
Abstract
The plastic waste crisis is among humanity’s most urgent challenges. However, widespread adoption of sustainable plastics is hindered by their often inadequate processing characteristics and performance. Here, we introduce a bio-inspired strategy for the modification of a representative high molar mass, biodegradable aliphatic polyester that helps overcome these limitations and remains effective at molar masses far greater than the entanglement molar mass. We use co-assembly of oligopeptide-based polymer end groups and a low molar mass additive to create a hierarchical structure characterized by regularly spaced nanofibrils interconnected by entangled polymer segments. The modified materials show a rubbery plateau at temperatures above their melting point, associated with strongly increased melt strength, extraordinary melt extensibility, improved dimensional stability, and accelerated crystallization. These thermomechanical property changes open up otherwise inaccessible processing routes and offer considerable scope for improving solid-state properties, thereby addressing typical shortcomings of sustainable alternatives to conventional plastics.
Similar content being viewed by others

Introduction
Plastics have transformed many aspects of our lives for the better, but the continued exponential growth in their production has led to a corresponding surge in plastic waste1,2. However, while the plastic waste crisis is one of humanity’s most pressing challenges, it remains a complex systemic problem with no single optimal solution1,3. Aliphatic polyesters nevertheless represent a highly promising materials class for a more sustainable plastics economy4,5,6 because they integrate multiple complementary features considered vital to any holistic strategy. They may often be sourced from renewable feedstocks, which is key to favorable life cycle assessment7,8, and a wide variety of grades are commercially available1, many of which are biodegradable, mitigating the consequences of leakage into the environment9. Aliphatic polyesters are also amenable to chemical recycling to monomer under mild conditions5,6,10,11, and may be designed to have properties similar to polyolefins12, which represent some two-thirds of plastics production worldwide but are non-biodegradable5.
However, the market share of more sustainable plastics remains low, as their implementation is often hindered by their failure to meet the processing and property requirements of their intended applications. Typical shortcomings include low melt strength, melt elasticity, melt extensibility (severely restricting processing windows), slow crystallization (implying unacceptably long cycle times in important industrial processes), and limited maximum use temperatures8. These may, to some extent, be addressed individually via established strategies involving, for instance, nucleating agents, chain extenders, long-chain branching, blending, or reinforcement13. More recently, covalent networks and vitrimers14,15 or supramolecular polymers based on thermoreversible self-assembly of multivalent, self-complementary hydrogen-bonding motifs have also been used to produce materials with rheological properties typical of well-entangled or cross-linked polymers combined with sharp transitions to low-viscosity melts, offering improved processing and recyclability16,17,18,19. The dynamic nature of these materials may also lead to remarkable self-healing properties20,21,22,23, shape-memory behavior24, and stimuli-responsiveness25,26. Moreover, ditopic hydrogen-bonded ligands27,28, including in particular β-sheet-forming oligopeptides or poly(amino acid)s29,30,31, provide additional mechanical reinforcement thanks to their bio-inspired, self-limiting aggregation into uniform, one-dimensional nanofibrils32,33. This has been used to tailor elasticity and toughness in thermoplastic elastomers29,34, as well as in biomedical scaffolds35 or structural guides for organic electronics36,37,38.
With some exceptions34,39, however, research into supramolecular polymers and networks has so far focused on supramolecular materials based on short-chain polymer segments, because self-assembly of the end groups becomes unfavorable as the polymer chain length increases40,41,42. This may preclude the use of polymer segments with molar masses well above their entanglement molar mass, which is a critical limitation, given that entanglement is fundamental to the mechanical properties of technologically relevant, high molar mass polymers43. The relative lack of interest in supramolecular assembly of high molar mass semicrystalline and glassy polymers44,45 may also stem from the prevailing notion that their strength and stiffness are dominated by the polymer matrix. Adequate melt processability and final properties are nevertheless indissociable when implementing new plastics technology. An effective supramolecular modification strategy for semicrystalline polymers with molar masses typical of commercial grades may, hence, be hugely beneficial for broadening their range of applications.
Here, we show that the co-assembly of polymer end groups and a matching low molar mass additive, both based on β-sheet-forming oligopeptides (Fig. 1), results in efficient, reproducible formation of a supramolecular network even for polymers with molar masses far greater than their entanglement molar mass. The silk-inspired46,47, hierarchical structure adopted by the oligopeptides is an essential design element in suppressing macroscopic phase separation of the additive in favor of a co-assembly of additive and polymer end groups into well-defined, regularly spaced, high-aspect-ratio, polymer-bridged, semirigid nanofibrils that are determinant for the melt properties. The nanofibrils act as physical cross-links, inducing rubber-like behavior with a plateau modulus on the order of 1 MPa at temperatures of up to 50 °C above the nominal polymer melting point, as required for many important industrial processing routes48, as well as strongly improved dimensional stability. Within this temperature window, the synergy between the dynamic and anisotropic cross-links and the entangled polymer results in enhanced low-strain elasticity, significant strain hardening, as well as exceptionally high melt extensibility of up to 3000%, opening up processing routes otherwise inaccessible to the base polymer, such as thermoforming, film blowing, and film melt-drawing.

a Supramolecular structure formation is decoupled from the molar mass of the polymer segment by co-assembly of monodisperse polymer end groups (blue) and a low molar mass additive (orange), both based on β-sheet-forming oligopeptides. The end-modified polymer alone shows limited network formation at high molar masses, and the additive phase separates from the non-modified polymer to form bulk precipitates. However, b co-assembly of the end groups and additives via multivalent amide hydrogen bonding gives rise to antiparallel β-sheet aggregates even when the polymer molar mass is high. c Inspired by nanostructure formation in silks46,47, the β-sheets are programmed to assemble further into semirigid nanofibrils with a uniform diameter, consisting of helically twisted stacks of a well-defined number of β-sheets, self-limited by helicity and polymer tethering29,32. d Polymer bridging results in a supramolecular network with a regular nanofibril spacing. The thermal stability, morphology, and semirigid nature of the nanofibrils are crucial to their packing and orientation during deformation at temperatures above the polymer melting point. e Extended-chain crystallization owing to stretching of the polymer segments bridging the nanofibrils may reinforce the network as it deforms, contributing to the exceptional melt extensibility. This allows the preparation of homogeneous, oriented films, whose morphology after cooling comprises aligned polymer-bridged nanofibrils and shish-kebab structures in the polymer matrix.
The nanofibrils also act as highly efficient nucleating agents, increasing polymer crystallization rates by over one order of magnitude compared with standard nucleating agents. This combination of properties facilitates macroscopic orientation and deformation-induced crystallization and, hence, tailoring of the solid-state morphology, which in turn results in novel room-temperature mechanical properties, such as a fivefold increase in yield strength. Given that the same concept can be applied to a wide range of other high molar mass polymers that are considered sustainable alternatives to common plastics, it may help to overcome the technical barriers to their broader adoption in consumer products.
Results and discussion
We have chosen poly(ε-caprolactone) (PCL) as a representative example of the family of aliphatic polyesters because, while commercial grades of PCL are generally petroleum-based, it can be sourced from renewable feedstock, it is chemically recyclable5, and its fast and complete biodegradation under various conditions has been well established49,50,51. At the same time, it is also a typical example of polymers whose important shortcomings with regard to processability and thermomechanical properties have severely limited their range of applications (Fig. S1). PCL with a number average molar mass, \({\bar{M}}_{{{{\rm{n}}}}}\), of 89,500 g/mol, which is about thirty times the entanglement molar mass, Me, of approximately 3000 g/mol52, has been modified with acetyl-l-alanyl-l-alanyl end groups (mPCL, 0.5 wt% end groups). Biodegradability measurements carried out according to standard procedures confirm that the known excellent biodegradability of PCL has not been altered by this end group modification (Fig. S1), while the dipeptide end groups are expected to be readily broken down by proteases abundant in soil and water53. These end groups are monodisperse, ditopic hydrogen bonding ligands that aggregate via trivalent self-complementary N–H···O = C hydrogen bonds into one-dimensionally extended, high aspect ratio, antiparallel β-sheet tapes, which are programmed to form helically twisted stacks that we shall refer to as well-defined nanofibrils (Fig. 1)29, in order to distinguish them from the less well defined hard domains typically seen in other systems. Thus, modified low molar mass PCL (mPCL20, \({\bar{M}}_{{{{\rm{n}}}}}=22,000\,{{{\rm{g}}}}/{{{\rm{mol}}}}\), 2 wt% end groups) indeed shows end-group association below 77 °C, and hence rubber-like behavior down to the PCL crystallization onset temperature of around 40 °C in cooling experiments, which is not seen in unmodified PCL with a similar molar mass. However, mPCL20 is highly brittle because \({\bar{M}}_{{{{\rm{n}}}}}\) is only about 7 Me, resulting in weak entanglement. In marked contrast, the high molar mass mPCL shows ductile behavior but no end-group association, so its properties are close to those of the corresponding unmodified PCL (Fig. S2).
This demonstrates the adverse effect of end-group dilution on end-group aggregation in high molar mass polymers. We address this problem by blending mPCL with the additive 2-octyldodecyl acetyl-l-alanyl-l-alanyl amide (A) that is based on the same supramolecular motif. As will be shown in what follows, the co-assembly of the monodisperse, ditopic mPCL end groups and the additive A results in well-defined nanofibrils in a region of the mPCL/A phase diagram (Fig. 2a, Figs. S3–5) that is absent from the phase diagram for reference blends of A with unmodified PCL with a similar molar mass and is also impossible to reproduce with monotopic self-assembling motifs such as ureidopyrimidone, for which the addition of a complementary additive leads to the break-up of the network16. The unmodified PCL/A blends show upper critical solution temperature behavior, i.e., an optically transparent, homogeneous melt over the entire range of A contents at sufficiently high temperatures, but precipitation of bulk additive domains on cooling (Fig. S4). While the behavior of mPCL/A resembles that of PCL/A at high A contents (Fig. S6), pressed specimens of mPCL/A are optically homogeneous at all temperatures prior to crystallization of the PCL when the A content is below about 5 wt% (Figs. 2b and S7). Room-temperature FTIR spectroscopy in this concentration range nevertheless demonstrates the additive and end groups to self-assemble quantitatively into extended antiparallel β-sheet-like aggregates, implying nanophase separation (Figs. 2c and S8). The solid bulk additive precipitates in both mPCL/A (at A contents greater than 5 wt%) and the PCL/A reference blends take the form of birefringent microfibers (Figs. 2d and S6–7) with chiral columnar mesophase structure and a characteristic intercolumnar spacing, dA, of 2.8 nm (Figs. 2e and S9). The corresponding SAXS Bragg reflection may hence be used to further evaluate the transition from precipitation in the form of nanofibrils, for which it is absent, to mixed regimes of microfibers and nanofibrils as the A content is increased in mPCL/A. Bulk additive precipitation is found to be fully suppressed in favor of nanofibrils at up to at least 2.5 wt% A, and this transition shifts to as much as 5 wt% A in specimens deformed during cooling (Figs. 2f and S10), consistent with optical microscopy and the concentration dependence of the additive dissociation temperatures, Td, determined by DSC (Figs. S3 and S5).

a The mPCL/A binary phase diagram constructed from DSC heating scans shows a new phase region (blue) at low A contents, where the co-assembly of A and mPCL end groups on cooling results in a supramolecular network of polymer-bridged nanofibrils (NF), as distinct from the solid bulk precipitates (As) observed at higher concentrations and in the PCL/A reference blends. b Within this phase region, hot-pressed mPCL/A appears homogeneous in bright-field optical micrographs (OM) and between cross-polarizers (POM) above the PCL melting point (the micrographs shown were recorded at 70 °C). However, the additive and end groups self-assemble quantitatively into β-sheet aggregates according to c room temperature Fourier transform infrared (FTIR) spectroscopy. d, e OM, POM, and atomic force microscopy (AFM) phase images from the PCL/A reference blends under the same conditions reveal bulk precipitates of A in the form of microfibers. These consist of a chiral columnar mesophase made up of close-packed columns of additive molecules helically twisted around the microfiber axis. The power spectrum (bottom left) of the inset in the AFM image shows the columns to have a periodic spacing, dA, of 2.8 nm parallel to the microfiber surfaces, consistent with a prominent 2D SAXS reflection (bottom right) observed for both pure A and PCL/A. f The normalized intensity of the SAXS reflection at 2.8 nm may be used to distinguish the nanophase-separated regime from regimes dominated by bulk additive precipitation. Bulk additive precipitation is suppressed at A contents of up to 2.5 wt% in mPCL/A cooled under quiescent conditions and at A contents of up to 5 wt% in specimens deformed during cooling. g AFM phase images of mPCL/A prepared by hot pressing at 80 °C and recorded prior to the onset of PCL crystallization after melting in situ show aligned, high-aspect-ratio nanofibrils homogeneously distributed over the entire specimen. (See Figs. S3–11 for details.).
AFM phase images of hot-pressed mPCL/A containing 2 and 5 wt% of A, and recorded prior to the onset of PCL crystallization after melting in situ, confirm bulk additive precipitates to be absent in this composition range and allow direct observation of the nanofibrils. These nanofibrils are homogeneously distributed in the PCL matrix with a regular interfibrillar spacing, dNF (Figs. 2g and S11), which is necessarily much larger than the intercolumnar spacing, dA, observed in solid bulk additive precipitates, and decreases from about 17.5 nm at 2 wt% A to 9.5 nm at 5 wt% A. Assuming the nanofibrils to be hexagonally packed and contain all the end groups and additive molecules, as implied by IR spectroscopy (Fig. 2c), this suggests each nanofibril comprises a helically twisted stack of about three β-sheet tapes (Fig. S12).
A well-defined nanofibril diameter follows from the controlled nature of superstructure formation in monodisperse chiral β-sheet-forming oligopeptides29,32. The helical twist induced in the β-sheet aggregates self-limits β-sheet stacking so that the resulting nanofibrils are constituted by a fixed number of stacked β-sheet tapes, such that the height of the stack corresponds approximately to the length of the oligopeptide. This superstructure formation also implies the oligopeptide nanofibrils to be semirigid, with persistence lengths far larger than for other types of 1D hydrogen-bonded aggregates, but smaller than for nanocrystals with a high degree of 3D order29,36,54,55. They hence align locally to give a nematic texture that orients readily under shear. The resulting morphologies are similar to those of cylindrical phases in microphase-separated ABA triblock copolymers, implying some 60% of the tethered PCL segments bridge adjacent nanofibrils56,57. The bridging segments and nanofibrils hence constitute a physical network, trapping entanglements above the PCL melting point, while the molar mass of the PCL segments is sufficiently high to ensure a stable response to large deformations in both the melt and the solid state. This hierarchical structure, which comprises at least four structural levels (the chiral oligopeptides form hydrogen-bonded β-sheet tapes, further stacked into polymer-bridged nanofibrils that in turn show nematic order) is reminiscent of certain key molecular and nanoscale features of silks46,47, and clearly distinguishes our system from previous examples of the use of low molar mass additives as supramolecular fillers in hydrogen-bonded polymers58,59. As will be discussed in what follows, this hierarchical structure is crucial to the remarkable solidification behavior and melt elasticity of mPCL/A, opening up new possibilities for melt processing without compromising solid-state performance.
Concerning solidification, the nanofibrillar network in mPCL/A is a far more effective nucleating agent for PCL crystallization than the bulk additive (whose performance is similar to standard nucleating agents), presumably because of both the high dispersion of the nanofibrils and tethering of the polymer chains60. Thus, the PCL crystallization half-time for mPCL/A (2 wt%) determined from isothermal DSC at 45 °C is about an order of magnitude shorter than for PCL/A (2 wt%), which contains only bulk A microfibers at this temperature, and 25 times shorter than for pure PCL (Figs. 3a and S13). However, the PCL matrix morphology remains spherulitic, and the nanofibrils have little effect on its crystallinity, which is about 40 wt% for all the materials after cooling from the melt at 10 °C/min (Fig. S13).

a PCL crystallization half times, τ1/2, from isothermal DSC measurements at 45 °C after cooling from the melt at 10 °C/min, show that polymer crystallization is about six times faster in mPCL than in PCL, up to twenty-five times faster in mPCL/A than in PCL, and about nine times faster in mPCL/A than in the corresponding PCL/A reference blends at low A contents. b According to oscillatory shear rheometry cooling scans (strain 0.5%, frequency 1 rad/s, cooling rate 1 °C/min), both mPCL/A (blue) and PCL/A (orange) exhibit rheological onset temperatures for aggregate formation, Tagg, defined as the temperature at which the melt storage modulus, G’, and loss modulus, G”, start to deviate significantly from those of pure PCL on cooling, that are in excellent agreement with the onset temperatures of the corresponding DSC exotherms (Fig. S3). However, at low A contents, rubbery elastic behavior (G’ > G”, light blue area) is only observed for mPCL/A. The corresponding rubbery plateau extends from the PCL crystallization temperature up to the rheological softening temperature, Ts (G’ = G”). Within this temperature window, G’ is about an order of magnitude higher for mPCL/A than for PCL/A. c, d Ts and the effective plateau modulus (G’ at 60 °C) increase systematically with A content up to about 5 wt% A. e Dogbone specimens of mPCL/A (5 wt%) remain dimensionally stable for several hours at 95 °C, whereas specimens of PCL or PCL/A fail within a few seconds. f A thermoformed mPCL/A (5 wt%) cup retains its shape when filled with boiling water, although it becomes transparent, indicating a loss of crystallinity in the PCL matrix. However, cups prepared from PCL and PCL/A fail within a few seconds. (See Figs. S13–15 for details).
Significant modifications to the melting behavior of mPCL/A are also apparent in the nanofibrillar regime, that is, for A contents below about 5 wt%. In oscillatory shear rheometry temperature sweeps, both mPCL/A and PCL/A show clear rheological onset temperatures for aggregate formation, Tagg, defined as the temperature where the behavior first deviates significantly from that of pure PCL on cooling (Fig. 3b). In each case, Tagg is close to the onset temperature of the corresponding DSC exotherms (Figs. S3 and S14). However, in marked contrast with the reference materials, the modified mPCL/A blends show a rubbery regime down to the lowest A contents investigated, extending from the PCL crystallization temperature up to a rheological softening temperature, Ts, defined here as the crossover temperature of the melt storage modulus, G’, and loss modulus, G”. Because the rubbery regime is only observed in the mPCL/A blends, it is inferred to be due to establishment of an elastically active network by the polymer-bridged nanofibrils. By comparison, the limited melt reinforcement by the bulk additive precipitates in PCL/A is attributed to their coarse dispersion and lack of coupling to the polymer matrix, so they act as non-interacting solid filler particles at low concentrations.
Ts in mPCL/A increases steeply with A content in the range of 0–3 wt%, and then levels off at about 110 °C as the A content approaches 5 wt%, that is, some 50 °C above the PCL melting point and around 70 °C above the crystallization onset temperature on cooling (Fig. 3c). The plateau modulus, defined here as G’ at 60 °C, also increases strongly with A content in this regime to reach a maximum of 0.94 MPa at 5 wt% A, before falling off again at higher A contents. This represents an increase of more than two orders of magnitude with respect to PCL, and by a factor of about six with respect to the corresponding PCL/A reference blends (Fig. 3d). The supramolecular network in mPCL/A is hence most effective at A contents close to 5 wt%, which was therefore chosen as the concentration for subsequent investigations.
The mPCL/A blends show markedly improved high-temperature dimensional stability in the nanofibrillar regime thanks to their elastic response well above the PCL melting point. Thus, suspended hot-pressed sheets of mPCL/A (5 wt%) retain their initial shape for hours at 95 °C, whereas reference PCL/A or pure PCL specimens fail within seconds (Figs. 3e and S15, Video V1). Similarly, thermoformed mPCL/A (5 wt%) cups (see below) filled with boiling water support the weight of the water for minutes, although their transparency indicates the PCL matrix to have melted. This is a remarkable achievement for a PCL-based formulation, as reflected by the behavior of a reference PCL/A cup, which disintegrates immediately when filled with boiling water (Figs. 3f and S15, Videos V2 and V3).
Low-strain melt elasticity is also seen in other supramolecular polymers and networks18 but rarely translates into useful tensile properties at large deformations because of the low polymer molar masses usually employed. In the present case, however, extensional rheometry at 70 °C and a constant logarithmic (Hencky) strain rate of 2 s−1, indicates a strongly non-linear response for mPCL/A (5 wt%), similar to that of conventional high molar mass polymers in the rubbery regime61,62. Thus, not only is the initial extensional viscosity of mPCL/A (5 wt%) over an order of magnitude greater than for PCL/A (5 wt%) but it also shows a local stress maximum or yield stress at 0.48 MPa and a nominal strain of 120%; pronounced strain hardening from about 610% nominal strain; unstable fracture within the gage length characterized by a well-defined planar fracture surface, an ultimate strength of 0.82 MPa, and an exceptionally high nominal strain-at-break of nearly 3000% (Figs. 4a and S16). In marked contrast, PCL/A exhibits unstable viscous necking beyond the stress maximum and the unmodified PCL cannot be tested at all under these conditions owing to its lack of mechanical stability. The total energy-at-break of mPCL/A (5 wt%) of 18 MJ/m3, is hence some 22 times that of PCL/A (5 wt%). The analogy with microphase-separated ABA triblock copolymers alluded to earlier is useful in interpreting the rheological behavior of mPCL/A, assuming the B block to be in the melt state and its molar mass to be very much greater than Me. In either case, if the molten polymer chains are covalently tethered to a solid cylindrical phase that moves affinely during deformation, their response is expected to be dominated by entanglement at low strains, while the cylindrical phase domains act essentially as a hard reinforcement63,64. This implies a plateau modulus of about 0.9 MPa for mPCL/A at low A contents, taking Me to be 3000 g/mol, which is consistent with the results for 2–5 wt% A (Fig. 3d) and thus suggests strong anchoring of the mPCL chain ends to the nanofibrils in this composition range. Prior to the onset of non-Gaussian strain-hardening, the shear modulus is predicted to tend towards that of a phantom network formed by the tethered polymer chains as the strain increases, assuming the network points corresponding to the chain ends remain immutable63. However, this does not account for the observed yielding and strain softening at strains above about 120% (Fig. 4a). Significant relaxation of loaded network strands must, therefore, take place, analogous to the onset of disentanglement in high molar mass linear polymers in the rubbery state61. In the present case, relaxation is assumed to involve shear of the nanofibrils and/or dynamic exchange of the end groups via (reversible) dissociation of the intermolecular hydrogen bonds65. In extensional flow, these processes are presumably accompanied by the alignment of the nanofibrils with the tensile axis and are in competition with the increased orientation of the polymer chains and non-Gaussian strain hardening as deformation proceeds (Fig. 4b)66,67. Moreover, molecular orientation may be further stabilized by extended-chain crystallization at large deformations, provided the deformation temperature is below the equilibrium melting point of PCL, which may be as high as 100 °C68. This behavior, and particularly the exceptionally high strain-at-break, attributed in large part to the length of the bridging chains and the dynamic nature of the nanofibrils acting as physical cross-links, cannot easily be reproduced by, for instance, conventional block or segmented copolymerization, which implies substantial modification to the solid-state and melt flow behavior of the base polymer. However, the proposed concept of a double network anchored by β-sheet nanofibrils covalently linked to extensible chain segments does mirror certain key features of silk materials, which may show extraordinary combinations of strength, ductility, and energy at break46,47.

a Nominal tensile stress-logarithmic (Hencky) strain curves from elongational viscosity measurements on mPCL/A (5 wt%) (blue) and PCL/A (5 wt%) at a constant logarithmic (Hencky) strain rate of 2 s–1 and a temperature of 70 °C, that is, above the nominal PCL melting point of 60 °C. mPCL/A (5 wt%) shows (1) an initially linear elastic response with Young’s modulus of 6.1 MPa, (2) a local stress maximum (yielding) at 0.48 MPa and a nominal strain of 120%, (3) homogeneous viscoplastic deformation, (4) strain hardening, and (5) unstable fracture with a well-defined planar fracture surface, resulting in ultimate strength of 0.82 MPa and a nominal strain-at-break of 2940%. PCL/A, on the other hand, shows unstable viscous necking. b We attribute the behavior of mPCL/A (5 wt%) to (1) deformation of an elastically active network made up of entangled PCL chains, reinforced and anchored by co-assembly of the end groups and the additive, (2) orientation of the network and partial relaxation of the anchored chains via dynamic end-group exchange between nanofibrils, and (3) longitudinal displacement of the nanofibrils and viscoplastic extension of the anchored chains, followed finally by strain-hardening, which may involve strain-induced crystallization of the extended PCL chains and fracture. c In consequence, mPCL/A (5 wt%) shows stable bubble formation during film blowing at 70 °C (0.1 bar overpressure), whereas PCL/A immediately fails. d Hot-pressed sheets of mPCL/A (5 wt%) may be thermoformed into self-supporting cups, whereas PCL/A materials can only be thermoformed with the aid of a paper support. e Melt-drawing of mPCL/A (5 wt%) results in homogeneous deformation up to large nominal draw strains, εm. (See Fig. S16 for details).
The extensibility and strain hardening of mPCL/A under tensile loading at temperatures above the polymer melting point, together with the accelerated PCL matrix crystallization, is highly relevant to industrial processing techniques that involve tensile or biaxial drawing in the melt state. These include film blowing, extrusion and injection blow molding, injection stretch blow molding, thermoforming, or foaming. Pronounced strain hardening, for instance, may stabilize melt deformation at a well-defined draw ratio, leading to a uniform wall thickness during blow molding, which is a crucial factor in the success of poly(ethylene terephthalate) (PET) in bottle manufacturing69. mPCL/A is, therefore, more compatible with this important class of processing techniques than unmodified PCL, which lacks the necessary melt strength. Hence, film-blowing of hot-pressed mPCL/A (5 wt%) sheets at 70 °C results in stable bubble formation, whereas unstable defects propagate in sheets of PCL and PCL/A immediately on application of the pressure (Fig. 4c). Similarly, thermoforming of hot-pressed mPCL/A (5 wt%) sheets may be used to manufacture cups with high feature fidelity that may easily be removed from the mold, are self-supporting, and whose wall thickness is in the range 30–90 µm (Figs. 4d and S16). On the other hand, PCL/A (5 wt%), can only be thermoformed with the aid of a paper support, and the resulting wall thickness varies from 30 to as much as 220 µm at the base of the cup.
It is also possible to produce homogeneous films with thicknesses of the order of 300 µm by isothermal or non-isothermal melt drawing hot-pressed mPCL/A (5 wt%) sheets to strains, εm, of up to 2000%, whereas melt drawing of pure PCL and PCL/A gives poorly defined, inhomogeneous filaments (Figs. 4e, 5a, and S17). FTIR spectroscopy (Fig. S18) and 2D WAXD and SAXS (Fig. S19) of mPCL/A (5 wt%) films melt drawn at 70 °C and a nominal strain rate of 2 s−1 to 500, 1000, or 2000%, indicate preferential orientation of the nanofibrils along the draw direction and the presence of PCL crystalline lamellae oriented perpendicular to the draw direction, and show strong meridional (hk0) diffraction peaks from the usual orthorhombic PCL crystal modification70, although drawing has little effect on the lamellar long period, which remains roughly unchanged at 15–18 nm.
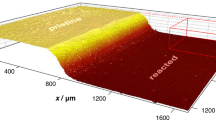
Furthermore, 2D synchrotron SAXS of mPCL/A (5 wt%) reveals an equatorial streak, which becomes more pronounced as εm increases, while the meridional scattering from the lamellae decreases (Figs. 5b and S20). This suggests the equatorial streak to be primarily due to extended-chain PCL crystals71, and indeed it is particularly evident in the non-isothermally drawn films (drawn and cooled simultaneously), in which strain-induced PCL crystallization is expected to be enhanced as the temperature decreases, consistent with an increase in the DSC peak melting point to 66 °C (Fig. 5c). Epitaxial overgrowth of lamellar PCL crystals on further cooling then results in shish-kebab structures, as also seen in AFM images of the film surfaces (Fig. 5d, e).

a Hot-pressed sheets of mPCL/A (5 wt%), PCL/A (5 wt%), and PCL melt drawn at 70 °C and a nominal strain rate of 2 s−1. mPCL/A (5 wt%) forms films with a uniform thickness, while PCL and PCL/A show viscous necking, resulting in inhomogeneous filaments. b In addition to the meridional scattering corresponding to the lamellar long period, 2D synchrotron SAXS patterns of melt-drawn films of mPCL/A (5 wt%), show a pronounced equatorial streak, which is particularly marked at high draw ratios and in films drawn under non-isothermal conditions. This combination of features is attributed to the formation of shish-kebab structures, that is, extended-chain polymer crystals oriented along the fiber axis and epitaxially nucleated lamellar polymer crystals oriented perpendicularly to the fiber axis. Accordingly, c DSC scans of the deformed specimens show a significantly higher melting transition in the first than in the second heating or than non-deformed PCL specimens. d AFM phase images of mPCL/A (5 wt%) non-isothermally melt-drawn to a nominal strain, εm, of 2000% together with selected grey-level profiles confirm the existence of a highly oriented morphology composed of nanofibrils (identified with finely spaced features observed locally, profile A–B), extended-chain PCL crystals (coarser fibrillar features with a spacing of about 25 nm, profile C–D), and lamellar PCL crystals growing epitaxially from the extended-chain crystal cores and stacked perpendicularly to the draw direction (average spacing of Lp = 18 ± 2 nm, profile E–F), as is schematically shown in (e). f Representative room temperature nominal stress-strain curves of melt-drawn mPCL/A (5 wt%) films show a strong increase in yield stress with increasing εm accompanied by a decrease in strain-at-break, which is particularly marked for non-isothermal drawing. g Young’s modulus vs. ultimate tensile strength for common packaging films (cf. Table S2) compared with the present results for PCL (orange) and mPCL/A (5 wt%) films at different εm after isothermal (blue) and non-isothermal (red) drawing. Error bars indicate the standard deviations.
The transition from a predominantly lamellar to a more extended-chain crystal morphology with increasing εm induces concomitant changes in the room-temperature tensile properties of the melt-drawn mPCL/A (5 wt%) films (Figs. 5f and S21, Table S1). The isothermally drawn films show a systematic increase in Young’s modulus and nominal yield stress with εm. Moreover, the highest yield stresses and tensile strengths are observed in the non-isothermally drawn films, albeit accompanied by a large reduction in the strain-at-break, suggesting increased molecular orientation. The fact that the strain-at-break in the isothermally drawn films remains above 200% for εm as high as 2000%, together with the limited work-hardening, indicates molecular relaxation during drawing, which offers considerable scope for further increases in tensile strength and stiffness through, for instance, optimized cold drawing72,73. Hence, while the performance of PCL itself is fundamentally limited by its low Tm and Tg, it is unrealistic to expect PCL-based materials to rival high-performance films of glassy polymers such as oriented poly(ethylene terephthalate) (PET), our approach renders it competitive with other ductile, commodity film-forming materials such as low-density polyethylene (LPDE), linear low-density polyethylene (LLDPE) and high-density polyethylene (HDPE), which dominate the flexible film packaging market (Fig. 5g).
In conclusion, we have demonstrated that the reversible co-assembly of polymer end groups based on ditopic and monodisperse oligopeptides and a corresponding low molar mass additive into a hierarchical structure of polymer-tethered nanofibrils remains effective for the supramolecular modification of polymers with molar masses well above their entanglement molar mass. This approach is, therefore, a highly promising means of addressing some of the most critical shortcomings of typical aliphatic polyesters with regard to their processing behavior and final performance without compromising their biodegradability (Fig. S1). The strong nucleating effect of the polymer-tethered nanofibrils on polymer crystallization should translate into reduced process cycle times, which is particularly important for polyesters, many of which crystallize comparatively slowly. Moreover, the supramolecular network of polymer-bridged nanofibrils gives rise to a new, extended rubbery plateau above the nominal polymer melting point with a tailorable plateau modulus of about 1 MPa. This provides significantly enhanced high-temperature dimensional stability and melt strength, as well as a remarkable degree of melt extensibility, with nominal strains-at-break approaching 3000%. A representative high molar mass, biodegradable aliphatic polyester (PCL) is hence rendered suitable for processing by, for instance, thermoforming, film blowing, or film drawing within a well-defined temperature window up to the dissociation temperature of the nanofibrils, while maintaining the flow behavior required for injection-based techniques above that temperature. We further show that the facilitated melt deformation to large strains and the concomitant changes in orientation and morphology lead to tensile strengths typical of conventional packaging film materials. Given that the concept of end-group and additive co-assembly is applicable beyond PCL74, our approach offers new perspectives for the manufacture of film and foam packaging from a wide variety of polymers currently regarded as sustainable alternatives to conventional plastics. However, possible effects on materials microstructure—and consequently on key performance parameters for sustainability—will need to be carefully evaluated in each case.
Methods
Blend preparation
For a typical mPCL/A and PCL/A blend about 1 g of mPCL or PCL, respectively, and the additive A were dissolved in 50–75 mL of freshly distilled dichloromethane (DCM). The solvent was removed with a rotary evaporator, and the remaining solid material was dried under a high vacuum overnight. For reproducible structure formation from a homogeneous melt state, all specimens of the mPCL/A blend and the PCL/A reference blends were subsequently heated to above the dissociation temperature, Td, of the aggregates and cooled to well below their association peak temperature at a cooling rate of 10 °C/min, unless stated otherwise in the figure captions. This thermal protocol was also applied to reference specimens of the pristine PCL, mPCL, and mPCL20 as part of their structural and thermomechanical characterization.
Differential scanning calorimetry
DSC measurements were performed using a Mettler Toledo DSC 3+ in a nitrogen atmosphere (flow rate 50 mL/min) with 2–8 mg specimens and a default scanning rate of 10 °C/min. Each material was typically subjected to at least two independent DSC measurements, and at least three heating and two cooling scans were applied in each case to check for reproducibility. Unless stated otherwise, only the linear baseline-corrected second heating and first cooling scans are shown. Phase diagrams were established from the observed dissociation temperatures, Td, whose concentration dependence and the corresponding binodals for liquid–liquid phase separation were described analytically using the Flory–Huggins approach, as described in the legend to Fig. S4. Isothermal experiments were conducted by heating the specimens to the homogeneous melt state (up to 200 °C depending on the composition), followed by cooling at 10 °C/min to the measurement temperature (45 °C) and maintaining this temperature until the end of the crystallization process.
Gel permeation chromatography
The number-average molar mass, \({\bar{M}}_{{{{\rm{n}}}}}\), the weight-average molar mass, \({\bar{M}}_{{{{\rm{w}}}}},\) and the dispersity, Đ, were determined by dissolving a 3–5 mg sample in 1 mL tetrahydrofuran and filtering the solution through a 0.220 μm Nylon filter before injection. The elution was performed in tetrahydrofuran at 40 °C at a flow rate of 1 mL/min, using an Agilent 1260 Infinity instrument incorporating the 390-MDS detector train equipped with a refractive index detector, one PSS SDV precolumn and either two PLgel 5 µm MIXED-C Analytical columns or two PSS SDV Analytical Linear XL columns. The calibration was performed with polystyrene standards with Mn in the range of 682–2,520,000 g/mol.
Infrared spectroscopy
Specimens for IR spectroscopy were prepared in the DSC by cooling a few milligrams of the material from the homogeneous melt state to room temperature at 10 °C/min. IR spectra of solid specimens were recorded in attenuated total reflectance (ATR) mode on a JASCO FT/IR 6300 spectrometer using the Miracle ATR accessory from PIKE. The polarized IR spectra of the melt-stretched PCL-based films were averages of 100 scans recorded in transmission mode using a Bruker Vertex 70v FTIR Spectrometer attached to a Bruker Hyperion 3000 microscope with an N2 cooled MCT detector incorporating a KBr beam splitter and a ZnSe-based wire grid polarizer.
Atomic force microscopy
Selected materials were cooled from the melt at 200–230 °C to ambient temperature at 10 °C/min. Flat surfaces were prepared by hot-pressing the specimens directly onto a steel AFM stub at the desired temperature (between the PCL matrix melting point and the dissociation temperature of the fibrils) with the aid of an amorphous Kapton™ H release film (DuPont) and a Linkham TMS600 hot stage. They were then cooled to room temperature at 10 °C/min, and the release film was removed. Images were obtained using an Asylum Research Cypher VRS AFM equipped with a heating stage and a laser-excited MikroMasch aluminum-coated NC14 probe (resonance frequency 160 kHz, and force constant 5 N/m) in intermittent contact mode, with a typical scanning frequency of 5 Hz and amplitude ratios of 0.3–0.5.
Optical microscopy
Specimens of a few milligrams were compressed between glass cover slides, heated to the homogeneous melt state (up to 200 °C depending on the composition), and then cooled at a nominal rate of 10 °C/min to the desired observation temperature using a Linkham TMS600 hot-stage. Images were recorded using an Olympus BX60 optical microscope equipped with crossed polarizers once the temperature had stabilized.
Shear rheology and extensional viscosity
Dynamic shear rheology was performed using a parallel plate TA Instruments ARES 2 rheometer. 8 mm diameter steel plates were used throughout, with the gap set to 0.8–1.5 mm. The blend specimens were loaded in the homogeneous melt state (at up to 200 °C depending on the composition) and cooled at 1 °C/min with a fixed radial frequency of 1 rad/s and a strain of 0.5%, while a higher strain of 5% was used for PCL. A standard test fixture from TA Instruments was used for the extensional viscosity measurements. Hot-pressed sheets were cut into 20 × 7 × 0.3 mm3 strips, placed in the test fixture, and held at the test temperature for 3 min before starting the measurement.
1D NMR spectroscopy
1H and 13C NMR spectra were obtained at 298 K using a Bruker Avance III 400 spectrometer at a frequency of 400 MHz and calibrated with respect to the residual solvent peaks of CDCl3 (7.26 ppm 1H NMR; 77.16 ppm 13C NMR). Chemical shifts are expressed in parts per million (ppm) (s = singlet, d = doublet, t = triplet, m = multiplet).
High-resolution mass spectrometry
HRMS spectra were recorded using a Waters Q-ToF Ultima with electrospray ionization (ESI).
Thermogravimetric analysis
TGA measurements were conducted on a PerkinElmer TGA 4000. The specimens (3–11 mg) were dried under a high vacuum for several days and heated in the TGA chamber from 30 to 900 °C at a scanning rate of 10 °C/min. The onset of decomposition was taken to be the temperature at which the mass loss first reached 10%.
Tensile testing (UTM)
Room temperature tensile tests were performed using a Zwick Roell 5 kN Universal Testing Machine (UTM) at a nominal extension rate of 10% of the initial length, L0 (17–23 mm), per minute with a pre-stress of 0.1 MPa. Dogbone specimens with a length of 4 cm and a gage length of 2 cm were prepared from the hot-pressed or melt-drawn films using a steel punch. The films had an average thickness of 330 µm (undrawn hot-pressed specimens of pristine PCL), 340 µm (undrawn hot-pressed specimens of PCL/A), 100 µm (mPCL/A (5 wt%) films melt-drawn to a strain, εm, of 500% at Tmelt = 70 °C), 50 µm (mPCL/A (5 wt%) films melt-drawn to a strain, εm, of 1000% at Tmelt = 70 °C), 30 µm (mPCL/A (5 wt%) films melt-drawn to a strain, εm, of 2000% at Tmelt = 70 °C), and 25 µm (mPCL/A (5 wt%) films drawn to 2000% while cooling from 70 °C to below the melting point of the PCL matrix). The width of the clamping zone was 1 cm, and that of the gage length was 0.5 cm. The specimens were mounted using standard 500 N clamps. Young’s modulus was calculated from the maximal slope of the nominal stress–strain curve in the linear elastic regime.
2D small and wide-angle X-ray scattering (SAXS/WAXS)
2D patterns were obtained using a Bruker D8 Discover Plus system with a TXS Cu rotating anode and a Göbel mirror for focusing. A 0.5 mm pinhole and a 0.3 mm collimator were placed in the primary beam path to select a beam of approximately 300 × 400 µm2 in area at the specimen position. A Cu Kα (λ = 1.54 Å) micro source was used in transmission mode with an Eiger2 500 K detector at 300 mm, calibrated using silver behenate. The intensity profiles were obtained by azimuthal or radial integration of the 2D patterns using Bruker Difrac Eva software. A uniaxial orientation distribution about a reference direction or “fiber axis” may be expressed as a series of even-order Legendre polynomials \({P}_{n}[\cos \vartheta ]\), where \(\vartheta\) is the angle between a unique axis characteristic of the orienting units and the fiber axis. Such distributions are conveniently characterized in terms of the coefficient of the second-order term in the series expansion
also known as Herman’s orientation factor. \(\left\langle {P}_{2}\right\rangle\) for the normals to a set of Bragg planes may be determined from 2D scattering patterns according to
where \({I}_{\psi }\) is the scattered intensity of the corresponding Bragg reflection at the azimuthal angle \(\psi\). Herman’s orientation factor takes the value of 1 for a specimen fully oriented parallel to the fiber axis, –1/2 for planar isotropic orientation perpendicular to the fiber axis, and 0 for isotropic specimens.
Synchrotron scanning small angle X-ray scattering
Scanning SAXS was performed at the cSAXS (X12SA) beamline at the Paul Scherrer Institut (PSI, Switzerland). The X-ray beam was monochromated by a fixed-exit double crystal Si (111) monochromator to 12.4 keV and focused to 20 × 7 µm2. To minimize the air scattering and absorption, a flight tube was placed between the specimen and the detector. The scattering signal was recorded by a Pilatus 2 M detector, and the transmitted beam was measured with a photodiode on a beamstop placed inside the flight tube. The specimen-to-detector distance was 7.12 m, calibrated using silver behanate. The exposure time was 0.1 s for all the specimens. The 2D scattering patterns were radially integrated to obtain 1D scattering curves in 64 azimuthal detector segments with the cSAXS Matlab analysis package. The azimuthal plots were calculated either from the radially integrated data or directly from the pixel intensities, depending on the measured intensity and statistics in the q-range of interest.
Hot-pressing
The specimens were placed in an aluminum mold with dimensions of 25 × 25 × 0.03 cm3 and a square (5 × 5 cm2) or circular (d = 7 cm) hole in the center, and placed between Teflon plates of lateral dimensions of 50 × 50 cm. A Lauffer Pressen UVL 5.0 laboratory press was loaded at 30 °C and evacuated to 1 mbar before heating to 200–230 °C at 10 °C/min. Once the desired melt temperature was reached, a pressure of 60 N/m was applied for 5 min, followed by cooling to ambient temperature at 10 °C/min.
Film melt drawing
Hot-pressed films of lateral dimensions of 5 × 5 cm2 were placed in the oven of the Zwick Roell 5 kN UTM using 500 N clamps and equilibrated at 70 °C for 5 min. The draw rate was set to 2 s−1, and the maximum extension was set to a melt strain, εm, of 500, 1000, or 2000%. Non-isothermally melt-drawn mPCL/A (5 wt%) was prepared by opening the oven door 5 s after drawing began.
Film blowing
Sheets with a thickness of 400 µm were prepared from mPCL/A (5 wt%) and PCL by hot-pressing at 200 °C. The sheet was placed on a film-blowing device with pressure control and placed in an oven at 70 °C for 20 min, during which the PCL matrix fully melted. A constant overpressure of about 0.1 bar was then applied, and the resulting deformation was recorded with a video camera.
Thermoforming
Circular hot-pressed specimens with a diameter of 7 cm and thickness of 0.3 mm were processed into a cup (30 mm in diameter and 35 mm in depth) above the melting point of the PCL matrix using a Formech HD686 vacuum former by heating for 20–30 s in an infrared oven and thermoforming after a delay of 0.4 s, with 100% plug assist, 50% vacuum power, 0% air pressure and 0% pre-stretch into the 40 °C mold. PCL/A (5 wt%) was thermoformed using paper support to avoid damage due to the direct application of the vacuum via the holes present in the steel mold, while mPCL/A (5 wt%) could be thermoformed without the need for support.
Heat stability testing
To test their heat stability, the thermoformed specimens were filled with boiling water, and their response was recorded with a video camera. Further stability tests were carried out by suspending dogbone specimens prepared from hot-pressed sheets using a steel punch for tensile testing in a Memmert VO49 vacuum oven at 95 °C.
Biodegradability testing
The biodegradability test was conducted according to ISO standard guidelines (ISO 14851) at 25 °C. The biochemical oxygen demand (BOD) was monitored while stirring over several days under aerobic conditions with a BOD meter using a temperature-controlled BOD reactor (Oxitop® with a 500 mL BOD reactor, WTW. Co. LTD). A powder specimen (initial weight about 30 mg) was placed in a BOD reactor, and 100 mL of natural seawater from Tokyo Bay (Tokyo, Japan) was added to the reactor as an inoculum, together with 0.1 mL of a mineral salt solution containing 8.50 g/L of KH2PO4, 21.75 g/L of K2HPO4, 67.00 g/L of Na2HPO4 × 12 H2O, 1.70 g/L of NH4Cl, 22.50 g/L of MgSO4 × 7 H2O, 27.50 g/L of CaCl2 and 0.25 g/L of FeCl3 × 6 H2O. The BOD biodegradability of the PCL-based materials was calculated by subtracting the BOD of the control from that of the test solution and dividing it by the theoretical oxygen demand of the test specimen. The BOD of the control was determined from an average of two blank tests using 100 mL of the natural seawater without the test specimen.
Materials and chemical synthesis
Details on the materials used and chemical synthesis procedures are given in the Supplementary Information.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author on request.
References
World Economic Forum, Ellen MacArthur Foundation & McKinsey & Company. The New Plastics Economy—Rethinking the Future of Plastics. (2016).
Geyer, R., Jambeck, J. R. & Law, K. L. Production, use, and fate of all plastics ever made. Sci. Adv. 3, e1700782 (2017).
Lau, W. W. Y. et al. Evaluating scenarios toward zero plastic pollution. Science 369, 1455–1461 (2020).
Stempfle, F., Ortmann, P. & Mecking, S. Long-chain aliphatic polymers to bridge the gap between semicrystalline polyolefins and traditional polycondensates. Chem. Rev. 116, 4597–4641 (2016).
Coates, G. W. & Getzler, Y. D. Y. L. Chemical recycling to monomer for an ideal, circular polymer economy. Nat. Rev. Mater. 5, 501–516 (2020).
Tang, X. & Chen, E. Y.-X. Toward infinitely recyclable plastics derived from renewable cyclic esters. Chemistry 5, 284–312 (2019).
Manker, L. P. et al. Sustainable polyesters via direct functionalization of lignocellulosic sugars. Nat. Chem. 14, 976–984 (2022).
Zhu, Y., Romain, C. & Williams, C. K. Sustainable polymers from renewable resources. Nature 540, 354–362 (2016).
Law, K. L. & Narayan, R. Reducing environmental plastic pollution by designing polymer materials for managed end-of-life. Nat. Rev. Mater. 7, 104–116 (2022).
Zhu, J.-B., Watson, E. M., Tang, J. & Chen, E. Y.-X. A synthetic polymer system with repeatable chemical recyclability. Science 360, 398–403 (2018).
Häußler, M., Eck, M., Rothauer, D. & Mecking, S. Closed-loop recycling of polyethylene-like materials. Nature 590, 423–427 (2021).
Eck, M. et al. Biodegradable high‐density polyethylene‐like material. Angew. Chem. Int. Ed. 62, e202213438 (2023).
Tsui, A., Wright, Z. C. & Frank, C. W. Biodegradable polyesters from renewable resources. Annu. Rev. Chem. Biomol. Eng. 4, 143–170 (2013).
Röttger, M. et al. High-performance vitrimers from commodity thermoplastics through dioxaborolane metathesis. Science 356, 62–65 (2017).
Brutman, J. P., Delgado, P. A., & Hillmyer, M. A. Polylactide vitrimers. ACS Macro Lett. 3, 607–610 (2014).
Sijbesma, R. P. et al. Reversible polymers formed from self-complementary monomers using quadruple hydrogen bonding. Science 278, 1601–1604 (1997).
Folmer, B. J. B., Sijbesma, R. P., Versteegen, R. M., van der Rijt, J. A. J. & Meijer, E. W. Supramolecular polymer materials: chain extension of telechelic polymers using a reactive hydrogen-bonding synthon. Adv. Mater. 12, 874–878 (2000).
Seiffert, S. & Sprakel, J. Physical chemistry of supramolecular polymer networks. Chem. Soc. Rev. 41, 909–930 (2012).
Voorhaar, L. & Hoogenboom, R. Supramolecular polymer networks: hydrogels and bulk materials. Chem. Soc. Rev. 45, 4013–4031 (2016).
Cordier, P., Tournilhac, F., Soulié-Ziakovic, C. & Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 451, 977–980 (2008).
Yanagisawa, Y., Nan, Y., Okuro, K. & Aida, T. Mechanically robust, readily repairable polymers via tailored noncovalent cross-linking. Science 359, 72–76 (2018).
Wang, S. & Urban, M. W. Self-healing polymers. Nat. Rev. Mater. 5, 562–583 (2020).
Chen, S. & Binder, W. H. Dynamic ordering and phase segregation in hydrogen-bonded polymers. Acc. Chem. Res. 49, 1409–1420 (2016).
Jiang, Z.-C. et al. Shape memory polymers based on supramolecular interactions. ACS Appl. Mater. Interfaces 9, 20276–20293 (2017).
Montero de Espinosa, L., Meesorn, W., Moatsou, D. & Weder, C. Bioinspired polymer systems with stimuli-responsive mechanical properties. Chem. Rev. 117, 12851–12892 (2017).
Yan, X., Wang, F., Zheng, B. & Huang, F. Stimuli-responsive supramolecular polymeric materials. Chem. Soc. Rev. 41, 6042 (2012).
Roosma, J., Mes, T., Leclère, P., Palmans, A. R. A. & Meijer, E. W. Supramolecular materials from benzene−1,3,5-tricarboxamide-based nanorods. J. Am. Chem. Soc. 130, 1120–1121 (2008).
Cantekin, S., Greef, T. F. Ade & Palmans, A. R. A. Benzene−1,3,5-tricarboxamide: a versatile ordering moiety for supramolecular chemistry. Chem. Soc. Rev. 41, 6125–6137 (2012).
Croisier, E. et al. A toolbox of oligopeptide-modified polymers for tailored elastomers. Nat. Commun. 5, 4728 (2014).
Matolyak, L. E., Keum, J. K., Voorde, K. M. Vde & Korley, L. T. J. Synthetic approach to tailored physical associations in peptide-polyurea/polyurethane hybrids. Org. Biomol. Chem. 15, 7607–7617 (2017).
Johnson, J. C., Wanasekara, N. D. & Korley, L. T. J. Influence of secondary structure and hydrogen-bonding arrangement on the mechanical properties of peptidic-polyurea hybrids. J. Mater. Chem. B 2, 2554–2561 (2014).
Aggeli, A. et al. Hierarchical self-assembly of chiral rod-like molecules as a model for peptide β-sheet tapes, ribbons, fibrils, and fibers. Proc. Natl Acad. Sci. USA 98, 11857–11862 (2001).
Levin, A. et al. Biomimetic peptide self-assembly for functional materials. Nat. Rev. Chem. 4, 615–634 (2020).
Tang, M. et al. Towards a supertough thermoplastic polyisoprene elastomer based on a biomimic strategy. Angew. Chem. Int. Ed. 57, 15836–15840 (2018).
Sato, K., Hendricks, M. P., Palmer, L. C. & Stupp, S. I. Peptide supramolecular materials for therapeutics. Chem. Soc. Rev. 47, 7539–7551 (2018).
Marty, R. et al. Hierarchically structured microfibers of ‘Single Stack’ perylene bisimide and quaterthiophene nanowires. ACS Nano 7, 8498–8508 (2013).
Hafner, R. J. et al. Unusually long-lived photocharges in helical organic semiconductor nanostructures. ACS Nano 12, 9116–9125 (2018).
Diegelmann, S. R., Gorham, J. M. & Tovar, J. D. One-dimensional optoelectronic nanostructures derived from the aqueous self-assembly of π-conjugated oligopeptides. J. Am. Chem. Soc. 130, 13840–13841 (2008).
Scavuzzo, J. J. et al. Supramolecular elastomers. Particulate β-sheet nanocrystal-reinforced synthetic elastic networks. Macromolecules 49, 2688–2697 (2016).
Hirschberg, J. H. K. K. et al. Supramolecular polymers from linear telechelic siloxanes with quadruple-hydrogen-bonded units. Macromolecules 32, 2696–2705 (1999).
Creton, C. et al. Microstructure and self-assembly of supramolecular polymers center-functionalized with strong stickers. Macromolecules 48, 8232–8239 (2015).
Colombani, O. et al. Attempt toward 1D cross-linked thermoplastic elastomers: structure and mechanical properties of a new system. Macromolecules 38, 1752–1759 (2005).
Porter, R. S. & Johnson, J. F. The entanglement concept in polymer systems. Chem. Rev. 66, 1–27 (1966).
van Beek, D. J. M., Gillissen, M. A. J., van As, B. A. C., Palmans, A. R. A. & Sijbesma, R. P. Supramolecular copolyesters with tunable properties. Macromolecules 40, 6340–6348 (2007).
Yamauchi, K., Kanomata, A., Inoue, T. & Long, T. E. Thermoreversible polyesters consisting of multiple hydrogen bonding (MHB). Macromolecules 37, 3519–3522 (2004).
Yarger, J. L., Cherry, B. R. & van der Vaart, A. Uncovering the structure–function relationship in spider silk. Nat. Rev. Mater. 3, 1–11 (2018).
Ling, S., Kaplan, D. L. & Buehler, M. J. Nanofibrils in nature and materials engineering. Nat. Rev. Mater. 3, 1–15 (2018).
Malkin, A. Y. A., Arinstein, A. & Kulichikhin, V. G. Polymer extension flows and instabilities. Prog. Polym. Sci. 39, 959–978 (2014).
Bartnikowski, M., Dargaville, T. R., Ivanovski, S. & Hutmacher, D. W. Degradation mechanisms of polycaprolactone in the context of chemistry, geometry and environment. Prog. Polym. Sci. 96, 1–20 (2019).
Agostinho, B., Silvestre, A. J. D., Coutinho, J. A. P. & Sousa, A. F. Synthetic (bio)degradable polymers—when does recycling fail? Green. Chem. 25, 13–31 (2023).
Rosenboom, J.-G., Langer, R. & Traverso, G. Bioplastics for a circular economy. Nat. Rev. Mater. 7, 117–137 (2022).
Gimenez, J., Cassagnau, P. & Michel, A. Bulk polymerization of ε-caprolactone: Rheological predictive laws. J. Rheol. 44, 527–547 (2000).
Wilkinson, A., Hill, P. W., Farrar, J. F., Jones, D. L. & Bardgett, R. D. Rapid microbial uptake and mineralization of amino acids and peptides along a grassland productivity gradient. Soil Biol. Biochem. 72, 75–83 (2014).
Jahnke, E., Lieberwirth, I., Severin, N., Rabe, J. P. & Frauenrath, H. Topochemical polymerization in supramolecular polymers of oligopeptide-functionalized diacetylenes. Angew. Chem. Int. Ed. 45, 5383–5386 (2006).
Tian, L. et al. Development of a robust supramolecular method to prepare well-defined nanofibrils from conjugated molecules. Chem. Sci. 3, 1512–1521 (2012).
Matsen, M. W. & Thompson, R. B. Equilibrium behavior of symmetric ABA triblock copolymer melts. J. Chem. Phys. 111, 7139–7146 (1999).
Watanabe, H. Slow dielectric relaxation of a styrene-isoprene-styrene triblock copolymer with dipole inversion in the middle block: a challenge to a loop/bridge problem. Macromolecules 28, 5006–5011 (1995).
Meijer, E. W., Wisse, E., Govaert, L. E. & Meijer, H. E. H. Unusual tuning of mechanical properties of thermoplastic elastomers using supramolecular fillers. Macromolecules 39, 7425–7432 (2006).
Ippel, B. D., Van Haaften, E. E., Bouten, C. V. C. & Dankers, P. Y. W. Impact of additives on mechanical properties of supramolecular electrospun scaffolds. ACS Appl. Polym. Mater. 2, 3742–3748 (2020).
Ming, Y., Zhou, Z., Hao, T. & Nie, Y. Molecular simulation of polymer crystallization under chain and space confinement. Phys. Chem. Chem. Phys. 23, 17382–17391 (2021).
Wang, Y. & Wang, S.-Q. Salient features in uniaxial extension of polymer melts and solutions: progressive loss of entanglements, yielding, non-gaussian stretching, and rupture. Macromolecules 44, 5427–5435 (2011).
Liu, G. & Wang, S.-Q. Entangled linear polymer solutions at high shear: from strain softening to hardening. Macromolecules 49, 9647–9654 (2016).
Rubinstein, M. & Panyukov, S. Elasticity of polymer networks. Macromolecules 35, 6670–6686 (2002).
Roos, A. & Creton, C. Effect of the presence of diblock copolymer on the nonlinear elastic and viscoelastic properties of elastomeric triblock copolymers. Macromolecules 38, 7807–7818 (2005).
Stukalin, E. B., Cai, L.-H., Kumar, N. A., Leibler, L. & Rubinstein, M. Self-healing of unentangled polymer networks with reversible bonds. Macromolecules 46, 7525–7541 (2013).
Pakula, T., Saijo, K., Kawai, H. & Hashimoto, T. Deformation behavior of styrene-butadiene-styrene triblock copolymer with cylindrical morphology. Macromolecules 18, 1294–1302 (1985).
Hamley, I. W. Structure and flow behaviour of block copolymers. J. Phys. Condens. Matter 13, R643 (2001).
Heck, B., Hugel, T., Iijima, M., Sadiku, E. & Strobl, G. Steps in the transition of an entangled polymer melt to the partially crystalline state. N. J. Phys. 1, 17 (1999).
Pham, X.-T., Thibault, F. & Lim, L.-T. Modeling and simulation of stretch blow molding of polyethylene terephthalate. Polym. Eng. Sci. 44, 1460–1472 (2004).
Hu, H. & Dorset, D. L. Crystal structure of poly(ε-caprolactone). Macromolecules 23, 4604–4607 (1990).
Kimata, S. et al. Molecular basis of the shish-kebab morphology in polymer crystallization. Science 316, 1014–1017 (2007).
Bauer, B. et al. Melt-spun, cross-section modified polycaprolactone fibers for use in tendon and ligament. Tissue Eng. Fibers 10, 23 (2022).
Selli, F., Erdoğan, U. H., Hufenus, R. & Perret, E. Mesophase in melt-spun poly(ϵ-caprolactone) filaments: Structure–mechanical property relationship. Polymer 206, 122870 (2020).
Haque, F. M. et al. Defining the macromolecules of tomorrow through synergistic sustainable polymer research. Chem. Rev. 122, 6322–6373 (2022).
Acknowledgements
The authors would like to thank Mitsubishi Chemical Corporation for financial support and Mettler Toledo for the generous provision of the equipment for thermal characterization. The authors further thank Dr. Shawn Koppenhoefer for video recording and photography. The authors acknowledge the Paul Scherrer Institute, Villigen, Switzerland, for the provision of synchrotron beamtime at the beamline cSAXS of the SLS. D.G. acknowledges support from the EPFL Fellows program co-funded by Marie Skłodowska-Curie Horizon 2020 Grant no. 291771; Y.H. acknowledges support from the EPFL Innovators program co-funded by Marie Skłodowska-Curie Horizon 2020 Grant no. 754354. S.T. and M.W. acknowledge funding by the SNSF BRIDGE program under the grant agreement no. 40B2-0_211501.
Author information
Authors and Affiliations
Contributions
H.F. conceived the original idea, developed the research concept, and designed the experiments with D.G. and S.H. S.H. and D.G. synthesized the materials. S.H. performed the elongational viscosity measurements. D.G. was responsible for DSC, optical microscopy, and rheology. A.S. carried out the film blowing. C.J.G.P. performed the AFM measurements and contributed to the development of the phase diagrams. Y.H. carried out the X-ray and thermoforming experiments and investigated the melt-stretched films. T.T. and M.L. carried out the synchrotron measurements. N.C. helped with the analysis of the X-ray data. O.O., S.T., M.W., and G.S. contributed to the structural and thermomechanical characterization of our materials. D.G., C.J.G.P., and H.F. analyzed the data and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
While working on this project, S.H. was employed by Mitsubishi Chemical Corporation, which also partly funded the research project. Additionally, H.F., D.G., C.J.G.P., O.O., and S.H. are inventors on the two related patent applications WO2020229594 and WO2019096365.
Peer review
Peer review information
Nature Communications thanks Shijun Li, Kazunori Sugiyasu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Görl, D., Haraguchi, S., Hryshunin, Y. et al. Supramolecular modification of sustainable high-molar-mass polymers for improved processing and performance. Nat Commun 16, 217 (2025). https://doi.org/10.1038/s41467-024-55166-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55166-1
This article is cited by
-
Dynamics of hydrogen-bonded end groups in bulk polymers revealed by solid-state NMR spectroscopy relaxation dispersion experiments
Communications Chemistry (2025)
-
Microcellular Foaming-derived Superlight Ultra-high Molecular Weight Poly(vinylidene fluoride) Foams for Outstanding Thermal Insulation Applications
Chinese Journal of Polymer Science (2025)
