
Abstract
This study focuses on optimizing catalytic activity in photocatalytic hydrogen evolution reaction by precisely designing and modulating the electronic structure of metal single atoms. The catalyst, denoted as PtSA@S-TFPT, integrates low-valence platinum single atoms into sulfur-containing covalent organic frameworks. The robust asymmetric four-coordination between sulfur and platinum within the framework enables a high platinum loading of 12.1 wt%, resulting in efficient photocatalytic hydrogen production activity of 11.4 mmol g−1 h−1 and stable performance under visible light. These outcomes are attributed to a reduced hydrogen desorption barrier and enhanced photogenerated charge separation, as indicated by density functional theory calculations and dynamic carrier analysis. This work challenges traditional notions and opens an avenue for developing low-valence metal single atom-loaded covalent organic framework catalysts to advance photocatalytic hydrogen evolution.
Similar content being viewed by others


Introduction
As a promising avenue toward sustainable energy production, hydrogen holds considerable potential due to its high-energy density and negligible emission of air pollutants. These characteristic positions hydrogen as a viable solution to address the current energy crisis and mitigate environmental challenges associated with conventional fossil fuels1,2,3,4. Among the various methods for hydrogen generation, photocatalytic water splitting emerges as an attractive option owing to its cost-effectiveness and environmentally friendly nature5. In contrast to conventional homogeneous and heterogeneous catalysts, single-atom catalysts (SACs) have garnered significant attention due to their remarkable atom-utilization efficiency, unique unsaturated coordination environments, and distinctive electronic structures, rendering them highly active in diverse catalytic reactions, notably the hydrogen evolution reaction (HER)6,7,8,9,10. Particularly noteworthy is the potential of low-valence single atoms immobilized on suitable substrates to optimize metal-carrier interactions and enhance adsorption/desorption kinetics, thereby promoting the efficiency of HER process11,12,13. Despite notable advancements in this field, challenges persist, including issues related to low metal loading and inadequate stability, often stemming from the intricate interplay between individual metal (or metalloid) atoms and defective supports (or metalloids)13,14,15,16,17. This complexity impedes a comprehensive understanding of HER mechanism, presenting an ongoing obstacle in the quest to develop universally applicable and straightforward methodologies for fabricating high-loading, stable, and precisely defined SACs in the low-valence state.
As a burgeoning class of porous organic crystalline materials, covalent organic frameworks (COFs) are assembled from organic building blocks composed exclusively of light elements, such as carbon, nitrogen, oxygen, boron, sulfur, etc, linked together by covalent bonds18,19,20,21. Distinguished by their exceptional properties, COFs exhibit high crystallinity, intrinsic porosity, structural regularity, functional versatility, design adaptability, and superior stability compared to other porous materials22,23,24,25. Capitalizing on the inherent advantages and structural adjustability of COFs, precise manipulation of the electronic structure and valence states of metal single atoms within the COF matrix can be achieved6,8,9,26,27,28,29. This presents an avenue for the controlled creation of low-valence SACs by tailoring COF chemical structures and single-atom loading protocols. While numerous instances of COFs hosting single-site metal atoms have been documented, a significant proportion of these metal sites predominantly exist in high-valence states due to strong coordination interactions with multiple adjacent electronegative elements (O/N/S)15. Despite the catalytic activity exhibited by SACs featuring high-valence metal atoms, their performance undergoes a notable decline during catalytic reactions, particularly in HER, where high-valence metal sites tend to undergo reduction, leading to the formation of metallic nanoparticles that compromise catalytic efficiency and stability30,31,32,33. Therefore, the ongoing pursuit of highly efficient and stable low-valence SACs integrated into COFs for HER remains a critical area of research focus.
In this study, we present the development of a catalyst designated as PtSA@S-TFPT, tailored for the photocatalytic HER, showcasing both high catalytic efficiency and remarkable stability. This achievement is realized through the incorporation of low-valence platinum (Pt) single atoms into sulfur-containing COFs. Utilizing methanol as a mild reducing agent facilitated the reduction of Pt to a low valence state, establishing robust asymmetric four-coordination with sulfur/oxygen moieties without inducing particle aggregation. Contrary to conventional assumptions regarding sulfur coordination that typically pose challenges to the catalytic activity of noble metals, PtSA@S-TFPT demonstrates exceptional photocatalytic hydrogen evolution performance, achieving a notable rate of 11.4 mmol g−1 h−1 and sustained stability over a duration of 3 × 3 h under visible light. The superior catalytic performance of PtSA@S-TFPT is further elucidated by density functional theory (DFT) calculations and dynamic carrier analysis, attributing this outcome to the presence of Pt single atoms, which effectively reduce the hydrogen desorption barrier and enhance photogenerated charge separation.
Results and discussion
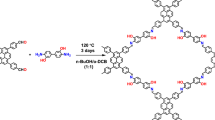
The schematic representation of the synthetic pathway for generating atomically dispersed Pt single atoms embedded within sulfur-rich porous COF skeleton is depicted in Fig. 1a. Initially, the synthesis involved the preparation of a sulfur-rich COF, designated as S-TFPT, through the reaction between 2,5-bis(2-(methylthio)ethoxy)benzene-1,4-dihydrazide (METPH) and 1,3,5-tris-(4-formyl-phenyl) triazine (TFPT) with an acid catalyst at 120 °C for a duration of 3 days, yielding yellow powder S-TFPT with an 85.0% yield (See Experimental Section for detailed procedures, Supplementary Figs. 1–2). Verification of the successful formation of S-TFPT was accomplished through Fourier transform infrared (FT-IR) spectroscopy analysis, as depicted in Supplementary Fig. 3. The appearance of a characteristic peak at 1609 cm−1 corresponding to hydrazone bond stretching vibration, alongside the significant attenuation of peaks associated with the stretching vibrations of N-H (3325 cm−1 and 3414 cm−1 in TFPT) and C = O (1705 cm−1 in METPH and 1658 cm−1 in TFPT), provides clear evidence for the formation of Schiff bases. Furthermore, solid-state 13C cross-polarization magic-angle-spinning (CP/MAS) nuclear magnetic resonance (NMR) spectrum confirms the successful synthesis of S-TFPT, with a distinct chemical shift observed for the hydrazone carbon at 159.9 ppm (Supplementary Fig. 4). Additionally, thermogravimetric analysis (TGA) analysis reveals the excellent thermal stability of S-TFPT up to 300 °C under N2 atmosphere conditions (Supplementary Fig. 5).

a Synthesis pathway of PtSA@S-TFPT. b Experimental, Pawley refined, and simulated PXRD patterns of S-TFPT and PtSA@S-TFPT. c Reconstructed structure of 14-layer hexagonal channel.
The crystallinity and topological structure of S-TFPT were characterized by powder X-ray diffraction (PXRD) measurement in conjunction with structural simulation and Pawley refinement techniques. Based on the geometric features and connectivity patterns of METPH and TFPT, three distinct stacking models, namely AA, AB, and ABC stacking crystal models, were built utilizing the Materials Studio software package (Supplementary Fig. 6a–f and Tables S1–3). Among these models, the eclipsed AA stacking configuration effectively reproduces most of diffraction positions and their intensities observed in the refined PXRD pattern of S-TFPT, as evidenced by negligible deviation (Rwp = 2.67% and Rp = 1.75%) (Fig. 1b and Supplementary Fig. 6g–i). The experimental diffraction peaks (Fig. 1b and Supplementary Fig. 7) observed for S-TFPT at 2θ values of 2.40°, 4.14°, 4.71°, 6.12°, and 26.62° are attributed to the Bragg diffractions originating from the (100), (110), (200), (210), and (001) crystallographic planes, respectively. Furthermore, Pawley refinement procedure yielded the determination of P6/m (no.175) unit cell parameters, with values of a = 46.4289 Å, b = 46.4289 Å, c = 3.3119 Å, α = β = 90°, and γ = 120° (Supplementary Table S1). Additionally, the Brunauer-Emmett-Teller (BET) surface area and pore size distribution of S-TFPT were calculated to be approximately 1033.10 m2 g−1 and 2.95 nm, respectively, which is in good agreement with the theoretical value of 2.97 nm (Supplementary Figs. 8-9). These results demonstrate the high porosity of S-TFPT and provide further validation of its crystal structure.
Notably, the presence of well-ordered and abundant sulfur-rich groups within S-TFPT confers inherent adsorption capabilities, serving as strategic sites for the controlled capture of metal single atoms while effectively hindering metal migration and aggregation (See details in Comparison Experiment Section and Supplementary Figs. 10–14). Encouraged by the advantageous attributes, we endeavored to synthesize a SAC of PtSA@S-TFPT. This synthetic route involved immersing S-TFPT in a methanol solution containing chloroplatinate hexahydrate (H2PtCl6•6H2O) at 60 °C for 1.5 h (Fig. 1a, c, See Experimental Section for detailed procedures). The resultant PtSA@S-TFPT exhibits analogous FT-IR (Supplementary Fig. 3), 13C CP/MAS NMR (Supplementary Fig. 4), TGA (Supplementary Fig. 5), and PXRD (Supplementary Fig. 7) to these of S-TFPT, indicating the preservation of both chemical and crystal structures. Moreover, TGA analysis reveals a high Pt loading capacity of 12.1 wt% in PtSA@S-TFPT (Supplementary Fig. 15). The observed reduction in specific surface area and pore size after post-metallization suggested partial occupation of the internal cavities and pores of S-TFPT by platinum species (Supplementary Fig. 8b). Subsequent X-ray photoelectron spectroscopy (XPS) analysis provided definitive evidence of the presence of platinum within PtSA@S-TFPT, with characteristic binding energy peaks at 76.6 and 73.3 eV for the 4 f doublet of Pt, indicative of partially positively charged Ptx+ (0 <x < 2) species in PtSA@S-TFPT (Supplementary Fig. 16a, b). Furthermore, a positive shift in the binding energy for the 2p doublet of sulfur following loading of Pt single atoms suggested coordination between sulfur and platinum elements, likely facilitated by electron transfer from sulfur to Pt2+ ions (Supplementary Fig. 16c).
Field-emission scanning electron microscopy and transmission electron microscope (TEM) techniques were employed to investigate the morphological characteristics of PtSA@S-TFPT. SEM (Supplementary Fig. 17) and TEM (Fig. 2a) images of PtSA@S-TFPT reveal a uniform nanobelt morphology with a widespread distribution of lattice spacing measuring 3.2 Å, as observed along the [001] crystallographic orientation. Besides, the aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) images unveil distinct bright spots (highlighted by red circles) characterized by focused sizes ranging from 0.1 to 0.2 nm within PtSA@S-TFPT (Fig. 2b and Supplementary Fig. 18). This observation provides compelling evidence for the presence of atomically dispersed Pt single atoms rather than Pt clusters or nanoparticles in PtSA@S-TFPT. Consistent with these findings, high-angle annular dark-field scanning transmission electron microscopy (HADDF-STEM) and energy-dispersive X-ray (EDX) mapping analyses of PtSA@S-TFPT demonstrate the homogeneous distribution of carbon (C), nitrogen (N), oxygen (O), sulfur (S), and platinum (Pt) elements, without discernible instances of element aggregations (Fig. 2c–h).

a A transmission electron microscopy image of PtSA@S-TFPT. Insert: fast fourier transform (FFT) from white box, and lattice spacing viewed along the [001] direction. b An AC-HAADF-STEM image of PtSA@S-TFPT. c AC HAADF-STEM image and (d–h) corresponding EDX mapping of PtSA@S-TFPT.
To further examine the dispersion state of Pt single atoms within PtSA@S-TFPT, the diffuse reflectance infrared Fourier transform spectroscopy was employed. As shown in Supplementary Fig. 19, the presence of a narrow and quasi-symmetrical band at ~2034 cm−1 is an indicative of linearly adsorbed CO on Pt single atoms, thereby corroborating the dispersion of Pt single atoms34. Furthermore, the normalized X-ray absorption near-edge structure (XANES) spectrum of PtSA@S-TFPT reveals that the electron state of Pt single atoms lay between that of Pt foil and PtO2, consistent with the findings in XPS analysis (Fig. 3a). Additionally, the Fourier-transformed (FT) k2-weighted R-space extended X-ray absorption fine structure (EXAFS) was conducted to further elucidate the coordination configuration of Pt single atoms. The dominant peak observed at 2.03 Å was phase-corrected to 2.11 Å, indicating that PtSA@S-TFPT is formed via Pt-S and Pt-O coordination patterns, with no evidence of Pt-Pt coordination (Fig. 3b, Supplementary Fig. 20, and Supplementary Tables S4 and S5). This absence of Pt-Pt coordination was further confirmed by the peaks corresponding to Pt foil and PtO2, consistent with the findings in the XPS analysis (Supplementary Figs. 16 and 21). Furthermore, the fitting analyses of EXAFS features (Fig. 3c–e) reveal that the Pt atom is bonded with two S atoms and two O atoms at an atomic distance of 2.07 Å (Pt-S) and 2.35 Å (Pt-O) within PtSA@S-TFPT. These above-mentioned findings provide compelling evidence for the presence of Pt single atoms and establishment of an asymmetric four-coordination between sulfur/oxygen and Pt single atoms within PtSA@S-TFPT. Using the same synthetic method, other metal SACs with low-valence state embedded in S-TFPT can also be prepared (See Experimental Section for detailed procedures and Supplementary Figs. 22–24).

a Experimental Pt L-edge XANES and (b) Pt L-edge EXAFS spectra of PtSA@S-TFPT, PtO2, and Pt foil excluding phase correction. Fourier transforms of EXAFS spectra in (c) k-space and (d) R-space with their corresponding best fitting results for PtSA@S-TFPT excluding phase correction. e Schematic model and calculated bond lengths of PtSA@S-TFPT (top: front view, bottom: side view).
Given the abundance of accessible active Pt sites, PtSA@S-TFPT holds considerable potential as a catalyst for the photocatalytic HER. To elucidate the exceptional HER catalytic performance of PtSA@S-TFPT, we conducted a controlled experiment involving the fabrication of Pt nanoparticles (PtNPs) embedded in S-TFPT, designated as PtNPs@S-TFPT (See Experimental Section for detailed procedures, Supplementary Figs. 25–33). The Pt contents in PtSA@S-TFPT and PtNPs@S-TFPT were measured using inductively coupled plasma spectrometry (ICP) and found to be nearly identical at 12.97 wt% and 11.92 wt%, respectively. UV-visible diffuse reflectance spectroscopy (UV-Vis DRS) and XPS valance band spectra of PtSA@S-TFPT, PtNPs@S-TFPT, and S-TFPT were conducted to investigate their optical properties. The UV-Vis DRS spectrum of PtSA@S-TFPT exhibits an apparent red shift compared to PtNPs@S-TFPT and S-TFPT, suggesting its pronounced visible-light responsiveness (Fig. 4a). The optical band gaps of PtSA@S-TFPT were calculated to be 2.30 eV, narrower than these of S-TFPT (2.77 eV) and PtNPs@S-TFPT (2.56 eV), indicating that Pt single atoms could effectively narrow the band gap, thereby enhancing light absorption capability (Supplementary Fig. 25). Furthermore, the conduction band (CB) and valence band (VB) positions of the PtSA@S-TFPT, estimated by XPS and optical band gaps analyses, were found to be conducive for both proton reduction and water oxidation reactions, suggesting a stronger driving force for proton reduction (Fig. 4b and Supplementary Fig. 26).

Diffuse reflectance UV/Vis spectra (a) and energy band positions (b) of PtSA@S-TFPT, PtNPs@S-TFPT, and S-TFPT. c Time course and cycling stability of photocatalytic H2 productions. d Diffuse reflectance UV/Vis spectrum of PtSA@S-TFPT and wavelength-dependent AQE of photocatalytic H2 production for PtSA@S-TFPT. e H2 evolution rate for state-of-the-art representative COFs. f Adsorption strength of hydrogen atoms on PtNPs@S-TFPT and PtSA@S-TFPT at the active site Pt. Adsorption model of hydrogen atoms on (g) PtNPs@S-TFPT and (h) PtSA@S-TFPT.
Motivated by these findings, the photocatalytic H2 production performance of PtSA@S-TFPT was evaluated under visible-light irradiation (λ > 420 nm) using triethanolamine (TEOA) as the sacrificial agent and CH3OH/H2O solution as solvents. Impressively, a long-term HER test over PtSA@S-TFPT exhibits an average rate up to 11.44 mmol g−1·h−1, more than eight times to that of PtNPs@S-TFPT. Moreover, after a 3 × 3 h cycle test, no significant decrease in HER performance was observed (Fig. 4c). The apparent quantum efficiencies (AQE) for the photocatalysts were measured at various wavelengths. The highest AQE of PtSA@S-TFPT could be calculated as 4.65% at 420 nm (Fig. 4d). The AQE decreased as the wavelength of irradiated light increased, consistent with the light absorption property of PtSA@S-TFPT (Supplementary Tables S6 and S7). Additionally, PXRD, AC-HAADF-STEM, S-TEM, TGA, XPS, and FT-IR analyses of the spent PtSA@S-TFPT catalysts show minimal changes and no nanoparticles were observed after photocatalysis (Supplementary Figs. 34–38). This further confirms the robust nature of PtSA@S-TFPT for photocatalysis. Furthermore, the catalytic HER ability of PtSA@S-TFPT was comparable with other prominent catalysts, demonstrating its excellent performance35,36,37,38,39,40,41,42,43,44,45,46,47,48 (Fig. 4e, Supplementary Tables S6 and S7). Theoretical investigations employing DFT calculations were conducted to elucidate the underlying factors contributing to the superior catalytic activity observed in PtSA@S-TFPT. The Gibbs free energy hydrogen adsorption (∆GH) serves as a critical descriptor for the HER activity. As shown in Fig. 4f, the energy barriers of 0.76 and −0.86 eV were identified for PtSA@S-TFPT and PtNPs@S-TFPT, respectively, suggesting that the rate-determining step involves desorption (Heyrovsky/Tafel reaction) for PtSA@S-TFPT and adsorption (Volmer reaction) for PtNPs@S-TFPT. Notably, in contrast to PtNPs@S-TFPT, where hydrogen adsorption occurs as depicted in Fig. 4g, PtSA@S-TFPT exhibits a moderate interaction with hydrogen atoms, as illustrated in Fig. 4h, which facilitates desorption. The lower energy barriers for H* generation observed in PtSA@S-TFPT are conducive to subsequent H2 evolution.
To elucidate the mechanisms underlying carrier separation and recombination, we investigated the properties of photogenerated carrier dynamics in the samples using transient photocurrent responses, electrochemical impedance spectroscopy (EIS), DFT calculations, photoluminescence (PL) spectra, and time-resolved PL spectra, respectively (Fig. 5). Notably, PtSA@S-TFPT demonstrates a substantial cathodic photocurrent, approximately 4 and 1.5 times greater than that of S-TFPT and PtNPs@S-TFPT, respectively (Fig. 5a, See the Electrochemical Studies section in the supplementary material for detailed methodology). This enhancement indicates that the presence of platinum single atoms significantly facilitates the effective separation of photogenerated carriers in PtSA@S-TFPT49,50. The accelerated electron-hole separation rate is further supported by the smaller semicircle radius observed in the EIS of PtSA@S-TFPT compared to S-TFPT and PtNPs@S-TFPT (Fig. 5b). Moreover, PL and time-resolved PL spectra further elucidated electron-hole pair recombination dynamics (Fig. 5c and Supplementary Fig. 39). PtSA@S-TFPT exhibits the lowest fluorescence intensity and longest average lifetime, indicating that electron-hole recombination is effectively inhibited, which correlates with improved catalytic performance51. To reveal the carrier separation mechanisms among these samples, we computed the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) distributions using DFT (See the DFT calculations section in the supplementary material for detailed methodology). The HOMO and LUMO of S-TFPT predominantly localize within the skeleton, exhibiting significant overlap and leading to inefficient separation of photogenerated carriers52,53 (Fig. 5d, e). In contrast, the introduction of Pt metals facilitates electron donation from parts of the S-TFPT skeleton to the metal sites, concentrating the LUMO of both PtNPs@S-TFPT and PtSA@S-TFPT around the platinum positions53,54 (Fig. 5f–i). The strong electron donation effect in PtNPs@S-TFPT resultes in suboptimal charge separation, with HOMO and LUMO confined within the nanoparticle fraction55 (Fig. 5f, g). Conversely, in PtSA@S-TFPT, characterized by low-valent and asymmetrically coordinated single atoms, the orbital distribution becomes highly non-overlapping, with the HOMO localizes on the Pt single atoms and the LUMO distributes across the S-TFPT framework (Fig. 5h, i). This arrangement effectively mitigates carrier recombination, thereby enhancing catalytic activity. Collectively, the unique heterojunction system created by integrating Pt single atoms into the S-TFPT framework accelerates electron-hole pair generation, enhances carrier transfer, effectively inhibits recombination, and ultimately improves photocatalytic hydrogen evolution activity.

a Transient photocurrent responses and (b) EIS Nyquist plots of S-TFPT, PtNPs@S-TFPT, and PtSA@S-TFPT. c PL spectra of S-TFPT, PtNPs@S-TFPT, and PtSA@S-TFPT, excited at 417 nm. Calculated HOMO (d, f, h) and LUMO (e, g, i) distributions for the model molecules: S-TFPT (d, e), PtNPs@S-TFPT (f, g), and PtSA@S-TFPT (h, i). (The yellow region indicates charge accumulation, while the cyan region indicates charge depletion. Isosurface value is 4 × 10−9 e/Bohr3).
In summary, we have successfully engineered a catalyst, PtSA@S-TFPT, with significantly enhanced catalytic activity for the photocatalytic HER. By incorporating low-valence platinum single atoms into sulfur-enriched COFs using methanol as a mild reducing agent, the resultant SAC demonstrates notable improvements in both photocatalytic efficiency and stability. The exceptional performance of PtSA@S-TFPT is primarily attributed to the presence of platinum single atoms, which reduce the hydrogen desorption barrier and promote the separation of photogenerated carriers. These findings are supported by transient photocurrent measurements, electrochemical impedance spectroscopy, DFT calculations, and photoluminescence spectra. This study challenges conventional paradigms and paves the way for the synthesis of low-valence metal single atom-loaded COFs, thus propelling the field of photocatalytic hydrogen evolution forward.
Methods
Synthesis of S-TFPT
A 10-mL Pyrex tube was initially loaded with 4,4′,4″-(1,3,5-Triazine-2,4,6-triyl)tribenzaldehyde (TFPT, 13.1 mg, 0.03 mmol), 2,5-bis(2-(methylthio)ethoxy)benzene-1,4-dihydrazide (METPH, 18.7 mg, 0.05 mmol), and a mixture of DMA/n-BuOH (v:v = 1:2, 0.5 mL). The resulting mixture was sonicated for 5 min to achieve a clear solution, followed by the addition of 0.40 mL of trifluoroacetic acid (TFA). Subsequently, the tube was rapidly cooled to 77 K using a liquid nitrogen bath and subjected to three freeze-pump-thaw cycles for degassing. The sealed tube was then evacuated under vacuum and heated at 120 °C for 3 days. Upon reaching room temperature, a yellow precipitate was obtained through centrifugation at 6000 rpm for 2 min, followed by washing with anhydrous acetone. The collected sample underwent a solvent exchange with anhydrous tetrahydrofuran thrice and was dried at 80 °C under vacuum for 12 h to yield a yellow powder (28 mg, 85% isolated yield). The infrared spectroscopy (IR) analysis (powder, cm−1) revealed characteristic peaks at 1663(m), 1609(w), 1504(s), 1416(m), 1354(m), 1312(w), 1200(m), 1084(w), 1015(w), 1203(m), 814(m).
Synthesis of PtSA@S-TFPT
33 mg of chloroplatinate hexahydrate (H2PtCl6•6H2O) was dissolved in 8 mL of 25 vt% methanol/aqueous solution using ultrasound. Subsequently, 60 mg of S-TFPT was immersed in the solution under light-free conditions. After soaking for 3 d, the resulting product was filtered and washed with methanol. The obtained powder was then immersed in 5 mL of methanol and cooled using liquid nitrogen at 77 K, followed by subjecting the mixture to three freeze-pump-thaw cycles. Upon sealing the tube, the mixture was heated at 60 °C for 1.5 h to enable in-situ Pt reduction. Finally, the resulting PtSA@S-TFPT product was collected by filtration and washed with anhydrous methanol.
Synthesis of PtNPs@S-TFPT
Typically, 33 mg of H2PtCl6•6H2O was dissolved in 8 mL of 25 vt% methanol/aqueous solution. Then, 60 mg of S-TFPT was immersed in the solution without exposure to light. Following a 3-day immersion period, the resultant product was filtered and rinsed with methanol. Subsequently, it was soaked in 2 mL of a 1 M NaBH4/methanol solution for 2 min, resulting in the formation of a dark yellow solid powder, designated as PtNPs@S-TFPT.
Photocatalysis
The photocatalytic H2 evolution test was conducted using a photocatalytic reaction setup (Labsolar 6 A; Beijing Perfect light Technology Co., Ltd). Specifically, 20 mg of PtSA@S-TFPT was dispersed in a solution containing 35 mL of deionized water and 5 mL of methanol. The suspension was sonicated for 1 h to ensure uniform dispersion before adding 5 mL of triethanolamine as the sacrificial agent. The mixture was stirred at 500 rpm and degassed using a vacuum pump. Xenon lamps (300 W) emitting light with wavelengths greater than 420 nm and an optical power density of 1 W/cm2 served as the illumination source. To prevent photothermal effects, a water circulation system maintained the samples at room temperature. The evolved H2 gas in the reactor headspace was collected automatically using a gas-tight syringe connected to a photocatalytic system and quantified using a gas chromatograph equipped with a thermal conductivity detector. To ensure consistency, the system was degassed for 30 min after each cycle of illumination, and the optical power density was recalibrated before the subsequent cycle of illumination.
Data availability
All data are available in the main text or the supplementary materials. Correspondence and requests for materials should be addressed to Yongwu Peng (ywpeng@zjut.edu.cn) or Gao Li (gaoli@dicp.ac.cn). Source data are provided with this paper.
References
Tian, X. et al. Transition metal nitride coated with atomic layers of Pt as a low-cost, highly stable electrocatalyst for the oxygen reduction reaction. J. Am. Chem. Soc. 138, 1575–1583 (2016).
Tian, X. et al. High-performance core-shell catalyst with nitride nanoparticles as a core: well-defined titanium copper nitride coated with an atomic Pt layer for the oxygen reduction reaction. ACS Catal. 7, 3810–3817 (2017).
Deng, Y. et al. Hierarchically open-porous carbon networks enriched with exclusive Fe-Nx active sites as efficient oxygen reduction catalysts towards acidic H2-O2 PEM fuel cell and alkaline Zn-air battery. Chem. Eng. J. 390, 124479 (2020).
Deng, Y. et al. Advanced atomically dispersed metal-nitrogen-carbon catalysts toward cathodic oxygen reduction in PEM fuel cells. Adv. Energy Mater. 11, 2101222 (2021).
Xu, X. et al. Leveraging metal nodes in metal-organic frameworks for advanced anodic hydrazine oxidation assisted seawater splitting. ACS Nano 17, 10906–10917 (2023).
Qiao, B. et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 3, 634–641 (2011).
Liu, G. et al. MoS2 monolayer catalyst doped with isolated Co atoms for the hydrodeoxygenation reaction. Nat. Chem. 9, 810–816 (2017).
Wang, A., Li, J. & Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2, 65–81 (2018).
Lin, L. et al. Low-temperature hydrogen production from water and methanol using Pt/α-MoC catalysts. Nature 544, 80–83 (2017).
Zhang, J. et al. Cation vacancy stabilization of single-atomic-site Pt1/Ni(OH)x catalyst for diboration of alkynes and alkenes. Nat. Commun. 9, 1002 (2018).
Wei, Z. W. et al. Reversed charge transfer and enhanced hydrogen spillover in platinum nanoclusters anchored on titanium oxide with rich oxygen vacancies boost hydrogen evolution reaction. Angew. Chem. Int. Ed. 133, 16758–16763 (2021).
Gao, R. et al. Pt/Fe2O3 with Pt-Fe pair sites as a catalyst for oxygen reduction with ultralow Pt loading. Nat. Energy 6, 614–623 (2021).
Wang, W. et al. Confining zero‐valent platinum single atoms in α‐MoC1−x for pH‐universal hydrogen evolution reaction. Adv. Funct. Mater. 32, 2108464 (2022).
Cao, D. et al. Volcano-type relationship between oxidation states and catalytic activity of single-atom catalysts towards hydrogen evolution. Nat. Commun. 13, 5843 (2022).
Wang, L. et al. Stabilizing low‐valence single atoms by constructing metalloid tungsten carbide supports for efficient hydrogen oxidation and evolution. Angew. Chem. Int. Ed. 62, e202311937 (2023).
Li, S. et al. Low‐valence Znδ+(0<δ<2) single‐atom material as highly efficient electrocatalyst for CO2 reduction. Angew. Chem. Int. Ed. 60, 22826–22832 (2021).
Jeong, H. et al. Controlling the oxidation state of Pt single atoms for maximizing catalytic activity. Angew. Chem., Int. Ed. 132, 20872–20877 (2020).
Yaghi, O. M. Reticular chemistry: molecular precision in infinite 2D and 3D. Mol. Front. J. 3, 66–83 (2019).
Song, Y., Sun, Q., Aguila, B. & Ma, S. Covalent organic frameworks: opportunities of covalent organic frameworks for advanced applications. Adv. Sci. 6, 1970011 (2019).
Nie, H. et al. Light‐controllable ionic conductivity in a polymeric ionic liquid. Angew. Chem. Int. Ed. 132, 5161–5166 (2020).
Chen, G. et al. Transparent, self-healing, and defogging wearable devices enabled by plasmonic silver nanoparticle-embedded covalent-organic framework nanosheets. ACS Mater. Lett. 6, 648–655 (2024).
Ran, L. et al. Engineering single-atom active sites on covalent organic frameworks for boosting CO2 photoreduction. J. Am. Chem. Soc. 144, 17097–17109 (2022).
Liu, Y. et al. Covalent-organic-framework-based composite materials. Chem 6, 3172–3202 (2020).
Cui, X. et al. Emerging covalent organic frameworks tailored materials for electrocatalysis. Nano Energy 70, 104525 (2020).
Chen, G. et al. Salt-assisted fabrication of a water-based covalent organic framework ink and its hybrid films for photothermal actuators. ACS Appl. Mater. Interfaces 15, 36804–36810 (2023).
Qu, Y. et al. Direct transformation of bulk copper into copper single sites via emitting and trapping of atoms. Nat. Catal. 1, 781–786 (2018).
Gu, J., Hsu, C.-S., Bai, L., Chen, H. M. & Hu, X. Atomically dispersed Fe3+ sites catalyze efficient CO2 electroreduction to CO. Science 364, 1091–1094 (2019).
Zhang, J. et al. Single platinum atoms immobilized on an MXene as an efficient catalyst for the hydrogen evolution reaction. Nat. Catal. 1, 985–992 (2018).
Zhao, L. et al. Cascade anchoring strategy for general mass production of high-loading single-atomic metal-nitrogen catalysts. Nat. Commun. 10, 1278 (2019).
Chen, C. et al. Zero‐valent palladium single‐atoms catalysts confined in black phosphorus for efficient semi‐hydrogenation. Adv. Mater. 33, 2008471 (2021).
Ma, T. et al. Crystalline lattice‐confined atomic Pt in metal carbides to match electronic structures and hydrogen evolution behaviors of platinum. Adv. Mater. 34, 2206368 (2022).
Fang, S. et al. Uncovering near-free platinum single-atom dynamics during electrochemical hydrogen evolution reaction. Nat. Commun. 11, 1029 (2020).
Jin, X. et al. Electron configuration modulation of nickel single atoms for elevated photocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 132, 6894–6898 (2020).
Sun, G. et al. Breaking the scaling relationship via thermally stable Pt/Cu single atom alloys for catalytic dehydrogenation. Nat. Commun. 9, 4454 (2018).
Stegbauer, L., Schwinghammer, K. & Lotsch, B. V. A hydrazone-based covalent organic framework for photocatalytic hydrogen production. Chem. Sci. 5, 2789–2793 (2014).
Vyas, V. S. et al. A tunable azine covalent organic framework platform for visible light-induced hydrogen generation. Nat. Commun. 6, 1–9 (2015).
Wang, X. et al. Sulfone-containing covalent organic frameworks for photocatalytic hydrogen evolution from water. Nat. Chem. 10, 1180–1189 (2018).
Pachfule, P. et al. Diacetylene functionalized covalent organic framework (COF) for photocatalytic hydrogen generation. J. Am. Chem. Soc. 140, 1423–1427 (2018).
Banerjee, T. et al. Single-site photocatalytic H2 evolution from covalent organic frameworks with molecular cobaloxime co-catalysts. J. Am. Chem. Soc. 139, 16228–16234 (2017).
Ghosh, S. et al. Identification of prime factors to maximize the photocatalytic hydrogen evolution of covalent organic frameworks. J. Am. Chem. Soc. 142, 9752–9762 (2020).
Li, L. et al. Sulfur-doped covalent triazine-based frameworks for enhanced photocatalytic hydrogen evolution from water under visible light. J. Mater. Chem. A 4, 12402–12406 (2016).
Kuecken, S. et al. Fast tuning of covalent triazine frameworks for photocatalytic hydrogen evolution. Chem. Commun. 53, 5854–5857 (2017).
Wang, K. et al. Covalent triazine frameworks via a low‐temperature polycondensation approach. Angew. Chem., Int. Ed. 56, 14149–14153 (2017).
Xie, J. et al. Efficient visible light-driven water oxidation and proton reduction by an ordered covalent triazine-based framework. Energy Environ. Sci. 11, 1617–1624 (2018).
Liu, M. et al. Crystalline covalent triazine frameworks by in situ oxidation of alcohols to aldehyde monomers. Angew. Chem. Int. Ed. 57, 11968–11972 (2018).
Haase, F., Banerjee, T., Savasci, G., Ochsenfeld, C. & Lotsch, B. V. Structure-property-activity relationships in a pyridine containing azine-linked covalent organic framework for photocatalytic hydrogen evolution. Faraday Discuss 201, 247–264 (2017).
Biswal, B. P. et al. Sustained solar H2 evolution from a thiazolo [5, 4-d] thiazole-bridged covalent organic framework and nickel-thiolate cluster in water. J. Am. Chem. Soc. 141, 11082–11092 (2019).
Wang, Y. et al. Facile construction of fully sp2-carbon conjugated two-dimensional covalent organic frameworks containing benzobisthiazole units. Nat. Commun. 13, 1–7 (2022).
Mi, Z. et al. Covalent organic frameworks enabling site isolation of viologen‐derived electron‐transfer mediators for stable photocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 60, 9642–9649 (2021).
He, T. et al. Integrated interfacial design of covalent organic framework photocatalysts to promote hydrogen evolution from water. Nat. Commun. 14, 329 (2023).
Liu, C. et al. Lateral functionalization of a one-dimensional covalent organic framework for efficient photocatalytic hydrogen evolution from water. J. Mater. Chem. A 12, 16063–16069 (2024).
Li, Q. et al. Modulated connection modes of redox units in molecular junction covalent organic frameworks for artificial photosynthetic overall reaction. J. Am. Chem. Soc. 145, 23167–23175 (2023).
Li, Y. et al. In situ photodeposition of platinum clusters on a covalent organic framework for photocatalytic hydrogen production. Nat. Commun. 13, 1355 (2022).
Li, J. et al. In situ construction of single‐atom electronic bridge on COF to enhance photocatalytic H2 production. Angew. Chem. Int. Ed. 63, e202411721 (2024).
Sun, Z. X., Sun, K., Gao, M. L., Metin, Ö. & Jiang, H. L. Optimizing Pt electronic states through formation of a schottky junction on non‐reducible metal-organic frameworks for enhanced photocatalysis. Angew. Chem. Int. Ed. 134, e202206108 (2022).
Acknowledgements
Y.P. thanks the financial support from the National Key Research and Development Project of China (2022YFE0113800), the National Natural Science Foundation of China (22375179), the start-up grant of Zhejiang University of Technology (2019125016829), and National Natural Science Foundation of China Basic Research Program for Young Students (Doctoral Candidates) (223B2117). G.X. and G.L. acknowledge the financial support from National Natural Science Foundation of China (51907173, 22172167, and 22178320). We would like to thank SSRF (BL05U, BL06B, and BL14W) beamlines for experimental data collection.
Author information
Authors and Affiliations
Contributions
L.C., G.C., and C.G. contributed equally to this work. Y.P. and G.L. conceived and designed the research project. L.C., G.C., and C.G. designed, synthesized, and characterized the materials. Y.Z. performed the theoretical calculations. Z.X. and J.L. assisted in the conduct of the experiment. G.Xu. helped with the manuscript discussion. G.L. and Y. P. co-wrote and revised the manuscript. All authors contributed to this work, read the manuscript, and all data are reported in the main text and supplemental materials.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Shu Seki, Yingjie Zhao and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, L., Chen, G., Gong, C. et al. Low-valence platinum single atoms in sulfur-containing covalent organic frameworks for photocatalytic hydrogen evolution. Nat Commun 15, 10501 (2024). https://doi.org/10.1038/s41467-024-54959-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54959-8
This article is cited by
-
Atomically dispersed Pt species anchored on Al3+-doped SrTiO3 for photocatalytic overall water splitting
Science China Materials (2026)
-
Unfolding the significance of metal oxides for photocatalytic sustainable water splitting combined with organic pollutants
Journal of Materials Science (2025)
-
2D sp2 carbon-conjugated covalent organic frameworks: photocatalytic platforms for solar energy conversion
Rare Metals (2025)
