
Abstract
Studies in SIV-infected macaques show that the virus reservoir is particularly refractory to conventional suppressive antiretroviral therapy (ART). We posit that optimized ART regimens designed to have robust penetration in tissue reservoirs and long-lasting antiviral activity may be advantageous for HIV or SIV remission. Here we treat macaques infected with RT-SHIV with oral emtricitabine/tenofovir alafenamide and long-acting cabotegravir/rilpivirine without (n = 4) or with (n = 4) the immune activator vesatolimod after the initial onset of viremia. We document full suppression in all animals during treatment (4-12 months) and no virus rebound after treatment discontinuation (1.5-2 years of follow up) despite CD8 + T cell depletion. We show efficient multidrug penetration in virus reservoirs and persisting rilpivirine in plasma for 2 years after the last dose. Our results document a type of virus remission that is achieved through early treatment initiation and provision of ultra long-lasting antiviral activity that persists after treatment cessation.
Similar content being viewed by others


Introduction
Antiretroviral therapy (ART) has drastically improved clinical outcomes for people living with HIV. However, ART is not curative and requires lifelong medication adherence to ensure viral suppression and limit progression to AIDS. The inability of ART to cure HIV is due to the rapid establishment of a virus reservoir predominantly in memory CD4 + T cells1. The T cell archive of replication-competent virus is the primary source of virus rebound in HIV-1-infected persons who discontinue ART and represents a significant challenge to achieving an HIV cure2. Attempts to achieve a functional or sterilizing cure have therefore focused on targeting the latent virus reservoir with various approaches, including gene-editing technology, “shock and kill” strategies to reactivate and eradicate the latent virus, and latency silencing3. In a minority of patients, early ART initiation has been associated with virus remission highlighting the potential of this strategy for HIV cure4,5,6,7.
Macaque models of sexual HIV infection have been used to study the impact of early treatment on reservoir formation and to precisely time reservoir seeding following rectal infection with SIVmac2518,9. Whitney et al. used a subcutaneous three-drug ART regimen of tenofovir (TFV) or tenofovir disoproxil fumarate (TDF), emtricitabine (FTC), and dolutegravir (DTG) initiated before or after the onset of acute viremia and continued for 6 months. Despite suppressive ART, the virus rebounded after ART discontinuation in all animals that initiated ART on day 3 after infection and before detectable viremia, or on days 7–14 post-infection during acute viremia8,9. These data demonstrate that the virus reservoir is seeded very early during the eclipse phase of infection and is particularly refractory to conventional suppressive ART. We posited that disruption of early reservoir formation may require different ART modalities that are more potent and have increased tissue penetration at reservoir sites. We designed a 4-drug ART regimen containing daily oral tenofovir alafenamide (TAF) and FTC, and intramuscular cabotegravir long-acting (CAB LA) and rilpivirine long-acting (RPV LA). TAF was selected over TDF for its improved dosing of lymphoid tissues and peripheral blood mononuclear cells (PBMCs) which results in ~ 6-fold higher concentrations of the pharmacologically active metabolite, TFV-diphosphate (TFV-DP)10,11,12. We administered TAF and FTC orally, as opposed to subcutaneously, to better recapitulate tissue biodistribution in humans13,14. CAB and RPV were selected for their added advantage of penetrating the central nervous system15. The long-acting formulations of CAB and RPV were chosen over oral formulations because of the long pharmacologic drug tail that persists after treatment which may enhance virus control. In humans, the apparent terminal elimination half-lives for CAB and RPV after a single intramuscular injection are 39–80 days and 91–196 days, respectively16. We tested the ability of early treatment with this regimen to achieve virus remission in macaques rectally infected with a pathogenic simian HIV isolate that contains the HIV-1 reverse transcriptase (RT-SHIV). Animals were treated after the onset of viremia for 1 year and monitored after treatment discontinuation for virus rebound and seroconversion for 2 additional years including a period of experimental CD8 + T cell depletion. In a second group of animals that also initiated treatment after the onset of viremia, we tested if the addition of the immune activator vesatolimod (VES) to a shorter 4-month treatment could also achieve remission. We document durable virus remission (defined as lack of detectable viremia in the absence of treatment) in all animals treated with this optimized ART regimen with or without VES despite CD8 + T cell depletion and detectable intact proviruses 1.5–2 years after treatment cessation.
Results
Durable remission with early ART
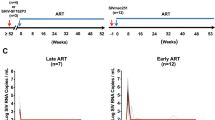
We infected 4 macaques intrarectally with a pathogenic RT-SHIV that contains the HIV-1 reverse transcriptase (RT)17,18,19. This chimeric virus was chosen because SIV RTs are not susceptible to non-nucleoside RT inhibitors (NNRTIs)20. We initiated treatment with daily FTC/TAF and monthly CAB LA/RPV LA after two consecutive SHIV RNA positive measurements in plasma (5 to 6 days after SHIV inoculation) (Fig. 1a). The median (range) plasma RNA levels at treatment initiation were 3.1 (2.3–3.8) log10 copies/mL with peak levels (3.4 (2.7–4.3) log10 copies/mL) seen 10 days post-infection (Fig. 1b). All 4 early ART (eART) treated macaques were virologically suppressed at day 19–28 post-infection and remained undetectable during treatment with FTC/TAF/CAB LA/RPV LA (months 1–6) and maintenance therapy with CAB LA/RPV LA (months 6–12) (Fig. 1c). In contrast, untreated animals (n = 4) infected with the same dose of RT-SHIV had high peak viremias (median = 6.9 (range = 6.7–8.1) log10 copies/mL) and virus set points that ranged between 3 and 7 log10 copies/mL (Fig. 1b). Two of the untreated controls were rapid progressors showing a drastic decline in CD4 + T cells to 44 and 275 cells/µL and progression to simian AIDS within 6 months. The remaining 2 controls had 540 and 1127 cells/µL at the time of necropsy at month 11. After ART discontinuation at month 12, none of the treated animals experienced virus rebound during a follow-up period that extended for almost 2 years (Fig. 1c).

a Experimental Design. Macaques infected with RT-SHIV (n = 4) were treated with daily tenofovir alafenamide/emtricitabine (FTC/TAF) and monthly intramuscular injections of cabotegravir and rilpivirine long-acting (CAB LA/RPV LA) 5 to 6 days after infection. Treatment was continued for six months followed by maintenance with CAB LA/RPV LA until month twelve. At month 30, macaques received an anti-CD8 monoclonal antibody MT807R1 to deplete CD8 + cells. b Acute SHIV viremia in treated (blue lines) and untreated (red lines) macaques. The thick lines denote median values. c SHIV RNA levels in treated (blue) and control (red) macaques during treatment (shaded blue area) and after treatment cessation. The crosses indicate the time of euthanasia. The horizontal dotted line denotes the limit of quantification (12.5 copies/mL). ART; antiretroviral therapy. Source data are provided as a Source Data file.
Lack of virus rebound after CD8+ T cell depletion
To further understand the degree of immune virus control after treatment cessation we depleted CD8 + T cells in the treated animals 17 months after treatment cessation when CAB was no longer detected in plasma and RPV levels were below plasma levels seen in humans receiving 600 mg of RPV LA every 4 weeks21. Treatment with the CD8 depletion antibody MT807R1 effectively depleted > 99.9% of CD8 + T cells in blood as illustrated by a decline in CD8 + T cell frequencies from 43.8% (range = 34.2–47.1) at baseline to 0.018% (range = 0.007–0.038) 18 days after MT807R1 treatment (Supplementary Fig. 1). The frequency of CD8 + T cells remained below 1% until day 48-60 post MT807R1 treatment followed by a gradual increase to 9.5% (range = 4.8–13.5). Despite the drastic reduction in CD8 + T cells, no virus rebound was observed in any of the treated animals within 4 months after depletion, however, one macaque (10–47) had a transient blip below the limit of quantification 3 months after MT807R1 treatment (Fig. 1c). Overall, these results suggest that CD8 + T cells are not the main contributor to the observed virus remission.
Limited serologic responses during and after treatment cessation
We next investigated antibody responses against gag (p27), pol (p66), env (gp140, gp130, gp41, gp36), and nef (Fig. 2). In 2 untreated control animals, responses to SHIV were characterized by a rapid increase in antibody levels against gp36, gp41, gp140, gp130, and p66 pol. One of the two untreated animals also mounted a measurable response to nef. In contrast, antibody responses in the 4 treated animals were predominantly against gp140. Two of the four treated macaques also showed a limited response to gp130 that waned over time. Antibody titers did not increase in any of the 4 animals after treatment cessation at month 12 except for one animal that showed a short and transient increase in antibody levels to nef at month 12 and another animal that showed an increase in the response to gp41 (Fig. 2). Overall, these data reflect very limited and non-evolving SHIV-specific humoral responses.

SHIV plasma IgG levels against gp140, gp130, gp36, gp41, p66, nef, and p27 were measured longitudinally using a Bio-Plex assay. Light blue lines denote the mean fluorescent intensity (MFI) for each treated animal; dark blue line indicates the median MFI values. Two untreated controls (macaques 09–19 and 10–87) are shown in red. Treatment in the early ART (eART) animals was initiated 5 to 6 days after infection. The shaded blue area represents the period of treatment with FTC/TAF/CAB LA/RPV LA (months 1-6) and CAB LA/RPV LA (months 6–12). The horizontal dotted line denotes cutoff values for each analyte. Source data are provided as a Source Data file.
Drug concentrations in plasma, PBMCs, and tissues
To better characterize the pharmacologic profile of the ART regimen we measured drug concentrations before and after treatment discontinuation and compared data to those in humans to provide clinical translation of findings. The concentrations of CAB and RPV in plasma and intracellular TFV-DP and FTC-TP in PBMCs are shown in Fig. 3. Median (range) CAB levels during the treatment phase (2400 ng/mL; 830–4980) were 13 times above the protein-adjusted 90% inhibitory concentration (PA-IC90 = 166 ng/mL) and within plasma levels seen in humans treated with 400 mg of CAB LA every month for 48 weeks (2740 ng/mL) (Fig. 3a)21. After the last injection, CAB concentrations remained above the clinical trough level for treatment of 4 × PA-IC90 for 5-6 additional weeks and declined below the PA-IC90 two months later (Fig. 3b). The median concentrations of RPV in plasma during treatment was 108 ng/mL (56–182), also within human levels achieved with 600 mg of RPV LA every month for 48 weeks (97.5 ng/mL) (Fig. 3a)21. After the last injection, RPV levels slowly declined although they remained about 3 times above the PA-IC90 value for RPV (12 ng/mL) for 2 years after treatment cessation (Fig. 3c).

a Median (range) concentrations of CAB and RPV in plasma during treatment. CAB concentrations were measured every week. RPV concentrations were measured 2–4 weeks after dosing. Each data point denotes the median CAB or RPV levels seen in the 4 treated animals in a given week (42 weeks for CAB and 13 weeks for RPV). b Median (range) concentrations of CAB in plasma in the 4 treated macaques during treatment (months 0–12) and after treatment cessation at month 12. The horizontal red line denotes the mean plasma levels seen in humans receiving 400 mg of CAB LA every 4 weeks for 48 weeks. c Median (range) concentrations of RPV in plasma in the 4 treated macaques during treatment (months 0–12) and after treatment cessation at month 12. The horizontal red line denotes the mean plasma levels seen in humans receiving 600 mg of RPV LA every 4 weeks for 48 weeks. The shaded blue area in panels b and c denotes the period of treatment. The dotted black lines denote the protein-adjusted IC90 values (PA-IC90) for CAB (166 ng/ml) and RPV (12 ng/ml). d Median (range) concentrations of TFV-DP in PBMCs measured 24 h post-dosing during the entire study and divided by periods of full adherence to oral FTC/TAF (moths 0–3.5) and periods of suboptimal adherence (months 3.5–6). e Median (range) concentrations of FTC-TP 24 post-dosing were also divided by periods of full adherence and suboptimal adherence. Each data point in panels d and e denotes the median TFV-DP or FTC-TP levels seen in the 4 treated macaques in a given week. f Ratios between dATP or dCTP and intracellular TFV-DP or FTC-TP in PBMCs, LNMCs, and tissues. Horizontal lines denote medians, and the error bars are ranges. Each LNMC data point denotes ratios obtained in 1–3 biopsies per animal. Each PBMC data point denotes ratios obtained in a given week during the period of full adherence to FTC/TAF. Each rectal data point denotes ratios obtained in 2-3 biopsies per animal. Source data are provided as a Source Data file.
In PBMCs, TFV-DP and FTC-TP concentrations varied between periods of full adherence (months 0–3.5) or suboptimal adherence (months 3.5 to 6) to oral FTC/TAF (Fig. 3d, e). During the period of full adherence to oral FTC/TAF, median TFV-DP levels were 1753 (275–2809) fmols/106 PBMCs, which is about 3.2 times higher than those observed in humans treated with 25 mg of TAF (525 fmols/106 PBMCs)12. Likewise, median FTC-TP concentrations were 573 (333–1528) fmols/106 PBMCs, approximately 3.6-fold lower than those observed in humans treated with 300 mg of FTC (2,060 fmols/106 PBMCs)12. During the period of suboptimal adherence to oral FTC/TAF, TFV-DP and FTC-TP concentrations were 329 (63–1688) and 306 (undetectable-964) fmols/106 PBMCs, respectively (Fig. 3d, e).
We also measured drug concentrations in lymphoid tissue, rectal tissues, and cerebrospinal fluid (CSF) (Table 1). Median concentrations of TFV-DP and FTC-TP in lymph node mononuclear cells (LNMCs) collected from axillary or inguinal lymph nodes were 367 (142–620) and 260 (below limit of quantification (BLQ)–383) fmols/106 LNMCs, respectively. Median concentrations of TFV-DP and FTC-TP in rectal tissues were 14.6 (5.3–28) fmols/mg of tissue and BLQ (BLQ-17) fmols/mg of tissue, respectively. When the analysis was done by the total amount of extracellular drug per mg of rectal tissue, the levels of FTC, TFV, CAB, and RPV were 1.48, 0.64, 0.60, and 0.45 ng/mg of tissue, respectively (Table 1). In CSF, CAB showed the highest concentrations (5.95 ng/mL) followed by RPV (4.06 ng/mL) and FTC (2.36 ng/mL). TFV was not detected in any of the CSF samples. The CSF-to-plasma ratios were 4 times higher for RPV than for CAB (2.8% and 0.6%, respectively).
To further define the penetration of CAB and RPV in tissue virus reservoirs, we measured CAB and RPV concentrations in lymphoid tissue, the lower gastrointestinal (GI) tract, and the brain collected at necropsy from a SHIV-infected female macaque treated with a single intramuscular injection of Cabenuva® (also administered at 50 mg/kg CAB LA and 200 mg/kg RPV LA). Supplementary Table 1 shows drug concentrations observed 3 weeks after injection. CAB and RPV achieved high concentrations in lymphoid tissues (median of 975 ng/g and 773 ng/g, respectively) and in the GI tract (medians of 695 ng/g and 666 ng/g, respectively). Consistent with the higher CSF-to-plasma penetration ratio, the median concentration of RPV in brain tissues was 5.8 times higher than that of CAB (340 ng/g compared to 58 ng/g).
Concentrations of dATP and dCTP in PBMCs and tissues
TFV-DP and FTC-TP compete with dATP and dCTP, respectively, as substrates for incorporation by reverse transcriptase. As a measure of the antiviral activity of FTC-TP and TFV-DP in cells, we calculated dATP/TFV-DP and dCTP/FTC-TP ratios in PBMCs and tissues. High ratios indicate a reduced likelihood of FTC-TP or TFV-DP to block reverse transcription22. Figure 3f shows a comparative analysis of dNTP/nucleotide ratios in PBMCs, LNMCs, and rectal tissues collected during treatment. In the case of dATP/TFV-DP, the lowest ratios were seen in LNMCs followed by PBMCs and rectal tissues (p = 0.0044 and p < 0.001 for comparisons between LNMC/PBMCs and PBMCs/rectal), suggesting that TFV-DP has the highest activity in lymphoid tissue and the least in rectal tissue. In contrast to dATP/TFV-DP, the ratio between dCTP/FTC-TP was similar in LNMCs and in PBMCs (p = 0.367). dCTP/FTC-TP ratios in the only two rectal biopsies with detectable FTC-TP were higher than in PBMCs or LNMCs.
Short ART with VES also results in virus remission
We next investigated in 4 macaques if the addition of the immunomodulator VES to a shortened eART regimen could also result in virus remission. eART with FTC/TAF/CAB LA/RPV LA was initiated after the onset of viremia, days 6–8 post-infection, and continued for 4 months. Supplementary Fig. 2 shows the adherence to oral FTC/TAF and the overall longitudinal concentrations of TFV-DP (median = 1162 (rage = 417–2093) fmols/106 PBMCs) and FTC-TP (median = 553 (246–914 fmols/106 PBMCs) during the entire treatment period. VES treatment was started two days after ART initiation and was given orally once a week for three months (Fig. 4a). Overall, the levels of virus replication during the first three weeks of treatment (as expressed by RNA copies/day*mL of plasma) were similar among macaques in the eART + VES and eART-only groups (median of 40 and 45, respectively, p = 0.686) (Fig. 4b). The median (range) plasma RNA level at treatment initiation (day 6–8) was 3.7 (3.0–4.3) log10 copies/mL with peak levels (4.2 (3.7–4.4) log10 copies/mL) seen 8–12 days post-infection (Fig. 4c). Viremia was rapidly suppressed (day 19–28) and maintained below the limit of detection during the 4 months of ART and following treatment discontinuation except for one animal (MAJ) that showed a virus blip 5 months after treatment cessation (9 months post infection) (Fig. 4c). No virus rebound was observed in any of the macaques following depletion of CD8 + T cells. As noted in animals that received ART without VES, SHIV IgG responses were limited to gp140 albeit these responses were delayed compared to the eART-only group (36.5 [33–40] vs 14 [14–17] days, p = 0.0286) (Fig. 4d). Supplementary Fig. 3 shows the kinetics of antibody responses against gag (p27) pol (p66), env (gp140, gp130, gp41, gp36) and nef seen in the 4 macaques treated with eART + VES.

a Treatment with FTC/TAF/CAB LA/RPV LA was initiated at day 6/8 post-infection and treatment with VES at day 8/11. FTC/TAF/CAB LA/RPV LA was administered for 4 months and VES for 3 months. Transient CD8 depletion was done 16 months after treatment initiation (or 12 months after treatment discontinuation). b Area under the curve values for SHIV RNA (AUC0-19days) in untreated controls (red) and macaques treated with eART only (blue) and eART + VES (green). Each data point reflects AUC values seen in one macaque. AUC0-19days values were similar among macaques in the eART + VES and eART-only groups (p = 0.686; two-sided Wilcoxon Rank Sum test). c SHIV infection dynamics during treatment and after discontinuation. The four untreated controls are shown in red for comparison. d SHIV plasma IgG levels against gp140 during acute infection in macaques treated with eART + VES (individual animals in light green; dark green denotes median), eART alone (individual animals in light blue; dark blue denotes median), and in two untreated controls (red lines). Source data are provided as a Source Data file.
Intact proviral DNA levels in PBMCs and tissues
Intact and defective proviral DNA levels were measured in PBMCs and tissues using the intact proviral DNA assay (IPDA)23. At day 21 after infection (or ~ 2 weeks after treatment initiation), the median intact proviral DNA level in PBMCs in the eART-treated macaques was 42 copies/106 PBMCs (range = 15–192) compared to 9392 (4416–15,439) copies/106 PBMCs in untreated animals (Table 2). Intact proviral DNA was also detected in one of the eART animals (macaque 10–47) 1 year after treatment cessation and 2 months after CD8 T cell depletion, a period that coincided with a transient viral blip in plasma (Fig. 1). In the eART+VES group, intact proviral DNA at day 21–25 after infection was only detected in one animal (macaque 42268) (Table 2).
At necropsy, none of the eART animals had detectable intact proviruses in PBMCs as opposed to the two untreated controls tested, which had 12,315 and 964 copies/106 PBMCs (Table 2). Interestingly, macaque 44346 treated with eART + VES had levels of intact provirus DNA at necropsy 1 year post-infection (480 copies/106 PBMCs) that were similar to untreated macaque 10–87 (964 copies/106 PBMCs). However, macaque 44346 remained aviremic, while macaque 10–87 exhibited virus loads of 4.4–5.5 log10 copies/mL during a period of 6 months, likely reflecting the role of RPV LA in maintaining long-term suppression.
We also measured intact proviral DNA at necropsy in lymph nodes (mesenteric, inguinal, or axillary) and spleen from the eART group, and mesenteric lymph nodes from the eART + VES group. The eART-treated animals had no detectable intact provirus in the spleen, inguinal, or axillary lymph nodes (Table 3). However, 2 of the 4 eART animals (macaques 10–47 and 28025) and 1 of the 4 eART + VES-treated animals (macaque 44346) had detectable intact provirus in mesenteric lymph nodes. As noted in PBMCs, macaque 44346 (eART + VES group) had a frequency of intact proviruses in mesenteric lymph nodes that was similar to that seen in untreated control macaque 10–87 (Table 3).
Discussion
Conventional ART regimens are designed to durably suppress viremia in blood but do not prevent HIV or SIV rebound after treatment discontinuation even when given early after infection8,9,24. We reasoned that alternative ART modalities are needed to increase the likelihood of virus remission. In this study, we document long-term post-treatment viral control in macaques treated at the onset of viremia with a suppressive regimen containing oral and long-acting antiretrovirals. Remission was observed in all study animals irrespective of the length of ART (4 months or 1 year) and was maintained for about 2 years until necropsy. These data contrast findings in a similar intrarectal model of infection with SIV in which early treatment with suppressive subcutaneous FTC, TDF, and DTG was not effective in achieving remission8,9. We show that acute infection dynamics and chronic viremias in untreated RT-SHIV infections were similar to those seen with SIV suggesting that differences in virus pathogenicity may not explain the different outcomes. Also, two of our untreated animals progressed to simian AIDS within only 6 months. We posit that the robust post-treatment control seen in our study reflects early treatment initiation, the use of an optimized ART regimen that better penetrates tissue reservoir sites, and the addition of long-acting drug formulations that provide prolonged antiviral activity after treatment cessation. We detail the pharmacologic advantages of the regimen by conducting extensive pharmacokinetic studies. We found that the concentrations of TFV-DP in PBMCs and lymphoid tissues were about 10-fold higher than those achieved with TDF highlighting the pharmacological advantage of TAF vs TDF in blocking infection in lymphoid tissues10. We also show dATP/TFV-DP ratios that are indicative of efficient RT inhibition22. We detect both CAB and RPV at high concentrations in lymphoid tissues and the GI tract. Importantly, we show that this regimen distributes two potent drugs, CAB and RPV, at inhibitory concentrations in CSF and brain tissues that are not provided by the conventional ART regimen with FTC, TDF, and DTG. Based on the in vitro EC90 values for RPV and CAB of 0.66 ng/mL and 0.34 ng/mL, respectively, the RPV and CAB concentrations in CSF were about 6 times and 17 times above the EC90, respectively. In brain tissues, RPV and CAB concentrations were ~ 500 and ~ 100 times above the in vitro EC90, respectively, highlighting the antiviral activity of RPV and CAB in brain tissues. Therefore, our pharmacokinetic observations point to an efficient drug distribution in virus reservoir sites at a very early stage of reservoir seeding. A second critical feature of this regimen is the inclusion of long-acting drugs that prolong antiviral activity for at least two years after treatment cessation. Overall, we show the capability of this novel ART modality to achieve durable virus remission.
We also note that our ART regimen is clinically relevant and easily translatable to humans. Antiretroviral drug doses were adjusted by allometric scaling due to the smaller size and faster metabolisms of macaques to better mimic clinical doses in humans. FTC and TAF were administered orally as in humans to better model drug biodistribution in tissues, and CAB LA and RPV LA were given by intramuscular injection. The addition of the NNRTI RPV also incorporates a new class of drugs that, to our knowledge has never been part of ART regimens in macaque models of remission and cure. This was significant since NNRTI-based regimens have been associated with delayed post-treatment virus rebound in some clinical studies25. More recently, RPV was also found to induce clearance of latently infected cells through pyroptotic cell death, a mechanism that could potentially reduce the size of the latent HIV reservoir26.
The use of the long-acting formulations of CAB and RPV was also important as these formulations have an extended pharmacological tail capable of maintaining therapeutic drug levels for several months after treatment discontinuation. We report here plasma CAB levels above the 4 × PA-IC90 for about 5-6 weeks after treatment cessation and RPV concentrations of about 3 × PA-IC90 for almost 2 years after the last injection, a length of time that would likely be extended in humans due to the longer CAB and RPV half-life27,28. In macaques, the clearance of CAB is much faster than in humans27. In humans, the estimated apparent terminal elimination half-life of RPV after a single intramuscular administration is 91–196 days16. Repeated RPV dosing results in significant drug accumulation29 as we also noted in macaques. It is possible that the persistence of RPV may have contributed to post-treatment control by providing continuous antiviral activity through pyroptotic cell death of infected cells and suppression of any residual virus replication26. This scenario is different from treatment of established infections which requires bimonthly or monthly dosing of long-acting RPV and CAB to maintain virus suppression21. Persisting RPV for ~ 2 years after the last dosing at subtherapeutic concentrations in our macaques should not be sufficient to prevent virus rebound under continuous residual virus replication. Therefore, the lack of virus rebound or emergence of RPV-resistant viremia in all animals despite almost two years of plasma exposure to RPV supports minimal or no residual virus replication and points to a disrupted virus reservoir, as illustrated by the 20 to > 1000-fold reduction in the frequency of intact proviruses seen in treated animals within weeks of infection. These observations open the possibility for novel HIV remission approaches based on the efficient control of a disrupted reservoir using infrequent RPV LA injections (or other long-acting antiretrovirals) given perhaps every 1-2 years. Because the 4 drugs in the tested regimen are clinically available, they can be explored in clinical studies such as treatment of early or late acute infection to assess their advantage over conventional ART in inducing remission.
Our study is a treatment study of infected macaques with demonstrated viremia and, thus, is fundamentally different from prevention studies with CAB LA as PrEP in uninfected macaques in which protected animals remained seronegative and negative for SHIV RNA in plasma and SHIV DNA in PBMCs27,30. Interestingly however, the dynamics of early infection seen in our study share some characteristics with those observed in people who received CAB LA for pre-exposure prophylaxis (PrEP) and become infected despite on-time injections31. These breakthrough infections are also characterized by transient low-level RNA and DNA detection and diminished or delayed antibody responses32. It will be interesting to understand if CAB LA PrEP failures are associated with some degree of reservoir disruption and virus remission.
The addition of the TLR7 agonist VES to our ART regimen also resulted in durable viral remission that extended for almost 2 years. We selected VES for its ability to enhance innate and adaptive immune responses by activating NK cells, CD4 + and CD8 + T cells33,34,35,36. CD4 + cells that are transitioning from an effector to a memory phenotype are thought to be a primary source of the latent HIV reservoir37,38,39. We hypothesize that VES activation may limit the number of CD4 T cells transitioning to a memory phenotype, in turn reducing the pool of reservoir cells. In addition, VES-accelerated CD8 + T cell responses might also provide opportunities for a more robust anti-HIV activity. In a recent study in macaques, early treatment with ARVs also favored the development of long-term CD8 + T cells with enhanced proliferative and SIV suppressive capacity40. In humans, VES has been studied as a latency-reversing agent and associated with decreases in intact proviral DNA and a modest effect on time to rebound41. In our study, we administered VES 2-3 days after ART initiation to minimize and purge reservoir formation from activated CD4 + T cells and found a reduction in intact proviruses in all four animals. Notably, none of the macaques showed virus rebound despite a period of treatment of only 4 months and subsequent depletion of CD8 + T cells 1 year later. It will be important to see if a shorter duration of ART treatment without VES would achieve a similar degree of virus remission and also to further investigate the effect of latency disruption during early infection.
Our study has a few limitations. Our findings in the rectal infection macaque model may not be generalizable to all routes of SHIV acquisition and vaginal, penile, and intravenous infections may require independent confirmation. Okoye et al. reported virus remission in animals infected with SIV intravenously and treated for almost two years with TDF, FTC, and DTG at day 4/542, but when treatment was initiated on day 6–12 almost all of the animals had viral rebound. Importantly, the same ART regimen initiated on day 3 post rectal virus challenge did not result in remission8,9. It is also not known if oral formulations of CAB and RPV lacking the long pharmacological tail might result in the same degree of post-treatment control. Additional studies will be required to assess these oral regimens and the importance of the pharmacologic drug tail. Likewise, it will be important to define if treatment initiated later in acute infection or after seroconversion would be as effective in maintaining post-treatment control as this would be the case for most people who initiate early HIV treatment. Lastly, although the degree and length of remission seen in our study are unprecedented, we do not know if remission would be maintained when RPV levels in plasma become undetectable.
Several important observations can be made from the analysis of intact proviral DNA. First, early treatment with FTC, TAF, CAB LA, and RPV LA with or without VES resulted in a significant reduction in the frequency of intact proviruses during acute infection. In contrast, Whitney et al. did not observe reductions in SIV DNA during early infection when FTC/TDF/DTG was initiated 7–10 days after infection8,9. These observations highlight the superiority of FTC, TAF, CAB LA and RPV LA combination in reducing the early virus reservoir. Second, despite the lack of detectable plasma viremia throughout the study, intact proviruses were detected in PBMCs and mesenteric lymph nodes from some of the treated animals 1.5–2 years after treatment cessation. Although it is not known if these proviruses would be capable of viral rebound, these findings suggest that RPV was sufficient to control the small virus reservoir established during early infection.
In summary, we show in macaques that a combination of early reservoir reduction, pharmacological drug optimization, and ultra-long-lasting antiviral activity that persists for years after treatment cessation can result in virus remission. Additional macaque studies with this ART combination are planned to investigate the window of treatment initiation (i.e., late acute infection or chronic infection) and treatment duration that will achieve virus remission. These results will inform new remission studies in humans using this ART combination and open a wider opportunity to test other combinations that provide ultra-long-lasting antiviral activity after treatment cessation43.
Methods
Ethics statement
All animal procedures received prior approval from the Institutional Animal Care and Use Committee (IACUC) of the Centers for Disease Control and Prevention and were conducted in a United States Department of Agriculture (USDA)-registered, Office of Laboratory Animal Welfare (OLAW)-assured, and AAALAC International-accredited animal facility in accordance with the Guide for the Care and Use of Laboratory Animals. Euthanasia was performed and confirmed by the attending veterinarian. A total of 13 Rhesus macaques (10 males, 3 females) were used in the study. At the end of the study, animals were anesthetized with 1 mg/kg ketamine and euthanized using intravenous injection of Pentobarbital euthanasia solution (> 100 mg/kg). Death was confirmed by monitoring the absence of heart rate and breathing. The procedure was performed in accordance with the CDC-Atlanta IACUC Policy on Euthanasia and Care Guidelines recommended by the American Veterinary Medical Association Guidelines on Euthanasia, 2020.
Viruses, antiretroviral drugs, and dosing
RT-SHIV was selected for the study since it contains the HIVHXBc2 RT that is sensitive to RPV. RT-SHIV was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH17,18,19. The stock was maintained in liquid nitrogen in individual 1 mL aliquots until use. TAF and FTC were gifts from MS Hetero Labs Limited and Laurus Labs, respectively. CAB LA and RPV LA were obtained from ViiV Healthcare and Janssen, respectively. TAF (1.5 mg/kg) and FTC (20 mg/kg) were given orally once a day mixed with food44. CAB LA (50 mg/kg) and RPV LA (200 mg/kg) were given intramuscularly once a month27,45. The macaque used in the terminal PK study received CAB LA (50 mg/kg) and RPV LA (200 mg/kg) from Cabenuva®.
Animals were trained to take TAF, and FTC mixed with food prior to study initiation46. Macaques were observed for medication consumption, and adherence was documented using medication diaries. Adherence in the eART group was nearly perfect (99.5% or 352/356 doses consumed) during the first 3.5 months of study with imperfect adherence only noted in one animal (30622) who ate 50–90% of the dose during the first week. After a sudden drop in adherence at month 3.5, numerous food contingencies for FTC/TAF delivery were tested, including juices, sugar cookies, and chocolate chip cookie dough but had limited success46. In an attempt to maintain effective TFV-DP and FTC-TP concentrations, we administered FTC/TAF twice a week by gavage. Twice weekly dosing by gavage and sporadic adherence to the drug mixed with food maintained detectable levels of FTC-TP and TFV-DP through the remainder of the treatment period.
Animals in the eART + VES group were also monitored for daily FTC/TAF intake. Given the drop in TFV-DP and FTC-TP levels observed in the eART group during periods of suboptimal adherence, we incorporated subcutaneous FTC/TAF dosing (20 mg/kg and 0.2 mg/kg, respectively) when the oral medications were not consumed. On the days of anesthesia, FTC and TAF were always administered by oral gavage.
Macaque study design
Rhesus macaques were evenly distributed according to MHC class I alleles with two Mamu-A*01 animals in the untreated control group (10–87 and 12C189), two Mamu-A*01 animals in the eART group (10–47 and 28025), and two Mamu-A*01 animals in the eART + VES group (44174 and 42268). All animals were challenged with 1 mL of RT-SHIV (103.3 TCID50) intrarectally. Four untreated macaques (3 males and 1 female; median weight = 13.7 kg (range = 11.5–15.4)) were used as controls. Treatment with TAF, FTC, CAB LA, and RPV LA in the eART group (4 males; median weight = 14.5 kg (range = 11.3–15.5)) was initiated after infection was confirmed by the detection of SHIV RNA in plasma, which occurred on day 5–8 post-inoculation. Macaques in the eART group received TAF/FTC/CAB LA/RPV LA for 6 months followed by a maintenance regimen with CAB LA and RPV L/A for an additional 6 months. Macaques in the eART + VES group (3 males and 1 female; median weight = 10.8 kg (range = 5.9–12.6)) received 4 months of FTC/TAF/CAB LA/RPV LA therapy initiated 6–8 days post-infection. VES treatment was added 2 days after ART initiation and was given orally once a week (0.15 mg/kg) for three months. All treated animals were monitored for virus rebound during 1.5 to 2 years after treatment cessation including a period of transient in vivo CD8 depletion at month 30 (eART group) or 16 (eART + VES group) with anti-CD8 antibody MT807R1 (one subcutaneous 10 mg/kg dose followed by three subsequent intravenous 5 mg/kg doses 3, 7, and 10 days later). The rhesus IgG1 recombinant anti-CD8 alpha [MT807R1] monoclonal antibody was engineered and produced by the Nonhuman Primate Reagent Resource (NIH Nonhuman Primate Reagent Resource Cat# PR-0817, RRID:AB_2716320). SHIV testing was done every 1-2 weeks to monitor for transient blips.
SHIV RNA assay
Plasma viral load was determined by an RT-PCR assay modified to increase sensitivity to 12.5 copies/mL by using a larger volume of plasma47. Briefly, 2.5 mL of macaque plasma was ultracentrifuged for 45 min at 43,000 rpm at 4 °C, and viral RNA was extracted using Qiagen Viral Mini Kit per the manufacturer’s protocol. Quantitative RT-PCR was performed to amplify a conserved region of the gag using a SuperScript III One-step RT-PCR kit.
Intact provirus DNA assay (IPDA)
Accelevir performed an SIV-adapted IPDA® to characterize and quantify persistent RT-SHIV proviruses. Samples with a cell input below ~ 104 cells were excluded from the analysis. Overall, genomic DNA was isolated from a median of 4.5 × 105 lymph nodes or peripheral blood mononuclear cells (range = 9.8 × 103, 9.2 × 105). Briefly, RNA-free genomic DNA was isolated using the QIAamp DNA Mini Kit (QIAGEN). DNA concentrations were determined by fluorometry (Qubit dsDNA BR Assay Kit, Thermo Fisher Scientific), and DNA quality was determined by ultraviolet-visible spectrophotometry (QIAxpert, Qiagen). Genomic DNA was then analyzed by the SIV-adapted IPDA®23. Operators performed testing under Accelevir standard operating procedures and were blinded to sample and study identifiers.
Analysis of intracellular FTC-TP, TFV-DP, and dNTPs
PBMCs were isolated from blood collected in BD Vacutainer® CPT™ Sodium Citrate Cell Preparation Tubes, and red blood cells were lysed with BioLegend RBC lysis buffer. Cell counts and viability were determined using a Guava Cell Counter and Cytosoft data acquisition and analysis software (version 6.02; Millipore Billerica, MA) or a Countess (Invitrogen). Three million viable cells were pelleted, washed with 1 mL of physiological saline, and resuspended in 500 µl ice-cold 80% methanol. Samples were vortexed for 1 min and immediately frozen at − 70 °C. Prior to analysis, cellular debris was pelleted, and supernatants were transferred to new tubes. Supernatants were dried down and reconstituted in 100 μl of 50 mM ammonium acetate buffer (pH 7.0), then centrifuged at 17,000 × g to remove insoluble particulates. Intracellular dATP, dCTP, TFV-DP, and FTC-TP concentrations were measured using an automated on-line weak anion-exchange (WAX) solid-phase extraction (SPE) method coupled with ion-pair (IP) chromatography-tandem mass spectrometry (MS-MS)48. TFV-DP and FTC-TP were monitored through 448 → 176, 488 → 230, 495 → 136, and 468 → 112 m/z fragments, respectively, with [13C5] adenine-labeled internal standards for each analyte. Calibration curves were generated from serially diluted standards over the range from 0.25 to 10 nM. The lower limit of quantification is 10 ng/mL for TFV-DP and 25 ng/mL for FTC-TP. All calibration curves had r2 values greater than 0.9948. In tissue homogenates, the limit of detection was 100 and 500 fmol/sample for TFV-DP and FTC-TP, respectively.
Analysis of FTC, TFV, CAB, and RPV in plasma and tissues
The concentrations of CAB in plasma and the concentrations of FTC and TFV in tissue homogenates were measured by HPLC-MS/MS. A cocktail of methanol containing internal standards was added to precipitate proteins. After a brief centrifugation and removal of protein precipitates, the supernatant was evaporated to near dryness and re-suspended with mobile phase A. An aqueous-acetonitrile mobile-phase gradient was used to elute the drugs from a C18 column (100*1 mm, Imtakt) and into the analyzer. Drug concentrations were calculated from a standard curve with a range of 0.5–2000 ng/mL using Analyst software. The LOQ for CAB in plasma is 10 ng/mL, and the limit of quantification (LOQ) for tissue FTC and TFV is 1 ng/sample. Plasma concentrations of CAB were measured weekly. Plasma concentrations of RPV were measured 4 (9 observations), 2 (2 observations), or 3 (2 observations) weeks after the previous injection. The concentrations of RPV in plasma, CSF, and tissues and the concentrations of CAB in CSF were measured at the Clinical Pharmacology and Analytical Chemistry (CPAC) Laboratory, University of North Carolina Center for AIDS Research. The LOQ for RPV in plasma is 1 ng/mL.
Cell surface staining and flow cytometry
PBMCs were stained for viability using the Zombie Yellow™ Fixable Viability Kit (Biolegend®) prior to cell staining with fluorochrome-conjugated antibodies as previously described49. Stained samples were run on an LSRII flow cytometer, and data was acquired using FACS DIVA software (BD Biosciences) and analyzed using FlowJo software (TreeStar, Inc.). The fluorochrome-conjugated antibodies are described in Supplementary Table 2.
Luminex assay for antibody responses
The Luminex assay was performed for the detection of IgG reactivity against MagPlex magnetic COOH microspheres (Luminex, Austin, TX) coupled with the following recombinant proteins: SIVmac239 gp140, SIVmac251 gp130, SIVmac251 gp41, SIVmac251 nef, SIVmac251 p27, HIV-1 p66, and HIV-2 gp36 (Immunodiagnostics, Inc., Woburn, MA) (Immune Technology, New York, NY). For sample addition and non-specific binding controls, coupling reactions were also performed using goat anti-human IgG (Invitrogen, Carlsbad, CA) and BSA (Sigma-Aldrich, St. Louis, MO), respectively. The coupling of proteins to the magnetic microspheres was performed as previously described50. All samples were tested in duplicate. A 1:50 dilution was performed on all plasma samples in preincubation buffer (PBS with 1% BSA, 0.5% polyvinylalcohol (PVA, Sigma-Aldrich), and 0.8% polyvinylpyrrolidone (PVP, Sigma-Aldrich)) as described51. Diluted samples were incubated for 15 minutes at room temperature on a titer plate shaker at 600–800 rpm. A working microsphere mixture was prepared by combining 1 × 105 microspheres/mL of each coupled microsphere set in assay buffer (PBS with 1% BSA). The microsphere mixture and prediluted plasma samples were combined in duplicate wells of a flat-bottom 96-well plate (Thermo Fisher Scientific, Waltham, MA) at a volume of 50 µL of each/well. The plate was incubated for 30 minutes on a shaker at 600–800 rpm with protection from light. The microspheres were washed 3 times with assay buffer using a BioTek ELx50 microplate strip washer with magnet (BioTek, Winooski, VT), resuspended in 100 µL of a 1:1000 dilution of phycoerythrin (PE)-labeled, goat anti-human IgG (Sigma Aldrich), vortexed for 30 s on a plate vortexer, followed by a 30 min incubation on a shaker at 600–800 rpm with protection from light. The microspheres were washed twice with assay buffer, then resuspended in 125 µL of assay buffer and analyzed on the Luminex 200 System (Luminex, Austin, TX). Median fluorescent intensity (MFI) values were generated for each sample and duplicate samples MFIs were averaged.
Positive control was created by pooling plasma from six SHIV-positive rhesus macaques and was included on each assay plate. The positive control was positive for all SIV/SHIV-specific analytes included in the testing panel. Cutoff values were calculated for each analyte using SHIV negative rhesus macaque plasma samples. The cutoff value for each analyte was determined using the following formula: average MFI value of the negative samples + 2 standard deviations.
Statistical analysis
Two-sided Wilcoxon Rank Sum tests were used to compare the medians between the two groups. We used the exact test for each of the comparisons and assumed that our data followed a non-normal curve due to small sample sizes. Medians and ranges (versus means and square roots) were compared for each group due to this assumption. Furthermore, due to the small sample sizes, we decided not to include any covariates in our comparisons. Data collection was performed using Microsoft Excel and imported into SAS software version 9.4 for analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information. Any additional requests for information can be directed to the corresponding author. The antibodies used for flow cytometry analysis are provided in Supplementary Table 2. Source data are provided in this paper.
References
Busman-Sahay, K., Starke, C. E., Nekorchuk, M. D. & Estes, J. D. Eliminating HIV reservoirs for a cure: the issue is in the tissue. Curr. Opin. HIV AIDS 16, 200–208 (2021).
Siliciano, J. D. & Siliciano, R. F. Recent developments in the effort to cure HIV infection: going beyond N = 1. J. Clin. Invest. 12610, 409–414 (2016).
Ndung’u, T., McCune, J. M. & Deeks, S. G. Why and where an HIV cure is needed and how it might be achieved. Nature 576, 397–405 (2019).
Shelton, E. M., Reeves, D. B. & Bender Ignacio, R. A. Initiation of antiretroviral therapy during primary HIV infection: effects on the latent HIV reservoir, including on analytic treatment interruptions. AIDS Rev. 23, 28–39 (2020).
Namazi, G. et al. The control of HIV after antiretroviral medication pause (CHAMP) study: posttreatment controllers identified from 14 clinical studies. J. Infect. Dis. 218, 1954–1963 (2018).
Sáez-Cirión, A. et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 9, e1003211 (2013).
Colby, D. J. et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat. Med. 24, 923–926 (2018).
Whitney, J. B. et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 512, 74–77 (2014).
Whitney, J. et al. Prevention of SIVmac251 reservoir seeding in rhesus monkeys by early antiretroviral therapy. Nat. Commun. 9, 5429 (2018).
Fletcher, C. V. et al. The lymphoid tissue pharmacokinetics of tenofovir disoproxil fumarate and tenofovir alafenamide in HIV-infected persons. Clin. Pharm. Ther. 108, 971–975 (2020).
Lee, W. A. et al. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob. Agents Chemother. 49, 1898–1906 (2005).
Thurman, A. R. et al. Safety and pharmacokinetics of a tenofovir alafenamide Fumarate-emtricitabine based oral antiretroviral regimen for prevention of HIV acquisition in women: a randomized controlled trial. EClinicalMedicine 36, 100893 (2021).
García-Lerma, J. G. et al. Prevention of rectal SHIV transmission in macaques by daily or intermittent prophylaxis with emtricitabine and tenofovir. PLoS Med. 5, e28 (2008).
Van Rompay, K. K. A. et al. Compared to subcutaneous tenofovir, oral tenofovir disoproxyl fumarate administration preferentially concentrates the drug into gut-associated lymphoid cells in simian immunodeficiency virus-infected macaques. Antimicrob. Agents Chemother. 56, 4980–4984 (2012).
Letendre, S. L. et al. Pharmacokinetics and antiviral activity of cabotegravir and rilpivirine in cerebrospinal fluid following long-acting injectable administration in HIV-infected adults. J. Antimicrob. Chemother. 75, 648–655 (2020).
Hodge, D., Back, D. J., Gibbons, S., Khoo, S. H. & Marzolini, C. Pharmacokinetics and drug-drug interactions of long-acting intramuscular cabotegravir and rilpivirine. Clin. Pharmacokinet. 60, 835–853 (2021).
North, T. W. et al. Suppression of virus load by highly active antiretroviral therapy in rhesus macaques infected with a recombinant simian immunodeficiency virus containing reverse transcriptase from human immunodeficiency virus type 1. J. Virol. 79, 7349–7354 (2005).
Soderberg, K. et al. A nucleotide substitution in the tRNA(Lys) primer binding site dramatically increases replication of recombinant simian immunodeficiency virus containing a human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 76, 5803–5806 (2002).
Uberla, K. et al. Animal model for the therapy of acquired immunodeficiency syndrome with reverse transcriptase inhibitors. Proc. Natl. Acad. Sci. USA 92, 8210–8214 (1995).
Balzarini, J., Weeger, M., Camarasa, M. J., De Clercq, E. & Uberla, K. Sensitivity/resistance profile of a simian immunodeficiency virus containing the reverse transcriptase gene of human immunodeficiency virus type 1 (HIV-1) toward the HIV-1-specific non-nucleoside reverse transcriptase inhibitors. Biochem. Biophys. Res. Commun. 211, 850–856 (1995).
Overton, E. T. et al. Long-acting cabotegravir and rilpivirine dosed every 2 months in adults with HIV-1 infection (ATLAS-2M), 48-week results: a randomised, multicentre, open-label, phase 3b, non-inferiority study. Lancet 396, 1994–2005 (2021).
García-Lerma, J. G. et al. Natural substrate concentrations can modulate the prophylactic efficacy of nucleotide HIV reverse transcriptase inhibitors. J. Virol. 85, 6610–6617 (2011).
Fray, E. J. et al. Antiretroviral therapy reveals triphasic decay of intact SIV genomes and persistence of ancestral variants. Cell Host Microbe 31, 356–372 (2023).
Crowell, T. A. et al. Impact of antiretroviral therapy during acute or early HIV infection on virologic and immunologic outcomes: results from a multinational clinical trial. AIDS 38, 1141–1152 (2024).
Li, J. et al. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS 30, 343–353 (2016).
Wang, Q. et al. CARD8 is an inflammasome sensor for HIV-1 protease activity. Science 371, eabe1707 (2021).
Andrews, C. D. et al. Long-acting integrase inhibitor protects macaques from intrarectal simian/human immunodeficiency virus. Science 343, 1151–1154 (2014).
Cranston, R. et al. A multiple dose phase 1 assessment of rilpivirine long acting in a model of preexposure prophylaxis against HIV. AIDS Res. Hum. Retroviruses 35, 794–804 (2019).
Neyens, M., Crauwels, H. M., Perez-Ruixo, J. J. & Rossenu, S. Population pharmacokinetics of the rilpivirine long-acting formulation after intramuscular dosing in healthy subjects and people living with HIV. J. Antimicrob. Chemother. 76, 3255–3262 (2021).
Radzio, J. et al. The long-acting integrase inhibitor GSK744 protects macaques from repeated intravaginal SHIV challenge. Sci. Trans. Med. 7, 270ra275 (2015).
Landovitz, R. J. et al. Cabotegravir for HIV prevention in cisgender men and transgender women. N. Engl. J. Med. 385, 595–608 (2021).
Marzinke, M. A. et al. Characterization of human immunodeficiency virus (HIV) infection in cisgender men and transgender women who have sex with men receiving injectable cabotegravir for HIV prevention: HPTN 083. J. Infect. Dis. 224, 1581–1592 (2021).
Boni, C. et al. TLR7 agonist increases responses of Hepatitis B virus-specific T cells and natural killer cells in patients with chronic hepatitis B treated with nucleos(t)ide analogues. Gastroenterology 154, 1764–1777 (2018).
Borducchi, E. N. et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature 563, 360–364 (2018).
Lim, S. Y. et al. TLR7 agonists induce transient viremia and reduce the viral reservoir in SIV-infected rhesus macaques on antiretroviral therapy. Sci. Transl. Med. 10, eaao4521 (2018).
Tsai, A. et al. Toll-like receptor 7 agonist GS-9620 induces HIV expression and HIV-specific immunity in cells from HIV-infected individuals on suppressive antiretroviral therapy. J. Virol. 91, e02166–02116 (2017).
Chavez, L., Calvanese, V. & Verdin, E. HIV Latency is established directly and early in both resting and activated primary CD4 T cells. PLoS Pathog. 11, e1004955 (2015).
Murray, A. J., Kwon, K. J., Farber, D. L. & Siliciano, R. F. The latent reservoir for HIV-1: How immunologic memory and clonal expansion contribute to HIV-1 persistence. J. Immunol. 197, 407–417 (2016).
Shan, L. et al. Transcriptional reprogramming during effector-to-memory transition renders CD4+ T cells permissive for latent HIV-1 infection. Immunity 47, 766–775 (2017).
Passaes, C. et al. Early antiretroviral therapy favors post-treatment SIV control associated with the expansion of enhanced memory CD8+ T-cells. Nat. Commun. 15, 178 (2024).
SenGupta, D. et al. The TLR7 agonist vesatolimod induced a modest delay in viral rebound in HIV controllers after cessation of antiretroviral therapy. Sci. Transl. Med. 13, eabg3071 (2021).
Okoye, A. A. et al. Early antiretroviral therapy limits SIV reservoir establishment to delay or prevent post-treatment viral rebound. Nat. Med. 24, 1430–1440 (2018).
Gupta, S. K. et al. Lenacapavir administered every 26 weeks or daily in combination with oral daily antiretroviral therapy for initial treatment of HIV: a randomised, open-label, active-controlled, phase 2 trial. Lancet HIV 10, e15–e23 (2023).
Massud, I. et al. Chemoprophylaxis with oral emtricitabine and tenofovir alafenamide combination protects macaques from rectal SHIV infection. J. Infect. Dis. 214, 1058–1062 (2016).
Melody, K. et al. Low frequency of drug-resistant variants selected by long-acting rilpivirine in macaques infected with simian immunodeficiency virus containing HIV-1 reverse transcriptase. Antimicrob. Agents Chemother. 59, 7762–7770 (2015).
Daly, M. B. et al. Training rhesus macaques to take daily oral antiretroviral therapy for preclinical evaluation of HIV prevention and treatment strategies. PLoS ONE 14, e0225146 (2019).
García-Lerma, J. G. et al. Intermittent prophylaxis with oral truvada protects macaques form rectal SHIV infection. Sci. Transl. Med. 2, 14ra14 (2010).
Kuklenyik, Z. et al. On-line coupling of anion exchange and ion pair chromatography for measurement of intracellular triphosphate metabolites of reverse transcriptase inhibitors. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 877, 3659–3666 (2009).
Swaims-Kohlmeier, A. et al. Proinflammatory oscillations over the menstrual cycle drives bystander CD4 T cell recruitment and SHIV susceptibility from vaginal challenge. EBioMedicine 69, 103472 (2021).
Curtis, K. A. et al. Development and characterization of a bead-based, multiplex assay for estimation of recent HIV type 1 infection. AIDS Res. Hum. Retroviruses 28, 188–197 (2012).
Waterboer, T., Sehr, P. & Pawlita, M. Suppression of non-specific binding in serological Luminex assays. J. Immunol. Methods 309, 200–204 (2006).
Acknowledgements
We thank Mara Sterling and Kenji Nishiura for their help in processing some of the macaque specimens, James L. Weed for training macaques to ingest FTC and TAF orally, Patrick Mills, and Rex Howard for performing some of the veterinary procedures, and Shanon Bachman, Frank Deyounks, Ryan Johnson, and Kristen Kelley for performing animal procedures. We also thank Hetero Drugs Ltd., and Laurus Labs Ltd for providing FTC and TAF. The following reagent was obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: RT-Simian-Human Immunodeficiency Virus, ARP-11342, contributed by Dr. Thomas North and Dr. Joseph Sodroski. The Anti-CD8 alpha [MT807R1] antibody used in this study was provided by the NIH Nonhuman Primate Reagent Resource (P40 OD028116). The findings and conclusions of this manuscript are those of the authors and do not necessarily represent the official views of the CDC. This work was funded with CDC intramural funds (to J.G.G.-L.). P.W. is a former Janssen employee (retired).
Author information
Authors and Affiliations
Contributions
M.B.D., P.W., W.S., W.H., and J.G.G.-L. were responsible for project conceptualization. M.B.D., C.D., A.H., D.R., S.R., A.S.K., S.S., J.M., J.C., D.K., K.C., and J.G.G.-L. performed the investigations. Formal analysis was done by M.B.D., A.S.K., G.K., Y.P., and J.G.G.-L. Data curation was done by M.B.D. and A.S.K. Funding acquisition and supervision by J.G.G.-L. and project administration by MBD and J.G.G.-L. The writing was done by M.B.D., W.H., and J.G.G.-L.
Corresponding author
Ethics declarations
Competing interests
J.G.G.-L., M.B.D., W.H., P.W., and W.S. are named in a US Patent Application (No. 18/002,494) entitled “Methods for achieving viral remission using long-acting antiretroviral agents”. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Courtney Fletcher, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Daly, M.B., Dinh, C., Holder, A. et al. SHIV remission in macaques with early treatment initiation and ultra long-lasting antiviral activity. Nat Commun 15, 10550 (2024). https://doi.org/10.1038/s41467-024-54783-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54783-0
