
Abstract
A chemical reagent to access methyl sulfones has been developed. Its reaction with various bis-nucleophiles leads to the rapid formation of previously unknown heteroaromatic methyl sulfones. Analogous strategy can also be used to construct alkyl-, CHF2-, CF3- and even bicyclo[1.1.1]pentane-containing derivatives. These compounds have been demonstrated to have a high potential for use in medicinal chemistry and coordination chemistry.
Similar content being viewed by others
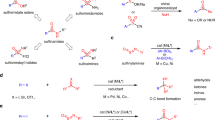
Introduction
Methyl sulfone (MeSO2) is a standard polar substituent used in chemistry together with methoxy, dimethylamino, acetoxy, and acetyl (Fig. 1). It is common in agrochemicals1, and can be found in structures of >30 drugs (substructure search at go.drugbank.com on 14.03.2024 revealed 31 bioactive compounds with MeSO2-substituent (used options: “approved” + “vet approved” + “investigational”)2,3. All of them are either aliphatic or aromatic compounds (Fig. 1, see also SI p. S7–S8). Heteroaromatic methyl sulfones, due to the lack of convenient and effective chemical approaches to them, are considerably much less investigated.

MeSO2-group in drug discovery projects, known approaches to methyl sulfones, and our reagent.
Two common approaches to heteroaromatic methyl sulfones exist. Both routes rely on the modification of the already installed functional groups: (a) oxidation of the thiomethyl group4,5,6; and (b) metal-mediated cross-coupling of halides/boronates with sodium sulfinate7,8,9,10,11,12,13 or sulfur dioxide/methyl iodide (Fig. 1)14,15,16,17,18,19,20,21,22,23,24,25,26,27.
Here, we present a principally trailblazing approach to methyl sulfones that relies on the 1,3-heterocycle disconnection logic with the developed reagent 1 (Fig. 1).
Results and discussion
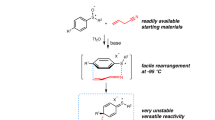
In our daily practice, we often use two standard approaches to methyl sulfones described above (Fig. 1). Recently, we received a request from a pharmaceutical company for a synthesis of pyrazole 2 (Fig. 2), which was described in a patent before28. In that method, bromide 3 was treated with nBuLi followed by the addition of (MeS)2. Due to its smell, the crude product (4) was used in the next oxidation reaction without further purification. Oxidation of the latter with m-chloroperoxybenzoic acid (mCPBA) gave 51 mg of the needed product 2 in 26% yield after a column chromatographic separation. It became obvious to us, that a principally distinctive approach to pyrazole 2 was needed. We decided to challenge the preparation of a reagent with the already installed MeSO2-substituent that could be easily converted into the needed product.

Patent approach vs this work. X-ray crystal structure of compound 1 (carbon—white, oxygen—red, nitrogen—blue, sulfur—green). Hydrogen atoms and PF6− anion are omitted for clarity. Ellipsoids are shown at a 30% probability level.
Having studied the literature, we brought our attention to vinamidinium salts that have been utilized to access substituted (hetero)aromatic compounds via a 1,3-disconnection logic29. In 1961, Arnold obtained 2-chlorovinamidinium perchlorate by reaction of chloroacetic acid under Vilsmeier–Haack conditions30,31. Later, Gupton and others extended this approach to obtain various vinamidinium perchlorates32,33,34,35. In 2000, Davies and colleagues developed safer vinamidinium hexafluorophosphate salts36.
Keeping this in mind, we decided to attempt the preparation of the previously unknown MeSO2-vinamidinium reagent 1 (Fig. 1). At the beginning, we were concerned that the activated methyl sulfone substituent would not be compatible with the harsh Vilsmeier-Haack conditions, but out of curiosity still decided to pursue that route. From the commercially available and inexpensive sodium methyl sulfinate (5), we initially obtained ester 6 (Fig. 2). The subsequent saponification afforded the carboxylic acid 7. After some optimization, we found that the Vilsmeier-Haack reaction of acid 7 at 90 °C led to the formation of the desired compound 1 in 58% yield. Compound 1 was a yellow crystalline solid stable at room temperature in air for at least one year. The structure of reagent 1 was confirmed by X-ray crystallographic analysis.
The subsequent reaction of reagent 1 with hydrazine hydrate in EtOH under reflux smoothly gave the desired pyrazole 2 (Figs. 2 and 3). Importantly, using this approach, we could easily obtain the product in a 171 g scale in a single run. Its structure was confirmed by X-ray crystallographic analysis.

Reaction conditions: compound 2: N2H4•H2O, EtOH, reflux, 12 h. Compound 8: MeNHNH2•H2SO4, K2CO3, DMF, 70 °C, 12 h. Compounds 9-12: N,N-bis-nucleophiles, Py, 90 °C, 12 h. Compound 13: guanidine acetate, K2CO3, MeCN, 60 °C, 12 h. Compound 14: urea, K2CO3, Py, 100 °C, 12 h. Compound 15: thiourea, NaOMe, MeOH, 50 °C, 12 h. The product was isolated as a sodium salt. Compound 17: S-Me thiourea hydroiodide, Py, rt, 12 h (compound 17 was obtained instead of expected 16). Compounds 18-30: N,N-bis-nucleophiles, Py, 100 °C, 12 h. X-ray crystal structures of compounds 8, 9, and 29 (carbon—white, oxygen—red, sulfur—green, bromine—orange, nitrogen—blue). Ellipsoids are shown at a 30% probability level. n.d.:- not determined.
Scope
Having finished the synthesis of pyrazole 2, we wondered if reagent 1 could be used to assemble other MeSO2-substituted heterocycles. At the outset, we tried various N,N-bis-nucleophiles. Reaction with methyl hydrazine and functionalized aromatic hydrazines cleanly gave the corresponding pyrazoles 8–12 in 48–90% yield (Fig. 3). Reaction with guanidine, urea, and thiourea led to the formation of pyrimidines 13–15 in 54–95% yield. Reaction with S-methyl thiourea, however, gave instead of the expected product 16, the dimethylamino derivative 17. The latter must have been formed via the initial formation of compound 16 and the subsequent SNAr-reaction with dimethylamine present in the reaction mixture. Reaction with substituted amidines gave pyrimidines 18–21 in 94–96% yield. Reaction of 1 with 2-chloroacetamide in pyridine gave instead of the expected product 22, the corresponding pyridinium salt 23. Cyclization of 1 with various amino NH-heterocycles produced bicyclic products 24–30.
It is important to note that this method worked efficiently well on milligram, gram, and even multigram scales (2, 13) without any significant change in the reaction yield. Most products were crystalline solids and could be purified easily by crystallization. For liquid/oil compounds the purification was performed by column chromatography. The molecular structures of products 8, 9, 12, and 29 were confirmed by X-ray crystallographic analysis.
Next, we tried various N,C-bis-nucleophiles (Fig. 4). The reaction of vinamidinium salt 1 with glycine derivates gave pyrroles 31–33 in 49–95% yield. Analogously, pyridones 34–36 and pyridines 37, 38 were synthesized in 46–92% yield. The reaction of salt 1 with various amino pyrazoles, amino isoxazoles, amino thiazoles, amino pyrroles, and amino thiophenes gave the corresponding bicyclic heterocycles 39–46 in 76–93% yield. It is worth noting that while the reaction of reagent 1 with methyl 5-amino-2-furoate in pyridine gave the desired bicyclic product 47, the analogous reaction in acetonitrile led to the selective formation of the non-cyclized compound 48. The reaction of 1 with electron-rich aminouracil, aminopyridine, and anilines gave bicyclic products 49–54 in 49–89% yield. The structure of compounds 34, 41, 42, and 45 was confirmed by X-ray crystallographic analysis.

Reaction conditions: compounds 31–47 and 49–56: N,N-bis-nucleophiles, Py, 90 °C, 12 h; compound 48: methyl 5-aminofuran-2-carboxylate, K2CO3, MeCN, 60 °C, 12 h. X-ray crystal structures of compounds 34, 41, 42, and 45 (carbon—white, oxygen—red, sulfur—green, bromine—orange, nitrogen—blue). Ellipsoids are shown at a 30% probability level. n.d. not determined.
Remarkably, despite the simplicity of the current approach to methyl sulfones, the preparation of all compounds 8–54 has never been described before by any methods.
The developed method for MeSO2-derivatives was not without limitations, however. It worked well only for the electron-rich systems. Not sufficiently activated para-bromo and para-methoxy anilines gave only the side products 55 and 56, correspondingly. From reagent 1 we also could not obtain products 57–60 as the formation of complex mixtures was observed in each case (Fig. 4; for more negative results see SI p. S38–S41).
Modifications
Representative modifications of the resulting methyl sulfones were undertaken to obtain various functionalized building blocks (compounds with one or two functional groups) for use in medicinal chemistry (Fig. 5A). Saponification of the ester group in products 28, 32, 33, 35, and 40 gave carboxylic acids a. Acidic cleavage of N-Boc group in 18–21 gave amines b. The reaction of pyrimidone 14 and pyridines 34, 35 with POCl3 afforded compounds with active chlorine atoms c. Alkylation of compound 15 with methyl iodide gave the previously inaccessible product 16. Finally, treatment of compound 50 with hydrobromic acid gave pyridone 50d, and its subsequent reaction with POCl3 afforded chloride 50e.

Group A. Reaction conditions: a LiOH, MeOH, H2O, rt, 12 h; b HCl in dry dioxane, rt, 12 h; c POCl3, reflux, 12 h; d (i) 48% aq. solution of HBr, reflux, 12 h, (ii) POCl3, reflux, 12 h; e POCl3, reflux, 12 h; f MeI, MeOH-H2O, rt, 12 h. Reaction conditions: g POCl3, KPF6, DMF, 90 °C, 2 h; h 61b–63b: N2H4•H2O, EtOH, reflux, 12 h; 64b: N2H4•H2O, MeONa, MeOH, 70 °C, 12 h; i guanidine acetate, Py, 100 °C, 12 h. Group C. Limitations of the method. Group D. Comparison of approaches to pyrimidines 14c and 18b. n.d. not determined.
Alkyl sulfones
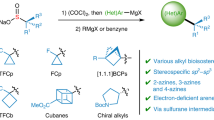
Having a practical and scalable procedure to reagent 1 in hand, we wondered if other alkyl sulfones could be obtained similarly. The treatment of ethyl (61) and isopropyl (62) carboxylic acids (see SI p. S49 and S52) under Vilsmeier-Haack conditions gave the desired reagents 61a, 62a (Fig. 5B). For difluoromethyl- and trifluoromethyl-containing compounds, we discovered that the tert-butyl esters 63 and 64 could be directly converted in one step into the vinamidinium salts 63a, 64a. The reaction of all four reagents 61a–64a with hydrazine hydrate gave pyrazoles 61b–64b in 25–71% yield37. The treatment of reagents 61a–64a with guanidine provided pyrimidines 61c–64c in 31–79% yield. These two representative reactions of reagents 61a–64a demonstrated that they could be widely used for the preparation of other sulfonylated derivatives similar to those obtained from reagent 1 (Figs. 4 and 5). The molecular structures of compounds 63b, 64a, and 64b were confirmed by X-ray crystallographic analysis.
Due to the discovery of the Brigatinib drug38, the P(O)Me2-substituent has recently become popular in chemistry39. We, therefore, decided also to attempt the preparation of the corresponding vinamidinium salt (Fig. 5C). Unfortunately, the reaction of carboxylic acid 65 under the previously used conditions led to the formation of a complex mixture, rather than the desired reagent 65a.
Unfortunately, attempts to extend this chemistry at other S(VI)-motifs (sulfonamides, SF5, sulfoximines) were unsuccessful at this point (see SI p. S61–S63 for more details).
Comparison with other methods
The reagent 1 developed herein can be used not only to make sulfones, but also to simplify the synthesis of the existing ones. The synthesis of pyrazole 2 was already discussed earlier (Fig. 2). Another example involves pyrimidine 14c, which was reported in a patent. It was obtained in milligram quantities in 7% total yield from bromide 66 (Fig. 5D)40. Using our method, pyrimidine 14c could be obtained from reagent 1 in a 53 g scale in a single run in 65% yield. Analogously, amine 18b was obtained previously in 41% yield from the nitrile 6741. Using reagent 1, this product could be obtained in a multigram amount in one run in 88% yield (Fig. 5D).
Bicyclo[1.1.1]pentanes
Recently, bicyclo[1.1.1]pentanes were introduced as saturated bioisosteres of the benzene ring42,43 Therefore, unconventional methods to access these molecules are of importance. Recently, an approach towards bicyclo[1.1.1]pentane-containing sulfones bearing a halogen atom at the bridgehead position, Hal-[1.1.1]-SO2R, was elaborated44. The reduction of the halogen atom in this system was problematic. Here, we have developed a reagent to synthesize the bridgehead non-substituted bicyclo[1.1.1]pentane sulfones: H-[1.1.1]-SO2R. An addition of [1.1.1]-propellane to methyl thioglycolate (68)45 followed by oxidation of the intermediate sulfide, and saponification of the ester group gave carboxylic acid 69. The Vilsmeier-Haack reaction of the latter gave the expected reagent 69a in 69% yield. Representative cyclizations of salt 69a with various bis-nucleophiles gave bicyclo[1.1.1]pentane-containing pyrazole 70, pyrimidine 71, and pyrrole 72. The structure of compound 70 was confirmed by X-ray crystallographic analysis (Fig. 6A).

Group A. Reaction conditions: bis-nucleophiles, Py, 90 °C, 12 h. X-ray crystal structure of compound 70 (carbon—white, oxygen—red, sulfur—green, bromine—orange, nitrogen—blue). Ellipsoids are shown at a 30% probability level. Group B. Reaction conditions: (D3-2): N2H4•HCl, CH3CN, 80 °C 12 h, 25%; (D3-73): (i) O-Methylisourea hemisulfate, D3-1, Py, 90 °C, 12 h; (ii) HBr (48%), reflux, 12 h, 99%; (D3-74): tert-butyl methylglycinate hydrochloride, D3-1, Py, 90 °C, 12 h, 96%.
Deuterated compounds
The developed approach also allowed the preparation of deuterated derivatives (Fig. 6B). From compound 68 in several steps the deuterated carboxylic acid D3-7 was easily obtained. Under the previously developed conditions, synthesis of the vinamidinium salt D3-1 was further achieved. Three representative reactions of the latter with various bis-nucleophiles conveniently afforded the desired deuterated heterocycles: pyrazole D3-2, pyrimidine D3-73, and pyrrole D3-74. Synthesis of other deuterated derivatives was also possible (see SI p. S69–S74).
Physicochemical properties
Having developed a general and practical approach to methyl sulfones, we also decided to experimentally investigate their physicochemical properties such as lipophilicity, water solubility, and metabolic stability.
In the initial stage, we synthesized three model amides 75–77 (see SI p. S75–S76; Fig. 7) to see the impact of the replacement of the hydrogen atom (75) with the methoxy (76) and the methyl sulfone (77) groups at the lipophilicity. We used two parameters: calculated (clogP) and experimental (logP) lipophilicities (clogP was calculated with ChemAxon Marvin Sketch (version 22.13)). According to both parameters, clogP, and logP, the replacement of the hydrogen atom (75) with the methoxy group (76) had a small effect on lipophilicity. At the same time, an analogous replacement with the MeSO2-substituent (77) led to a dramatic decrease in lipophilicity by ca. 1.2 logP units.

Compounds 75–77; antibacterial drugs Sulfadiazine, Sulfameter with the MeSO2-containing analog 78, neuroleptic drug Tiapride with its aza-analog 79. Sol.: the experimental kinetic solubility in phosphate-buffered saline, pH 7.4 (µM). logP: the experimental distribution coefficient in n-octanol/water. Reliable logP values could be obtained within a range of 1.0–3.0. logD (7.4): the experimental distribution coefficient in n-octanol/phosphate-buffered saline, pH 7.4. Reliable logD values could be obtained within a range of 1.0–4.5. clogP: the calculated lipophilicity. CLint: the experimental metabolic stability in human liver microsomes (µL min−1 mg−1). t1/2 (min): the experimental half-time of a metabolic decomposition in human liver microsomes. *Parameter should be considered as approximate due to the high stability of compounds.
Next, we studied the physicochemical properties of antibacterial drugs Sulfadiazine, Sulfameter, and their MeSO2-containing analog 78 (see SI p. S394–S418; Fig. 7). All three compounds showed high solubility in water: 397 µM (Sulfadiazine) vs 361 µM (Sulfameter); 391 µM (78). All three compounds were also metabolically stable, outside the sensitivity range of the experimental method, CLint (mg min−1 μL−1): 0–2. Replacement of the hydrogen atom (Sulfadiazine) with the MeSO2-substituent (78) also led to a dramatic decrease of the lipophilicity by ca. 1.2 clogP units: 0.39 (Sulfadiazine) vs −0.8 (78). An effect on the experimental lipophilicity, logD, was less pronounced, due to the values outside the senility range of the experimental method: −0.8 (Sulfadiazine) vs <−1 (78).
In addition, from the previously obtained MeSO2-derivative 35c (Fig. 5A), we synthesized compound 79 (see SI p. S78–S79; Fig. 7)—an aza-analog of the neuroleptic drug Tiapride. The structure of compound 79 was confirmed by X-ray crystallographic analysis. We also measured the physico-chemical properties of Tiapride and its analog 79, and they happened to be very similar. Both compounds had high water solubility, and high metabolic stability (Fig. 7).
As a summary, in model compound 75, and the antibacterial drug Sulfadiazine, the replacement of the hydrogen atom with the MeSO2-substituent led to a dramatic decrease of lipophilicity by >1 logP/logD units.
Biological activity
Pharmacological studies of the efficacy of Tiapride in animals have demonstrated that its activity is due to the selective blockade of dopamine D2 and D3 receptors in the limbic areas of the brain. Therefore, we used a catalepsy model to study its activity. For this purpose, female CD-1 mice of three groups were intraperitoneally injected with Tiapride (200 mg/kg), its analog 79 (200 mg/kg), or Vehicle. After 90 min, the presence of catalepsy was tested using Catalepsy bar tests. To conduct the test, the mouse was placed with its front paws on a horizontal bar with a diameter of 5 mm and raised to a height of 4 cm above the working surface. The catalepsy score (the time during which each mouse maintained this position—from the moment it placed paws on the bar until the moment it was removed from the bar) was manually recorded with a stopwatch up to a maximum of 1 min. The average time of three consecutive tests was used for analysis.
As a result, the catalepsy score was significantly higher in the Tiapride dosing group compared to the Vehicle-treated group. At the same time, the injection of 79 did not have a similar effect—the catalepsy score values remained at the level of the Control Vehicle-treated group. This fact may indicate the absence of binding to dopamine receptors D2 and D3 of the modified molecule 79, which is necessary for inducing catalepsy in mice (see SI p. S419–S425).
Application in coordination chemistry
Many of the N-heterocyclic compounds noted above could serve as good ligands for metal ions. To illustrate one such utility, pyrazolates of silver(I) were investigated, which represent an important class of compounds with interesting features such as argentophilic Ag•••Ag contacts and useful applications, including antibacterial properties46,47,48,49. The survey shows that silver complexes of 3,5-disubstituted pyrazoles are the most common46,47,48,49, while the binary silver complexes of 3,5-nonsubstituted pyrazoles are very limited50,51,52. Here we illustrate the successful use of pyrazoles 2 and 64b in silver(I) chemistry.
The treatment of a mixture of pyrazole 2 and AgNO3 in methanol with triethylamine afforded {[2’]Ag}3 (2’ = deprotonated 2) as a white solid in 90% yield. Complex {[64b’]Ag}3 (64b’ = deprotonated 64b) was obtained from pyrazole 64b analogously in methanol-acetonitrile mixture in 85% yield. These compounds are insoluble in dichloromethane, toluene, and acetone, but somewhat soluble in tetrahydrofuran and dimethyl sulfoxide. Unfortunately, they did not provide crystalline material suitable for single-crystal X-ray structure determination. Based on the literature analogy50,51, as well as mass spectroscopic data of {[2’]Ag}3 and {[64b’]Ag}3, we believe that they exist in the typical trinuclear form48.
To further confirm the identity of {[2’]Ag}3 and {[64b’]Ag}3, we prepared their phosphine complexes by treating their suspension with PPh3 in dichloromethane. These reactions produced air-stable {[2’]Ag(PPh3)}2 and {[64b’]Ag(PPh3)}2, respectively, in essentially quantitative yield. Both complexes were soluble in common organic solvents. X-ray crystallographic analyses revealed that they are dinuclear species (Fig. 8; see SI p. S383–S394). These molecules sit on a center of inversion and feature planar six-membered Ag2N4 cores. The Ag-P distances of {[2’]Ag(PPh3)}2 and {[64b’]Ag(PPh3)}2 are similar at 2.3713(4) and 2.3703(3) Å, respectively (see SI p. S383). Overall, MeSO2- and CF3SO2-substituted pyrazoles afford silver complexes in excellent yields and could serve as effective ligand supports for many metal ions producing useful products53.

Molecular structures of {[2’]Ag(PPh3)}2 (left) and {[64b’]Ag(PPh3)}2 (right) (carbon—white, oxygen—red, sulfur—green, nitrogen—blue, phosphorus—pink, silver—gray, fluorine—light green). Ellipsoids are shown at a 50% probability level. Hydrogen atoms have been omitted for clarity.
In this work, we have developed a chemical reagent 1 to access methyl sulfones. Its reaction with various N,N- and N,C- bis-nucleophiles leads to the formation of the previously unknown methyl sulfones. This transformation works efficiently on a milligram, gram, and even multigram scale. Analogous strategy can also be used to construct alkyl-, CHF2−, CF3−, deuterated, and even bicyclo[1.1.1]pentane-containing derivatives. These compounds have been comprehensively characterized. They also have been demonstrated to have a high potential for use in medicinal chemistry and coordination chemistry.
Methods
Synthesis of N-(3-(dimethylamino)-2-(methylsulfonyl)allylidene)-N-methylmethanaminium hexafluorophosphate (1)
POCl3 (450 mL, 4.82 mol, 3.10 equiv) was added dropwise at −5 °C (ice-salt bath) to a solution of 2-(methylsulfonyl)acetic acid (215.00 g, 1.56 mol, 1.00 equiv) in DMF (730 mL, 9.44 mol, 6.05 equiv). Then the cooling bath was removed, and the reaction mixture was gradually heated to 90 °C (oil bath with a thermocouple). The intensive gas evolution was observed. Heating was continued until complete gas evolution (ca. 2 h), then the reaction mixture was cooled to room temperature. Potassium hexafluorophosphate (KPF6) (350.00 g, 1.90 mol, 1.22 equiv) was dissolved in distilled water (ca. 4.5 L) upon stirring, crushed ice was added (ca. 1.5 kg), and the reaction mixture was added in portions to the obtained solution. After stirring for 30 min, the formed precipitate was filtered and washed with cold water (3 × 500 mL). The final product was dried at 60 °C (oil bath with a thermocouple) in vacuo (~14 mmHg). Yield: 315.00 g, 0.90 mol, 58%, yellow solid, m.p. = 151–152 °C.
Synthesis of 4-(methylsulfonyl)-1H-pyrazole (2)
Vinamidinium salt 1 (575.00 g, 1.64 mol, 1.00 equiv) was suspended in EtOH (3 L), and hydrazine hydrate (250 mL, 4.92 mol, 3.00 equiv) was added at once at room temperature. Then the reaction mixture was heated under reflux (oil bath with a thermocouple) overnight. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was dried in vacuo (14 mmHg, 90 °C). The final product was purified by column chromatography (SiO2, CH2Cl2/MeOH, 10:1, Rf = 0.56). Yield: 170.00 g, 1.164 mol, 71%, white solid, m.p. = 102–103 °C.
NMR spectra were analyzed with MestreNova (11.0.3–18688).
Research animals
All experimental animal procedures performed were approved by the local Institutional Animal Care and Use Committee of BIENTA LTD (BACUC, Approval number: EN-EFF-AP-250624) and in adherence with the European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes. Local ethical committees are authorized to approve and monitor compliance with humane, ethical, moral, and legal principles regarding experimental animals when conducting scientific experiments in accordance with the order of the Ministry of Education and Science, Youth and Sports of Ukraine No. 416/20729 dated March 16, 2012.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Experimental data and characterization data for all new compounds prepared during these studies are provided in the Supplementary Information of this manuscript. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Center, under deposition numbers 2292252 (for 1), 2292249 (for 2), 2338739 (for 8), 2338736 (for 9), 2342487 (for 12), 2342771 (for 29), 2342486 (for 34), 2338735 (for 41), 2338738 (for 42), 2338737 (for 45), 2292253 (for 64a), 2292250 (for 63b), 2292251 (for 64b), 2292254 (for 70), 2389191 (for 79), 2334367 ({[2’]Ag(PPh3)}2), and 2334368 ({[64b’]Ag(PPh3)}2). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All data are available from the corresponding author upon request. Source data are provided. Source data are provided in this paper.
References
The Pesticide Manual. (ed MacBean, C.) (British Crop Production Council, 2012).
Jung, M. & Lindsay, V. N. G. One-pot synthesis of strain-release reagents from methyl sulfones. J. Am. Chem. Soc. 144, 4764–4769 (2022).
Li, Y., Li, D., Zheng, T., Li, H. & Ren, X. Double-addition reaction of aryl methyl sulfones with N-tert-butylsulfinyl imines: diastereoselective and concise synthesis of 2-sulfonylated 1,3-diamines. Chem. Eur. J. 14, 14986–14990 (2020).
Cho, C.-H., Neuenswander, B. & Larock, R. C. Diverse methyl sulfone-containing benzo[b]thiophene library via iodocyclization and palladium-catalyzed coupling. J. Comb. Chem. 12, 278–285 (2010).
Fukuda, N. & Ikemoto, T. Imide-catalyzed oxidation system: sulfides to sulfoxides and sulfones. J. Org. Chem. 75, 4629–4631 (2010).
Gamelas, C. A. et al. Selective and mild oxidation of sulfides to sulfoxides or sulfones using H2O2 and CpMo(CO)3Cl as catalysts. Tetrahedron Lett. 49, 4708–4712 (2008).
Baskin, J. M. & Wang, Z. An efficient copper catalyst for the formation of sulfones from sulfinic acid salts and aryl iodides. Org. Lett. 4, 4423–4425 (2002).
Zhu, W. & Ma, D. Synthesis of aryl sulfones via L-proline-promoted cui-catalyzed coupling reaction of aryl halides with sulfinic acid salts. J. Org. Chem. 70, 2696–2700 (2005).
Ma, D. et al. A new class of amide ligands enable Cu-catalyzed coupling of sodium methanesulfinate with (hetero)aryl chlorides. Chin. J. Chem. 35, 1661–1664 (2017).
Zhao, J. et al. A class of amide ligands enable Cu-catalyzed coupling of (hetero)aryl halides with sulfinic acid salts under mild conditions. J. Org. Chem. 83, 6589–6598 (2018).
Beaulieu, C., Guay, D., Wang, Z. & Evans, D. A. A mild and efficient new synthesis of aryl sulfones from boronic acids and sulfinic acid salts. Tetrahedron Lett. 45, 3233–3236 (2004).
Kar, A. et al. A general copper-catalyzed sulfonylation of arylboronic acids. Org. Lett. 9, 3405–3408 (2007).
Huang, F. & Batey, R. A. Cross-coupling of organoboronic acids and sulfinate salts using catalytic copper(II) acetate and 1,10-phenanthroline: synthesis of aryl and alkenylsulfones. Tetrahedron 63, 7667–7672 (2007).
Shavnya, A., Coffey, S. B., Smith, A. C. & Mascitti, V. Palladium-catalyzed sulfination of aryl and heteroaryl halides: direct access to sulfones and sulfonamides. Org. Lett. 15, 6226–6229 (2013).
Richards-Taylor, C. S., Blakemore, D. C. & Willis, M. C. One-pot three-component sulfone synthesis exploiting palladium-catalysed aryl halide aminosulfonylation. Chem. Sci. 5, 222–228 (2014).
Hethcox, J. C. et al. Nickel-catalyzed sulfonylation of aryl bromides enabled by potassium metabisulfite as a uniquely effective SO2 surrogate. Angew. Chem. Int. Ed. 62, e202217623 (2023).
Kin, P., Lo, T., Chen, Y. & Willis, M. C. Nickel(II)-catalyzed synthesis of sulfinates from aryl and heteroaryl boronic acids and the sulfur dioxide surrogate DABSO. ACS Catal. 9, 10668–10673 (2019).
Wang, M., Zhao, J. & Jiang, X. Aryl methyl sulfone construction from eco-friendly inorganic sulfur dioxide and methyl reagents. ChemSusChem 12, 3064–3068 (2019).
Yuan, G. et al. Copper-catalyzed aerobic oxidation and cleavage/formation of C–S bond: a novel synthesis of aryl methyl sulfones from aryl halides and DMSO. Chem. Commun. 48, 7513–7515 (2012).
Yang, Y. et al. Copper-catalyzed S-methylation of sulfonyl hydrazides with TBHP for the synthesis of methyl sulfones in water. Green. Chem. 19, 112–116 (2017).
Douglas, J. J., Buttar, D., Locke, K. & Turner, A. An intramolecular diels–alder approach to the isoindolinone core of AZD8154. Org. Pros. Res. Dev. 28, 2284–2295 (2024).
Liu, N.-W., Liang, S. & Manolikakes, G. Recent advances in the synthesis of sulfones. Synthesis 48, 1939–1973 (2016).
Joseph, D., Idris, M. A., Chen, J. & Lee, S. Recent advances in the catalytic synthesis of arylsulfonyl compounds. ACS Catal. 11, 4169–4204 (2021).
Zeng, D., Wang, M., Deng, W.-P. & Jiang, X. The same oxygenation-state introduction of hypervalent sulfur under transition-metal-free conditions. Org. Chem. Front. 7, 3956–3966 (2020).
Wang, M. & Jiang, X. The same oxidation-state introduction of hypervalent sulfur via transition-metal catalysis. Chem. Rec. 21, 3338–3355 (2021).
Teng, S., Shultz, Z. P., Shan, C., Wojtas, L. & Lopchuk, J. M. Asymmetric synthesis of sulfoximines, sulfonimidoyl fluorides, and sulfonimidamides enabled by an enantiopure bifunctional S(VI) reagent. Nat. Chem. 16, 183–192 (2024).
Shultz, Z. P., Scattolin, T., Wojtas, L. & Lopchuk, J. M. Stereospecific α-(hetero)arylation of sulfoximines and sulfonimidamides. Nat. Synth. 1, 170–179 (2022).
Botella, G. M., Harrison, B. L., Robichaud, A. J., Salituro, F. G. & Beresis, R. T. 19-Nor C3,3-disubstituted C21-N-pyrazolyl steroids and methods of use thereof. WO2014169833A1, 131 (2014).
Lloyd, D. & McNab, H. Vinamidines and vinamidinium salts-examples of stabilized push-pull alkenes. Angew. Chem. lnt. Ed. 15, 459–468 (1976).
Arnold, Z. Synthetic reactions of dimethylformamide. xii. Formylation of some carboxylic acids and their derivatives. Collect. Czech. Chem. Commun. 26, 3051–3058 (1961).
Arnold, Z. Note on the formylation of chloro- and bromoacetic acid. Collect. Czech. Chem. Commun. 30, 2125–2127 (1965).
Gupton, J. T., Krolikowski, D. A., Yu, R. H. & Riesinger, S. W. Application of 2-substituted vinamidinium salts to the synthesis of 2,4-disubstituted pyrroles. J. Org. Chem. 55, 4735–4740 (1990).
Gupton, J. T., Riesinger, S. W., Shah, A. S., Gall, J. E. & Bevirt, K. M. The preparation and some reactions of 2-(arylsu1fonyl)vinamidinium salts. J. Org. Chem. 56, 976–980 (1991).
Hoffmann, M. G. A new route to 1,5-disubstituted 4-arylsulfonylpyrazoles by lithiation of 1-methyl-4-arylsulfonylpyrazoles. Tetrahedron 51, 9511–9518 (1995).
Yamanaka, H., Takekawa, T., Morita, K., Ishihara, T. & Gupton, J. T. Preparation of novel 13-trifluoromethyl vinamidinium salt and its synthetic application to trifluoromethylated heterocycles. Tetrahedron Lett. 37, 1829–1832 (1996).
Davies, I. W. et al. An efficient preparation of vinamidinium hexafluorophosphate salts. J. Org. Chem. 65, 4571–4574 (2000).
Das, P., Gondo, S., Tokunaga, E., Sumii, Y. & Shibata, N. Anionic triflyldiazomethane: generation and its application for synthesis of pyrazole-3-triflones via [3 + 2] cycloaddition reaction. Org. Lett. 20, 558–561 (2018).
Huang, W. S. et al. Discovery of brigatinib (AP26113), a phosphine oxide-containing, potent, orally active inhibitor of anaplastic lymphoma kinase. J. Med. Chem. 59, 4948–4964 (2016).
Finkbeiner, P., Hehn, J. P. & Gnamm, C. Phosphine oxides from a medicinal chemist’s perspective: physicochemical and in vitro parameters relevant for drug discovery. J. Med. Chem. 63, 7081–7107 (2020).
Fox, R. M., Harris, P. A., Holenz, J., Seefeld, M. A. & Zhou, D. Heterocyclic amides as kinase inhibitors. WO2019130230A1, 72 (2019).
Yamamoto, S. et al. Heterocyclic compounds with an ror(gamma)t modulating activity. WO2018030550A1, 353 (2018).
Stepan, A. F. et al. Application of the Bicyclo[1.1.1]pentane motif as a nonclassical phenyl ring bioisostere in the design of a potent and orally active γ-secretase inhibitor. J. Med. Chem. 55, 3414–3424 (2012).
Mykhailiuk, P. K. Saturated bioisosteres of benzene: where to go next? Org. Biomol. Chem. 17, 2839–2849 (2019).
Pickford, H. D. et al. Rapid and scalable halosulfonylation of strain-release reagents. Angew. Chem. Int. Ed. 62, e202213508 (2023).
Semmler, K., Szeimies, G. & Belzner, J. Tetracyclo[5.1.0.01,6.02,7]octane, a [1.1.1]propellane derivative, and a new route to the parent hydrocarbon. J. Am. Chem. Soc. 107, 6410–6411 (1985).
Galassi, R., Rawashdeh-Omary, M. A., Dias, H. V. R. & Omary, M. A. Homoleptic cyclic trinuclear d10 complexes: from self-association via metallophilic and excimeric bonding to the breakage thereof via oxidative addition, dative bonding, quadrupolar, and heterometal bonding interactions. Comments Inorg. Chem. 39, 287–348 (2019).
Halcrow, M. A. Pyrazoles and pyrazolides-flexible synthons in self-assembly. Dalton Trans. 12, 2059–2073 (2009).
Zheng, J., Lu, Z., Wu, K., Ning, G.-H. & Li, D. Coinage-metal-based cyclic trinuclear complexes with metal-metal interactions: theories to experiments and structures to functions. Chem. Rev. 120, 9675–9742 (2020).
Schmidbaur, H. & Schier, A. Argentophilic interactions. Angew. Chem. Int. Ed. 54, 746–784 (2015).
Masciocchi, N. et al. The multiphase nature of the Cu(pz) and Ag(pz) (Hpz = Pyrazole) systems: selective syntheses and ab-initio X-ray powder diffraction structural characterization of copper(I) and silver(I) pyrazolates. J. Am. Chem. Soc. 116, 7668–7676 (1994).
Bertolotti, F., Maspero, A., Cervellino, A., Guagliardi, A. & Masciocchi, N. Bending by faulting: a multiple scale study of copper and silver nitropyrazolates. Cryst. Growth Des. 14, 2913–2922 (2014).
Kandel, S., Stenger-Smith, J., Chakraborty, I. & Raptis, R. G. Syntheses and X-ray crystal structures of a family of dinuclear silver(I)pyrazolates: assessment of their antibacterial efficacy against P. aeruginosa with a soft tissue and skin infection model. Polyhedron 154, 390–397 (2018).
Nímia, H. H. et al. Comparative study of Silver Sulfadiazine with other materials for healing and infection prevention in burns: a systematic review and meta-analysis. Burns 45, 282–292 (2019).
Acknowledgements
P.M. is grateful to Dr. S. Shishkina (IOC, Kyiv) for the X-ray studies, Dr. D. Bylina (Enamine) for HRMS measurements, Mr. Artem Skreminskyi (Enamine) for logP measurements, and Dr. Y. Holota (Bienta) for the help with ADME measurements.
Author information
Authors and Affiliations
Contributions
Y. P., A. A. T., H. V. R. D., and P. K. M. designed the experiments. Y. P., V. R., S. B., A. B., I. P., V. Q. H. P., H. V. R. D., I. V. S., O. G., and Y. V. D. conducted and analyzed the experiments described in this report. P. K. M., I. V. S., and H. V. R. D. prepared this paper for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare the following competing interests: Y. P., V. R., S. B., A. B., Y. V. D., A. A. T., I. V. S., and P. K. M. are employees of a chemical supplier Enamine. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Bing Gao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Poplavskyi, Y., Ripenko, V., Bova, S. et al. A reagent to access methyl sulfones. Nat Commun 16, 1132 (2025). https://doi.org/10.1038/s41467-024-55027-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55027-x
