
Abstract
The functionalized polycycle with densely contiguous tertiary stereocenters is a formidable challenge in synthesizing the parvistemoline family of Stemona alkaloids. We herein report their catalytic, asymmetric total syntheses in 13–14 steps from commercially available 2-(methoxycarbonyl)-pyrrole, featuring the development and deployment of an Ir/Pd-synergistically-catalyzed allylation of α-non-substituted keto esters with secondary aryl-substituted alcohols, stereodivergently accessible to four stereoisomers. Using chiral Pd-enolate and Ir π-allyl complex under neutral conditions, no epimerization occurs. Additionally, the other two adjacent stereogenic centers can be installed diastereoselectively by Zn(BH4)2-promoted reduction and Krische’s Ir-catalyzed 2-(alkoxycarbonyl)allylation. Oxy-Michael addition delivered the fused tetrahydrofuran-γ-lactone scaffold. At the later stage, hydrogenation or oxidation of pyrrole moiety furnished groups of tetrahydropyrrole and pyrrolidone. Finally, vinylogous Mannich reaction of an in situ generated iminium ion or Krische’s Ir-catalyzed 2-(alkoxycarbonyl)allylation of aldehyde installed the monocyclic lactone for parvistemonine (2) and didehydroparvistemonine (3), respectively.
Similar content being viewed by others

Introduction
The alkaloids obtained from plants belonging to the Stemonaceae family have been found to treat numerous disorders, including pertussis, pulmonary tuberculosis, and bronchitis, in China and other East Asian countries for a long history1,2,3,4,5. Because of the unique chemical structure bearing a plenitude of complex stereochemistry and potent bioactivities, these alkaloids have attracted significant attention from the synthetic community with numerous reported strategies6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31. In the 1990s, Xu and co-workers isolated three unusual alkaloids of this family32,33, named parvistemoline (1), parvistemonine (2), and didehydroparvistemonine (3), from the roots of Stemona parviflora collected from Hainan Island, southern China (Fig. 1a). The structures of these alkaloids were elucidated by a combination of spectroscopic techniques and chemical conversion of parvistemonine (2) to didehydroparvistemonine (3) with Ag2O oxidation. These alkaloids are structurally unique, featuring a fused tetrahydrofuran-γ-lactone scaffold with densely contiguous tertiary stereocenters. Since their isolation, the chemical synthesis of this Stemona alkaloid subgroup has remained elusive. To our knowledge, only one report concerning their synthetic efforts was previously outlined by us34. In that paper, although we had assembled the simplified tricyclic core and installed three stereocenters through the execution of asymmetric catalysis and chiral auxiliary, the ensuing efficient elaboration of complex targets was far from trivial. Compared to the significant advances made to the syntheses for other family members, the synthetic strategy toward parvistemoline alkaloids is still nascent. The functionalized polycycle with up to seven, densely contiguous tertiary stereocenters is a formidable challenge in synthesizing the parvistemoline family of Stemona alkaloids. Additionally, for parvistemonine (2), trans-substituted tetrahydropyrrole poses an extra synthetic challenge.

a the parvistemoline family of Stemona alkaloids. b other representative natural products.
The contiguous tertiary stereocenters are ubiquitous structural motifs in natural products. Natural products produced by all organisms, including bacteria, fungi, plants, and animals, demonstrate this complexity in stereochemistry, as shown in Fig. 1b (4-9)35. Therefore, the design of catalytic, asymmetric reactions that proceed with high stereoselectivity and ultimately can be utilized in natural product synthesis is an essential goal in chemical synthesis. However, traditional asymmetric reactions of stereocontrolling two stereogenic centers can be realized by a single chiral catalyst, generating only one of the stereoisomers. Notably, it frequently encounters a vexing problem when such a highly stereoselective reported reaction (for example, with >20:1 dr) would be executed in a total synthesis endeavor; it turns out that only the minor, inaccessible diastereomer that can match the chirality of the natural product. In this context, a new concept of stereodivergent dual catalysis was introduced recently by Carreira36,37,38,39, which entails the simultaneous use of two distinct chiral catalysts to furnish products with complete control over the configuration of two chiral centers. Four stereoisomers can be prepared from the same starting materials and identical conditions by employing the four possible catalyst permutations in pairs. Over the past few years, many efforts have been devoted to the synthesis of all the stereoisomers of the target product using dual catalysis, in which bimetallic catalysis has gained increasing interest40,41.
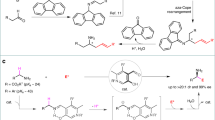
α-Alkylation of a β-keto ester is a frequently used reaction for carbon-carbon bond formation. However, this approach is often limited to controlling one stereogenic center when extended to its asymmetric variant. As shown in Fig. 2a, transition metals such as Pd, Rh, and Ir can catalyze an allylic alkylation of ketone ester42,43,44,45. Nonetheless, the product easily racemizes under acid or basic conditions, providing an almost 1:1 diastereomeric mixture. Aware of this limitation, in 2022, Tanaka and Kitamura reported an ingenious solution in which chiral Pd-enolate and chiral Ru π-allyl species synergistically catalyzed dehydrative allylation of β-keto esters (Fig. 2b)46. As nearly neutral conditions were used, this reaction gives a high level of diastereo- and enantioselectivity. Drawing inspiration from Tanaka and Kitamura’s success in using chiral Pd-enolate, we hypothesized that if successful, the chiral Pd-enolate would synergistically catalyze an analogous allylic substitution of chiral Ir π-allyl complex to afford the same results (Fig. 2c). It is noteworthy that unlike the linear allylic alcohol activated by the Ru catalyst, herein, we envisioned using branched secondary allylic alcohol, which is usually easier to prepare through vinyl group addition to the aldehydes. In contrast, some commercially unavailable linear allylic alcohols need multiple steps to prepare, i.e., through the HWE reaction of an aldehyde and then reduction to alcohol. Especially in our case of the ensuing total synthesis project, this holds true because the starting material 13 is readily available47, while its counterpart of linear allylic alcohol is far more challenging to synthesize due to the presence of an ester group on the pyrrole core. If we were successful in developing Ir/Pd-dual-catalyzed allylation, the allylated product, as shown in Fig. 2d, has three contiguous tertiary centers and would be accessible to eight stereoisomers if the β-keto group is diastereoselectively reduced using two complementary reducing agents. Given the multiple functionalities possessing three stereogenic centers that can be manipulated at will, we speculated that this valuable Ir/Pd-dual-catalyzed allylation might be one of the powerful reactions utilized in natural product synthesis.

a Previous reports using Pd, Rh, and Ir catalysis fail to control the α-carbon of β-keto esters. b Recent report by Tanaka & Kitamura employing Pd/Ru synergetic catalysis controls two vicinal chiral centers. c This work investigates unknown Ir/Pd-dual catalyzed allylation of secondary aryl-substituted alcohols. d Potential utilities of the allylated product in organic synthesis.
Herein, we describe the development and deployment of this Ir/Pd-synergistically-catalyzed allylation of α-non-substituted keto ester with secondary aryl-substituted alcohols that plays a crucial role in completing the total syntheses of three parvistemoline alkaloids.
Results and discussion
Development of Ir/Pd-dual-catalyzed allylic alkylation of β-keto esters
To test our Ir/Pd-dual-catalyzed allylic alkylation of β-keto ester, we chose p-methyl-α-vinylbenzyl alcohol (10a) as the model substrate to initiate exploration of the stereoselective allylic alkylation of tert-butyl 3-oxobutanoate (11). Considering the robustness of Carreira’s Ir/(phosphoramidite, olefin) catalyst system in catalyzing the asymmetric allylation of a diverse array of nucleophiles36,37,38,39,48,49, we adopted the standard conditions for Ir-catalyzed allylation, namely, the electrophilic Ir π-allyl complex formed by the addition of secondary alcohol 10a to an in situ generated catalyst from a phosphoramidite olefin ligand (S)-L and [Ir(cod)Cl]2, and paying attention to the nucleophilic partner.
As shown in Table 1, treatment of the above electrophilic Ir π-allyl complex with the addition of tert-butyl 3-oxobutanoate (11) and Pd((R)-BINAP)(H2O)2(OTf)2 in THF at 50 °C for 1 h was found to be optimal. Under these conditions, allylated β-keto ester 12a was obtained in 86% yield with 20:1 dr and >99% ee (entry 1). Changing the solvent to toluene, the reaction was performed with decreased yield but still with excellent selectivities (entry 2). Lowering the temperature or using Pd((R)-SEGPHOS)(H2O)2(OTf)2 instead of Pd((R)-BINAP)(H2O)2(OTf)2, somewhat lower yields were observed; however, the selectivities unaffected (entries 3, 4). Other chiral catalysts, for example, Ni((R)-BINAP)(H2O)2(OTf)2, Cu((S,S)-t-butyl-box)(H2O)2(OTf)2, and the complexes consisting of Cu(II)(OTf)2 or Cu(I)(CH3CN)4BF4 and (S,Sp)-iPr-Phosferrox show little or no diastereoselectivity and the observed yield or enantioselectivity is inferior to that obtained for Pd-ligand complexes (entries 5-8). Interestingly, employing Zn(OTf)2 instead of chiral Pd complex or using a combination of racemic Ir complex along with chiral Pd catalyst, both cases provided decreased yield and similarly poor diastereoselectivity; however, the ee values are still good (entries 9, 10). These results demonstrated that one of the catalysts, chiral Pd or Ir complex, can be equally responsible for the enantioselectivity. (for more detailed conditions screening, see Table S1 and Table S2).
With optimized conditions in hand, we set out to explore the scope of our reaction. As summarized in Fig. 3, a wide range of secondary alcohols made from aryl and hetero-aryl groups with or without substituents afforded the corresponding allylated β-keto esters 12 in good yields with high diastereo-, regio- and enantioselectivity. Compared to p-methyl-α-vinylbenzyl alcohol (10a), the phenyl group and o-methyl substituted phenyl substrates gave the products with slightly deceased yields but high selectivities (12b and 12c). Halogenated aromatic substrates bearing o-, m-, and p-substituents were well tolerated, affording allylated products (12d-12i) in good yields and unanimously excellent stereoselectivity. Notably, o-bromo phenyl substrate could provide the product 12g in synthetically useful yield, a crystalline compound whose structure, including the absolute configuration, was verified by X-ray crystallographic analysis (CCDC 2286423). Carbamate p-NHBoc is compatible, providing 12j in good yield and excellent selectivity. Subsequently, electro-rich substrates with different methoxy substitutions uniformly gave the products in high yields with superb stereoselectivity (12k-12m). In addition, other aromatics and heteroaromatic, such as benzodioxole, naphthalene, furan, thiophene, and indole (12n-12r), proved suitable substrates for the reaction. Importantly, the generality of β-keto esters 11 was also briefly surveyed. As shown in Fig. 3, the R group of β-keto esters 11 could be linear or branched alkyl substitutes (12s-12u). Other groups such as cyclopropyl, cyclopentyl, phenyl, and benzyl were tolerated, affording the corresponding products 12v-12y in good yields with synthetically useful diastereoselectivity and high enantioselectivity. We were interested in finding that 12z, which could not be accessed by Tanaka and Kitamura’s Pd/Ru protocol, could be efficiently obtained in this study. Therefore, this methodology constitutes a complementary option to Pd/Ru catalysis for the allylation of β-keto esters. However, we found one limitation of this reaction where α-substituted β-keto ester (12aa and 12ab) did not react efficiently under the optimal conditions. Predictably, aliphatic-substituted secondary alcohol failed to induce the desired substitution (12ac).

a Unless otherwise noted, all reactions were performed on a 0.2 mmol scale under the standard condition (see SI). Yield of the isolated products (diastereomeric mixture) after purification by chromatography. The dr and bl were determined by 1H NMR analysis of the crude reaction mixture (filtration through a short pad of celite). The ee was determined by HPLC analysis. b [Ir(cod)Cl]2 (0.08 equiv), (S)-L (0.32 equiv), Pd((R)-BINAP)(H2O)2(OTf)2 (0.16 equiv) and THF (1.0 M) were used.
Considering two chiral catalysts’ configurations might influence the reaction’s outcome through matched-mismatched effects, we continued to investigate the reaction by changing Carreira’s iridium ligand from (S)-L to (R)-L and using identical Pd((R)-BINAP)(H2O)2(OTf)2. As shown in Fig. 4, the generality conforms with that of Fig. 3; a series of typical secondary alcohols prepared from the aromatic group with electro-neutral, electro-rich, and electro-poor substituents, and some hetero-aryl groups were briefly surveyed, affording the corresponding syn-allylated β-keto-esters in good yields with a high degree of enantio-, regio- and diastereoselectivity. In contrast to the anti-products obtained in Fig. 3, there is a slight decrease in their diastereoselectivity in Fig. 4, which could be attributed to the mismatched catalyst system in Ir-(R)-L and Pd-(R)-L compared to the matched Ir-(S)-L and Pd-(R)-L catalyst system. Notably, increasing the steric of R group of the β-keto esters 11, the diastereoselectivity fell sharply (syn-12s and syn-12v). To evaluate the Ir/Pd two catalysts’ synergetic effect on the reaction, we further examined all permutations. As outlined in Fig. 5, p-methoxyl-α-vinylbenzyl alcohol (10 m) was selected as the substrate, and it was found that the substitution with 11 proceeded smoothly, four stereoisomers of 12 m synthesized stereodivergently. Gratifyingly, the catalyst system’s matched-mismatched effects were minimized in the transition state, which was supported by the fact that diastereoselectivity is unanimously high (dr >10:1) for organic synthetic preparation.

All reactions were performed under the same conditions in Fig. 3 except for using (R)-L.

Ar = p-methoxyphenyl.
We next sought to interrogate the mechanism of this reaction. Based on the recent mechanistic studies of Ir-catalyzed enantioselective allylic substitution enabled by Carreira ligand49, as outlined in Fig. 6, we proposed that coordination of allylic alcohol 10 to iridium gives η2 complex I, which undergoes TfOH-promoted oxidative addition to form η3-allyl iridium complex II. Meanwhile, in the palladium catalytic cycle, Pd catalyst IV reacts with β-keto ester 11, generating its palladium enolate V accompanied by TfOH liberation, which promotes the transformation of η2 complex I to η3-allyl iridium complex II in the iridium catalytic cycle. Nucleophilic attack of chiral palladium enolate complex V on the allyl fragment affords product complex III and regenerates Pd catalyst. Finally, displacement of the allylated product with allylic alcohol 10 completes the catalytic cycle. To minimize the nonbonded interaction, as can be seen in the structures of V and V*, it is the methyl group on the carboxylate that approaches the phenyl ring indicated in yellow of the palladium enolate complex V rather than the bulky tBu group in V*. The bulkiness of the tBu group on the ester is essential. In our cases of syn-12s and syn-12v, the poor diastereoselectivity arises from the fewer differences in steric repulsion for the ethyl or cyclopropyl group with the tBu group.

Ar group includes aryl and hetero-aryl groups with or without substituents.
Asymmetric total synthesis of parvistemoline alkaloids
Having established the critical methodology, we turned our attention to the initial interest: asymmetric total synthesis of parvistemoline alkaloids. As shown in Fig. 7, our synthesis began with pyrrole allylic alcohol 13, which is accessible in a two-step, one-pot reaction from commercial 2-(methoxycarbonyl)-pyrrole47. Considering the functional complexity of 13 compared to other substrates in Fig. 3, adjusting the previously optimized conditions was required. Gratifyingly, doubling the catalyst loading and decreasing the temperature to −20 °C could deliver the keto ester 14 in 79% yield with high levels of diastereo- and enantioselectivity. Reducing the ketone group of 14 using Zn(BH4)2 furnished erythro-3-hydroxy-2-substituted alcohol 15 diastereoselectively in 92% yield. The high diastereoselectivity was attributed to the highly coordinating ability of Zn to carbonyl oxygens via Zimmerman-Traxler model. Notably, if reduced by K-selectride, threo-3-hydroxyl diastereomer of 15 would be produced in high selectivity through the Felkin-Ahn model. Theoretically, our acyclic stereocontrolled synthesis of eight possible isomers of 15 with three contiguous tertiary chiral centers could be realized50,51,52,53,54,55,56,57,58,59,60,61,62. To this end, as outlined in Fig. 8, we first investigated the synthesis of four stereoisomers of 14. Gratifyingly, in spite of the increased functional complexity of substrate 13 compared to 10 m, all diastereomers of 14 were obtained parallelly in high yields with high degrees of diastereo- and enantioselectivity under identical conditions, varying only with the pairwise Ir and Pd catalyst combinations. On the basis of four intermediates of 14, accordingly, eight stereoisomers of 15 were smoothly synthesized by choosing out a reducing agent between Zn(BH4)2 and K-selectride.

Zn(BH4)2 Zinc borohydride, Grubbs 2nd Grubbs catalyst second generation, HBpin pinacolborane, DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene, TBSOTf tert-Butyldimethylsilyl trifluoromethanesulfonate, TMSOTf Trimethylsilyl trifluoromethanesulfonate.

K-selectride potassium tri-sec-butyl borohydride.
To close the seven-membered ring, an RCM reaction of 15 employing Grubbs’ second-generation catalyst delivered 16 in 88% yield (Fig. 7). Transformation of 16 to the TBS-protected carboxylic acid 17 was accomplished in 85% yield by a one-pot reaction in which furnishing the corresponding TBS silyl ether by addition of TBSOTf/Et3N and then cleavage of tert-butyl ester by TMSOTf. Subsequent reduction of the carboxylic acid to aldehyde has been achieved through a newly reported methodology where pinacol borane is used as a reducing agent and triflyl pyridinium as an activating reagent for carboxylic acid63. The resulting aldehyde 18 was converted to α-exo-methylene γ-butyrolactone 20 stereospecifically through an Ir-catalyzed 2-(alkoxycarbonyl)allylation under the conditions developed by Krische where (S)-Ir-tol-BINAP as a catalyst and acrylic ester 19 as an allylic metal surrogate64,65,66,67,68. However, the chemical yield of 20 is modest due to incomplete conversion of the substrate 18 and the formation of byproducts; in contrast, the same reaction of 18 with antipodal chiral catalyst (R)-Ir-to-BINAP led to epi-20 selectively in 80% yield (the difference in reactivity of two Krische catalysts shows a potential mismatched effect on (S)-Ir-tol-BINAP with chiral aldehyde 18, see S41 and S42 of Supplementary Information). Given the high diastereoselectivity controlled by the chiral catalyst in this step, at this point, it is noteworthy that by changing the chiral catalysts and by adopting two complementary procedures for reduction of the ketone (i.e., Zn(BH4)2 or K-selectride), these four contiguous tertiary stereogenic centers in 20 could be installed at will. Consequently, any of the 16 stereoisomers of 20 would be accessed theoretically. Ruthenium-catalyzed isomerization of the exo-methylene of 20 to an endocyclic double bond afforded 3-methyl furanone intermediate (not shown), to which was added hydrochloric acid to remove TBS protecting group, providing alcohol 21 in 85% yield in a one-pot reaction. Treatment of 21 with DBU, an intramolecular oxy-Michael addition proceeded smoothly to give a 3:1 mixture in 90% combined yield, favoring the desired 22a. The minor diastereomer 22b, differentiating in configuration at α-methyl group of lactone, proved to be a crystalline solid, and thus, single crystal X-ray analysis was employed to unambiguously assign the illustrated stereochemistry of the newly formed fused tetrahydrofuran-γ-lactone scaffold (CCDC 2394438). Overall, building upon the efficient transition-metal-catalyzed reactions, the challenging tetracyclic framework of parvistemoline alkaloids has been prepared in eight steps.
With tetracycle 22a in hand, as shown in Fig. 9, we continued our total synthesis by hydrogenating the olefin in the seven-membered ring. The modified procedure using a 1:1 mixture of Pd/C and Pearlman’s catalyst gave 23 better results than the conventional method using a single catalyst. Subsequent global reduction of the lactone and methyl ester of 23 with DIBAL-H furnished corresponding lactol and primary alcohol, respectively, which was oxidized with the Ley-Griffith protocol and gave aldehyde 24 in good yield. 24 is a crystalline compound whose structure was confirmed by X-ray crystallography (CCDC 2394439). With pyrrole-substituted aldehyde 24 in hand, using rhodium-catalyzed decarbonylation of aldehyde conditions, pyrrole 25 could be obtained in 78% yield. Thus obtained pyrrole 25 was oxidized using freshly purified m-CPBA under Dai’s conditions23 to give the dihydro-2H-pyrrol-2-one intermediate, which was further reduced with Pd/C catalyzed hydrogenation, smoothly providing parvistemoline (1). As anticipated, our synthetic product is spectroscopically and analytically identical to those of the natural source.

DIBAL-H Diisobutylaluminum hydride, m-CPBA meta-chloroperoxybenzoic acid, Rh/Al2O3 Rhodium on alumina, DCM Dichloromethane.
Our synthetic journey continued to complete didehydroparvistemonine (3). To this end, as shown in Fig. 9, installing the α-exo-methylene γ-butyrolactone attached to the pyrrole core using Krische’s Ir-catalyzed 2-(alkoxycarbonyl)allylation of aldehyde 24 afforded 26 in 3:1 ratio favoring C18 (S) isomer in 87% combined yield. 26 is unstable and used for the next step as soon as possible. Subsequently, Takaya’s asymmetric hydrogenation conditions69 were used to deliver didehydroparvistemonine (3) along with its C18 (R) isomer in quantitative combined yield. The synthetic sample of 3 was purified with HPLC, whose NMR data are in complete agreement with those of the isolation paper; however, the magnitude of optical rotation exhibits a noticeable difference despite the identical sign. This difference could be explained by the following two factors. First, we found that a didehydroparvistemonine sample could not dissolve well in methanol but was completely dissolved in CHCl3. Consequently, we measured the optical rotation in CHCl3. Second, as we elucidated previously, a partial epimerization in solution occurs at C18 attached to the pyrrole core, which was usually unnoticed by isolation authors before we recently rationalized during the total synthesis47. Fortunately, didehydroparvistemonine (3) proved to be a crystalline compound, and then a single-crystal X-ray analysis was employed to assign absolute stereochemistry (CCDC 2286427).
With parvistemoline (1) in hand, inspired by Chida-Sato’s synthesis of stemonine19,24, an iridium-catalyzed hydrosilylation was used. As shown in Fig. 9, treatment of parvistemoline (1) with Vaska complex [IrCl(CO)(PPh3)2] and tetramethyldisiloxane (TMDS) generated an enamine intermediate (not shown), subsequent one-pot addition of 2-siloxyfuran 27 and 2-nitrobenzoic acid provided pentacyclic 28 and epi-28, which were separated by flash chromatography on silica gel, in 36% combined yield. Notably, treatment of epi-28 with DBU failed to effectively isomerize to 28 mainly because a malign epimerization occurred at the site of tetrahydrofuran-γ-lactone. Hydrogenation of tetracycle 22a under forcing conditions using Rh/Al2O3 with 15 bar of hydrogen gas in the presence of sulfuric acid, which was used to activate pyrrole by generating the iminium salt, afforded a pair of pyrrolidines 29a and 29b in 90% combined yield, slightly favoring 29b. In this operation, both the olefin of the seven-membered ring and pyrrole moiety were reduced thoroughly. The newly formed two stereocenters of pyrrolidine 29a were revealed by ROESY correlation experiments, while 29b, fortunately, was a crystalline compound whose structure was secured through single-crystal X-ray analysis (CCDC 2286428). In addition, undesired 29b could be cycled by facile oxidation to pyrrole and then resubmitted to hydrogenation.
Considering the modest yield on the vinylogous Mannich reaction employing iridium-catalyzed hydrosilylation, we pursued an alternative approach at this junction. Hydrolysis of methyl ester of 29a with 48% hydrobromic acid followed by treatment with oxalyl chloride in the presence of DMF delivered acid chloride intermediate 30, which decomposes into the iminium salt8,70. Analogously, to the resultant iminium was added 2-siloxyfuran 27, and much better results occurred, providing pentacyclic 28 and epi-28 in 68% overall yield. Finally, using Rh/Al2O3 with 5 bar of hydrogen gas19, stereoselective hydrogenation of 28 quantitively yielded parvistemonine (2). Its spectral properties fully matched the natural isolate. Additionally, we converted this material into didehydroparvistemonine (3) through MnO2 oxidation24. Because the latter’s chemical structure was unambiguously established by X-ray crystallography, this oxidation further solidifies our conclusion on the synthesis of title alkaloids.
In summary, we have accomplished the total synthesis of three parvistemoline family members of Stemona alkaloids: (-)-parvistemoline (1), (+)-parvistemonine (2), and (+)-didehydroparvistemonine (3) in 13-14 steps from commercial 2-(methoxycarbonyl)-pyrrole. One of the highlights is the development and deployment of an Ir/Pd-synergistically-catalyzed allylation of α-non-substituted keto esters with secondary aryl-substituted alcohols, successfully controlling the absolute stereochemistry of two tertiary centers on demand, one of them being susceptible to racemization and otherwise difficult to control. Based on this methodology, the challenging other two contiguous tertiary stereocenters were installed diastereoselectively by Zn(BH4)2-promoted reduction and Krische’s Ir-catalyzed 2-(alkoxycarbonyl)allylation, consecutively. Other features include oxy-Michael addition delivering the fused tetrahydrofuran-γ-lactone scaffold, hydrogenation or oxidation of pyrrole moiety furnishing groups of tetrahydropyrrole and pyrrolidone, respectively, and vinylogous Mannich reaction of an in situ generated iminium ion or Krische’s Ir-catalyzed 2-(alkoxycarbonyl)allylation of aldehyde installing the monocyclic γ-butyrolactone for parvistemonine (2) and didehydroparvistemonine (3). Strategies developed herein would apply to other complex natural products.
Method
General procedure for Ir/Pd-catalyzed allylic alkylation
In a 5 ml round-bottom flask, [Ir(cod)Cl]2 (2.7 mg, 2 mol%) and (S)-L or (R)-L (8.1 mg, 8 mol%) were dissolved in THF (400 μL, 0.5 M) under an atmosphere of nitrogen. The mixture was vigorously stirred for 15 min. To the resulting solution were added sequentially allylic alcohol 10 (0.2 mmol, 1.0 equiv), β-keto ester 11 (0.3 mmol, 1.5 equiv), and Pd((R)-BINAP)(H2O)2(OTf)2 (8.5 mg, 4 mol%). The reaction was stirred at 50 °C and monitored by TLC. After the reaction ended, the crude reaction mixture was diluted by 10 mL petroleum ether/EtOAc (10:1) mixture and filtered through a short-pad of celite with petroleum ether/EtOAc (10:1, 3 × 10 mL). The filtrate was concentrated in vacuo and purified by flash chromatography to give the desired product 12 (note: to prevent epimerization of product 12, flash chromatography was performed as fast as possible).
Data availability
The X-ray crystallographic data for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC) under deposition numbers 2286423 (12g), 2394438 (22b), 2394439 (24), 2286427 (3), and 2286428 (29b). Copies of these data can be accessed free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within this article and its Supplementary Information file. Data supporting the findings of this manuscript are also available from the authors upon request. The experimental procedures and characterization of all new compounds are provided in Supplementary Information.
References
Pilli, R. A. & Ferreira de Oliveira, M. C. Recent progress in the chemistry of the Stemona alkaloids. Nat. Prod. Rep. 17, 117–127 (2000).
Pilli, R. A., Rosso, G. B. & Ferreira de Oliveira, M. C. The Stemona alkaloids. In: Cordell G. A. (ed.) The Alkaloids, 77−173 (Elsevier, 2005).
Pilli, R. A., Rosso, G. B. & Ferreira de Oliveira, M. C. The chemistry of Stemona alkaloids: an update. Nat. Prod. Rep. 27, 1908–1937 (2010).
Iwata, T. & Shindo, M. Synthesis, stereochemical stability, and biological activity of stemonamine and its related Stemona alkaloids. Heterocycles 98, 349–377 (2019).
Olivier, W. J., Henneveld, J. S., Smith, J. A., Hawkins, B. C. & Bissember, A. C. Strategies for the synthesis of Stemona alkaloids: an update. Nat. Prod. Rep. 39, 2308–2335 (2022).
Williams, D. R., Brown, D. L. & Benbow, J. W. Studies of Stemona alkaloids. Total synthesis of (+)-croomine. J. Am. Chem. Soc. 111, 1923–1925 (1989).
Wipf, P., Kim, Y. & Goldstein, D. M. Asymmetric total synthesis of the Stemona alkaloid (-)-stenine. J. Am. Chem. Soc. 117, 11106–11112 (1995).
Martin, S. F. & Barr, K. Vinylogous Mannich reactions. The asymmetric total synthesis of (+)-croomine. J. Am. Chem. Soc. 118, 3299–3300 (1996).
Kende, A. S., Smalley, T. L. & Huang, H. Total synthesis of (±)-isostemofoline. J. Am. Chem. Soc. 121, 7431–7432 (1999).
Jacobi, P. A. & Lee, K. Total synthesis of (±)- and (-)-stemoamide. J. Am. Chem. Soc. 122, 4295–4303 (2000).
Wipf, P., Rector, S. R. & Takahashi, H. Total synthesis of (-)-tuberostemonine. J. Am. Chem. Soc. 124, 14848–14849 (2002).
Bruggemann, M. et al. Total synthesis of (±)-didehydrostemofoline (asparagamine A) and (±)-isodidehydrostemofoline. J. Am. Chem. Soc. 125, 15284–15285 (2003).
Wipf, P. & Spencer, S. R. Asymmetric total syntheses of tuberostemonine, didehydrotuberostemonine, and 13-epituberostemonine. J. Am. Chem. Soc. 127, 225–235 (2005).
Frankowski, K. J., Golden, J. E., Zeng, Y., Lei, Y. & Aube, J. Syntheses of the Stemona alkaloids (±)-stenine, (±)-neostenine, and (±)-13-epineostenine using a stereodivergent Diels-Alder/Azido-Schmidt reaction. J. Am. Chem. Soc. 130, 6018–6024 (2008).
Zhao, Y.-M., Gu, P., Tu, Y.-Q., Fan, C.-A. & Zhang, Q. An efficient total synthesis of (±)-stemonamine. Org. Lett. 10, 1763–1766 (2008).
Wang, Y., Zhu, L., Zhang, Y. & Hong, R. Bioinspired and concise synthesis of (±)-stemoamide. Angew. Chem. Int. Ed. 50, 2787–2790 (2011).
Chen, J., Chen, J., Xie, Y. & Zhang, H. Enantioselective total synthesis of (-)-stenine. Angew. Chem. Int. Ed. 51, 1024–1027 (2012).
Bardaji, N. et al. Flexible approach to Stemona alkaloids: total syntheses of (-)-stemospironine and three new diastereoisomeric analogs. Org. Lett. 14, 4854–4857 (2012).
Yoritate, M. et al. Unified total synthesis of stemoamide-type alkaloids by chemoselective assembly of five-membered building blocks. J. Am. Chem. Soc. 139, 18386–18391 (2017).
Ma, K., Yin, X. & Dai, M. Total syntheses of bisdehydroneostemonine and bisdehydrostemoninine by catalytic carbonylative spirolactonization. Angew. Chem. Int. Ed. 57, 15209–15212 (2018).
Connelly, R. L., Knowles, J. P. & Booker-Milburn, K. I. A rapid synthetic approach to the ABCD core of the Stemona alkaloids. Org. Lett. 21, 18–21 (2019).
Hou, Y. et al. Asymmetric total syntheses and biological studies of tuberostemoamide and sessilifoliamide A. Org. Lett. 21, 2952–2956 (2019).
Yin, X., Ma, K., Dong, Y. & Dai, M. Pyrrole strategy for the γ-lactam-containing Stemona alkaloids: (±)-stemoamide, (±)-tuberostemoamide, and (±)-sessilifoliamde A. Org. Lett. 22, 5001–5004 (2020).
Soda, Y. et al. Unified total synthesis of pentacyclic stemoamide-type alkaloids. Org. Lett. 22, 7502–7507 (2020).
Huang, X.-Z., Gao, L.-H. & Huang, P.-Q. Enantioselective total syntheses of (+)-stemofoline and three congeners based on a biogenetic hypothesis. Nat. Commun. 11, 5314–5323 (2020).
Olivier, W. J., Bissember, A. C. & Smith, J. A. Unified total syntheses of (±)-sessilifoliamides B, C, and D. Org. Lett. 23, 3437–3441 (2021).
Guo, Z. et al. Tailored synthesis of skeletally diverse Stemona alkaloid through chemoselective dyotropic rearrangements of β-lactones. Angew. Chem. Int. Ed. 60, 14545–14553 (2021).
Cao, F. et al. Synthesis of the proposed structures of parvistemoamide and their transformations to stemoamide derivatives. Org. Lett. 23, 6222–6226 (2021).
Olivier, W. J., Lucas, N. T., Bissember, A. C. & Smith, J. A. Revised structures of dehydrostenines A and B: total syntheses of (±)-dehydrostenine A and structure assigned to dehydrostenine B. Chem. Eur. J. 27, 15382–15386 (2021).
Olivier, W. J. et al. Exploring cyclization strategies to access Stemona alkaloids: Subtle effects influencing reactivity in intramolecular Michael additions. Org. Lett. 23, 8494–8498 (2021).
Wang, X. et al. Asymmetric total synthesis of four Stemona alkaloids. Org. Lett. 25, 2213–2217 (2023).
Lin, W., Ying, B., Tang, Z., Xu, R. & Zhong, Q. Structure of parvistemonine. Huaxue Xuebao 48, 811–814 (1990).
Lin, W., Xu, R. & Zhong, Q. Chemical studies on Stemona alkaloids. I. Studies on new alkaloids of Stemona parviflora Wright C. H. Huaxue Xuebao 49, 927–931 (1991).
Zheng, Y., Yang, H.-D., Wei, K. & Yang, Y.-R. Synthetic studies toward parvistemoline using asymmetric Ir/amine-catalyzed allylation. J. Org. Chem. 86, 6025–6029 (2021).
Nicolaou, K. C. & Chen, J.-S. Classics in Total Synthesis III–Further Targets, Strategies, Methods (Wiley-VCH, 2011).
Krautwald, S., Sarlah, D., Schafroth, M. A. & Carreira, E. M. Enantio- and diastereodivergent dual catalysis: α-allylation of branched aldehydes. Science 340, 1065–1068 (2013).
Krautwald, S., Schafroth, M. A., Sarlah, D. & Carreira, E. M. Stereodivergent α-allylation of linear aldehydes with dual iridium and amine catalysis. J. Am. Chem. Soc. 136, 3020–3023 (2014).
Schafroth, M. A., Zuccarello, G., Krautwald, S., Sarlah, D. & Carreira, E. M. Stereodivergent total synthesis of Δ9-tetrahydrocannabinols. Angew. Chem. Int. Ed. 53, 13898–13901 (2014).
Krautwald, S. & Carreira, E. M. Stereodivergence in asymmetric catalysis. J. Am. Chem. Soc. 139, 5627–5639 (2017).
Huo, X., Li, G., Wang, X. & Zhang, W. Bimetallic catalysis in stereodivergent synthesis. Angew. Chem. Int. Ed. 61, e202210086 (2022).
Wei, L., Fu, C., Wang, Z.-F., Tao, H.-Y. & Wang, C.-J. Synergistic dual catalysis in stereodivergent synthesis. ACS Catal. 14, 3812–3844 (2024).
Trost, B. M. & Van Vraken, D. L. Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 96, 395–422 (1996).
Mohr, J. T. & Stoltz, B. M. Enantioselective Tsuji allylations. Chem. Asian J. 2, 1476–1491 (2007).
Lu, Z. & Ma, S. Metal-catalyzed enantioselective allylation in asymmetric synthesis. Angew. Chem. Int. Ed. 47, 258–297 (2008).
Cheng, Q. et al. Iridium-catalyzed asymmetric allylic substitution reactions. Chem. Rev. 119, 1855–1969 (2019).
Le, T. P., Tanaka, S., Yoshimura, M., Sato, K. & Kitamura, M. Stereodivergent dehydrative allylation of β-keto esters using a Ru/Pd synergistic catalyst. Nat. Commun. 13, 5876 (2022).
Deng, Y., Liang, X., Wei, K. & Yang, Y.-R. Ir-catalyzed asymmetric total syntheses of bisdehydrotuberostemonine D, putative bisdehydrotuberostemonine E and structural revision of the latter. J. Am. Chem. Soc. 143, 20622–20627 (2021).
Rössler, S. L., Petrone, D. A. & Carreira, E. M. Iridium-catalyzed asymmetric synthesis of functionally rich molecules enabled by (phosphoramidite, olefin) ligands. Acc. Chem. Res. 52, 2657–2672 (2019).
Rössler, S. L., Krautwald, S. & Carreira, E. M. Study of intermediates in iridium-(phosphoramidite, olefin)-catalyzed enantioselective allylic substitution. J. Am. Chem. Soc. 139, 3603–3606 (2017).
Huo, X., He, R., Zhang, X. & Zhang, W. An Ir/Zn dual catalysis for enantio- and diastereodivergent α-allylation of α-hydroxyketones. J. Am. Chem. Soc. 138, 11093–11096 (2016).
Huo, X., Zhang, J., Fu, J., He, R. & Zhang, W. Ir/Cu dual catalysis: enantio- and diastereodivergent access to α,α-disubstituted α-amino acids bearing vicinal stereocenters. J. Am. Chem. Soc. 140, 2080–2084 (2018).
Wei, L., Zhu, Q., Xu, S.-M., Chang, X. & Wang, C.-J. Stereodivergent synthesis of α,α-disubstituted α-amino acids via synergistic Cu/Ir catalysis. J. Am. Chem. Soc. 140, 1508–1513 (2018).
Jiang, X., Boehm, P. & Hartwig, J. F. Stereodivergent allylation of azaaryl acetamides and acetates by synergistic iridium and copper catalysis. J. Am. Chem. Soc. 140, 1239–1242 (2018).
He, Z.-T., Jiang, X. & Hartwig, J. F. Stereodivergent construction of tertiary fluorides in vicinal stereogenic pairs by allylic substitution with iridium and copper catalysts. J. Am. Chem. Soc. 141, 13066–13073 (2019).
Zhang, Q. et al. Stereodivergent coupling of 1,3-dienes with aldimine esters enabled by synergistic Pd and Cu catalysis. J. Am. Chem. Soc. 141, 14554–14559 (2019).
He, R. et al. Stereodivergent Pd/Cu catalysis for the dynamic kinetic asymmetric transformation of racemic unsymmetrical 1,3-disubstituted allyl acetates. J. Am. Chem. Soc. 142, 8097–8103 (2020).
Huo, X. et al. Stereodivergent Pd/Cu catalysis for asymmetric desymmetric alkylation of allylic geminal dicarboxylates. CCS Chem. 4, 1720–1731 (2022).
He, Z., Peng, L. & Guo, C. Catalytic stereodivergent total synthesis of amathaspiramide D. Nat. Synth. 1, 393–400 (2022).
Lu, R., Zhang, Q. & Guo, C. Catalytic stereodivergent allylic alkylation of 2-acylimidazoles for natural product synthesis. Nat. Commun. 14, 8118 (2023).
You, C., Shi, M., Mi, X. & Luo, S. Asymmetric α-allylic allenylation of β-ketocarbonyls and aldehydes by synergistic Pd/chiral primary amine catalysis. Nat. Commun. 14, 2911 (2023).
Qi, J. et al. Simultaneous dual Cu/Ir catalysis: stereodivergent synthesis of chiral β-Lactams with adjacent tertiary/quaternary/tertiary stereo-centers. ACS Catal. 13, 2555–2564 (2023).
Zhang, J. et al. Synergistic Pd/Cu-catalyzed 1,5-double chiral inductions. J. Am. Chem. Soc. 146, 9241–9251 (2024).
Chen, D. et al. Triflylpyridinium enables rapid and scalable controlled reduction of carboxylic acids to aldehydes using pinacolborane. Angew. Chem. Int. Ed. 62, e202215168 (2023).
Kim, I. S., Ngai, M. Y. & Krische, M. J. Enantioselective iridium-catalyzed carbonyl allylation from the alcohol or al-dehyde oxidation level using allyl acetate as an allyl metal surrogate. J. Am. Chem. Soc. 130, 6340–6341 (2008).
Kim, I. S., Ngai, M. Y. & Krische, M. J. Enantioselective iridium-catalyzed carbonyl allylation from the alcohol or aldehyde oxidation level via transfer hydrogenative coupling of allyl acetate: Departure from chirally modified allyl metal reagents in carbonyl addition. J. Am. Chem. Soc. 130, 14891–14899 (2008).
Han, S. B., Han, H. & Krische, M. J. Diastereo- and enantioselective anti-alkoxyallylation employing allylic gem-dicarboxylates as allyl donors via iridium-catalyzed transfer hydrogenation. J. Am. Chem. Soc. 132, 1760–1761 (2010).
Montgomery, T. P., Hassan, A., Park, B. Y. & Krische, M. J. Enantioselective conversion of primary alcohols to α-exo-methylene γ-butyrolactone via iridium-catalyzed C-C bond-forming transfer hydrogenation: 2-(alkoxycarbonyl)allylation. J. Am. Chem. Soc. 134, 11100–11103 (2012).
Kim, S. W., Zhang, W. & Krische, M. J. Catalytic enantioselective carbonyl allylation and propargylation via alcohol-mediated hydrogen transfer: merging the chemistry of Grignard and Sabatier. Acc. Chem. Res. 50, 2371–2380 (2017).
Ohta, T., Miyake, T., Seido, N., Kumobayashi, H. & Takaya, H. Asymmetric hydrogenation of olefins with aprotic oxygen functionalities catalyzed by BINAP-Ru(II) complexes. J. Org. Chem. 60, 357–363 (1995).
Dean, R. T., Padgett, H. C. & Rapoport, H. A high-yield regio-specific preparation of iminium salts. J. Am. Chem. Soc. 98, 7448–7449 (1976).
Acknowledgements
Financial support is provided by the National Natural Science Foundation of China (U23A2082, 22171274), the Chinese Academy of Sciences (ZDBS-LY-SM030), and the Yunling Scholars Special Project of Yunnan Province (XDYC-YLXZ-2023-0028). X.L. is grateful to the Youth Innovation Promotion Association of CAS.
Author information
Authors and Affiliations
Contributions
Y.-R.Y. directed the project and wrote the manuscript. Y.-R.Y. and X.L. conceived the synthetic route and analyzed the results. X.L., Q.-H.D., H.-F.Y., J.-T.Y., Y.D., and L.S. conducted the experimental work. W.K. assisted Y.-R.Y. in the project investigation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liang, X., Ding, QH., Yang, JT. et al. Total syntheses of the parvistemoline alkaloids enabled by stereocontrolled Ir/Pd-catalyzed allylic alkylation. Nat Commun 15, 10812 (2024). https://doi.org/10.1038/s41467-024-55111-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55111-2
