genomic position (number of reads aligning to the position), positions of Single Nucleotide Variants (SNVs), insertions, and deletions.
Identifying deletions leading to LPYPQILLL peptide
To identify all deletions leading to the generation of the LPYPQILLL CD8 + T cell epitope (frame+1), the base files generated from all SARS-CoV-2 NCBI sequencing libraries were queried using an in-house python 3.11-based algorithm. Briefly, all deletions of one or more amino acids were searched between the frame+1 stop codon directly preceding the epitope and the start of the LPYPQILLL epitope (genomic positions 23,623 and 23,693, respectively). All deletions ending prior to the stop codon as well as after the epitope were omitted. While all lengths of deletions were included in our exhaustive, comprehensive deletion analysis, only those leading to the appropriate frame shift (frame+1) were considered in the context of the LPYPQILLL peptide. To account for putative sequencing errors, three distinct thresholds were applied prior to analyses: deletions identified in at least 2 reads; 50 reads; and 100 reads.
Epitope conservation analysis using EpiTrack
A multiple sequence alignment (MSA) comprising all GISAID SARS-CoV-2 entries (n = 14.6 M sequences) and using Wuhan-1 (NC_045512.2) as reference was downloaded from GISAID, along with all corresponding metadata on 10/24/2023. All subsequent data processing and analyses were performed using EpiTrack (see Code Availability). GISAID entries without metadata, with incomplete year/month sampling dates, or associated with non-human hosts were omitted. The resulting dataset was used in all subsequent analyses. For all MHCvalidator-identified peptides of interest, the nucleotide sequence of the peptide was extracted from all SARS-CoV-2 sequence of the MSA (when available) and translated. The resulting sequence was defined as an “alternative peptide” if the amino acid sequence differed from that of the wuhan-1 reference sequences. The number of occurrences of all distinct alternative peptides as well as the unmutated peptide were determined. Only alternative epitopes identified in at least 10 GISAID entries were considered in subsequent analyses. As a computational control, all analyses were repeated on a set of end-to-end 9-mers spanning the entire SARS-CoV-2 canonical proteome. For all epitopes of interest (CD8+ epitopes identified from SARS-CoV-2-infected cells, n = 10; BNT162b4 mRNA vaccine CD8+ epitopes, n = 6) as well as for all control 9-mers, the epitope-specific rate of mutation was expressed as the number of alternative peptides found across the GISAID database divided by the total number of GISAID sequences for which the epitope had sequencing coverage.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry JY immunopeptidomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD052187. Other mass spectrometry datasets for this study were acquired from the following public repositories: mono-allelic cell line data - MassIVE: MSV000080527 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=a67baac756f5421faf51c5d4bac3005f); BNT162b4 data - MassIVE repository: MSV000091008 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=88caf6334d494a72b3c8d5e9988a64e6); SARS-CoV-2 infections - PRIDE repository: PXD025499. Source data are provided with this paper.
Code availability
MHCvalidator is available on the CaronLab GitHub page: https://github.com/CaronLab/mhc-validator. Instructions on how to run MHCvalidator in its different configurations are explained in this page. The scripts pertaining to the intra-host deletion analyses, along with the list of NCBI libraries used, as well as the mutational dynamics analyses are available on the HussinLab GitHub page: https://github.com/HussinLab/EpiTrack. Source code are also available on Zenodo: MHCvalidator (https://zenodo.org/records/13736549) and EpiTrack (https://zenodo.org/records/13738788)136.
References
Le, T. T. et al. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 19, 305–306 (2020).
Watson, O. J. et al. Global impact of the first year of COVID-19 vaccination: A mathematical modelling study. Lancet Infect. Dis. 22, 1293–1302 (2022).
Cao, Y. et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 602, 657–663 (2022).
Wang, Q. et al. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 186, 279–286.e8 (2023).
Jian, F. et al. Further humoral immunity evasion of emerging SARS-CoV-2 BA.4 and BA.5 subvariants. Lancet Infect. Dis. 22, 1535–1537 (2022).
Sette, A. & Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 184, 861–880 (2021).
Wherry, E. J. & Barouch, D. H. T cell immunity to COVID-19 vaccines. Science 377, 821–822 (2022).
Diniz, M. O., Maini, M. K. & Swadling, L. T cell control of SARS-CoV-2: When, which, and where? Semin. Immunol. 70, 101828 (2023).
Heitmann, J. S. et al. A COVID-19 peptide vaccine for the induction of SARS-CoV-2 T cell immunity. Nature 601, 617–622 (2021).
Tandler, C. et al. Long-term efficacy of the peptide-based COVID-19 T cell activator CoVac-1 in healthy adults. Int. J. Infect. Dis. 139, 69–77 (2023).
Heitmann, J. S. et al. Phase I/II trial of a peptide-based COVID-19 T-cell activator in patients with B-cell deficiency. Nat. Commun. 14, 5032 (2023).
Arieta, C. M. et al. The T-cell-directed vaccine BNT162b4 encoding conserved non-spike antigens protects animals from severe SARS-CoV-2 infection. Cell 186, 2392–2409 (2023).
Nathan, A. et al. Structure-guided T cell vaccine design for SARS-CoV-2 variants and sarbecoviruses. Cell 184, 4401–4413.e10 (2021).
Hamelin, D. J. et al. The mutational landscape of SARS-CoV-2 variants diversifies T cell targets in an HLA supertype-dependent manner. Cell Syst. 13, 143–157 (2021).
Caron, E. et al. An open-source computational and data resource to analyze digital maps of immunopeptidomes. Elife 4, e07661 (2015).
Kapoor, S., Maréchal, L., Sirois, I. & Caron, É. Scaling up robust immunopeptidomics technologies for a global T cell surveillance digital network. J. Exp. Med. 221, e20231739 (2024).
Caron, E. et al. Analysis of major histocompatibility complex (MHC) immunopeptidomes using mass spectrometry. Mol. Cell Proteom. 14, 3105–3117 (2015).
Kubiniok, P. et al. Understanding the constitutive presentation of MHC class I immunopeptidomes in primary tissues. iScience 25, 103768 (2022).
Kovalchik, K., Hamelin, D. & Caron, E. Generation of HLA allele-specific spectral libraries to identify and quantify immunopeptidomes by SWATH/DIA-MS. Methods Mol. Biol. 2420, 137–147 (2021).
Parker, R. et al. Mapping the SARS-CoV-2 spike glycoprotein-derived peptidome presented by HLA class II on dendritic cells. Cell Rep. 35, 109179 (2021).
Knierman, M. D. et al. The human leukocyte antigen class II immunopeptidome of the SARS-CoV-2 spike glycoprotein. Cell Rep. 33, 108454 (2020).
Weingarten-Gabbay, S. et al. Profiling SARS-CoV-2 HLA-I peptidome reveals T cell epitopes from out-of-frame ORFs. Cell 184, 3962–3980 (2021).
Nagler, A. et al. Identification of presented SARS-CoV-2 HLA class I and HLA class II peptides using HLA-peptidomics. Cell Rep. 35, 109305 (2021).
Pan, K. et al. Mass spectrometric identification of immunogenic SARS-CoV-2 epitopes and cognate TCRs. Proc. Natl. Acad. Sci. USA 118, e2111815118 (2021).
Gomez-Zepeda, D. et al. Thunder-DDA-PASEF enables high-coverage immunopeptidomics and is boosted by MS2Rescore with MS2PIP timsTOF fragmentation prediction model. Nat. Commun. 15, 2288 (2023).
Weingarten-Gabbay, S. et al. The HLA-II immunopeptidome of SARS-CoV-2. Cell Rep. 43, 113596 (2023).
Purcell, A. et al. Mapping the immunopeptidome of seven SARS-CoV-2 antigens across common HLA haplotypes. https://doi.org/10.21203/rs.3.rs-3564516/v1 (2023).
El-Baky, N. A., Amara, A. A. & Redwan, E. M. HLA-I and HLA-II peptidomes of SARS-CoV-2: A review. Vaccines 11, 548 (2023).
Nelde, A. et al. Increased soluble HLA in COVID-19 present a disease-related, diverse immunopeptidome associated with T cell immunity. iScience 25, 105643 (2022).
Becerra-Artiles, A. et al. Immunopeptidome profiling of human coronavirus OC43-infected cells identifies CD4 T-cell epitopes specific to seasonal coronaviruses or cross-reactive with SARS-CoV-2. PLoS Pathog. 19, e1011032 (2023).
Purcell, A. W., Ramarathinam, S. H. & Ternette, N. Mass spectrometry–based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 14, 1687–1707 (2019).
Sirois, I., Isabelle, M., Duquette, J. D., Saab, F. & Caron, E. Immunopeptidomics: Isolation of mouse and human MHC class I- and II-associated peptides for mass spectrometry analysis. J. Vis. Exp. https://doi.org/10.3791/63052 (2021).
Eng, J. K., McCormack, A. L. & Yates, J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 (1994).
Eng, J. K., Jahan, T. A. & Hoopmann, M. R. Comet: An open-source MS/MS sequence database search tool. Proteomics 13, 22–24 (2012).
Kim, S. & Pevzner, P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277 (2014).
Kong, A. T., Leprevost, F. V., Avtonomov, D. M., Mellacheruvu, D. & Nesvizhskii, A. I. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 14, 513–520 (2017).
Muntel, J. et al. Surpassing 10000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Mol. Omics 15, 348–360 (2019).
Xin, L. et al. A streamlined platform for analyzing tera-scale DDA and DIA mass spectrometry data enables highly sensitive immunopeptidomics. Nat. Commun. 13, 3108 (2022).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007).
Keller, A., Nesvizhskii, A. I., Kolker, E. & Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 (2002).
Shteynberg, D. et al. iProphet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell Proteomics 10, M111.007690 M111.007690 (2011).
Ma, K., Vitek, O. & Nesvizhskii, A. I. A statistical model-building perspective to identification of MS/MS spectra with PeptideProphet. BMC Bioinform. 13, S1 (2012).
Käll, L., Canterbury, J. D., Weston, J., Noble, W. S. & MacCoss, M. J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 (2007).
The, M., MacCoss, M. J., Noble, W. S. & Käll, L. Fast and accurate protein false discovery rates on large-scale proteomics data sets with Percolator 3.0. J. Am. Soc. Mass Spectr. 27, 1719–1727 (2016).
Jeong, K., Kim, S. & Bandeira, N. False discovery rates in spectral identification. BMC Bioinform. 13, S2 (2012).
Declercq, A. et al. MS2Rescore: Data-driven rescoring dramatically boosts immunopeptide identification rates. Mol. Cell Proteom. 21, 100266 (2022).
Li, K., Jain, A., Malovannaya, A., Wen, B. & Zhang, B. DeepRescore: Leveraging deep learning to improve peptide identification in immunopeptidomics. Proteomics 1900334. https://doi.org/10.1002/pmic.201900334 (2020).
Wilhelm, M. et al. Deep learning boosts sensitivity of mass spectrometry-based immunopeptidomics. Nat. Commun. 12, 3346 (2021).
Yang, K. L. et al. MSBooster: improving peptide identification rates using deep learning-based features. Nat. Commun. 14, 4539 (2023).
Liao, H. et al. MARS an improved de novo peptide candidate selection method for non-canonical antigen target discovery in cancer. Nat. Commun. 15, 661 (2024).
Peters, B., Nielsen, M. & Sette, A. T cell epitope predictions. Annu Rev. Immunol. 38, 123–145 (2020).
Albert, B. A. et al. Deep neural networks predict class I major histocompatibility complex epitope presentation and transfer learn neoepitope immunogenicity. Nat. Mach. Intell. 5, 861–872 (2023).
O’Donnell, T. J., Rubinsteyn, A. & Laserson, U. MHCflurry 2.0: Improved pan-allele prediction of MHC class I-presented peptides by incorporating antigen processing. Cell Syst. 11, 42–48.e7 (2020).
O’Donnell, T. J. et al. MHCflurry: Open-source class I MHC binding affinity prediction. Cell Syst. 7, 129–132.e4 (2018).
Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, W449–W454 (2020).
Nilsson, J. B. et al. Accurate prediction of HLA class II antigen presentation across all loci using tailored data acquisition and refined machine learning. Sci. Adv. 9, eadj6367 (2023).
Jurtz, V. et al. NetMHCpan-4.0: Improved Peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 199, 3360–3368 (2017).
Andreatta, M. et al. MS‐Rescue: A computational pipeline to increase the quality and yield of immunopeptidomics experiments. Proteomics 19, 1800357 (2019).
Granados, D. P. et al. Impact of genomic polymorphisms on the repertoire of human MHC class I-associated peptides. Nat. Commun. 5, 3600 (2014).
Bichmann, L. et al. MHCquant: Automated and reproducible data analysis for immunopeptidomics. J. Proteome Res. 18, 3876–3884 (2019).
Kovalchik, K. A. et al. MhcVizPipe: A quality control software for rapid assessment of small- to large-scale immunopeptidome data sets. Mol. Cell Proteom. 21, 100178 (2021).
Zeng, W.-F. et al. AlphaPeptDeep: a modular deep learning framework to predict peptide properties for proteomics. Nat. Commun. 13, 7238 (2022).
Falk, K., Rötzschke, O., Stevanovic, S., Jung, G. & Rammensee, H.-G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351, 290–296 (1991).
Vizcaíno, J. A. et al. The Human Immunopeptidome Project: a roadmap to predict and treat immune diseases. Mol. Cell Proteom. 19, 31–49 (2020).
Yang, S. et al. Antigenicity and infectivity characterisation of SARS-CoV-2 BA.2.86. Lancet Infect. Dis. 23, e457–e459 (2023).
Wang, Y. et al. Intra-host variation and evolutionary dynamics of SARS-CoV-2 populations in COVID-19 patients. Genome Med. 13, 30 (2021).
Li, J. et al. Two-step fitness selection for intra-host variations in SARS-CoV-2. Cell Rep. 38, 110205 (2022).
Pathak, A. K. et al. Spatio-temporal dynamics of intra-host variability in SARS-CoV-2 genomes. Nucleic Acids Res. 50, 1551–1561 (2022).
Liu, Y. et al. Rescuing low frequency variants within intra-host viral populations directly from Oxford Nanopore sequencing data. Nat. Commun. 13, 1321 (2022).
Gu, H. et al. Within-host genetic diversity of SARS-CoV-2 lineages in unvaccinated and vaccinated individuals. Nat. Commun. 14, 1793 (2023).
Sarkizova, S. et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 38, 199–209 (2020).
Finkel, Y. et al. The coding capacity of SARS-CoV-2. Nature 589, 125–130 (2021).
Davidson, A. D. et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 12, 68 (2020).
Kim, D. et al. The architecture of SARS-CoV-2 transcriptome. Cell 181, 914–921.e10 (2020).
Francisco, R. et al. HLA supertype variation across populations: new insights into the role of natural selection in the evolution of HLA-A and HLA-B polymorphisms. Immunogenetics 67, 651–663 (2015).
Nantel, S. et al. Symptomatology during previous SARS-CoV-2 infection and serostatus before vaccination influence the immunogenicity of BNT162b2 COVID-19 mRNA vaccine. Front Immunol. 13, 930252 (2022).
Ferretti, A. P. et al. Unbiased screens show CD8+ T cells of COVID-19 patients recognize shared epitopes in SARS-CoV-2 that largely reside outside the spike protein. Immunity 53, 1095–1107.e3 (2020).
Shomuradova, A. S. et al. SARS-CoV-2 epitopes are recognized by a public and diverse repertoire of human T cell receptors. Immunity 56, 1245–1257 (2020).
Schulien, I. et al. Characterization of pre-existing and induced SARS-CoV-2-specific CD8+ T cells. Nat. Med 27, 78–85 (2021).
Alter, G. et al. Immunogenicity of Ad26.COV2.S vaccine against SARS-CoV-2 variants in humans. Nature 596, 268–272 (2021).
Saini, S. K. et al. SARS-CoV-2 genome-wide T cell epitope mapping reveals immunodominance and substantial CD8+ T cell activation in COVID-19 patients. Sci. Immunol. 6, eabf7550 (2021).
Omasits, U., Ahrens, C. H., Müller, S. & Wollscheid, B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30, 884–886 (2014).
Calis, J. J. A. et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol. 9, e1003266 (2013).
Mayer, R. L. et al. Immunopeptidomics-based design of mRNA vaccine formulations against Listeria monocytogenes. Nat. Commun. 13, 6075 (2022).
Mayer, R. L. & Impens, F. Immunopeptidomics for next-generation bacterial vaccine development. Trends Microbiol 29, 1034–1045 (2021).
Ovsyannikova, I. G., Johnson, K. L., Bergen, H. R. & Poland, G. A. Mass spectrometry and peptide-based vaccine development. Clin. Pharm. Ther. 82, 644–652 (2007).
Bettencourt, P. et al. Identification of antigens presented by MHC for vaccines against tuberculosis. NPJ Vaccines 5, 2 (2020).
D’Souza, M. P. et al. Casting a wider net: Immunosurveillance by nonclassical MHC molecules. PLoS Pathog. 15, e1007567 (2019).
Qu, Z. et al. Structure and peptidome of the bat MHC cass I molecule reveal a novel mechanism leading to high-affinity peptide binding. J. Immunol. 202, 3493–3506 (2019).
Wen, B., Li, K., Zhang, Y. & Zhang, B. Cancer neoantigen prioritization through sensitive and reliable proteogenomics analysis. Nat. Commun. 11, 1759 (2020).
Gessulat, S. et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 16, 509–518 (2019).
Degroeve, S. & Martens, L. MS2PIP: A tool for MS/MS peak intensity prediction. Bioinformatics 29, 3199–3203 (2013).
Zhou, X.-X. et al. pDeep: Predicting MS/MS spectra of peptides with deep learning. Anal. Chem. 89, 12690–12697 (2017).
Meier, F. et al. Deep learning the collisional cross sections of the peptide universe from a million experimental values. Nat. Commun. 12, 1185 (2021).
Racle, J. et al. Machine learning predictions of MHC-II specificities reveal alternative binding mode of class II epitopes. Immunity 56, 1359–1375 (2023).
Müller, M. et al. Machine learning methods and harmonized datasets improve immunogenic neoantigen prediction. Immunity 56, 2650–2663.e6 (2023).
Motozono, C. et al. The SARS-CoV-2 Omicron BA.1 spike G446S mutation potentiates antiviral T-cell recognition. Nat. Commun. 13, 5440 (2022).
Hanada, K., Yewdell, J. W. & Yang, J. C. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature 427, 252–256 (2004).
Delong, T. et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 351, 711–714 (2016).
Paes, W. et al. Contribution of proteasome-catalyzed peptide cis-splicing to viral targeting by CD8+ T cells in HIV-1 infection. Proc. Natl Acad. Sci. USA 116, 244748–24759 (2019).
Tran, M. T. et al. T cell receptor recognition of hybrid insulin peptides bound to HLA-DQ8. Nat. Commun. 12, 5110 (2021).
Saab, F. et al. RHybridFinder: An R package to process immunopeptidomic data for putative hybrid peptide discovery. Star. Protoc. 2, 100875 (2021).
Shao, W. et al. The SysteMHC Atlas project. Nucleic Acids Res. 46, D1237–D1247 (2017).
Huang, X. et al. The SysteMHC Atlas v2.0, an updated resource for mass spectrometry-based immunopeptidomics. Nucleic Acids Res. 52, D1062–D1071 (2024).
Shao, W., Caron, E., Pedrioli, P. & Aebersold, R. Methods Mol. Biol. 2120, 173–181 (2020).
Tabb, D. L., McDonald, W. H. & Yates, J. R. DTASelect and contrast: Tools for assembling and comparing protein identifications from Shotgun proteomics. J. Proteome Res. 1, 21–26 (2002).
Racine, É. et al. The REinfection in COVID‐19 Estimation of Risk (RECOVER) study: Reinfection and serology dynamics in a cohort of Canadian healthcare workers. Influenza Other Resp. 16, 916–925 (2022).
Nantel, S. et al. Comparison of Omicron breakthrough infection versus monovalent SARS-CoV-2 intramuscular booster reveals differences in mucosal and systemic humoral immunity. Mucosal Immunol. 17, 201–210 (2024).
Zhao, J. et al. SARS-CoV-2 specific memory T cell epitopes identified in COVID-19-recovered subjects. Virus Res. 304, 198508 (2021).
Skelly, D. T. et al. Two doses of SARS-CoV-2 vaccination induce robust immune responses to emerging SARS-CoV-2 variants of concern. Nat. Commun. 12, 5061 (2021).
Mizukoshi, E. et al. Peptide vaccine-treated, long-term surviving cancer patients harbor self-renewing tumor-specific CD8+ T cells. Nat. Commun. 13, 3123 (2022).
Fournelle, D. et al. Intra-host viral populations of SARS-CoV-2 in immunosuppressed patients with hematologic cancers. bioRxiv 2022.10.19.512884. https://doi.org/10.1101/2022.10.19.512884 (2022).
Dersh, D., Hollý, J. & Yewdell, J. W. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat. Rev. Immunol. 21, 116–128 (2020).
Naranbhai, V. et al. T cell reactivity to the SARS-CoV-2 Omicron variant is preserved in most but not all individuals. Cell 185, 1041–1051.e6 (2022).
Motozono, C. et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 29, 1124–1136.e11 (2021).
Dolton, G. et al. Emergence of immune escape at dominant SARS-CoV-2 killer T cell epitope. Cell 185, 2936–2951.e19 (2022).
Stanevich, O. V. et al. SARS-CoV-2 escape from cytotoxic T cells during long-term COVID-19. Nat. Commun. 14, 149 (2023).
Sette, A., Sidney, J. & Grifoni, A. Pre-existing SARS-2-specific T cells are predicted to cross-recognize BA.2.86. Cell Host Microbe 32, 19–24.e2 (2023).
Müller, T. R. et al. Memory T cells effectively recognize the SARS-CoV-2 hypermutated BA.2.86 variant. Cell Host Microbe 32, 156–161.e3 (2024).
Meusser, B., Hirsch, C., Jarosch, E. & Sommer, T. ERAD: the long road to destruction. Nat. Cell Biol. 7, 766–772 (2005).
Purcell, A. W., Croft, N. P. & Tscharke, D. C. Immunology by numbers: quantitation of antigen presentation completes the quantitative milieu of systems immunology! Curr. Opin. Immunol. 40, 88–95 (2016).
Stutzmann, C. et al. Unlocking the potential of microfluidics in mass spectrometry-based immunopeptidomics for tumor antigen discovery. Cell Rep. Methods 3, 100511 (2023).
Thadani, N. N. et al. Learning from prepandemic data to forecast viral escape. Nature 622, 818–825 (2023).
Hulstaert, N. et al. ThermoRawFileParser: Modular, scalable, and cross-platform RAW file conversion. J. Proteome Res. 19, 537–542 (2020).
Adusumilli, R. & Mallick, P. Proteomics, Methods and Protocols. Methods Mol. Biol. 1550, 339–368 (2017).
Kraemer, A. I. et al. The immunopeptidome landscape associated with T cell infiltration, inflammation and immune editing in lung cancer. Nat. Cancer 4, 608–628 (2023).
Kina, E. et al. Breast cancer immunopeptidomes contain numerous shared tumor antigens. J. Clin. Investig. 134. https://doi.org/10.1172/jci166740 (2023).
Courcelles, M. et al. MAPDP: a cloud-based computational platform for immunopeptidomics analyses. J. Proteome Res 19, 1873–1881 (2020).
Andreatta, M., Alvarez, B. & Nielsen, M. GibbsCluster: Unsupervised clustering and alignment of peptide sequences. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx248 (2017).
Bassani-Sternberg, M. & Gfeller, D. Unsupervised HLA peptidome deconvolution improves ligand prediction accuracy and predicts cooperative effects in peptide–HLA interactions. J. Immunol. 197, 2492–2499 (2016).
Shahbazy, M. et al. MHCpLogics: An interactive machine learning-based tool for unsupervised data visualization and cluster analysis of immunopeptidomes. Brief. Bioinform. 25, bbae087 (2024).
Gfeller, D. et al. The length distribution and multiple specificity of naturally presented HLA-I ligands. J. Immunol. 201, 3705–3716 (2018).
Sidney, J. et al. Measurement of MHC/peptide interactions by gel filtration or monoclonal antibody capture. Curr. Protoc. Immunol 18, Unit 18.3 (2023).
Kovalchik, K. A., et al. Machine learning-enhanced immunopeptidomics advances T-cell epitope discovery for COVID-19 vaccines, Zenodo, https://doi.org/10.5281/zenodo.13736548 (MHCvalidator) and https://doi.org/10.5281/zenodo.13738767 (EpiTrack), 2024.
Acknowledgements
This work was supported by start-up funding from Yale School of Medicine (EC) as well as funding from the Fonds de recherche du Québec – Santé (FRQS) (EC), the Cole Foundation (EC), CHU Sainte-Justine and the Charles-Bruneau Foundations (EC), Canada Foundation for Innovation (EC), the National Sciences and Engineering Research Council (NSERC) (#RGPIN-2020-05232) (EC), the Canadian Institutes of Health Research (CIHR) (#174924, #172712) (EC, JGH), the CIHR Coronavirus Variants Rapid Response Network (CoVaRR-Net) (#ARR-175622) (EC) and a NSERC Discovery Grant (MLA). IRIC proteomics facility is a Genomics Technology platform funded in part by the Canadian Government through Genome Canada (PT). IEDB is supported by 75N93019C00001/AI/NIAID NIH HHS/United States (AS). KK is a recipient of IVADO’s postdoctoral scholarship (#4879287150) (KK). DJH is a recipient of the Hydro-Quebec doctoral and FRQS fellowships (DJH). JGH is FRQS Junior 2 research scholar (JGH). We gratefully acknowledge the GISAID Initiative and the generous contribution of all data contributors to both GISAID and NCBI, including the authors, their laboratories that collect the specimens and generated the genetic sequence and metadata on which part of this research is based. This work was completed thanks to computational resources provided by Digital Research Alliance of Canada, particularly Narval and Beluga clusters.
Author information
Authors and Affiliations
Contributions
Conceptualization: K.A.K., D.J.H., M.L.A., J.G.H., and E.C.; Machine-Learning: K.A.K., P.K., and M.L.A.; Data Curation and Bioinformatic Analysis: K.A.K., P.K., D.J.H., F.M., R.P., and J.C.G.; HLA typing: B.P., S.M.S., and M.S.; T cell experiments: B.B.; Investigation: K.A.K., D.J.H., P.K., I.S., B.B., B.G., R.P., Z.W., J.S., E.B., M.C., S.K.S., J.C.G., M.S., V.R., C.S., L.M., M.A.S., H.D., J.G.H., M.L.A., and E.C.; Writing – Original Draft: K.A.K. and E.C.; Writing – Review & Editing: K.A.K., D.J.H., P.K., I.S., B.G., B.B., R.P., Z.W., B.P., S.M.S., J.S., E.B., S.K.S., J.C.G., S.K., M.W., C.S., L.M., A.S., P.T., M.A.S., H.D., J.G.H., M.L.A., and E.C.; Supervision: J.G.H., M.L.A., and E.C.; Funding Acquisition: E.C.
Corresponding authors
Ethics declarations
Competing interests
E.C. and I.S. are co-founders of Neomabs Biotechnologies Inc. PT is a co-founder of Epitopea. A.S. is a consultant for Alcimed, Gritstone, Darwin Health, EmerVax, Gilead Sciences, Guggenheim Securities, Link University, RiverVest Venture Partners, and Arcturus. La Jolla Institute for Immunology has filed for patent protection for various aspects of T cell epitope and vaccine design work. All other authors declare no competing interests.
Inclusion & Ethics Statement
This research prioritizes inclusivity by engaging diverse populations, particularly underrepresented groups. All participants provided informed consent, and their confidentiality was safeguarded in accordance with ethical guidelines. The contributions of all team members were acknowledged, and potential biases were actively addressed. We aim to conduct research that advances knowledge while respecting the rights and dignity of all individuals involved.
Peer review
Peer review information
Nature Communications thanks David Gfeller and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kovalchik, K.A., Hamelin, D.J., Kubiniok, P. et al. Machine learning-enhanced immunopeptidomics applied to T-cell epitope discovery for COVID-19 vaccines. Nat Commun 15, 10316 (2024). https://doi.org/10.1038/s41467-024-54734-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54734-9
