
Abstract
Circuit-based biomarkers distinguishing the gradual progression of Lewy pathology across synucleinopathies remain unknown. Here, we show that seeding of α-synuclein preformed fibrils in mouse dorsal striatum and motor cortex leads to distinct prodromal-phase cortical dysfunction across months. Our findings reveal that while both seeding sites had increased cortical pathology and hyperexcitability, distinct differences in electrophysiological and cellular ensemble patterns were crucial in distinguishing pathology spread between the two seeding sites. Notably, while beta-band spike-field-coherence reflected a significant increase beginning in Layer-5 and then spreading to Layer-2/3, the rate of entrainment and the propensity of stochastic beta-burst dynamics was markedly seeding location-specific. This beta dysfunction was accompanied by gradual superficial excitatory ensemble instability following cortical, but not striatal, preformed fibrils injection. We reveal a link between Layer-5 dendritic vulnerabilities and translaminar beta event dysfunction, which could be used to differentiate symptomatically similar synucleinopathies.
Similar content being viewed by others


Introduction
The accumulation of misfolded α-synuclein (αSyn) in cortical Lewy bodies is a hallmark of Parkinson’s disease dementia (PDD) and dementia with Lewy bodies (DLB), together known as Lewy Body Dementias. Here, fibrils comprising misfolded αSyn propagate trans-neuronally through anatomically interconnected networks, amplifying disease pathology by templating aggregation across molecularly defined cell-types1,2,3,4,5,6. How circuit activity across these vulnerable pathways manifests at the earliest stages of αSyn insult and evolves as a function of PDD and DLB progression remains unknown. Unraveling such “circuit-level biomarkers” is critical given the symptomatically similar end-stage behavioral deficits observed across the two forms of the disease, for which, at present, no biomarker exists that reliably differentiates between the two.
PDD and DLB are characterized not only by motor deficits but also by cognitive decline, albeit not in the same order7,8. In PDD, Lewy pathology is synonymous with striatal dysfunction facilitated by neurodegeneration of dopaminergic neurons in the substantia-nigra pars compacta (SNc)8, with cortical dysfunction occurring subsequently9. In contrast, in DLB, motor dysfunction occurs within a year of cognitive decline, affecting cortical areas first10. These features suggest that cortical vulnerabilities are not necessarily a late-stage effect, as proposed in the Braak et al., hypothesis11, but could serve as a critical top-down mediator of motor dysfunction, a notion gaining traction9,12. Despite this, it remains unclear how cell-to-cell transmission of misfolded αSyn occurs under PDD and DLB pathogenesis and impacts activity at the cellular, multicellular, and circuit levels9. As the motor cortex is vulnerable across both PDD and DLB7,8, the influence of αSyn pathology on recurrent translaminar connections is likely to be significant13, wherein the activity of even a single neuron can result in a non-linear change within the network4,14. This framework agrees well with the work of Nemani et al., who showed that modest overexpression of αSyn, albeit in the striatum, does not lead to stark toxicity-related changes. Instead, it impairs glutamate and dopamine release, which could lead to network dysfunction and eventual cognitive change—a feature that could be widespread during the prodromal-phase in the cortex15,16. Yet, we know little about the relationship between LB formation and the evolution of functional circuit changes, and whether such shifts are intrinsic to the neurons or a product of functionally connected ensembles of recurrent neural populations. To dissect such changes, one must measure and correlate dynamic activity patterns, including spatial and temporal properties, to pathology spread4,17. Such changes could be assayed through cellular-level ensemble dynamics, layer-specific spiking dynamics, and spike-field coherences, which we combinatorially highlight in this study.
Functional neural populations, termed ensembles18,19,20,21, represent stable multicellular activity maps that predict perception, action, and behavior better than the sum of their constituent neurons19,20. In this regard, a recent study using two-photon calcium imaging reported cortical dysfunction across superficial layers under striatal αSyn pathology load17. The study highlighted cortical hyperactivity and possible excitation-inhibition imbalance 9 months post-αSyn-preformed-fibril (αSyn PFFs) administration but did not capture the evolution of translaminar circuit changes or ensemble-level pattern differences. A similar study by the same authors showed that striatal αSyn PFF injection results in the loss of spines on pyramidal Layer 5 (L5) neurons in the anatomically connected somatosensory cortex (S1)22. Together these studies suggest a cortical pathway for Lewy pathology spread via L5 projection neurons to Layer-2/3 (L2/3) neurons23,24, highlighting the need for mapping recurrent translaminar circuit changes. Such changes in deep-layer neurons may contribute to a local network amplification of pathophysiology that leads to impairments at the ensemble level in superficial layers—a feature that potentially reflects the effects of pathology on integration across ascending and descending pathways24,25, highlighting selective synaptic vulnerabilities within sensorimotor cortices25,26,27,28. Thus, key differences in bidirectional L5-L2/3 coupling as well as eventual L2/3 ensemble dynamics could lead to unique biomarkers distinguishing different forms of synucleinopathies.
From an electrophysiological perspective, robust task- or stimulus-driven beta dynamics are observed in physiological brain states and are linked to motor preparation29 and action29, with more recent studies expanding their involvement to encompass a broader range of cognitive functions, such as working memory30,31,32, long-term encoding33,34, and decision-making35,36. In the cortex, translaminar network synchronization shifts between superficial and deep cortical layers, with slower beta oscillations driven by deep-layer neurons and superficial layers dominated by high-frequency gamma oscillations30,32, suggesting that the encoding and maintenance of cortical state is dependent on precisely timed nested oscillatory dynamics31. In PD and related dementias, there is strong correlative evidence linking elevated subcortical beta-band activity at rest and beta-power changes in response to stimulation37,38. Beta expression is thus primed to strongly predict healthy and pathological motor performance39,40,41. Recently, such beta activity changes were also observed in the olfactory bulb of mice seeded with αSyn PFFs42, suggesting that aberrant beta may not be solely relegated to subcortical motor areas, but might also be highly prevalent in cortical regions impacted by vulnerable pathways43. This aspect could be critical, especially in delineating between PD and DLB progression. However, a question that remains unanswered is whether beta and its pathologically driven amplification mechanisms16,44 could serve as reliable biomarkers of disease progression.
Evidence from computational modeling and experimentation suggests that the after-hyperpolarization and M-type potassium currents of layer 5 (L5) pyramidal neurons are sufficient to drive the firing of these neurons at beta frequencies45. Computational evidence, backed by human EEG and MEG recordings46 suggests that, in the motor cortex of both mice and humans, transient beta events41,47,48 waves that last ~50 ms—emerge from the integration of bottom-up and top-down inputs along the distal and proximal dendrites of cortical pyramidal neurons. This is significant because these bursts could reflect the hyperexcitability of multiple neurons associated with αSyn pathology49 in a pathway-specific manner. Unraveling the cortical biophysical mechanism of these aberrant beta bursts, as well as their specific spatiotemporal interactions in healthy and diseased cortical networks, will help elucidate key biomarkers differentiating across similar syndromes that share characteristic Lewy deposits8.
In this study, we injected αSyn PFFs in the dorsal striatum and deep-layer motor cortex to induce phosphorylated, aggregated inclusions resembling human LBs2,50. We assayed translaminar patterns of motor cortical activity and behavior across a 3- and 12 month pathological timeline—respectively—and revealed how αSyn PFFs propagating through two distinct pathways generate separable biomarkers within recurrent circuits. First, as a function of seeding location (cortical vs. striatal) and over 12 weeks of Lewy pathology spread, we show that there is diverging phase entrainment between L2/3 and L5 relegated only to beta, and not gamma or theta frequencies. Second, we reveal that cortical seeding destabilizes neuronal ensembles, a feature absent in the brains of mice subjected to striatal seeding. We discovered that such destabilization is delicately tied to shifting patterns in top-down integration, reflected in the form of moment-to-moment bursts of beta transients (β-events). Mechanistically, we show that this increase in β-event power is tightly modulated by apical L5 dendritic Ca2+ currents, a critical marker of top-down modulation, and that upregulation of N-Methyl-D-Aspartate (NMDA) phosphorylation could drive aberrancies in β-events in both pathological models. Importantly, we describe how such patterns of cortical circuit dysfunction manifested before behavioral deficits, which were observed 1 year after injections under both seeding strategies. Our observations strongly suggest that translaminar spike-field patterns coupled with functional neural ensemble dynamics in the superficial cortex serve as powerful biomarkers and can be used “combinatorially” to differentiate between prodromal-phase synucleinopathies.
Results
Characterizing the early spread of Lewy pathology across vulnerable pathways
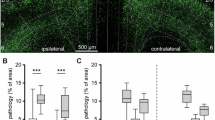
To explore the extent of pathogenic spread of Lewy bodies and its contribution to circuit dysfunction and prodromal-phase pre-motor deficits, we injected recombinant αSyn-PFFs (Supplementary Fig. S1a, S1b) in L5 of the mouse primary motor cortex (M1) or dorsal striatum (Fig. 1A, left,), and conducted a pathological assessment over a period of 12 weeks post-injection (wpi) (Fig. 1A, right). We compared the pathological burden of aSyn from both strategies to monomer synuclein injections in both regions in a separate cohort of mice. The injection of αSyn PFFs in either the cortex or striatum triggered the formation of intracellular Lewy body- and Lewy neurite-like aggregates within M1 (Fig. 1B, C). Interestingly, we also observed the emergence of αSyn pathology in various regions projecting to the motor cortex and striatum such as the sensorimotor cortex after 12 wpi (Fig. 1C, Supplementary Fig. S1c). Following M1 seeding, we noted apparently higher pathological expression in layer 5 of primary and secondary motor cortex (percentage of pSer129-αSyn+ neurons = 15.03 ± 8.38% and 14.13 ± 7.54%, respectively) and layer 4 of primary somatosensory cortex (8.33 ± 8.02%) in comparison to their more superficial layer 2/3 (L2/3) counterparts (12.54 ± 6.26 %, 12.21 ± 6.83%, 3.69 ± 2.11%; L2/3 of M1, M2, and S1 respectively). This was accompanied by a clear lack of pSer129-αSyn pathology in the dorsal striatum (2.48 ± 2.22%) (Fig. 1C, D). In contrast, dorsal striatal seeding led to a strong pathological spread in the striatum (percentage of pSer129-αSyn+ neurons = 26.58 ± 12.74%), followed by a comparable pathological burden in L5 of both primary and secondary motor cortex (percentage of pSer129-αSyn+ neurons = 15.74 ± 10.09 %, 14.63 ± 8.04%). Moreover, L4 and L2/3 of S1 showed a larger burden compared to cortical seeded (p < 0.001), with a comparable spread in M1 L5 and M2 L2/3. (Fig. 1C, D). Notably, although the percentage of neurons that stained positive for pSer129-αSyn was markedly greater in the L2/3 and L5 regions of mice injected in the cortex with αSyn PFFs versus αSyn monomer (monomer control), we observed no difference in overall somatic density across layers 12 weeks after injections between the two groups of animals.(Fig. 1E, F) (L2/3 monomer: 2093 ± 750, L2/3 PFF: 2072 ± 1128, L5 monomer: 2134 ± 417, L5 PFF: L2141 ± 864 neurons per mm2; p = 0.35, 0.22 for layer 2/3 and layer 5 respectively, n = 4 mice), suggesting a lack of early-stage cellular atrophy and death consistent with other reports5,17,51.

A Schematic of αSyn PFF injection into the primary motor cortex or dorsal striatum in separate cohorts of mice (left). Pathological assessment of evolving αSyn aggregate spread over a 12 week timeline (right). B Representative image showing pSer129-αSyn+ Lewy body- and Lewy neurite-like aggregates in Layer 5 of the primary motor cortex in a single cortically seeded mouse. Scale bar = 20 µm. C Pathological pSer129-αSyn+ neurons in select brain regions 12 weeks post-injection in both the cortex and striatum (VL, lateral ventricle), Scale bar: 200 µm. D Quantification of pSer129-αSyn+ neurons (% of total NeuN+ cells) in gross anatomical regions (n = 288, 392, 588, 370 neurons sampled across 4 slices across 4 mice (2 mice for cortical seeding and 2 mice for striatal seeding) for each region of M1, M2, S1, and STR respectively, one-way ANOVA with Bonferroni comparison test, **p < 10−2, ***p < 10−4 data shown as mean ± sd). E Comparison of pSer129- αSyn + neuron counts in PFF- versus monomer-injected mice 12 weeks post injection in the cortex (n = 4 slices, 340 neurons sampled, 4 mice, two-sided Mann–Whitney U-test **p < 10−7, ***p < 10−10, data shown as mean ± sd). F Density of NeuN-expressing neurons compared across L2/3 and L5 layers in PFF- and monomer-injected mice 12 weeks post injection in the cortex (n = 4 slices, 244 regions sampled, two-sided Mann-Whitney U-test p = 0.5, p = 0.11 for L2/3 and L5 respectively, data shown as mean ± sd). G Progressive accumulation of pSer129-αSyn+ puncta in primary motor cortex and dorsal striatum from 2 to 12 weeks post injection. Scale bar: 100 µm. H Comparison of pSer129+ neuron counts across weeks in the M1 region of mice injected with αSyn PFFs in primary motor cortex (n = 987 sampled neurons, 10 slices across 5 mice, 2 slices per week data shown as mean ± sem). I Comparison of pSer129+ neuron counts across weeks in the M1 region of mice injected with αSyn PFFs in dorsal striatum (n = 1020 sampled neurons, 10 slices across 5 mice, 2 slices per week). In (H, I) red and blue traces correspond to the % of pSer129+ neurons in L2/3 and L5, respectively for each seeding strategy; one-way ANOVA with Bonferroni comparison test, *p < 10−2, **p < 10−5, data shown as mean ± sem.
To characterize Lewy body spread across cortical layers over the 3 month period, we performed immunohistochemistry every 2 weeks of sections prepared from the primary motor cortex of mice injected with αSyn PFFs in the cortex or striatum (see Methods). We observed a gradual increase in the burden of pS129-αSyn aggregates across all layers in M1 of animals injected in either location, with the highest density focused in L5 and subsequently spreading to L2/3 (Fig. 1G, see Supplementary Fig. S1 for additional slices with counterstaining). Notably, we observed that significant Lewy pathology (consisting of obvious, somatic Lewy-like inclusions) did not appear in the M1 L2/3 region of cortically seeded mice until 6 wpi (percentage of pSer129-αSyn+ neurons = 10.7 ± 3.51%; p < 0.001) (Fig. 1H). Moreover, the appearance of pS129-αSyn aggregates was delayed (and the aggregate burden was reduced) in the M1 L2/3 region of striatally seeded mice compared to cortically seeded animals (percentage of pSer129-αSyn+ neurons = 6.49 ± 2.13% versus 13.22 ± 5.15%, p < 0.01) at 12 wpi (Fig. 1I). These findings suggest that the gradual spread in αSyn pathology differed slightly in cortically versus striatally seeded mice, but still consisted of a strong retrograde transynaptic progression of Lewy- like inclusions in M1, ending in similar pathological staging across layers. The continued propagation of αSyn aggregates across different brain regions also led to a heavy pathological load throughout the brain 12 months post-injection (Supplementary Fig. S1d). In addition, we observed little to no observable behavior changes (Supplementary Fig. S2a–d) involving locomotion in the case of striatally and cortically seeded mice within the first 3 months of αSyn-PFF seeding, whereas clear behavioral deficits were exhibited by animals in both groups 12 months post-injection (Supplementary Fig. S2e–i) (24 cortically injected and 22 striatally injected mice).
Electrophysiology reveals layer-specific vulnerabilities as a result of αSyn pathology
To investigate the effects of differing αSyn pathology on recurrent motor circuitry during active behavior52, we performed high-density electrophysiological recordings using silicon probes (see Methods) across layers of M1 in awake αSyn PFF-seeded and monomer seeded mice. Locomotory movements are the behavioral correlates of active motor preparation and execution23. Thus, mice were habituated on a running wheel 1 week before experiments and engaged in bouts of sustained running during recordings (Fig. 2A) (see Methods). We generated a velocity-triggered window to quantify periods of active locomotion. Using spectral decomposition, we analyzed the local field potential (LFP) during this period and identified frequency bands of theta (8–12 Hz), beta (12–30 Hz), and gamma (30–90 Hz) activity (Fig. 2B). We observed a significant increase in LFP power across these frequency bands in mice injected in the cortex or striatum with αSyn PFFs versus age-matched αSyn monomer (monomer control) at 12 weeks post-PFF injection (wpi) for both treatment groups (Fig. 2C, left, middle, n = 5 mice, Cortical Monomer n = 2, Cortical PFF n = 3, Striatal Monomer n = 2, Striatal PFF n = 3). Specifically, beta LFP power significantly increased by 12 wpi, highlighting a distinct change in cortical beta-band activity induced by αSyn pathology (Fig. 2C, right). During this 3-month period, no significant motor dysfunction was observed (Supplementary Fig. S2a–d). However, changes in exploratory behavior, rearing, and motor control were observed 6 months and 1 year after injection (Supplementary Fig. S2e–i). Investigating LFP alone reflects network-wide changes induced by αSyn pathology and does not capture the precise temporal contribution of underlying circuits. We thus measured putative excitatory single units across cortical layers and found that these showed rate-dependent modulation during locomotion (Fig. 2D). Analysis of putative excitatory single units revealed a gradual increase in mean firing rates in the brains of cortically and striatally seeded animals (Fig. 2E). However, while initially the spike rates in L5 were higher in comparison to L2/3, the rate of increase in L2/3 was rapid and steep towards the 12 week mark, suggesting an entrainment and amplification by L5. Specifically, cortical seeding drove a substantial increase in the mean firing rates of L2/3 neurons compared to L5 neurons during active locomotion (Two-way unbalanced ANOVA with Bonferroni comparison test, p < 0.01; N = 958 units (43 well-defined units per animal per timepoint); n = 22 Mice CTX; see figure legend for exact mice across weeks) (Fig. 2E, left). In striatally seeded animals, we observed a similar increase in firing rates over the 12 week period, but in this case, L2/3 seemed to linearly track the trend in L5 (Fig. 2E, right). Importantly, in comparison to cortical seeding, a slightly lower response of mean firing rates across both L2/3 and L5 neurons (Two-way unbalanced ANOVA with Bonferroni comparison test, p < 0.01; N = 709 units (35 well-defined units per animal per timepoint); n = 20 striatally seeded mice in total; see figure legend for exact mice across weeks) was observed. Notably, under all monomer conditions, where monomeric mice at 2 and 12 weeks after injection were pooled, firing rates of L2/3 pyramidal neurons were less than L5 (average firing rate during locomotion = 19.3 ± 5.1 L2/3 spikes/s; 39.4 ± 6.3 L5 spikes/s; p < 0.01).

A Schematic of experimental setup. B Representative CSD and wideband (1–100 Hz) LFP during initiated and running epochs averaged across trials from a single mouse (n = 407 trials). C (Left, middle) Power distribution of broadband LFP across cortical and striatal pathological weeks (n = 1083 trials, n = 5 mice, Cortical Monomer n = 2, Cortical PFF n = 3, Striatal Monomer n = 2, Striatal PFF n = 3). Note the shaded region corresponding to beta band (13–30 Hz). (One-way Anova with Bonferroni comparison test, n = 1040 trials). (Right) Changes of integrated beta LFP across weeks in cortically seeded mice F (4, 19) = 5.35, p = 0.007 (n = 18 mice, 3, 4, 4, 3, 4 across each two-week time point, i.e. every two weeks starting at week 2 and ending at week 12, respectively). D Representative single unit response of L2/3 and L5 pyramidal neurons during rest and running periods from a single monomer mouse (n = 11 trials). E Firing rate during active running for each layer across weeks in mice injected with αSyn PFFs in the cortex (left) FMon*PFF (5, 20) = 9.83, p = 3.7 × 10−9 or striatum (right) FMon*PFF (5, 20) = 15.12, p = 3.3 × 10−6. (Two-way unbalanced ANOVA with Bonferroni comparison test, *p < 0.01, **p < 0.001, ***p < 0.0001). F Schematic of spike field coherence (left). Beta spike-field coherence and phase calculated across depth (n = 2 cortical monomer-injected mice, 67 trials) (right). G Beta ITPC across depths and pathological weeks between monomer and cortical seeded PFF (left) FL23Mon*L23PFF (5,20) = 19.42, p = 8.66 × 10−09, FL5Mon*L5PFF (5,20) = 63.77, p = 1.42 × 10−09 and striatal seeded mice (right), FL23Mon*L23PFF(5, 18) = 17.6, p = 0.0024, FL5Mon*L5PFF (5, 18) = 8.99, p = 0.0004. (Two-way unbalanced ANOVA with Bonferroni comparison test, *p < 0.01, **p < 0.001, ***p < 0.0001). Data shown on box plots with 25th, median and 75th percentiles and with whiskers indicating min and max values, n = 22 cortically injected mice in total. Monomer Cortex n = 4 mice (2 mice at wpi and 2 mice at 12 wpi), Cortical PFF n = 18 mice (3, 4, 4, 3, 4 PFF mice across each two-week time point, i.e. every two weeks starting at week 2 and ending at week 12, respectively). n = 20 striatally injected mice in total. Striatal monomer n = 4 mice (2 mice at 2 wpi and 2 mice at 12 wpi) Striatal PFF n = 16 mice (3, 3, 3, 3, 4 PFF animals across each two-week time point, i.e. every two weeks starting at week 2 and ending at week 12, respectively), wpi weeks post-injection.
Firing patterns of neurons are heavily influenced by phase synchronization to the local-field potential (LFP). Therefore, we employed spike-field coherence53,54 (Fig. 2F, Supplementary Fig. S3a) to couple LFP and single-unit activity across cortical layers (see Methods). We focused on beta LFP and calculated the spike-field coherence and phase across cortical depths (Fig. 2F). To quantify the preference of single units to beta LFP, we looked at the beta inter-trial phase coherence (ITPC)53,54. Under monomer conditions pooled at 2 and 12 weeks, L5 neurons exhibited stronger beta ITPC than L2/3 neurons (ITPC during locomotion = 0.12 ± 0.03 for L5 and 0.06 ± 0.02 for L2/3; p < 0.05), in line with the theory that beta predominantly targets deep cortical layers45,55 (Fig. 2F, Supplementary Fig. S3b). Strikingly, in mice with cortically seeded αSyn pathology, we observed a shift in beta ITPC across weeks; superficial L2/3 ITPC was lower than L5 but then rapidly changed alongside L5 beginning at the 6-week time point, suggesting a gradual entrainment (Fig. 2G, left). However, in striatally seeded mice, increases in beta ITPC were more apparent in L2/3 at earlier timepoints relative to monomeric injections. This was followed by a larger increase of L5 ITPC in comparison to L2/3 12 weeks after seeding (Two-way unbalance ANOVA with Bonferroni comparison test, *p < 0.01, **p < 0.001, ***p < 0.0001, n = 22 Mice CTX, n = 20 Mice STR) (Fig. 2G, right). Such gradual changes in ITPC were primarily relegated to the beta and not gamma nor theta-bands for both seeding locations (Supplementary Fig. S3c–f). Importantly, such changes in spike rates or beta-ITPC were not observed when we compared monomeric cortical and striatal injected mice to un-injected mice under the same behavior (Supplementary Fig. S4a–d). Remarkably, the disruption observed under both seeding strategies in overall spike-LFP coherence emerged earlier than changes in broadband LFP power (Fig. 2C, right), indicating that the ITPC is a much more sensitive metric of pathology progression compared to gross LFP changes and can serve as a key marker of αSyn-induced circuit alterations in the motor cortex16. The rate of change in beta-ITPC across weeks shows that the measure carries within it a seeding location-dependence. Given that L5 recurrently modulates L2/3 via monosynaptic projections25,27, specifically via Layer 5A, it suggests that the L5 to L2/3 coupling might be particularly vulnerable to αSyn aggregates and a potential driver of L2/3 dysfunction.
The differences in the beta-ITPC profiles between animals injected with PFFs in the cortex versus striatum could be explained as follows -: (i) propagation of pathology must occur over a greater distance to impact L5 when administered in the striatum; and (ii) PFF injection at the two sites would be expected to affect non-identical circuit pathways, due to difference in seeding sites (Fig. 1D, H, and I), across the L5- > L2/3 axis26, thereby leading to differences in ITPC perturbation.
Finally, given that coherency of single units to LFPs is critical for both local and global synchronization of neuronal activity, the circuit alterations observed over the prodromal-phase could have significant implications for information processing and communication between different brain regions and cortical networks16,53,56 in pathologically similar forms of disease.
Superficial network hyperactivity patterns are pathway-specific
Fundamentally, networks of cortical pyramidal neurons in L2/3 exhibit sparse representation of sensory stimuli, allowing the cortex to nonlinearly represent sensory information in a distributed and efficient manner57,58. Based on the observation that αSyn PFF seeding in the cortex or striatum led to distinct beta-band ITPC profiles that progressively amplified in superficial layers over 12 weeks, we examined the extent to which this trend impacted cellular activity patterns and functional ensemble dynamics. To address this, we employed two-photon (2 P) calcium imaging with GCaMP6s expression under the control of the Thy-1 promoter (see Methods) in separate cohorts of mice (of either sex) injected with αSyn PFFs in the cortex or striatum, respectively (Fig. 3A, left). We imaged population activity of L2/3 pyramidal neurons in the motor cortex across 12 weeks and mapped their spatial and temporal correlations as a function of cortical and striatal seeding19,20, a feature not captured in our silicon probe recordings (Fig. 3A, right) (n = 9034 L2/3 pyramidal neurons from 20 cortically seeded mice; see figure legend for exact Ns across weeks, n = 7509 L2/3 pyramidal neurons from 20 striatal-seeded mice; see figure legend for exact mice across weeks).

A Schematic of experimental setup. B Denoised calcium transients in GCaMP6s expressing L2/3 neurons of monomer (green traces, bottom) and PFF-injected mice (blue, top). C Running evoked fluorescent activity and encoder trace in monomer animal under a representative running trial. D–I Pairwise analysis of data obtained under cortical and striatal PFF seeding models. D, E Low-dimensional neural state space representing transitions of rest and active running states 12 weeks post seeding in primary motor cortex or striatum of representative cortical monomer mouse (left) (n = 33 trials and n = 23 trials, respectively) and calcium event (right). Each row represents a neuron and each raster point represents a measure of neural activity. Percentage of neurons coactive within a given frame (bottom inset, right plots in D, E). Highly coactive regions above chance values are marked by a dashed line (See methods and Supplementary Fig. S5). F–I Pairwise analysis of neuronal activity. F, H αSyn PFF injection into motor cortex M1 (F, G) and dorsal striatum (H and I). Representative pairwise response from mice injected in the cortex (F) or striatum (H) with monomer control or aSyn PFFs 12 weeks post injection (‘12 wpi’) (n = 566 neurons, 4 mice). G, I Pairwise synchronicity values of evoked Ca2+ population responses across cortical. G FMon*PFF (5, 19) = 10.81, p = 5.18 × 10−6 and striatal (I) FMon*PFF (5, 18) = 13.11, p = 1.043 × 10−7, αSyn pathology compared to shuffled data (See methods). n = 9034 neurons from 20 cortically injected mice total. Cortical monomer n = 4 (2 mice at 2 wpi and 2 mice at 12 wpi), Cortical PFF mice n = 16 mice (3, 3, 3, 3, 4 PFF animals across each 2-week time point, i.e. every 2 weeks starting at week 2 and ending at week 12, respectively). n = 7509 neurons from 20 striatal mice total, Striatal monomer n = 4 (2 mice at 2 wpi and 2 mice at 12 wpi). Striatal PFF mice n = 16 mice (3, 3, 3, 3, 4 PFF animals across each 2-week time point, i.e. every 2 weeks starting at week 2 and ending at week 12, respectively). *p < 0.01, **p < 0.001, ***p < 0.0001, Two-way unbalance ANOVA with Bonferroni correction. J Schematic of evolved cortical networks based on pairwise activity and the emergence of cortical ensembles that are functionally dependent on time (FOV, field of view). (K) Representative traces of the two most stable ensembles across the behaving epoch from a single mouse (left) and their spatial arrangement (right). L–O α–Syn PFF injection into motor cortex M1 (L, M) and dorsal striatum (N, O). L, N Ensemble of neurons after mathematically decomposing data from binarized matrix under cortical and striatal seeding. M, O Probability of ensemble recruitment response across αSyn pathology under cortical FMon*PFF (5,19) = 13.62, p = 1.37 × 10−5, and striatal seeding model FMon*PFF (5,18) = 0.24, p = 0.94 (*p < 0.01, **p < 0.001, ***p < 0.0001, two-way unbalanced ANOVA with Bonferroni correction). n = 1950 ensembles from 20 cortically seeded mice, Cortical monomer n = 4 mice (2 mice at 2 wpi and 2 mice at 12 wpi), Cortical PFF n = 16 mice, n = 1871 ensembles from 20 striatally seeded mice, Striatal monomer n = 4 mice (2 mice at 2 wpi and 2 mice at 12 wpi), Striatal PFF n = 16 mice, data shown on box plots with 25th, median and 75th percentiles and with whiskers indicating min and max values, wpi weeks post-injection.
We quantified Ca2+ dynamics in motor cortex L2/3 (see Methods), across both PFF and monomer-injected mice (monomer experiments performed and pooled at 2- and 12 week timepoints only) (Fig. 3B, C) during spontaneous bouts of running. Because of the relatively large number of neurons imaged, a clear transition of cortical dynamics could be observed during locomotion, which could be projected into a lower-dimensional state-space (Fig. 3D, E). We then extracted pairwise correlations (based on synchronized Ca2+ activity) triggered by spontaneous bouts of locomotion (see Methods). Using this metric, we observed a gradual yet progressive increase in pairwise correlations for mice injected with αSyn PFFs in either seeding location (Fig. 3F–I). Notably, pairwise synchronicity extracted from the covariance maps (Fig. 3F, H) obtained for cortically seeded mice (Fig. 3G) reflected a marked difference as early as 4 weeks post-injection in comparison to striatally seeded, where a difference was seen only 8 weeks after PFF administration (Fig. 3I). These differences were significantly above monomer-injected mice for both cortically (n = 184 neurons, 33,850 pairwise connections (per FOV); n = 20 cortically seeded mice in total; see figure legend for exact Ns across weeks; p < 0.001; Two-way unbalance ANOVA with Bonferroni comparison test) and striatally seeded mice (n = 140 neurons,19,600 pairwise connections (per FOV); n = 20 striatally seeded mice in total; see figure legend for exact mice across weeks). While an increase in cortical pairwise activity alone is not surprising, our results highlight possible changes in cellular and circuit mechanisms critical for motor control and learning. Specifically, distinct pathway-specific hyperactivity patterns, observed from changes in spiking activity (Fig. 2E, G), can influence and alter computations requiring sparse L2/3 coding, for example, during motor learning59,60, in a manner that varies with pathology propagation across vulnerable pathways. Central to this line of thinking would be the disruption of neuronal ensemble dynamics, which we examine below.
While pairwise hyperactivity trends (Fig. 3G, I) are informative in conveying how any two neurons are coactive, we wondered if there was a starker difference in the activity patterns that could help differentiate pathway-specific progression. It is not clear, from average pairwise synchrony alone, how groups of 3 or more neurons coordinate activity within a dysfunctional network. Indeed, throughout locomoting bouts, a subset population of multiple coactive neurons, termed neuronal ensembles, emerged19. Ensemble dynamics reflect flexible spatial emergences and can signify a mixed representation of sensory features (Fig. 3J)61. Importantly, stable reactivation of neuronal ensembles is a critical feature of healthy brains and has been linked to motor learning21,59,60. To identify these ensembles, we measured population activity as multi-dimensional vectors corresponding to the degree of similar temporal activity across populations of neurons (see Methods and Supplementary Fig. S5a–d). We searched this multidimensional vector (i.e., time) space to mathematically extract various ensemble combinations, which were projected onto a 2D Euclidean space to reveal their spatio-temporal structure (Fig. 3K).
To test whether specific features of ensembles were altered in response to αSyn pathology, we quantified ensemble stability – the probability that neurons within an ensemble reactivate together (see Methods, Fig. 3L–O). The strength of co-activation of neurons within an ensemble, termed a connection, was critical to define the stability of the ensemble (Fig. 3L, N, Supplementary Fig. S5d). Interestingly, under cortical seeding we discovered a reduction in ensemble stability over the 12 week period in contrast to the progressive increase in pairwise synchronicity observed for cortically seeded mice (Fig. 3M) (Two-way unbalance ANOVA with Bonferroni comparison test, p < 0.001; n = 40 ensembles (per FOV); N = 49 FOV; n = 20 cortically seeded mice in total; see figure legend for exact mice across weeks). However, when we applied the same analysis to striatally seeded mice, we observed no such change in ensemble stability (Fig. 3O) (Two-way unbalance ANOVA with Bonferroni comparison test, p = 0.94, N = 69 ensembles (per FOV); N = 55 FOV; n = 20 striatally seeded mice in total; see figure legend for exact mice across weeks). A gradual reduction of ensemble stability implies that the probability for the same neurons within an ensemble to reactivate decreases across the pathological timeline. A comparison of pairwise network activity (synchronicity) and ensemble stability reveals a dichotomous relationship in mice with evolving αSyn pathology that is only found in cortically (but not striatally) seeded animals. Notably, the change in ensemble stability reflected a difference in the number of functional ensembles that could be formed. For animals injected with αSyn PFFs across both locations, we observed a significant decrease in the number of detected ensembles over time (Supplementary Fig. S5e, S5g). However, the number of functional connections was only different in mice subjected to cortical PFF seeding, suggesting a difference in temporal activity of cortical ensembles not found in striatally seeded animals. (Supplementary Fig. S5f, S5h). Importantly, throughout the 12 week period, there was no change in the number of cells or the number of cells per ensemble being imaged across mice (Supplementary Fig. S5i–k), suggesting that the effects seen at the population level and the differences observed as a function of seeding location were due to local circuit impairments. This was further substantiated when we compared functional pairwise activity and ensemble stability of monomeric mice to uninjected GCaMP mice (Supplementary Fig. S6a–d.) which revealed similar pairwise and ensemble activity across mice. Overall, the presence of sparse yet strong pairwise synchronicity and optimal ensemble activity is a hallmark of a healthy L2/320, and the phenomenon of systematic deviation in ensemble stability seems to only arise as a function of pathology.
Overall, the change in pairwise hyperactivity measured via two-photon calcium imaging in L2/3 (Fig. 3G, I) tracks the L5 to L2/3 entrainment observed in the electrophysiological measures (Fig. 2E and G) across weeks, eventually leading to seeding location-specific ensemble dysfunction in L2/3. Is there a possible connection between the hyperactivity and dysfunction observed in the superficial L2/3 layer and a pathology-induced effect in the L5 layer? This could be due to their recurrent connectivity. Additionally, the differences seen in ensemble stability may be due to pathway-specific vulnerabilities that are tied to L5 connectivity. Is there a way to identify such an effect in L5?
Translaminar beta dynamics indicate distinct pathway-specific L5 vulnerability
The disruption of supragranular spatiotemporal neural ensemble dynamics may have significant implications for recurrent translaminar activity. Current understanding across the cortical column posits that the interaction between bottom-up sensory processing and top-down feedback mechanisms generates flexible sensory representations through the coordination of gamma-beta interactions (Fig. 4A), with gamma and beta oscillation predominantly generated by superficial and deep-layer neurons, respectively55. This coordination allows for efficient sensory processing under attentional demands32,55,62,63. Also, beta oscillations have been implicated in the synchronization of activity across neural ensembles, highlighting their crucial role in cortical processing30. Is there a delineating feature of beta that can match the superficial ensemble dynamics differences as a function of seeding locations? We posit that such a marker, possibly driven by L5, alongside ensemble dynamic differences in superficial layers will serve as a powerful handle to differentiate between symptomatically similar synucleinopathies

A Potential mechanism of narrow-band beta LFP information flow. Beta LFP is modulated by deep layer cortical activity via feedback while gamma dominates in superficial layer via feedforward inputs. B Example response of beta/gamma LFP power during voluntary running in monomeric versus αSyn PFF animals. C Filtered beta LFP across velocity triggered trials. Blue traces correspond to putative bursts of beta transients with compiled waveforms of β-events (n = 11 trials). D Examples of peristimulus beta band activity across two trials. Asterisks mark detected β-event waveforms. E Exemplar β-event spectrogram. F β-event amplitude and duration in representative recording of one monomer mouse (n = 992 events). G, H Z-scored β-event amplitude compared to monomeric control animals (indicated by gray box) across depth and pathological timeline for cortical seeded mice (G) FsupMon*PFF (5, 20) = 8.78, p = 3.15 × 10−7, FdeepMon*PFF (5, 20) = 13.11, p = 8.76 × 10−11 and striatal seeded mice (H) FsupMon*PFF (5, 18) = 6.78, p = 0.0002, FdeepMon*PFF (5, 18) = 7.04, p = 0.0002 (*p < 0.01, **p < 0.001, ***p < 0.0001, two-way unbalanced ANOVA with Bonferroni correction); Cortically seeded: 1242 ± 302 β-events per animal per timepoint. n = 22 cortically injected mice in total. Cortical monomer n = 4 mice (2 mice at 2 wpi and 2 mice at 12 wpi), Cortical PFF mice n = 18 mice (3, 4, 4, 3, 4 animals across each 2-week time point, i.e. every 2 weeks starting at week 2 and ending at week 12, respectively). Striatally seeded: 1312 ± 401 β-events per animal per timepoint. n = 20 striatally injected mice in total, Striatal monomer n = 4 mice (2 mice at 2 wpi and 2 mice at 12 wpi), Striatal PFF mice n = 16 mice (3, 3, 3, 3, 4 PFF animals across each 2-week time point, i.e. every 2 weeks starting at week 2 and ending at week 12, respectively), wpi weeks post-injection, data shown as median with first and third quartiles. I Histogram distribution of β-event duration and number of events per trial across weeks for cortical (top) and striatal (bottom) seeded mice. Dashed line indicates significance in distribution compared to shuffled surrogates. Distribution of beta power and duration show high variability of β-event behavior. J Schematic of proposed emerging changes in narrow-band beta LFP dynamics. Aberrant deep layer beta dynamics may drive superficial beta dynamics via feedback (left). Normalized amplitude of β-event profiles for two example mice of cortically seeded (top) and striatally seeded (bottom) with corresponding depth. K Experimental schematic of dual LFP-Intracellular recordings (right). Intracellular recording from putative L5 and L2/3 pyramidal neurons in primary motor cortex (top trace). Wideband filtered LFP (4–40 Hz) (bottom trace). L LFP beta spectrogram and spiking response of L5 and L2/3 pyramidal neuron. Detected β-events is noted by white asterisk. M Vm and extracellular β-event -triggered coherence for L2/3 and L5 pyramidal neurons. (n = 10 neurons, 7 L5 neurons, 3 L2/3 neurons, p = 0.00045, ***p < 0.0005, one-sided Kruskal-Wallis, data shown on box plots with 25th, median and 75th percentiles and with whiskers indicating min and max values). N Extracellular β-event triggered subthreshold response of L5 and L2/3. Average β-event waveform (dash line), average subthreshold response from L5 pyramidal neurons (blue trace), n = 7 neurons, and subthreshold response to layer 2/3 neurons (black trace, n = 3 neurons). O β-event triggered AP bursting probability from L5 and L2/3 pyramidal neurons (N = 2406 β-events across 243 trials, p = 5.5 × 10−45, Kruskal-Wallis). P Intracellular-frequency response during bursting of L5 pyramidal neuron.
We analyzed the power distribution of beta and gamma across the cortical depth. Indeed, and consistent with previous studies on motor and working memory in non-human primates32,55,64,65, we observed that gamma amplitude was largest in the superficial layers (at depths up to 250 µm, yellow trace), while the beta power occurred in deeper layers (at depths >450 µm, blue trace) (Fig. 4B). Strikingly, in the brains of mice injected with αSyn PFF, there was a modest increase in beta power distribution across both seeding locations.
Recent work has also revealed that beta LFP is not a continuous, sustained rhythm, but rather a transient surge in amplitude that occurs throughout individual trials via beta bursts47,48 (Fig. 4C, D). These bursts have been linked to the initiation and maintenance of movement, as well as the suppression of unwanted movements29,66, and disruptions in beta burst activity have been associated with movement disorders such as Parkinson’s disease41,49. Therefore, we next asked if moment-to-moment features of cortical beta can reflect changing translaminar dynamics in response to αSyn pathology initiated from different seeding locations. We found that stochastic beta bursts in the motor cortex emerge as stereotypical waveforms across trials (Fig. 4D, E), typically lasting 40–100 ms, and fluctuating in waveform amplitude (Fig. 4F). These beta bursts span the beta frequency spectrum with a preference towards high beta (Fig. 4E). Here on, we term these stochastic bursts “beta events” (β-event)48. To quantify the effect of evolving αSyn pathology on β-events, we compared the β-event amplitude in the brains of mice injected with aSyn PFFs in the cortex or striatum, as well as monomer controls (Fig. 4G–I) (n = 22 cortically seeded mice in total, n = 20 striatally seeded mice in total; see figure legend for exact mice across weeks). With cortical seeded pathology, as opposed to the broadband beta-ITPC measures across the pathological timeline (Fig. 2G) in which we found an initial increase in amplitude in L5 followed by an L2/3 entrainment, we found a significant increase in β-event amplitude in L2/3 of M1 that preceded a more modest increase in L5 towards the 12 week timepoint (Fig. 4G) (Two-way unbalance ANOVA with Bonferroni comparison test, p < 0.001; (1242 ± 302 β-events per animal per timepoint); n = 22 cortically seeded mice in total; see figure legend for exact mice across weeks). Under striatal seeding however, the β-event trend tracked the rate previously observed with beta-ITPC in which L5 increased similarly to L2/3 over the 12 week period (Fig. 4H) (Two-way unbalance ANOVA with Bonferroni comparison test, p < 0.001; (1312 ± 401 β-events per animal per timepoint); n = 20 striatally seeded mice in total; see figure legend for exact mice across weeks). We also noted significant differences in the number of β-events per trial but not the duration of the waveform across weeks and against monomer-injected animals for both seeding locations (Fig. 4I). Importantly, beta events were distributed along the cortical depth for both seeding locations, but with a stronger preference to initiate towards the superficial layers of M1 (Fig. 4J) in cortically seeding animals.
The results across Fig. 1 thru Fig. 4J lead to a distinct translaminar circuit marker of disease progression across different seeding locations. Specifically, as a function of seeding location (ie. seeding in deep layer cortex but not striatum), we observed both the firing rate and beta-ITPC (spike-field coherence) increase first in L5, subsequently entrain L2/3 (Fig. 2E, G), and this entrainment coincides with L2/3 excitatory ensemble instability (Fig. 3M, O). Importantly, we show that the β-power increase in superficial layers occurs only during pathology while primarily being generated in deeper layers of the cortex in monomer controls (Fig. 4B). Analyzing beta band activity in the form of stochastic moment to moment bursts at the single trial level revealed an increase in β-event amplitude in L2/3 in comparison to L5 under cortical seeding while the opposite was observed under striatal seeding with L5 and L2/3 modestly increasing together (Fig. 4G, H). Combined, these results strongly suggest that β-event generators share a common translaminar locus possibly intrinsic to L5 neurons48 (Fig. 4J).
To elucidate the mechanism underlying β-event generation, we performed intracellular recordings, albeit separately, across putative L5 neurons and L2/3 neurons (at depths of 450–700 µm and 100–250 µm, respectively) in awake locomoting mice, while simultaneously measuring LFP recordings in superficial layers (at depths of 150–250 µm) (Fig. 4K). Spike-beta-event coherence measurements revealed a strong link between L5 bursting and extracellularly recorded beta-events, while L2/3 cells revealed a much weaker relationship (Fig. 4L, M) (Layer 5 = 0.58 ± 0.08; Layer 2/3 = 0.19 ± 0.05; p < 0.0001; N = 10 neurons). L5 neurons also displayed a more prominent membrane voltage oscillation compared to L2/3 neurons, indicative of a larger depolarization drive within the peristimulus β-event window (Fig. 4N, Layer 5 membrane potential change = 0.81 ± 0.12 mV; Layer 2/3 membrane potential change = 0.04 ± 0.007 mV; p < 0.01; n = 6 neurons). Additionally, L5 neurons were significantly more likely to exhibit intracellular bursts alongside extracellular beta events (Fig. 4O; 2406 beta events across all neurons). Examining the frequency components present in the intracellular recordings further revealed a broad beta-band response during L5 bursting (Fig. 4P). These findings together suggest that extracellular β-events may reflect the translaminar currents responsible for L5 burst mode signaling while conversely, a significantly lower effect is present for L2/3 in the primary motor cortex.
To further substantiate the link between L5 dendritic activity and the generation of translaminar β-events we performed biophysical modeling using morphologically realistic L5 neurons (Supplementary Fig. S7a, see Methods). With excitation inputs (driven at Poisson rates) across the basal and distal apical dendrites, β-events were indeed generated in the presence of large apical dendritic currents denoted by a somatic burst (Supplementary Fig. S7b). Strikingly, β-event amplitude was observed to be severely reduced under conditions of NMDA and voltage-gated Ca2+ channel activity block along the L5 apical dendrite (Supplementary Figs. S7d–f). We further verified this experimentally via application of baclofen, a GABAB agonist, along the calcium rich zone (depth of 150–250 µm) of L5 neurons (see Methods), which reduced β-event power significantly (Supplementary Fig. S8a, S8b), while application of NMDA agonist (see Methods) along the same region reduced the β-event rate appreciably (Supplementary Fig. S8c, S8d). Beta event dynamics we observed experimentally were largely absent when we performed the same set of simulations on biophysically modeled L2/3 neurons (Supplementary Fig. S9a–e). Notably, the translaminar profile and beta event amplitude were diminished compared to L5 neurons (Supplementary Fig. S9d, S9e, p < 0.005, Manns-Whitney U-test). These data suggest that NMDA channel function may be implicated in driving changes of beta event dynamics across a pathological timeline. To assess this, we conducted a phospho-proteomic analysis of the sensorimotor cortex in aSyn PFF versus monomer injected (monomer control) animals, which revealed differences in levels of phosphoproteins involved in NMDA function and localization (Supplementary Fig. S10). Specifically, we found increased Grin2b phosphorylation in the cortices of mice injected intrastriatally with αSyn PFFs versus monomer, which we further confirmed with immunohistochemical analysis (Supplementary Fig. S11).
β-events power as a function of pathology coincides with aberrant dendritic excitability
Finally, to corroborate that L5 dendritic activity is indeed aberrant in vivo during high β-event power, we performed αSyn PFFs seeding across both dorsal striatum and in the primary motor cortex in Rpb4-cre mice which expressed cre-flox reporter primarily in L5 pyramidal neurons (Fig. 5A, B). Viral injection of GCaMP6s-mRuby allowed for structural reconstruction and functional imaging of L5 tuft dendrites for mice under monomeric and αSyn PFF pathology under cortical or striatal seeding (Fig. 5C). We imaged L5 tuft dendrites (Fig. 5B, C) (at depths 40 µm−150 µm from the surface) in all 4 cohorts of mice after 6 weeks post-seeding (based on the timeline of L5 dynamics shown in Fig. 2 and Fig. 4) under locomotion (Fig. 5D). L5 tuft dendrites showed clear differences in pairwise population activity across dendrites during rest and locomotory states (rest: 0.07 ± 0.07; run: 0.23 ± 0.17; p < 0.001; n = 821 dendrites; 5 monomer mice) (Fig. 5E). When comparing dendritic populations, αSyn PFF injected mice exhibited higher pairwise synchronicity than monomer mice (Fig. 5F, G) across the four treatment groups. Notably, distinct and significant changes in L5 dendritic connectivity (Fig. 5H, top, denoting synchronicity) were observed in mice injected with αSyn PFF in the cortex (monomer: 0.014 ± 0.01, PFF: 0.034 ± 0.02,3264 dendritic segments; p < 0.001; n = 21 FOVs across 7 mice cortically injected mice) and striatum (monomer: 0.0137 ± 0.006, PFF: 0.02 ± 0.01, 2939 dendritic segments; p < 0.001; n = 19 FOVs across 6 striatally injected mice), with the striatal PFF seeding showing a slightly less but still significance change in pairwise connectivity compared to cortical PFF seeding (p < 0.001, one-way ANOVA, 8 mice) (Fig. 5H, bottom). These data support our observation that pathological changes in β-events are closely tied to L5 dendritic aberrancy. Moreover, there is a significant difference in the dendritic activity patterns evoked by αSyn PFF seeding at different locations, suggesting that L5 pyramidal neurons may play a critical role in differences seen across the translaminar circuit during the evolution of αSyn pathology.

A Experimental schematic and injection strategy for dendritic imaging during αSyn pathology. B Representative images of L5 dendrites in M1 motor cortex immunohistochemically (left) and in vivo calcium imaging (right) (scale bars = 100 µm and 20 µm, respectively from single FOV and brain slice, respectively.). C Fluorescence traces of dendritic tuft activity for single field of view of monomer (green) and cortically seeded mice (magenta). D Average dendritic population response during motor initiation from example mouse (n = 182 dendrites, 2 FOV from cortical monomer mouse). E Pairwise population response during rest and running states (n = 801 dendrites, 7 FOV, 4 mice total, 2 mice for each monomer group). F Example pairwise plot for each pathological condition (n = 281 dendritic segments, 4 mice). G Sørenson-Dice dendritic synchronicity map, observed for mice injected with monomeric aSyn (‘Monomer) or aSyn PFFs (note increased connectivity in the case of cortically seeded PFF animals). H Dendritic population synchronicity observed for mice in each treatment group during locomotion. (n = 13 animals, Cortical monomer n = 3 mice, Cortical PFF n = 4 mice, Striatal monomer n = 2 mice, Striatal PFF n = 4 mice, *p < 0.01, **p < 0.001, ***p < 0.0001, One-way ANOVA, data shown on box plots with 25th, median and 75th percentiles and with whiskers indicating min and max values). I Pathological outcomes of cortical and striatal seeding lead to differing cellular and subcellular responses.
Collectively, our results suggest that burst-mode activity in L5 neurons that is mediated by NMDA and Ca2+ signaling is a crucial element in the creation of β-events. Additionally, it appears that the abnormal phosphorylation of NMDA-related proteins could be a significant factor in the increased amplification of β-event power that is indicative of dendritic abnormalities in the local cortical circuits of the sensorimotor cortex in mice with aSyn pathology. The differences in β-event amplitude between seeding locations further suggest that L5 dendritic channels are a vulnerable target, which not only helps generate a pathway-specific translaminar signature of disease progression (Fig. 5i), but could also serve as therapeutic targets for treating synaptopathies.
Discussion
In this study, we introduced αSyn PFFs into the dorsal striatum and deep-layer motor cortex of mice, resulting in the formation of inclusions similar to those found in the brains of individuals with PDD and DLB11,12. Over a period of 3 months (Fig. 1), we then examined the motor cortical activity patterns in awake locomoting mice and elucidated how circuit patterns reveal distinct and differentiable biomarkers. We discovered that there is a competition between cortical L2/3 and L5 excitatory activity in terms of phase entrainment to beta frequencies (Fig. 2), but not gamma or theta, despite the similarities in spread of Lewy pathology induced by PFF injection across the two sites. Specifically, with cortical seeding, L5 activity initially dominates, but L2/3 subsequently outpaces L5 beta-phase-entrainment. With striatal seeding, we found that L2/3 entrainment by L5 in the cortex is weaker in comparison. We also observed that cortical seeding destabilizes superficial L2/3 ensembles (Fig. 3), which is not the case with striatal seeding. Importantly, this destabilization is reflected by changes in beta-band dynamics across superficial layers, and analyzing these dynamics in a moment-to-moment fashion reveals a seeding location-dependent increase in stochastic β-event dynamics (Fig. 4). Importantly, β-event dynamics was discovered to be tightly locked to apical L5 dendritic Ca2+ currents and aberrancies were reflected at this level of dendritic signaling (Fig. 5). Markedly, these distinct changes in cortical circuit function occurred before behavioral dysfunctions observed at later timepoints (Supplementary Fig. S2). The fact that beta dynamics are primarily driven by the feedback-layers in deep cortex55, a feature we clearly observed (Fig. 4B), and that top-down feedback to L5 neurons targets apical dendritic activity in the cortex25,67, suggests a mechanism in which top-down control of motor output might be a potent target of synuclein dysfunction during the prodromal phase.
αSyn pathology progressively spreads between anatomically connected regions and this spread may result in different stressors among distinct neuronal networks over time12,17,43. Here, we show that both striatal and cortical seeding of αSyn PFFs leads to widespread inclusion formation across the cortex (Fig. 1A–F), although with distinct circuit hyperactivity patterns possibly governed by anatomical connectivity. Surprisingly, the nature of circuit dysfunction is at odds with previous reports. For example, acute application of αSyn PFFs in vitro was shown to result in impaired synaptogenesis and reduced activity4,51, while in vivo injection of αSyn PFFs in the striatum led to (i) cortical hyperactivity attributed to a net change in excitation-inhibition balance exacerbated by gabaergic neuron loss, but with minimal Lewy neurite formation in the cortex after 9 months17,68, or (ii) a decrease in synapse and spine density as well as spontaneous excitatory post-synaptic current frequency in L5 of the somatosensory cortex51. Our results (Fig. 1G, H), show that the motor cortex is indeed able to exhibit marked Lewy pathology and that intricate recurrent circuits in vivo might actually amplify circuit dysfunction (Fig. 2. and Fig. 3), before full-scale neurodegeneration.
With high-density electrophysiological recordings (Fig. 2A, B) we observed distinct differences in spike-field entrainment, specifically in the beta-frequency (Fig. 2C–G), as a result of cortical versus striatal αSyn-PFF seeding across over a 12 week period. Notably, a L5-dependent entrainment of spike-field coherence in L2/3 was observed in response to both cortical and striatal αSyn-PFF seeding (Fig. 2E, G), but with distinct differences in rates of increase. This result has important implications within the context of recurrent cortical and cortical-basal ganglia circuits and strongly underscores cortical L5 neurons as a vulnerable node between differing pathological seeding strategies9,13,28. Studies in non-human primate models have proposed that beta-frequency dynamics are driven by deep-layer neurons reflecting top-down feedback, with superficial layers driving gamma-band activity reflecting feedforward signaling31,55—a result we recapitulated in this study (Fig. 4A, B). Moreover, we found that beta-frequency dynamics were switched (with greater β-event localization in superficial layers) and heavily modulated under both striatal and cortical αSyn-PFF seeding, yet with distinct differences. What are the circuit metrics that enable such differences?
Cortical microcircuits are hierarchically organized through feedforward, feedback, and lateral connections13. In the primary motor cortex, the L2/3 → L5A/B connection is the strongest, accounting for one-third of the total synaptic current in the circuit, followed by the reciprocal L5A → L2/3 connection as the second largest25. Therefore, strong recurrent inputs between superficial and deep layers can likely amplify any modulation in excitability due to pathophysiology26. This is highly likely given that we observed, for both cortical and striatal seeding, an initial increase in L5 beta ITPC followed by entrainment in L2/3 (Fig. 2E, G).
The differences in spike-field entrainment between mice injected intracortically versus intrastriatally could reflect the greater distance over which the propagation of pathology must occur to affect L5 when PFFs are administered in the striatum, or the likelihood that seed injection at the two sites influences non-identical circuit pathways across the L5- > L2/3 axis given where they are seeded (Fig. 1G–I). For example, Layer 5A (intratelencephalic neurons projecting to the striatum) and Layer 5B neurons (pyramidal tract neurons) will be impacted in different ways across the two seeding locations, thereby impacting L2/3 differently. Notably, under cortical seeding, both Layer 5A and 5B could have a direct insult, while under striatal seeding layer 5B could be delayed in comparison to L5A due to multiple synaptic delays.
In line with changes at the circuit level, cell-type specific inhibition can play a major role in shaping local pathological changes as a function of aSyn uptake. Recent studies have described the role of changes in excitation-inhibition balance in L2/3 as a critical foundation for optimal sensory coding61,69, including the role of somatostatin interneurons (SOM + ) in controlling L2/3 beta synchrony70,71. Given that these adapting inhibitory interneurons receive a strong excitatory drive from both deep-layer pyramidal neurons and laterally from within L2/369, αSyn PFF induced changes in L5 excitatory activity observed in this study could further amplify and alter superficial beta synchrony both via anterograde signaling (L5→SOM+ and L5→L2/3) and retrograde propagation (via PFF uptake by L2/3 axons)23,69. Future experiments should focus on disentangling such connections in the context of αSyn pathology.
By monitoring the average pairwise level (correlations) measured via calcium dynamics and (tabulated as the mean correlation between any two cells) in L2/3 of mice injected with αSyn PFFs at both seeding locations, we observed an apparent increase in network synchronicity (Fig. 3A–I) mirroring the rate of change in firing rates over the 12-week period observed with high-density electrophysiology probes (Fig. 2E and G). Yet, where these seeding strategies diverged was at the ensemble level (Fig. 3J, K), where local persistent multi-neuronal activity patterns were measured across time19. Cortical but not striatal seeding of αSyn PFFs revealed a systematic decrease in ensemble stability (Fig. 3L–O), highlighting the gradual inability for ensembles to reliably activate within their respective groups during sensorimotor behavior, which in this study was reaching a set speed of locomotion. Importantly, this decrease was not a simple consequence of single-neuron activity level changes or functional connectivity, since neither unit spike rate, beta spike-field coherence, nor pairwise correlation was a good predictor of ensemble stability. Instead, our results likely reflect a disorganization of interconnected neuronal dynamics relegated to cortical-originating pathology—synonymous with dementia with Lewy body staging. This disorganization may arise from a local loss of inhibition1,17,72 or impaired inhibitory drive as previously mentioned, which can dynamically alter spatio-temporal activation patterns of multiple neurons in L2/3, thus influencing L5 dynamics23,25,55. Our results support a model in which the pervasiveness of pathology is not an indicator of how disease propagation sculpts multi-neuronal activity patterns but rather that the variability of αSyn pathology can impact specific vulnerable recurrent synaptic pathways across the L2/3-L5 axis in a connectivity-dependent manner4,17,43, giving rise to distinct spatio-temporal activity patterns that amplify circuit dysfunction (Fig. 5I). The fact that the maximal change in spike-field coherence occurred at beta frequencies (Fig. 2C and G) suggests that top-down control of motor cortical dynamics is impacted in a pathway-specific manner possibly via amplification through the L5→L2/3 recurrent circuit13,25,27, and could be an early-stage marker of disease progression across vulnerable cortical pathways9,12,73. This work also highlights the potential interplay of how cortical dysfunction can act as stressors to downstream target regions involved in motor function including the generation of aberrant beta oscillations in human patients.
Mechanistically, such beta dysfunction manifests in the form of translaminar stochastic β-events48 (Fig. 4A–F) with amplitudes that grow with time over the prodromal phase (Fig. 4G–J). We show that these β-events are strongly modulated by L5 pyramidal neurons, specifically thick-tufted tract neurons and not L2/3 pyramidal neurons (Fig. 4K–P). This is not surprising, given that L5B thick-tufted pyramidal neurons receive strong contextual feedback across their distal dendrites from higher-order areas67,74, are strongly modulated by L5 SOM+ Martinotti cells69, and receive inputs from L5A neurons (which project to the striatum)25,26, L2/3 pyramidal neurons, and the thalamus24. The combination of these inputs can drive complex bursts in L5B neurons through glutamatergic and voltage-gated calcium channels74,75. The presence of NMDA-driven nonlinearities in the distal tuft of L5 dendrites plays a crucial role in dendritic electrogenesis and influences the regulation of calcium-induced bursting76, a potential vulnerability. Indeed, we observed increased dendritic coactivity across L5 neurons67,74,75 in PFF-injected mice (Fig. 5A–G). Importantly, NMDA currents last about 50 ms–80 ms which is approximately one beta period76. Using a compartmentalized model (Supplementary Fig. S7, 9), pharmacological controls (Supplementary Fig. S8), and phosphoproteome analysis (Supplementary Figs. S10, 11), we conclude that β-events are strongly tied to dendritic spiking and increased phosphorylation of NMDA-receptors, and is present within 3 months in both striatally and cortically seeded mice. This is in large agreement with previous studies that have reported an early impact of αSyn on glutamatergic neurotransmission77 and calcium-influx in neurons4. Specifically, aberrant αSyn impacted the subunit composition and function of both NMDARs and AMPARs77,78,79. However, the implications of such alterations were not explored within cortical microcircuit activity. Our results unambiguously establish that glutamatergic receptor function is significantly altered in the brains of mice injected with αSyn PFFs in both seeding locations and triggers aberrant L5 dendritic population activity in a location-dependent manner. Impaired somatic output resulting from this dendritic vulnerability can also alter L2/3 ensemble activity through recurrent connections. Importantly, these subcellular changes can be traced through β-events and can lead to demarcated behavioral changes. Thus, these findings support the idea that NMDA receptor hyperfunction and resultant aberrant L5 dendritic calcium spikes may be an early consequence in the propagation of αSyn pathology and provides a synaptic signature of such dysfunction in the form of β-events, much before full-scale neurodegeneration.
Summary
In summary, our results highlight a mechanism by which αSyn pathology originating from either the dorsal striatum or primary motor cortex, characteristic of pathology propagation in PDD and DLB, can initiate distinct translaminar cortical circuit dysfunction. Critically, our results show that L5 pyramidal neurons are vulnerable targets, and play a key role in generating pathologically aberrant β-events, a potential marker of top-down input-dependent dysfunction, and drives superficial cortical ensemble stability. Our findings suggest that this cellular vulnerability is uniquely positioned to act as a stressor leading to recurrently amplified local cortical activity, and sets a path towards future investigations centered on how temporal and spatial activity in the cortex can combinatorically inform on pathway-dependent Lewy pathology propagation. Our work also highlights the need to further explore the role of NMDA receptor and calcium channel dysfunction with regard to early-stage αSyn-induced changes within cortical microcircuits and investigate potential therapeutic strategies targeting these channels to mitigate the downstream effects of αSyn pathology on behavior.
Methods
All procedures were in accordance with protocols set forth by the NIH, Purdue Institutional Animal Care and Use Committee (IACUC), and the Purdue Laboratory Animal Program (LAP). Mice were housed in a 12 h:12 h light:dark cycle (temperature, 22-23 °C; humidity, 40%). For all recordings, we used mice with a C57BL/6 J (The Jackson Laboratory, #000664) background kept on a 12 h light/dark cycle in conventional housing with unrestricted access to food and water. Male and female mice were used in approximately equal numbers. Surgical procedures were carried out aseptically under 1–2% isoflurane anesthesia. Carprofen and dexamethasone was administered subcutaneously. Custom titanium headplate was attached to the skull using dental cement (Parkell Metabond). Mice were allowed to recover in their home cages for 3 days. Mice underwent habituation of head fixation and running on a 6-inch rotating wheel for at least 1 week before experiments were performed.
Production of preformed fibrils
Mouse recombinant αSyn was purified as described previously by Zhang et al. and Volpicelli et al.50,80. The expression of untagged protein variants was induced using isopropyl β-d-1-thiogalactopyranoside (IPTG) at 37 °C using E. coli BL21-DE3 cells transformed with pT7-7-αSyn plasmids. αSyn in the bacterial lysate was concentrated using an ammonium sulfate salting-out method and enriched via a boiling step. The boiled supernatant containing αSyn was passed through a HiLoad 16/600 Superdex 200 size exclusion column (Cytiva) and a HiPrepQ HP 16/10 (Cytiva) or DEAE (GE Healthcare Bio-Sciences) anion exchange column sequentially. Fractions enriched with αSyn were pooled, and endotoxin was removed using the Pierce high-capacity endotoxin removal resin (Thermo Fisher Scientific) so that residual endotoxin levels were below 0.5 endotoxin units (EU) per mL81. The protein was dialyzed against phosphate-buffered saline (PBS; pH 7.4) and stored at −80 °C for 6–12 months.
αSyn PFFs was generated by filtering 500 μL of a 5 mg/mL monomeric αSyn solution through a 0.22 μm filter and incubating the filtered protein solution in 1.5 mL sterile microcentrifuge tubes at 37 °C with agitation at 1,000 rpm for 7 days. After fibrillization, αSyn fibrils were concentrated by centrifuging the fibril suspension at 13,000 x g for 10 min and removing the supernatant containing the unfibrillized, monomeric protein. Pelleted fibrils were re-suspended in 250 μL of Dulbecco’s phosphate-buffered saline (DPBS; Cytiva). An aliquot of the fibril suspension (5 μL) was incubated with 8 M guanidine hydrochloride for 1 h at 22 °C to dissociate fibrils into monomers. The αSyn concentration was determined via absorbance measurements at 280 nm using a nanodrop instrument, with an extinction coefficient of 7450 M−1 cm−1. αSyn fibrils were stored at −80 °C as 25 μL of aliquots at a concentration of 5 mg/mL. Prior to injection, αSyn fibrils were sonicated in ethanol-sterilized sonicating tubes (Active Motif) using a cup horn sonicator (Qsonica q700) at 30% power (~100 W/s), with a cycle of 3 s on and 2 s off, for a total ‘on’ time of 15 min, while keeping the bath temperature constant between 5 and 15 °C.
Injections
Mice of both sexes (3–6 months) were injected with PFFs and subsequently analyzed over a period of 12 weeks. Animals were anesthetized using isoflurane with the animal properly secured on the stereotaxic frame. A suspension of sonicated αSyn PFFs (1.5 μL, 5 mg/mL) or a solution of control. Monomeric protein and PFFs was injected in primary motor cortex ± 1.8 mm lateral and 0.3 mm anterior to bregma, 0.7 mm from dura and striatum ± 2 mm lateral and 1 mm posterior to bregma, 2.5 mm from dura at a constant flow rate of 0.1 μL/min using a 10 μL syringe (Hamilton) fitted with a 33-gauge needle with a 45 angled tip. Equal volume was administered between monomeric and PFF injected mice. Total volume was 1 and 1.5 μL for cortical and striatal injections, respectively. The needle was left in place for 5 min post-infusion to avoid any backflow. Injected animals were administered 5 mg/kg carprofen in normal saline immediately post-surgery and 2 days following the surgery.
Probe recordings
A small craniotomy (<1 mm) was made over the primary motor cortex (0.4 mm AP, 1.5 mm ML). A 64-channel electrode (Masmanidis Lab, UCLA) was inserted perpendicular to the surface of the pia using a micromanipulator (Sensapex 4-uMP, Finland). The microelectrode was positioned 1 mm into the cortex and was allowed to settle for 20 min before recordings began. Signals were digitized at a bandwidth of 0.1–10 kHz and sampled at 20 kHz (Intan RHD, USA).
Intracellular recordings in vivo
Pipettes for intracellular recording (100–150 MΩ were pulled from borosilicate glass, 1 mm OD, outer diameter, 0.5 mm ID; Sutter Instruments, CA, USA), using a P-1000 micropipette puller (Sutter Instruments, CA, USA) and filled with 1 or 3 m potassium acetate. Current clamp signals from sharp pipettes were filtered by a 4-pole Bessel filter (4 kHz) and acquired at 20 kHz using a Digidata 1440. Tip potential offsets were zeros upon brain entry and prior to cell entry. The electrode was slowly advanced using a 3-axis manipulator (Luigs-Neuimann, Germany) until an increase in electrode impendence was observed. Break-in was achieved through typical “buzzing” procedure82.
Two-photon imaging
For two-photon imaging, a 3 mm cranial window was slowly drilled over the motor cortex (0.4 mm AP, 1.5 mm ML). To avoid excessive tissue heating, the skull was moistened with 0.9% sodium chloride. The dura was left intact for L2/3 imaging. Animals were imaged using two-photon microscopes (Sutter MOM, Sutter Instruments, CA, USA), and Ti:Sapphire laser (Spectra X3, Spectra-Physics, CA, USA) at 920 nm through a 16x water immersion objective (Nikon, NA: 0.8, WD:3 mm). Resonance galvanometers were controlled through MATLAB ScanImage (Vidrio tech, MBF, USA). Each imaging session lasted 50–70 min with an acquisition rate of 30.048 Hz under resonance scanning, 512 × 512 pixels, and typically with a field of view of 400 × 400 um. For L2/3 imaging, animals expressing GCaMP6s under the Thy1-promoter was used. For dendritic imaging of L5 pyramidal neurons, 400 nl of flex-gcamp6s-mRuby (Addgene, Massachusetts, USA) was injected in the primary motor cortex (0.1 mm AP, 1.5 mm ML).
Pharmacology
An injection micropipette was filled with 100 µM baclofen or 500 µM APV in standard extracellular saline. The pipette was inserted into M1 near the silicon probe and placed at a depth of ~200 µm beneath the pial surface. The drug was delivered by gentle pressure injection. Injections (100 nl each) were made every 20 min throughout the recording session. Sessions with no injections or saline injections followed the same time course as during baclofen and APV injections.
Signal processing
For LFP analysis, data was down sampled to 1024 Hz and bandpass filtered using a Butterworth filter from 1 to 200 Hz. Theta (4–12 Hz), Beta (12–30 Hz), and Gamma (30–90 Hz) spectrograms were extracted using a linear regression filter83. Spearman correlation was calculated across all channels, LFP above 90% similarity was considered for the analysis84. A parent LFP signal was derived by taking the median of the correlated LFPs to calculate spike-field coherence (see below). From here, locomotion-triggered value was obtained using a sliding window 2 s before and after locomotion onset.
Beta event detection
Β-events are detected based on their LFP spectral characteristics. Event candidates are determined if their power is 3 standard deviations above mean beta power, event profile is >20 ms, and <250 ms. Parameters were chosen based on the robustness of detection across pathological weeks. This routine is applied for each electrode across depth per trial.
Spike sorting
After the recording session, we subjected the electrical signals to processing, including the sorting of action potentials from individual neurons. This sorting was accomplished using Kilosort (https://github.com/MouseLand/Kilosort)85, followed by a meticulous manual review in Phy2 (https://github.com/cortex-lab/phy). Neurons were evaluated based on waveform characteristics, stability of firing rates observed throughout the recording session, and auto-correlograms. Spiking analysis and spike-field coherence was conducted on neurons exhibiting sustained activity throughout the entire recording session. Average neural activity for each animal per pathological timepoint were smoothed by calculating the mean spiking activity across multiple trials per recording and convolving by a Gaussian filter with a full-width at half-maximum (FWHM) width of 10 ms. The depth at which each spike occurred was determined based on the electrode that recorded the maximum waveform amplitude.
Spike-field coherence
Spike-field coherence of beta LFP was analyzed using a combination of custom coding routines and Chronux86, an open-source MATLAB package for analyzing spike-field coherence. Peristimulus beta LFP and single units was extracted using the same locomotion onset window described above. A three-dimensional array was generated for each trial containing the frequency response, time, and coherent (phase-vector) value. Phase inter-trial synchronization was derived using inter-trial phase clustering defined as
where n is the number of trials, k is the phase angle of the LFP in which a given spike occurs.
Image analysis
Imaging datasets were imported as Tiff stacks into ImageJ (v1.5) for batch preprocessing. High frequency denoising was done using a median filter and Kalman stack filter. Motion correction and fluorescence extraction of neurons were done using NormCorr (https://github.com/flatironinstitute/NoRMCorre) and CaImAn, (https://github.com/flatironinstitute/CaImAn-MATLAB), respectively87,88. Features extracted were refined through a convolved neural network (CNN) classifier and manual curation. Lastly, calcium spike rasters were generated by thresholding the first derivative of ΔF/F at 3 standard deviations above the baseline fluorescence activity. This spike matrix was used for subsequent population analysis described below.
Pairwise calcium analysis
A N x T binary spike matrix was constructed, where N denotes the number of neurons and T denotes the total number of frames for each calcium video. Each row (N) represents spiking events of a neuron. Coactive events where quantified by binning fluorescence traces to binary spiking events. SØrenson-Dice correlation (SDC) was used to quantify synchronous firing events of individual neurons89,90. Unique neuronal pairs identified through the binary matrix (N x T) are linearized and filtered through a function:
This metric is bound between 0 and 1 (where a value of 1 denotes that neuron pairs are identical). Thus, high cell-cell similarities represented high coactivation probabilities at any given frame between cells during periods of active locomotion. To calculate average pairwise synchronicity across pathological weeks, a bootstrapping procedure was used wherein each individual neuron’s spike activity was circularly shifted by random amounts of time. This was done 1000 times across all neurons and the significant pairwise connection was calculated based on a 99% cutoff to the shuffled distribution.
Ensemble detection and analysis
To detect cortical ensembles, we again carried out a bootstrapping procedure—1000 times across all neurons to determine periods of highly coactive activity patterns, which was reflected by the 99% percentile of fraction of total neurons activate in a given frame. Once defined, we computed the cosine similarity across the population using binned timed vectors to quantify periods of reactivation within similar neurons. The time component of the binary vector was binned every 150 ms (5 acquisition frames) and event spikes were summed to produce a time vector ti = (Ni, Ti), where Ni is the summed event spikes of time bin Ti. This vector series represents temporal changes of grouped neurons. To correlate changes of network states, we constructed a similarity index using a cosine-similarity function, defined as the similarity index between vector pairs ti and tj as20:
To extract the most informative cortical ensembles across pathological times points, principle components were extracted from cosine similarity maps using singular value decomposition (SVD)19,20. SVD allows for the identification of components that contribute the most to a given network state. The SVD of a matrix can be represented as the multiplication of 3 decomposed vectors U, Σ, VT. Since the multiplication of UVT yields an identity matrix, the SVD of our vectorized matrix M was defined as M = VVTΣ, where Σ contains eigenvalues which represent modes of matrix M. Typically >90% of the overall network activity can explained from 4–8% of the total dimension of the data whose reactivation time became candidates for ensembles.
To refine neurons involved in the ensembles, a between neuron “similarity index” as described above was calculated. The proportion of neurons within an ensemble reactivation was defined as the ensemble stability.
Neuron simulations of Layer 5 dynamics and beta events
Morphologically detailed biophysical L5 pyramidal neuron model was implemented in the NEURON simulation environment with python (3.7) interface91. Excitatory synapses and inhibitory synapses (1200 excitatory and 300 inhibitory synapses) were randomly distributed on the basal and apical dendrites, with an AMPAR to NMDAR ratio of 1:2. The synaptic conductance was simulated with a double-exponential kinetic with following rise and decay time constant: τ1_AMPA = 0.1 ms, τ2_AMPA = 2 ms, τ1_NMDA = 2 ms, τ2_NMDA = 75 ms, τ1_GABA = 1 ms, τ2_GABA = 5 ms. The fully-open synaptic conductance is: GAMPA = 0.2nS, GNMDA = 0.4nS, GGABA = 0.8nS. The reversal potential for excitatory and inhibitory synapses is 0 mV and −75 mV respectively. For NMDAR, a sigmoidal voltage dependent term was implemented92.
Each synapse receives a presynaptic Poisson spike train with a lognormal distribution of average firing rate (µ = 0.8–1, σ = 1). Single trials of 20 s simulation was used for statistical analysis of beta-event amplitude. For apical NMDA-R block, the NMDA-R conductance was reduced by 50% compared to the control condition to maintain spike generation. We applied the same condition when investigating the effect of voltage gated calcium channels in beta event generation. To observe the effect of decrease apical inhibition to beta event amplitude, the inhibitory input rate to apical dendrites was decreased 5-fold. The local field potential was computed from the transmembrane current of each compartment along the neuron using LFPy93. The simulated LFP was subsequently filtered at 12–30 Hz and β-events were detected using the same procedure described above.
Brain tissue preparation for phosphoproteomic analysis
Brain tissue samples (~15–20 mg) isolated from αSyn PFF- or monomer-injected mice were resuspended in 300 μL of 25 mM ammonium bicarbonate (ABC) supplemented with protease and phosphatase inhibitors and homogenized in a Precellys Evolution tissue homogenizer using soft-tissue homogenizer CK14 tubes (Bertin Technologies SAS, France). The total protein concentration was then measured using the bicinchoninic acid (BCA) assay (Pierce Chemical Co., USA). The Bligh and Dyer (B & D) extraction method was then performed using lysate volumes equivalent to 25 μg of total protein. Four volumes of cold (−20 °C) methanol were then added to the water-methanol layer, and samples were centrifuged at 17,200 x g for 10 min. The supernatant was removed, and pelleted proteins were dried in a vacuum centrifuge. The protein samples were resuspended in 10 μL 8 M urea solution supplemented with 10 mM DTT and incubated for 1 h at 37 °C. Next, 10 μL of alkylation reagent mixture (97.5% acetonitrile (ACN), 0.5% triethyl phosphine, 2% iodoethanol) was added, and samples were incubated for 1 h at 37 °C. After alkylation, samples were dried in a vacuum centrifuge and subsequently resuspended in 80 μL of 0.025 μg/μL trypsin (Pierce Chemical Co., USA). The digestion was carried out using a bar cycler (50 °C, 60 cycles; 50 s at 20 kPSI and 10 s at 1 ATM). Peptides were then desalted with a C18 silica MicroSpin column (The Nest Group Inc, USA). Phosphopeptide enrichment was then performed with PolyMac spin tips (Tymora Analytical, USA), following the manufacturer’s recommendations for phosphoproteomic analysis.
Mass spectrometry analysis
Peptides were separated using the UltiMate 3000 RSLC nano HPLC system (Thermo Fisher Scientific) connected to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). Peptide separation was carried out in a PepMap trap column (50 mm × 300 mm ID, with a 5 μm particle size and 100 Å pore size), followed by an Acclaim PepMap C18 silica column (500 mm × 75 μm ID, with a 2 μm particle size and 100 Å pore size). Samples were eluted using a gradient between two buffers, buffer A (0.1% formic acid in water) and buffer B (0.1% formic acid in 80% ACN). Samples were injected into the trap column for 5 min, using a flow rate of 5 μL/min. Then, the flow rate was set to 150 nL/min, and samples were separated in the analytical column using a 160 min gradient. Briefly, mobile phase B was set to 2% and increased to 27% for 110 min, after which the gradient was increased to 40% B within 15 min. The percentage of mobile phase B was then increased to 100% in 10 min, and held constant for an additional 10 min, after which the mobile phase B concentration was reset to 2%.
MS analysis was performed with the orbitrap detector, with a MS1 resolution of 60,000 and MS2 resolution of 15,000. Quadrupole isolation was set to “True”. The scan range was set to 375–1600 m/z. The Radio Frequency (RF) lenses were set to 30%, Automatic Gain Control (AGC) target was set to “Standard”, with a maximum injection time “Auto”, and 1 microscan. A dynamic exclusion duration of the 60 s was used, with the exclusion of isotopes. Data was collected using Data-Dependent Acquisition (DDA) mode with cycle time of 3 s, between scans. The precursor ion (MS1) intensity threshold was set to 5.0 × 103 and peptides were fragmented using high energy collisional dissociation (HCD) with 30% collision energy. MS/MS data were also collected using “Orbitrap” as a detector, with AGC target “Standard”, maximum injection time “Auto” and 1 microscan and isolation window of 1.2 m/z. The procedures listed above is consistent across previously published works94,95,96,97.
Bioinformatics analysis
Raw LC-MS/MS data were searched against the UniProt98 Mus musculus processed using the Maxquant platform99 (Ver 2.0.3.1; www.maxquant.org). Searches were performed using trypsin as an enzyme allowing up to 2 missed cleavages. Variable modifications were set for “methionine oxidation” and for phosphoproteomics “STY phosphorylation” and “carbamidomethyl” as a fixed modification. Other search parameters and data filtering were similar as described in ref. 100. A downstream analysis of Maxquant search results was performed using Perseus computational platform101. Raw data files were filtered based on contaminants, and intensity values were log 2-transformed. Protein/peptide entries present in >70% of the samples were selected for downstream analysis. Data imputation was performed for the replacement of missing values and data quality validation. A two-sample t-test was performed on the samples with permutation-based false discovery rate (FDR) correction. Heatmaps were generated in MATLAB, and sample clustering based on principal component analysis was performed utilizing MetaboAnalyst 5.0 (http://www.metaboanalyst.ca/).
Behavior assays
To evaluate the effects of transmissible αSyn-synuclein pathology on motor, cognitive learning and memory, and emotional behavior, mice were tested on a comprehensive behavior for 2 weeks2. The order of tests was randomized, and all tests were conducted by an experimenter blinded to treatment group. All tests were conducted between 08:00 and 4:00 in the lights-on cycle. Mice were habituated to the testing room 1 h before tests, and the apparatus were cleaned with 70% ethanol in between animals to minimize odor cues.
Open field test
To assess general activity, locomotion, and anxiety, mice were tested in the open field. Mice were individually placed in the center and allowed to freely explore for 15 min while the trial was videotaped. Subsequent video scoring was completed by an observer blind to treatment groups using Bonsai and custom coded MATLAB scripts to analyze exploration index and rearing time. Exploration index was calculated as the time each mouse spends in a defined area within the arena (a value of −1 denotes very high occupation in a single region while 1 denotes brief occupation in a single region).
Wire hang test
The wire hang test of motor function was conducted to observe mouse grip strength. The mice were placed onto a 24-gauge steel wire 60 cm above a Plexiglas case. The wire was lightly shaken to cause the animals to grip the wires and then released. The latency of mice to fall off the steel wire was measured, and average values were computed from two trials (10 min apart).
Statistics
For comparisons across two or more unpaired groups, the single-sided, Kruskal–Wallis ANOVA test was used. Multiple (3 or more) groups were compared using a Two-way unbalanced ANOVA. We conducted nested statistics of neurons->mouse->treatment group to report statistical values. In cases where this yielded significance (p < 0.01), post-hoc Bonferroni test was reported for comparisons between pairs of groups. In the case of paired groups, a Wilcoxon-Mann-Whitney U-test was used.
Immunohistochemistry (IHC)
Animals were euthanized by an overdose with isoflurane and perfused transcardially with cold PBS. Brains were removed quickly after decapitation and post-fixed overnight in the same fixative, and then cryoprotected in water containing 30% (w/v) sucrose at 4°C. Coronal sections (40 μm thick) were prepared from the brains of perfused mice using a frozen sliding microtome (Thermo Scientific Microm HM430). Sections were stored at -20°C in cryoprotectant (30% w/v sucrose and 30% v/v ethylene glycol in 0.1 M phosphate buffer) prior to IHC. Sections were blocked with a solution of 10% (v/v) normal donkey serum in PBS supplemented with Triton-X100 (0.3% v/v) (PBST) for 90 min and then incubated with primary antibody solution prepared in PBST supplemented with 1% (v/v) normal donkey serum overnight at 4°C. After washing in PBS, sections were incubated with secondary antibodies conjugated with Alexa fluorophores (Jackson ImmunoResearch Laboratories, 1:500 dilution) for 90 min at 22°C and then washed 3 x 10 min in PBS. The tissues were mounted on slides, allowed to dry overnight, and sealed with a coverslip using DPX mounting media (cat. no. 13512, Electron Microscopy Sciences). Primary antibodies used in this study were: anti-HuC/HuD (cat. no. A21271, Invitrogen, 1:50), anti-NeuN (cat. no. CTX00837, GeneTex, 1:1000) anti-pSer129-aSyn (EP1536Y) (cat. no. ab51253, Abcam, 1:500), anti p-Ser129-aSyn (81a) (cat. no. MABN826, Millipore Sigma, 1:500), anti-GRIN2B (cat. no. 54739, Signalway Antibody, 1:100), and anti-p-Ser1303-GRIN2B (cat. no. 11821, Signalway Antibody, 1:100). Quantitation of pathology from IHC sections was done using fluorescence analysis algorithms of annotated brain regions.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Calcium imaging and electrophysiological processed datasets generated in this study have been deposited in the Zenodo database under the accession code: 10.5281/zenodo.14029419. The mass spectrometry proteomics data pertaining to this study have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD058091. We have also uploaded phosphopeptide spectra to MS Viewer. These can be accessed using the key mbua9pixto. Data generated in this study are provided in Source Data file. Source data are provided with this paper.
Code availability
Code and example MATLAB scripts corresponding to figures, analysis of cortical ensembles, and detection of beta events are available at: https://github.com/HammadFKhan/SiteSpecificLewyPathologyCircuits.
References
Henrich, M. T. et al. Determinants of seeding and spreading of α-synuclein pathology in the brain. Sci. Adv. 6, eabc2487 (2020).
Luk, K. C. et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953 (2012).
Gonzalez-Rodriguez, P., Zampese, E. & Surmeier, D. J. Selective neuronal vulnerability in Parkinson’s disease. Prog. Brain Res. 252, 61–89 (2020).
Volpicelli-Daley, LauraA. et al. Exogenous α-Synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71 (2011).
Osterberg, V. R. et al. Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep. 10, 1252–1260 (2015).
Estaun-Panzano, J., Arotcarena, M. L. & Bezard, E. Monitoring α-synuclein aggregation. Neurobiol. Dis. 176, 105966 (2023).
Aarsland, D. et al. Parkinson disease-associated cognitive impairment. Nat. Rev. Dis. Prim. 7, 47 (2021).
Aldridge, G. M., Birnschein, A., Denburg, N. L. & Narayanan, N. S. Parkinson’s disease dementia and dementia with Lewy bodies have similar neuropsychological profiles. Front Neurol. 9, 123 (2018).
McGregor, M. M. & Nelson, A. B. Circuit mechanisms of Parkinson’s disease. Neuron 101, 1042–1056 (2019).
Irwin, D. J., Lee, V. M. & Trojanowski, J. Q. Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 14, 626–636 (2013).
Braak, H., Ghebremedhin, E., Rüb, U., Bratzke, H. & Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 318, 121–134 (2004).
Foffani, G. & Obeso, J. A. A cortical pathogenic theory of Parkinson’s disease. Neuron 99, 1116–1128 (2018).
Kiritani, T., Wickersham, I. R., Seung, H. S. & Shepherd, G. M. Hierarchical connectivity and connection-specific dynamics in the corticospinal-corticostriatal microcircuit in mouse motor cortex. J. Neurosci. 32, 4992–5001 (2012).
London, M., Roth, A., Beeren, L., Häusser, M. & Latham, P. E. Sensitivity to perturbations in vivo implies high noise and suggests rate coding in cortex. Nature 466, 123–127 (2010).
Nemani, V. M. et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79 (2010).
Goldberg, J. A., Rokni, U., Boraud, T., Vaadia, E. & Bergman, H. Spike synchronization in the cortex/basal-ganglia networks of Parkinsonian primates reflects global dynamics of the local field potentials. J. Neurosci. 24, 6003–6010 (2004).
Blumenstock, S. et al. Cortical circuit dysfunction in a mouse model of alpha-synucleinopathy in vivo. Brain Commun. 3, fcab273 (2021).
Carrillo-Reid, L., Yang, W., Bando, Y., Peterka, D. S. & Yuste, R. Imprinting and recalling cortical ensembles. Science 353, 691–694 (2016).
Carrillo-Reid, L., Miller, J. E., Hamm, J. P., Jackson, J. & Yuste, R. Endogenous sequential cortical activity evoked by visual stimuli. J. Neurosci. 35, 8813–8828 (2015).
Hamm, J. P., Peterka, D. S., Gogos, J. A. & Yuste, R. Altered cortical ensembles in mouse models of schizophrenia. Neuron 94, 153–167.e158 (2017).
Peters, A. J., Chen, S. X. & Komiyama, T. Emergence of reproducible spatiotemporal activity during motor learning. Nature 510, 263–267 (2014).
Blumenstock, S. et al. Seeding and transgenic overexpression of alpha-synuclein triggers dendritic spine pathology in the neocortex. EMBO Mol. Med. 9, 716–731 (2017).
Pluta, S. R., Telian, G. I., Naka, A. & Adesnik, H. Superficial layers suppress the deep layers to fine-tune cortical coding. J. Neurosci. 39, 2052–2064 (2019).
Hooks, B. M. et al. Organization of cortical and thalamic input to pyramidal neurons in mouse motor cortex. J. Neurosci. 33, 748–760 (2013).
Weiler, N., Wood, L., Yu, J., Solla, S. A. & Shepherd, G. M. G. Top-down laminar organization of the excitatory network in motor cortex. Nat. Neurosci. 11, 360–366 (2008).
Shepherd, G. M. Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci. 14, 278–291 (2013).
Onodera, K. & Kato, H. K. Translaminar recurrence from layer 5 suppresses superficial cortical layers. Nat. Commun. 13, 2585 (2022).
Guo, L. et al. Dynamic rewiring of neural circuits in the motor cortex in mouse models of Parkinson’s disease. Nat. Neurosci. 18, 1299–1309 (2015).
Khanna, P. & Carmena, J. M. Beta band oscillations in motor cortex reflect neural population signals that delay movement onset. elife 6, e24573 (2017).
Buschman, T. J., Denovellis, E. L., Diogo, C., Bullock, D. & Miller, E. K. Synchronous oscillatory neural ensembles for rules in the prefrontal cortex. Neuron 76, 838–846 (2012).
Buschman, T. J. & Miller, E. K. Top-down versus bottom-up control of attention in the prefrontal and posterior parietal cortices. Science 315, 1860–1862 (2007).
Bastos, A. M., Loonis, R., Kornblith, S., Lundqvist, M. & Miller, E. K. Laminar recordings in frontal cortex suggest distinct layers for maintenance and control of working memory. Proc. Natl Acad. Sci. USA 115, 1117–1122 (2018).
Roopun, A. K. et al. A beta2-frequency (20–30 Hz) oscillation in nonsynaptic networks of somatosensory cortex. Proc. Natl Acad. Sci. USA 103, 15646–15650 (2006).
Pfurtscheller, G. & Aranibar, A. Evaluation of event-related desynchronization (ERD) preceding and following voluntary self-paced movement. Electroencephalogr. Clin. Neurophysiol. 46, 138–146 (1979).
Leventhal, D. K. et al. Basal ganglia beta oscillations accompany cue utilization. Neuron 73, 523–536 (2012).
Bastos, A. M. et al. Canonical microcircuits for predictive coding. Neuron 76, 695–711 (2012).
Holt, A. B. et al. Phase-dependent suppression of beta oscillations in Parkinson’s disease patients. J. Neurosci. 39, 1119–1134 (2019).
Liu, C. et al. Closing the loop of DBS using the beta oscillations in cortex. Cogn. Neurodyn. 15, 1157–1167 (2021).
Lee, J. H., Whittington, M. A. & Kopell, N. J. Top-down beta rhythms support selective attention via interlaminar interaction: a model. PLoS Comput. Biol. 9, e1003164 (2013).
Feingold, J., Gibson, D. J., DePasquale, B. & Graybiel, A. M. Bursts of beta oscillation differentiate postperformance activity in the striatum and motor cortex of monkeys performing movement tasks. Proc. Natl Acad. Sci. USA 112, 13687–13692 (2015).
Jan, R. W. β-bursts reveal the trial-to-trial dynamics of movement initiation and cancellation. J. Neurosci. 40, 411 (2020).
Kulkarni, A. S. et al. Perturbation of in vivo neural activity following α-synuclein seeding in the olfactory bulb. J. Parkinsons Dis. 10, 1411–1427 (2020).
Henderson, M. X. et al. Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat. Neurosci. 22, 1248–1257 (2019).
McCarthy, M. et al. Striatal origin of the pathologic beta oscillations in Parkinson’s disease. Proc. Natl Acad. Sci. USA 108, 11620–11625 (2011).
Kramer, M. A. et al. Rhythm generation through period concatenation in rat somatosensory cortex. PLoS Comput. Biol. 4, e1000169 (2008).
Shin, H., Law, R., Tsutsui, S., Moore, C. I. & Jones, S. R. The rate of transient beta frequency events predicts behavior across tasks and species. Elife 6, e29086 (2017).
Bonaiuto, J. J. et al. Laminar dynamics of high amplitude beta bursts in human motor cortex. Neuroimage 242, 118479 (2021).
Sherman, M. A. et al. Neural mechanisms of transient neocortical beta rhythms: converging evidence from humans, computational modeling, monkeys, and mice. Proc. Natl Acad. Sci. USA 113, E4885–E4894 (2016).
Cagnan, H. et al. Temporal evolution of beta bursts in the parkinsonian cortical and basal ganglia network. Proc. Natl Acad. Sci. USA 116, 16095–16104 (2019).
Volpicelli-Daley, L. A., Luk, K. C. & Lee, V. M. Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 9, 2135–2146 (2014).
Pérez-Acuña, D., Shin, S. J., Rhee, K. H., Kim, S. J. & Lee, S. J. α-Synuclein propagation leads to synaptic abnormalities in the cortex through microglial synapse phagocytosis. Mol. Brain 16, 72 (2023).
Heindorf, M., Arber, S. & Keller, G. B. Mouse motor cortex coordinates the behavioral response to unpredicted sensory feedback. Neuron 99, 1040–1054.e1045 (2018).
Fries, P. Rhythms for cognition: communication through coherence. Neuron 88, 220–235 (2015).
Cohen, M. X. Analyzing Neural Time Series Data Theory and Practice (Cohen 2014) 1st edn, Vol. 600 (The MIT Press, 2014).
Bastos, A. M., Lundqvist, M., Waite, A. S., Kopell, N. & Miller, E. K. Layer and rhythm specificity for predictive routing. Proc. Natl Acad. Sci. USA 117, 31459–31469 (2020).
Stringer, C. et al. Spontaneous behaviors drive multidimensional, brainwide activity. Science 364, eaav7893 (2019).
Polack, P. O., Friedman, J. & Golshani, P. Cellular mechanisms of brain state-dependent gain modulation in visual cortex. Nat. Neurosci. 16, 1331–1339 (2013).
Stringer, C., Pachitariu, M., Steinmetz, N., Carandini, M. & Harris, K. D. High-dimensional geometry of population responses in visual cortex. Nature 571, 361–365 (2019).
Masamizu, Y. et al. Two distinct layer-specific dynamics of cortical ensembles during learning of a motor task. Nat. Neurosci. 17, 987–994 (2014).
Terada, S. I., Kobayashi, K. & Matsuzaki, M. Transition of distinct context-dependent ensembles from secondary to primary motor cortex in skilled motor performance. Cell Rep. 41, 111494 (2022).
Rigotti, M. et al. The importance of mixed selectivity in complex cognitive tasks. Nature 497, 585–590 (2013).
Fries, P. Neuronal gamma-band synchronization as a fundamental process in cortical computation. Annu Rev. Neurosci. 32, 209–224 (2009).
Bastos, A. M. et al. Visual areas exert feedforward and feedback influences through distinct frequency channels. Neuron 85, 390–401 (2015).
Cardin, J. A. et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 459, 663–667 (2009).
Sohal, V. S., Zhang, F., Yizhar, O. & Deisseroth, K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702 (2009).
Diesburg, D. A., Greenlee, J. D. W. & Wessel, J. R. Cortico-subcortical β burst dynamics underlying movement cancellation in humans. eLife 10, e70270 (2021).
Takahashi, N., Oertner, T. G., Hegemann, P. & Larkum, M. E. Active cortical dendrites modulate perception. Science 354, 1587–1590 (2016).
Wu, Q. et al. α-Synuclein (αSyn) preformed fibrils induce endogenous αSyn aggregation, compromise synaptic activity and enhance synapse loss in cultured excitatory hippocampal neurons. J. Neurosci. 39, 5080–5094 (2019).
Naka, A. et al. Complementary networks of cortical somatostatin interneurons enforce layer specific control. Elife 8, e43696 (2019).
Chen, G. et al. Distinct inhibitory circuits orchestrate crtical beta and gamma band oscillations. Neuron 96, 1403–1418.e1406 (2017).
Veit, J., Hakim, R., Jadi, M. P., Sejnowski, T. J. & Adesnik, H. Cortical gamma band synchronization through somatostatin interneurons. Nat. Neurosci. 20, 951–959 (2017).
Karnani, M. M., Agetsuma, M. & Yuste, R. A blanket of inhibition: functional inferences from dense inhibitory connectivity. Curr. Opin. Neurobiol. 26, 96–102 (2014).
Vinding, M. C. et al. Reduction of spontaneous cortical beta bursts in Parkinson’s disease is linked to symptom severity. Brain Commun. 2, fcaa052 (2020).
Larkum, M. E., Zhu, J. J. & Sakmann, B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature 398, 338–341 (1999).
Hill, D. N., Varga, Z., Jia, H., Sakmann, B. & Konnerth, A. Multibranch activity in basal and tuft dendrites during firing of layer 5 cortical neurons in vivo. Proc. Natl Acad. Sci. USA 110, 13618–13623 (2013).
Larkum, M. E., Nevian, T., Sandler, M., Polsky, A. & Schiller, J. Synaptic integration in tuft dendrites of layer 5 pyramidal neurons: a new unifying principle. Science 325, 756–760 (2009).
Tozzi, A. et al. Alpha-synuclein produces early behavioral alterations via striatal cholinergic synaptic dysfunction by interacting with GluN2D N-methyl-D-aspartate receptor subunit. Biol. Psychiatry 79, 402–414 (2016).
Shrivastava, A. N. et al. Differential membrane binding and seeding of distinct α-synuclein fibrillar polymorphs. Biophys. J. 118, 1301–1320 (2020).
Diógenes, M. J. et al. Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J. Neurosci. 32, 11750–11762 (2012).
Zhang, S. et al. Post-translational modifications of soluble α-synuclein regulate the amplification of pathological α-synuclein. Nat. Neurosci. 26, 213–225 (2023).
Polinski, N. K. et al. Best practices for generating and using alpha-synuclein pre-formed fibrils to model Parkinson’s disease in rodents. J. Parkinsons Dis. 8, 303–322 (2018).
Jayant, K. et al. Flexible nanopipettes for minimally invasive intracellular electrophysiology in vivo. Cell Rep. 26, 266–278.e265 (2019).
Belluscio, M. A., Mizuseki, K., Schmidt, R., Kempter, R. & Buzsáki, G. Cross-frequency phase-phase coupling between θ and γ oscillations in the hippocampus. J. Neurosci. 32, 423–435 (2012).
Senzai, Y., Fernandez-Ruiz, A. & Buzsáki, G. Layer-specific physiological features and Interlaminar Interactions in the primary visual cortex of the mouse. Neuron 101, 500–513.e505 (2019).
Pachitariu, M., Steinmetz, N., Kadir, S., Carandini, M. & Harris Kenneth D. Kilosort: Realtime spike-sorting for extracellular electrophysiology with hundreds of channels. bioRxiv https://doi.org/10.1101/061481 (2016).
Bokil, H., Andrews, P., Kulkarni, J. E., Mehta, S. & Mitra, P. P. Chronux: a platform for analyzing neural signals. J. Neurosci. Methods 192, 146–151 (2010).
Giovannucci, A. et al. CaImAn an open source tool for scalable calcium imaging data analysis. Elife 8, e38173 (2019).
Pnevmatikakis, E. A. & Giovannucci, A. NoRMCorre: An online algorithm for piecewise rigid motion correction of calcium imaging data. J. Neurosci. Methods 291, 83–94 (2017).
Sasaki, T., Matsuki, N. & Ikegaya, Y. Metastability of active CA3 networks. J. Neurosci. 27, 517–528 (2007).
Carrillo-Reid, L. et al. Encoding network states by striatal cell assemblies. J. Neurophysiol. 99, 1435–1450 (2008).
Hay, E., Hill, S., Schürmann, F., Markram, H. & Segev, I. Models of neocortical layer 5b pyramidal cells capturing a wide range of dendritic and perisomatic active properties. PLoS Comput Biol. 7, e1002107 (2011).
Jahr, C. E. & Stevens, C. F. Voltage dependence of NMDA-activated macroscopic conductances predicted by single-channel kinetics. J. Neurosci. 10, 3178–3182 (1990).
Hines, M. L., Davison, A. P. & Muller, E. NEURON and python. Front Neuroinform. 3, 1 (2009).
Zembroski, A. S., Buhman, K. K. & Aryal, U. K. Proteome and phosphoproteome characterization of liver in the postprandial state from diet-induced obese and lean mice. J. Proteom. 232, 104072 (2021).
Kim, S. Q. et al. Global landscape of protein complexes in postprandial-state livers from diet-induced obese and lean mice. Biochem. Biophys. Res. Commun. 629, 40–46 (2022).
Kim, S. Q. et al. Multi-omics approach reveals dysregulation of protein phosphorylation correlated with lipid metabolism in mouse non-alcoholic fatty liver. Cells 11, 1172 (2022).
Glatter, T. et al. Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion. J. Proteome Res. 11, 5145–5156 (2012).
UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489 (2021).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Mohallem, R. & Aryal, U. K. Nuclear phosphoproteome reveals prolyl isomerase PIN1 as a modulator of oncogene-induced senescence. Mol. Cell. Proteom. 23, 100715 (2024).
Tyanova, S. et al. The perseus computational platform for comprehensive analysis of (prote) omics data. Nat. Methods 13, 731–740 (2016).
Acknowledgements
Proteomic experiments were performed at the Purdue Proteomics Facility, Bindley Bioscience Center at Purdue University. We thank Allison Schaser, Scott Pluta and members of the NanoNeurotechnology lab for in-depth discussions. This work was funded by the Showalter Award to K.J, Ralph E. Powe Junior Faculty Enhancement Award to K.J., the Human Frontiers Science Program (HFSP) grant (RGY 0069) to K.J., Purdue Institute for Integrative Neuroscience seed grant to K.J. This work is also supported by the National Institutes of Health, New Innovator Award NIH DP2MH136494 to K.J. This work is based upon efforts supported by EMBRIO Institute, contract #2120200, a National Science Foundation (NSF) Biology Integration Institute to K.J., grants from the NSF (1937986-CBET) and the Branfman Family Foundation to J.-C.R., NIH T-32 Training grant (5T32DC016853) and NSF Graduate Fellowship (DGE-1842166) to H.F.K.
Author information
Authors and Affiliations
Contributions
H.F.K. designed, performed, and analyzed experiments. S.D. designed/optimized the synuclein seeding strategy, designed and performed histological and proteomic analysis. A.N.S. assisted with proteomic analysis and injections. S.X. and S.Y. assisted with biophysical modeling and microscope build-out. X.C. assisted with experiments early in the study. U.K.A. performed detailed proteomic analysis. T.L.K-.U., J-.C.R., and K.J. advised on experiments. J.-C.R. and K.J. conceived the study. K.J. designed experiments and oversaw the project. H.F.K. and K.J. wrote the paper with contributions and inputs from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Pengyi Yang, who co-reviewed with Di Xiao; Martin Larsen and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khan, H.F., Dutta, S., Scott, A.N. et al. Site-specific seeding of Lewy pathology induces distinct pre-motor cellular and dendritic vulnerabilities in the cortex. Nat Commun 15, 10775 (2024). https://doi.org/10.1038/s41467-024-54945-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54945-0
