
Abstract
Global food security depends heavily on a few staple crops, while orphan crops, despite being less studied, offer the potential benefits of environmental adaptation and enhanced nutritional traits, especially in a changing climate. Major crops have benefited from genomics-based breeding, initially using single genomes and later pangenomes. Recent advances in DNA sequencing have enabled pangenome construction for several orphan crops, offering a more comprehensive understanding of genetic diversity. Orphan crop research has now entered the pangenomics era and applying these pangenomes with advanced selection methods and genome editing technologies can transform these neglected species into crops of broader agricultural significance.
Similar content being viewed by others
Introduction
As of 2023, it was estimated that more than 780 million individuals, representing at least 10.5% of the global population, face conditions of hunger1. Currently three major crop species; rice, maize and wheat provide over half of the calories consumed globally2. While major staple crops have been extensively bred for high yields and specific adaptive traits, this intensive selection has led to reduced genetic diversity, potentially limiting their adaptability to rapidly changing climate conditions3. With an increasing population and the predicted impact of climate change on crop production, particularly in regions that already suffer food insecurity4, the prevalence of hunger is expected to rise. This poses significant challenges in achieving the United National Zero Hunger Goal by 20305. Orphan crops, also known as underutilised, neglected, minor, indigenous or niche crops, are typically cultivated in specific, often limited, geographic regions under low-input, resource-scarce conditions6. Unlike major staple crops, orphan crops receive minimal attention in terms of research, breeding, and commercial investment. Despite this, orphan crops often have significant agronomic advantages and hold substantial economic importance for smallholder farmers, demonstrating resilience to diverse environmental conditions and possessing high photosynthetic efficiency7. Additionally, orphan crops provide valuable nutritional benefits, playing an essential role in food systems by supporting local diets and meeting regional food demands. For example, pearl millet and cowpea are known for their heat and drought tolerance8,9, lupin for its high protein content10, chickpea as an affordable protein source for low-income countries11 and foxtail millet for its rich fibre and mineral content12. Thus, accelerating the breeding of orphan crops is a promising avenue to improve equitable food and nutrition access globally.
Recent advances have led to the generation of high-quality reference genome assemblies and the identification of functional genes and adaptive alleles in orphan crops13. More than 80 genome assemblies are now available for orphan crops across various families, including species from Fabaceae, Poaceae, Amaranthaceae, Euphorbiaceae and Dioscoreaceae. Population-wide genomic resequencing studies, though not yet widespread among orphan crops, have been conducted in some species such as pearl millet14, buckwheat15, lablab16 and lupin17. These efforts have enabled the dissection of evolutionary histories, the analysis of population structure, and the identification of causal genes associated with agronomic traits, together accelerating genomics-based breeding of orphan crops. However, a single reference genome assembly is inadequate in representing the genetic diversity of a species, limiting its effectiveness in genomics based breeding18. To address this reference bias, pangenomes have been developed that more accurately represent the genomic diversity of a species. By applying pangenomics, researchers can uncover the extensive, and often unexplored, genetic diversity within orphan crops and their wild relatives, paving the way for the accelerated application of genomics for crop improvement19,20,21.
In this Review, we summarise the three major approaches for pangenome construction and their advantages and limitations for crop improvement. We discuss the genomic advances in orphan crop breeding, including genomic selection and genome editing, while recognising the limitations of using a single reference genome. We highlight the advantages of using pangenomes as references and summarise the key findings from recent pangenomic studies in orphan crops. Lastly, we discuss how the incorporation of multi-omics datasets, machine learning and genome-editing approaches can support pangenomics-driven molecular breeding. The integration of these approaches offers the potential to reposition historically underutilised crops with untapped potential to crops of core agricultural importance, facilitating the production of novel crop varieties for the sustainable intensification and diversification of global food production.
Pangenomics principles, construction and applications for orphan crop improvement
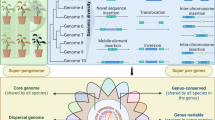
A pangenome comprises core genes present in all individuals, dispensable genes that are only found in some individuals and private genes that unique to a single individual. Many pangenomes focus on the gene content of a species, however with the increased availability of high-quality genome assemblies and graph pangenome methods, pangenomes increasingly capture a wider range of structural variations including chromosomal rearrangements (inversions and translocations)22. Pangenome studies have highlighted the impact of domestication and breeding on genome content, with a reduction in genome size and gene number per individual often observed in domesticated species compared to their wild relatives23,24. The types of genes lost during domestication and breeding include genes associated with disease resistance and abiotic stress, that may impose a yield penalty in modern agricultural systems where these factors are controlled or not present25. Knowledge of orphan crop pangenome content together with an understanding of which genes are lost during domestication of related species can help guide the improvement of these orphan crops while maintaining valuable disease resistance and stress tolerance traits26.
Advances in sequencing technologies have given rise to three primary methods for pangenome construction (Fig. 1 and Table 1): de novo assembly and comparison; reference genome-based iterative assembly; and graph-based pangenome construction22. The de novo assembly method entails constructing genome assemblies from selected representative accessions and comparing them to delineate shared and variable genomic regions. The advent of long-read sequencing technologies from companies including Pacific Biosciences and Oxford Nanopore Technologies have significantly enhanced the quality and completeness of plant genome assemblies, enabling a comprehensive analysis of structural variations between individuals27. HiFi sequencing is capable of generating high-quality orphan crop reference genome assemblies because it combines long read lengths with exceptional accuracy and relatively low cost. The accurate long reads (10–25 kb) can span complex and repetitive regions of the genome, enabling the generation of more complete and contiguous assemblies. Additionally, with the introduction of the latest SPRQ chemistry, HiFi sequencing can achieve higher throughput with lower costs. This affordability can benefit laboratories in lower-income countries by making orphan crop genome sequencing more accessible. However, the costs remain prohibitive for large scale population studies. For some orphan crop species with repetitive, heterozygous and polyploid genomes, such as quinoa and sweet potato, accurate whole genome alignment is challenging, and focusing on gene-centric comparisons can simplify genome complexity.

a de novo assembly and comparison; (b), reference genome-based iterative assembly; and (c), graph-based pangenome construction.
The iterative assembly approach for pangenome construction starts with a “map first, then assemble” strategy by aligning reads to a selected reference genome, then assembling unaligned reads into non-reference contigs that are then integrated into a linear pangenome reference 28. This is a very cost-effective approach for population scale analysis by enabling the accurate identification of genetic makers such as SNPs, small insertions/deletions and gene PAV, because as little as 10x Illumina short read sequence coverage is sufficient for each individual. Relatively cheap short read data can also be aligned to pangenome graphs to understand which paths through the graph represent the genomic structure of the individual sequenced. However, this method is limited by the challenge of accurately placing the newly assembled contigs within the genome. An alternative “assemble first, then map” strategy involves the de novo assembly of individual genomes followed by constructing a linear pangenome by mapping these assemblies to a reference genome29.
Recently, the graph-based pangenome approach has emerged as an alternative to linear-format plant pangenomes30. A graph-based pangenome is an advanced representation of genomic information that captures the genetic diversity within a species by using graph structures instead of traditional linear sequences, allowing for visualisation of structural variations such as inversions and duplications31. Graph-based methods enable more accurate representation of genomic variation, enhancing read alignment and capturing missing heritability in trait association studies, but are limited by the complexity involved in efficient graph pangenome construction and interpretation of graph-based structures32. An optimised approach may include a combination of graph pangenome construction from a representative subset of individuals, followed by iterative mapping of short reads from a large population to capture rare genes and assess genomic variation across the broader population, providing both large scale and structurally detailed information for a species.
Current state of genomic studies in orphan crops
Orphan crops are often central to local diets and agricultural practices, particularly in regions of Africa, Asia, and Latin America33. Many of these crops, such as finger millet (Eleusine coracana), tef (Eragrostis tef) and quinoa (Chenopodium quinoa), have a rich history of cultivation and traditional use. Despite their potential to enhance equitable food access and resilience in agricultural systems, genomic studies on these crops have been relatively sparse. However, advances in sequencing technologies, particularly reducing costs, have started to narrow this gap, enhancing our understanding of orphan crop genetic diversity and potential for improvement.
Early orphan crop genome sequencing efforts targeted crops such as sorghum34, pigeonpea35, and chickpea11, followed by a polyploid genome assembly for quinoa36. In the past few years, telomere-to-telomere (T2T) reference genomes for cassava37, foxtail millet12, and sorghum38 have been generated and applied to understand the genomic basis of adaptive and agronomic traits. These genome assemblies serve as foundational references for genomic improvement. For example, the genome sequence of the chickpea variety CDC Frontier identified more than 28,269 genes, including candidate genes for important agronomic traits such as disease resistance and deep rooting-based drought tolerance11. Varshney et al. further sequenced 90 chickpea accessions and provided insight into the molecular evolution of the two main market types of chickpeas, desi and kabuli, by identifying genomic regions that have undergone selective sweeps or are under balancing selection11. This analysis helped identify candidate genes that could be harnessed for enhancing disease resistance and regulating seed size. Similarly, the reference genome sequence of pigeonpea facilitated the identification of gene families associated with drought tolerance, supporting the development of improved varieties that enhance equitable food access in semi-arid tropical regions35.
As more than 90 percent of crop research funding is focused on major crops39, there is potential for cross-species comparisons, leveraging existing resources and knowledge of major crops to improve agronomic traits in orphan crops through the identification of homologous genes. For example, the sorghum orthologue of the maize domestication gene TB1 regulates tillering in both sorghum40 and pearl millet41, demonstrating the conserved nature of genetic mechanisms across different species that can be exploited for orphan crop improvement. The buckwheat orthologue FdMYB44 of the Arabidopsis gene AtMYB44 plays an important role in regulating flavonoid accumulation42. Furthermore, rice orthologues of the shattering gene SH1 and the grain size controlling gene GW5 retain their function in foxtail millet43. Conversely, insights into recently identified genes associated with nutritional content or stress tolerance in orphan crops offer potential for improving these traits in major crops. For example, key genes associated with heat and drought tolerance, such as Purple acid phosphatases PAP18 44 and Heat shock proteins HSP708 and HSP9045, have been characterised in pearl millet. Such genes represent novel genetic resources that can be utilised to bolster climate-resilience in staple crops, an approach that may be essential in maintaining productivity in uncertain future climate scenarios. Through genomic dissection of stress tolerance and comparative genomics, Islam et al. transferred the pearl millet glutathione peroxidase (PgGPX) gene into rice using transgenic approaches46, enhancing salt and drought tolerance. However, it is important to note that current comparative genomics studies of orphan crops have predominantly been performed at the single species and single reference level (Table 1).
Crop wild relatives and landraces are recognised for harbouring genes associated with agronomic traits, including abiotic and biotic stress tolerance, that have been reduced or lost in modern crops through domestication and selective breeding. Genome sequence analysis of these crop wild relatives supports the identification of these lost genes and enables their assessment for being brought back into modern germplasm. For example, genome sequencing of 994 pearl millet lines distinguished four major genetic clusters, confirming the West African origin of the species and identifying genomic regions showing reduced diversity in cultivated germplasm14. While population-scale resequencing provides insights into the genetic profiles of domesticated and agronomic traits, most studies have relied on a single reference genome and focused on single nucleotide polymorphism (SNP) markers (Table 1), overlooking variation that is present in non-reference genome regions, including structural variants, that could contribute to additional phenotypic variability, thereby underestimating heritability47. This limited genetic perspective not only reduces the resolution of trait association studies but also biases comparative genomic studies, potentially leading researchers to mistakenly infer the presence or absence of genes in a species based solely on their occurrence in the single reference genome.
Genomic selection is an efficient approach for predicting complex traits and informing breeding decisions that involves the use of genome-wide markers to estimate the genetic potential of individuals within a breeding population without requiring phenotypic data for every individual. Genomic selection has been widely applied in major crops such as maize48 and wheat49, as well as orphan crops including pearl millet14, common bean50, pigeonpea51 and cassava52. For example, Varshney et al. performed a comprehensive resequencing of 994 pearl millet lines, significantly improving the accuracy of genomic selection for grain yield prediction13. Their strategy incorporated both additive and dominance genetic effects to predict hybrid performance, identifying 170 high-potential hybrid combinations. Among these combinations, 11 have already been successfully used to develop high-yielding varieties, with the remaining 159 offering promising candidates for future hybrid development. However, single reference genome based genomic selection still faces several limitations, including genetic marker representation bias, the overlooking of structural variants, and imputation errors due to missing genotype information. Therefore, it is important to develop pangenomes to enable comprehensive marker identification and enhance the precision of genomic selection.
CRISPR/Cas9 technology has transformed our ability to validate gene function and introduce desirable traits in orphan crops. Orphan crops are often more stress-tolerant compared to major crops; however, traits related to yield and grain quality in these crops may require improvement. Gene editing technology has facilitated the refinement of orphan crops, through the targeted modification of selected genes7. For example, Lemmon et al. used CRISPR-Cas9 to modify domestication genes associated with plant architecture, flower production, and fruit size in groundcherry, demonstrating the potential of domesticating and redesigning orphan crops53. Similarly, the application of CRISPR/Cas9 genome editing successfully targeted the SEMIDWARF-1 (SD-1) gene orthologue in tef, resulting in the development of semidwarf tef plants that exhibit enhanced resistance to lodging54. This technology has also been applied to modify traits including photosynthesis in broomcorn millet55, seed shattering in green foxtail56, and disease resistance in cassava57. Moreover, genes identified in orphan crops can be targeted in major crops. For instance, by identifying the gene responsible for the biosynthesis of the allelochemical 2,4-dihydroxy-7-methoxy-1,4-benzoxazin-3-one (DIMBOA) in the genome of cockspur grass (Echinochloa crus-galli)58 and applying gene editing technology, DIMBOA-resistant rice cultivars were developed59, reducing the need for herbicide application. However, a lack of comprehensive genomic information for many orphan crops underscores the need for pangenomes that can facilitate the identification of gene targets for editing60.
Progress and findings of pangenome studies in orphan crops
Advances in DNA sequencing technologies and analysis methods have accelerated pangenome studies in orphan crops (Supplementary Data 1). Recent studies include foxtail millet43, pearl millet8, broomcorn millet61, pigeonpea62, chickpea63,64, mung bean65, common bean66, sorghum67,68, lupin69,70 and cassava71. Due to the cost of generating de novo genome assemblies and issues with genome assembly quality, early pangenome studies on orphan crops relied on the reference genome-based iterative assembly approach. A study encompassing 3366 chickpea accessions captured genetic diversity and characterised the population structure across cultivated chickpea and its wild progenitors64, revealing insights into species divergence and migration, and identifying superior haplotypes and novel alleles linked to important yield related agronomic traits, such as plant height, seed weight, days to maturity and yield. Similarly, a pangenome study based on the mung bean reference genome of variety Vrad_JL7 together with 354 accessions identified PAVs under selection that are important for the regulation of flowering65. The PAV-based GWAS in this study identified a 136 kb insertion on chromosome 4 associated with colour traits, with its presence correlating with purple coloration in buds, flowers, young stems, and petioles, and its absence resulting in green or yellow hues in these tissues. Cortinovis et al.66 constructed the first common bean pangenome by assembling four high-quality genomes and resequencing 339 accessions. Their findings indicate that partial or complete gene loss was a key adaptive change, with selection signatures linked to wild differentiation and domestication, contributing to a smaller pangenome in the Andean population compared to the Mesoamerican gene pool. Hufnagel et al.69 characterised PAVs within a white lupin pangenome, uncovering genetic variation between landraces and wild species. Ethiopian landraces exhibited greater genetic differentiation compared to other landraces and possessed a greater number of newly identified genes. However, Ethiopian landraces also lacked a number of genes related to abiotic stress tolerance that are present in the reference genome, including nine tandemly duplicated homologues of AtFRO2 on chromosome 17 that potentially contribute to adaptation to the iron-rich soils of the Ethiopian highlands.
Recent advances in graph-based pangenomes have enhanced the detection and characterisation of haplotypes across populations of chickpea63, foxtail millet12, pearl millet8, broomcorn61 and sorghum68. The construction of the Cicer graph-based pangenome revealed genetic variation and superior haplotypes affecting flowering time, vernalisation and disease resistance, with associated molecular markers that can be applied in chickpea breeding63. Graph-based pangenome analysis of foxtail millet facilitated the identification of a yield-associated gene, and defined a 366 bp PAV in its promoter region that suppresses the expression of SiGW3, enhancing grain weight in domesticated foxtail millet varieties43. A graph-based pangenome for pearl millet provided a comprehensive map of SVs, supporting trait association in this species. SVs between heat-resistant and heat-sensitive materials lead to differential expression of 11 endoplasmic reticulum (ER)-related genes, with additional SVs in other ER-related genes associated with heat stress adaptation8. Comparative genomic analysis of 11 pearl millet genomes and ten other genomes including rice, barley, sorghum and Arabidopsis revealed a significant expansion of the RWP-RK domain transcription factor family in pearl millet. Functional analysis further validated that the RWP-RK transcription factor PMF0G00024.1 positively regulates heat tolerance by transactivating two ER-related genes: one encoding an immunoglobulin protein (PMA2G00107.1) and the other encoding an oligosaccharyl transferase complex (PMA4G03758.1).
Pangenomes surpass single-reference genomes for orphan crop studies
Pangenomics outperforms traditional single-reference genomics by offering more comprehensive insights into genetic diversity (Fig. 2 and Supplementary Data 2). Single reference SNP-based analyses generally capture single-nucleotide variation across the population, providing insights into fine-scale genetic differentiation. In contrast, pangenomics PAV-based analyses, which focus on the presence or absence of gene or genomic sequences, can reveal broader structural variation patterns and population-specific adaptations. For example, studies in green millet56, mung bean65 and white lupin69 indicate that differences in population structure and clustering patterns associated with adaptation process are observed between SNP- and PAV-based phylogenetic analyses. Pangenomes enable cross-genus analyses and the translation of knowledge for crop improvement. For example, the comparative analysis of six legume species pangenomes, including the orphan crops chickpea, pigeonpea, and groundnut, identified conserved and expanded gene families associated with nodulation as well as unique gene families that contribute to oil biosynthesis in groundnut and soybean72. Pangenomes are also required for efficient genome editing as knowledge of gene PAV and copy number is required prior to modification. Capturing intraspecific or interspecific diversity through a pangenome is also important for gene and genome editing. It allows for the design of cultivar-specific sgRNAs and facilitates the translation of characterised mutagenesis sites from major crops to orphan crops73. Moreover, pangenomes facilitate more efficient and precise genomic selection by allowing accurate haplotype construction and supplying an expanded set of SNP and SV markers74. These additional markers enrich the training dataset and enhance the power of statistical models in genomic prediction. Pangenome based genomic selection has been successfully applied in sorghum and chickpea. Jensen et al.75 constructed a sorghum pangenome haplotype graph utilising 24 founder lines. They employed this graph to impute over 2700 training data samples with sequence coverage as low as 0.01×, resulting in a relatively low genotype error rate (3–5.9%). Moreover, using haplotype-based pangenomic selection, the accuracy of chickpea trait prediction significantly increased64. These studies demonstrate that a pangenome-based genomic selection strategy can reduce sequencing and genotyping costs, as well as making genomic selection more feasible and accurate for application in larger breeding populations.

Since a pangenome can capture the complete genetic diversity of a species and identify different types of genomic variations, it offers multiple advantages in genomic analyses and molecular breeding. a The pangenome serves as a powerful framework for multi-omics studies, facilitating more comprehensive analyses of differential gene expression, regulatory networks, post-translational modifications and protein interactions through transcriptomic, proteomic, metabolomics and epigenomic approaches. (b) With additional SNPs and SVs markers, a pangenome can provides further insights into population-wide variation and the selection patterns acting on different populations, such as crop wild relatives and domesticated species. c Pangenome-wide association studies utilising PAVs and SVs uncovers missing heritability, efficiently pinpointing functional genes associated with complex traits. d Pangenomes facilitate more efficient and precise genomic selection by enabling accurate haplotype construction and providing an expanded set of SNP and SV markers, thereby enhancing the dissection of complex traits and informing breeding decisions. e Pangenome-wide cross-genus comparison among different species allows the translation of knowledge between major crops and orphan crops. f New genetic variations and genes captured by pangenomes can be used as novel target sites for gene/genome editing, increasing the efficiency of the editing process of targeted trait.
The application of pangenomes for molecular breeding in orphan crops
Diverse germplasm resources stored in genebanks have long been recognized as foundational material for plant breeding. Traditionally, genebanks have been instrumental in safeguarding genetic resources. However, recent advances have enabled the integration of pangenome assemblies, genetic markers, and phenotypic profiles, providing new opportunities to enhance germplasm management and utilization in orphan crop breeding76,77. For example, genotyping of each accession can provide accurate molecular passport data to classify genebank germplasm, avoiding duplicates that occur when two seed lots originating from the same original seed lot are maintained as different accessions. In addition, genebank resources can support the mining of superior alleles and accelerate pre-breeding decisions and processes78, addressing the challenges of orphan crops, such as the limited genetic knowledge available for practical breeding. A major benefit of pangenome assemblies across diverse accessions, combined with large-scale genotyping of tens of thousands of individuals available in the genebank resources, is the ability to make informed selections of promising genotypes for pre-breeding. This process benefits from detailed phenotypic characterization of accessions with fixed genotypes, enabling the incorporation of superior alleles into modern varieties. Such targeted genetic improvements are especially valuable for orphan crops, extending beyond yield enhancement to improve resistance to pests and diseases and adaptability to changing climates79, that align with global food production trends.
Besides, by leveraging the genetic diversity information in a pangenome with multi-omics data, it is possible identify genes and alleles associated with desirable traits in major and orphan crops. Pangenomics based transcript analysis can provide population-wide insights into gene expression patterns, alternative splicing and the regulatory mechanisms of gene expression across multiple accessions with different conditions, tissues, or developmental stages. These patterns may be overlooked in transcriptome studies that use a single reference individual. This approach has been applied to explore the role of alternative splicing in rice cold tolerance, leading to the identification of the rice catalase OsCATC gene conferring enhanced cold tolerance80. The recently developed VG RPVG bioinformatics pipeline supports the combination of genomic variation data and transcript annotation to construct a spliced graph-based pangenome and directly quantify haplotype-specific transcript expression81. In addition, pangenomes can also serve as a reference for investigating panepigenomic data. This approach has been utilised in Arabidopsis82 and maize83 to reveal functional DNA methylation, non-coding elements and regulatory networks involved in plant growth and development. Panepigenomic studies are valuable as they provide insights into how gene expression is regulated across different varieties, uncovering epigenetic modifications that contribute to stress responses, phenotypic plasticity, and developmental processes84. The growth of direct methylation sequencing using the latest Oxford Nanopore Technologies and Pacific Bioscience technology will lead to the increased availability of this data. Pangenome-wide proteomic studies are also important, shedding light on post-translational modifications and protein interactions that drive key physiological traits85. While these pan-omics approaches have not yet been extensively utilised in the study of orphan crops, they show significant potential to deepen our comprehension of the molecular mechanisms influencing the adaptation and performance of these crops.
The application of artificial intelligence can enhance the accuracy and sensitivity of SV detection and genotyping across pangenomes. The availability of hundreds of haplotype-resolved pangenome assemblies and more than a thousand resequenced accessions, such as those generated in foxtail millet12, can serve as a standard for establishing ‘ground truth’ genotype sets that are required for training and fine-tuning machine learning SV detection models. Convolutional neural networks (CNNs) are adept at recognising SV breakpoint patterns from short-read sequencing data, and the integration of CNNs into bioinformatics pipelines, such as SVision86 and DeepSV87, enhances the accuracy and sensitivity of SV detection and genotyping across population-level resequencing datasets. Comprehensive pan-SV maps constructed through this approach have increased the resolution of molecular markers, providing cultivar-specific or additional targeted sites for genome editing, and enhanced the fine mapping of genes. By integrating genebank resources, multi-omics data, machine learning and gene/genome editing, pangenomics-based molecular breeding can accelerate the improvement of orphan crops and help to develop new crop varieties with enhanced traits.
Challenges and opportunities in orphan crop pangenomics
Compared to major crop species, there are several aspects that need to be considered when constructing and applying pangenomes for orphan crops. The lack of prior knowledge on crop diversity can make selecting individuals for pangenome construction challenging, and it may be cost effective to perform an initial diversity study to assess how best to select diverse individuals. This study could be relatively simple and separate from pangenome analysis using low-cost technology such as simple sequence repeats on crude DNA extractions or a subset of SNPs. More advanced approaches may make use of SNP arrays from related species. An alternative approach would be to combine the diversity study with the pangenome analysis by generating low coverage (10-20x) short read sequence data for a large number of individuals. This data could be used to directly assess diversity using kmer based methods such as MASH88 or mapped to a reference to call SNPs. While the cost of generating this data is relatively high, it can be reused in pangenome construction using the iterative mapping and/or graph-based pangenome approaches, providing detailed population-wide information on both SNPs and gene PAV. Due to its superior ability to represent genomic information and capture genetic diversity, the graph-based approach is emerging as the standard for pangenome studies.
Currently, the vg toolkit89 is the most widely used approach for constructing genetic variation graphs by integrating genetic variants stored in a VCF format file, generated through whole-genome comparisons, into a reference genome. Minigraph90 or Minigraph-Cactus91 is also an efficient tool for mapping sequences from multiple genomes to generate a reference-based pangenome. PGGB92 has emerged as a novel approach to overcome reference bias by allowing any genome in the dataset to serve as the reference. However, this tool is resource-intensive and time-consuming for large orphan crop genomes, often requiring multiple computational trials to optimize parameters. In addition, visualization tools such as Bandage93 and ODGI94 enable the display of nodes and SVs in a graph layout. Nevertheless, the majority of genome analysis tools are currently tailored for linear sequence formats, and significant challenges persist in the application of graph-based pangenomes due to the lack of appropriate tools. Variations stored in graph-based formats are not directly compatible with widely used tools such as VCFtools95 and so require linearisation of the graph prior to analysis. This highlights the need for the development of scalable software and data structures designed for graph-based pangenome analysis.
Despite advances in pangenomics, it has only recently been applied in crop breeding. Many initial developments were focused on genetic perspectives, leading to a usability gap for breeders. To address this, breeding data should be integrated into a pangenome framework alongside genetic knowledge, supported by visualization and analysis tools tailored to meet breeding applications. Breeders for major crops currently use genome references for trait association, haplotype dissection and selection, with pangenome references gradually replacing the single reference genomes. Orphan crops that lack single reference genome assemblies may circumvent the use of single reference genomes and proceed directly to use pangenome resources for trait dissection and the identification of favourable haplotypes for selection. The relatively low economic value of some orphan crops makes it a challenge both to secure suitable funds to even construct a pangenome as well as develop the capacity to exploit the constructed pangenome for crop improvement. In these cases, there may be more cost-effective ways to improve crop productivity using more traditional breeding and/or investment in agronomic practices. There is also a balance that needs to be made between establishing local genomics capability in these low- and middle-income countries where orphan crops are predominantly grown, and collaborating with established laboratories in developed countries that have advanced resources and expertise. While charitable foundations and international organizations play a role in funding research on orphan crops, the disparities in purchasing power can make outsourcing pangenome construction to commercial providers economically prohibitive. Building local capacity is important for sustainable progress and empowerment of local scientists. Particular challenge for applying genomic selection in orphan crops is that they often suffer from limited data and a lack of people with crop improvement experience compared to applications in major crops. One solution is to advocate for a global cooperation that involves contributing data resources, and expertise across various levels and disciplines, thereby overcoming resource limitations and promoting equitable advancements in orphan crop improvement.
There appears to be a promising path for the translation of knowledge from major crops to orphan crops, applying technology and knowledge that was developed through significant long term investment in major crops6. This application of proven technology greatly reduces the cost hurdle of establishing pangenomic approaches for the improvement of orphan crops. One aspect that is rarely addressed is how the knowledge from orphan crops can be reciprocally applied for the improvement of major crops. Plant research to date has focused on a small number of models and crop species, and there remains much to be learnt from studying a wider range of crop species, particularly when these have been relatively recently domesticated or possess novel or interesting phenotypes such as enhanced disease resistance or environmental adaptation. Given the importance of these crops, there is a sound economic reason to improve our understanding of orphan crops for the enhancement of both major and orphan crop species through pangenome supported breeding programs.
References
FAO, I. & UNICEF. WFP and WHO. 2019. The state of food security and nutrition in the world. FAO. (2021).
Shiferaw, B. et al. Crops that feed the world 10. Past successes and future challenges to the role played by wheat in global food security. Food security 5, 291–317 (2013).
Cullis, C. & Kunert, K. J. Unlocking the potential of orphan legumes. J. Exp. Bot. 68, 1895–1903 (2017).
Battisti, D. S. & Naylor, R. L. Historical warnings of future food insecurity with unprecedented seasonal heat. Science 323, 240–244 (2009).
Burlingame, B. & Dernini, S. Biodiversity And Sustainable Diets United Against Hunger 3–5 November 2010 FAO Headquarters, Rome. (2012).
Ye, C.-Y. & Fan, L. Orphan crops and their wild relatives in the genomic era. Mol. plant 14, 27–39 (2021).
Yaqoob, H. et al. Integrating genomics and genome editing for orphan crop improvement: a bridge between orphan crops and modern agriculture system. GM Crops Food 14, 1–20 (2023).
Yan, H. et al. Pangenomic analysis identifies structural variation associated with heat tolerance in pearl millet. Nat. Genet. 55, 507–518 (2023).
Liang, Q. et al. A view of the pan‐genome of domesticated cowpea (Vigna unguiculata [L].Walp Plant Genome 17, e20319 (2024).
Abraham, E. M. et al. The use of lupin as a source of protein in animal feeding: Genomic tools and breeding approaches. Int. J. Mol. Sci. 20, 851 (2019).
Varshney, R. K. et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246 (2013).
He, Q. et al. A complete reference genome assembly for foxtail millet and Setaria-db, a comprehensive database for Setaria. Mol. Plant 17, 219–222 (2024).
Chapman, M. A., He, Y. & Zhou, M. Beyond a reference genome: pangenomes and population genomics of underutilized and orphan crops for future food and nutrition security. N. Phytol. 234, 1583–1597 (2022).
Varshney, R. K. et al. Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nat. Biotechnol. 35, 969–976 (2017).
Zhang, K. et al. Resequencing of global Tartary buckwheat accessions reveals multiple domestication events and key loci associated with agronomic traits. Genome Biol. 22, 23 (2021).
Njaci, I. et al. Chromosome-level genome assembly and population genomic resource to accelerate orphan crop lablab breeding. Nat. Commun. 14, 1915 (2023).
Wang, P. et al. Whole-genome assembly and resequencing reveal genomic imprint and key genes of rapid domestication in narrow-leafed lupin. Plant J. 105, 1192–1210 (2021).
Bayer, P. E., Golicz, A. A., Scheben, A., Batley, J. & Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 6, 914–920 (2020).
Khan, A. W. et al. Super-pangenome by integrating the wild side of a species for accelerated crop improvement. Trends Plant Sci. 25, 148–158 (2020).
Shorinola, O. et al. Integrative and inclusive genomics to promote the use of underutilised crops. Nat. Commun. 15, 320 (2024).
Li, W. et al. Plant pan-genomics: recent advances, new challenges, and roads ahead. J. Genet Genomics 49, 833–846 (2022).
Hu, H., Li, R., Zhao, J., Batley, J. & Edwards, D. Technological Development and Advances for Constructing and Analyzing Plant Pangenomes. Genome Biol. Evol. 16, evae081 (2024).
Bayer, P. E. et al. Sequencing the USDA core soybean collection reveals gene loss during domestication and breeding. Plant Genome 15, e20109 (2022).
Petereit, J. et al. Pangenomics and Crop Genome Adaptation in a Changing Climate. Plants (Basel) 11, 1949 (2022).
Derbyshire, M. C., Newman, T. E., Thomas, W. J., Batley, J. & Edwards, D. The complex relationship between disease resistance and yield in crops. Plant Biotechnol. J. 22, 2612–2623 (2024).
Hu, H. et al. Plant pangenomics, current practice and future direction. Agri. Commun. 2, 100039 (2024).
Amarasinghe, S. L. et al. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 21, 30 (2020).
Golicz, A. A. et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 7, 13390 (2016).
Wang, J. et al. A pangenome analysis pipeline provides insights into functional gene identification in rice. Genome Biol. 24, 19 (2023).
Edwards, D. & Batley, J. Graph pangenomes find missing heritability. Nat. Genet. 54, 919–920 (2022).
Eizenga, J. M. et al. Pangenome Graphs. Annu Rev. Genomics Hum. Genet 21, 139–162 (2020).
Zhou, Y. et al. Graph pangenome captures missing heritability and empowers tomato breeding. Nature 606, 527–534 (2022).
Kumar, B., Singh, A. K., Bahuguna, R. N., Pareek, A. & Singla‐Pareek, S. L. Orphan crops: a genetic treasure trove for hunting stress tolerance genes. Food Energy Security 12, e436 (2023).
Paterson, A. H. et al. The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556 (2009).
Varshney, R. K. et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 30, 83–89 (2011).
Jarvis, D. E. et al. The genome of Chenopodium quinoa. Nature 542, 307–312 (2017).
Xu, X. D. et al. Telomere-to-telomere assembly of cassava genome reveals the evolution of cassava and divergence of allelic expression. Hortic. Res 10, uhad200 (2023).
Liu, F. et al. Unravelling sorghum functional genomics and molecular breeding: past achievements and future prospects. J. Genet. Genomics. https://doi.org/10.1016/j.jgg.2024.07.016 (2024).
Alston, J. M. & Pardey, P. G. Public funding for research into specialty crops. HortScience 43, 1461–1470 (2008).
Mace, E. S. & Jordan, D. R. Location of major effect genes in sorghum (Sorghum bicolor (L.). Moench. Theor. Appl. Genet. 121, 1339–1356 (2010).
Remigereau, M. S. et al. Cereal domestication and evolution of branching: evidence for soft selection in the Tb1 orthologue of pearl millet (Pennisetum glaucum [L.] R. Br.). PLoS One 6, e22404 (2011).
He, M. et al. Comparison of buckwheat genomes reveals the genetic basis of metabolomic divergence and ecotype differentiation. N. Phytol. 235, 1927–1943 (2022).
He, Q. et al. A graph-based genome and pan-genome variation of the model plant Setaria. Nat. Genet 55, 1232–1242 (2023).
Reddy, C. S., Kim, K. M., James, D., Varakumar, P. & Reddy, M. K. PgPAP18, a heat-inducible novel purple acid phosphatase 18-like gene (PgPAP18-like) from Pennisetum glaucum, may play a crucial role in environmental stress adaptation. Acta physiologiae Plant. 39, 1–10 (2017).
Reddy, P. S. et al. Molecular characterization and expression of a gene encoding cytosolic Hsp90 from Pennisetum glaucum and its role in abiotic stress adaptation. Gene 474, 29–38 (2011).
Islam, T., Manna, M. & Reddy, M. K. Glutathione peroxidase of Pennisetum glaucum (PgGPx) is a functional Cd2+ dependent peroxiredoxin that enhances tolerance against salinity and drought stress. PLoS One 10, e0143344 (2015).
Hu, H. et al. Unravelling inversions: Technological advances, challenges, and potential impact on crop breeding. Plant Biotechnol. J. 22, 544–554 (2024).
Zhao, Y. et al. Accuracy of genomic selection in European maize elite breeding populations. Theor. Appl. Genet 124, 769–776 (2012).
Huang, M. et al. Genomic selection for wheat traits and trait stability. Theor. Appl. Genet. 129, 1697–1710 (2016).
Keller, B. et al. Genomic Prediction of Agronomic Traits in Common Bean (Phaseolus vulgaris L.) Under Environmental Stress. Front Plant Sci. 11, 1001 (2020).
Bohra, A., Saxena, K. B., Varshney, R. K. & Saxena, R. K. Genomics-assisted breeding for pigeonpea improvement. Theor. Appl. Genet 133, 1721–1737 (2020).
Wolfe, M. D. et al. Prospects for Genomic Selection in Cassava Breeding. Plant Genome 10, https://doi.org/10.3835/plantgenome2017.03.0015 (2017).
Lemmon, Z. H. et al. Rapid improvement of domestication traits in an orphan crop by genome editing. Nat. Plants 4, 766–770 (2018).
Plaza-Wüthrich, S. & Tadele, Z. Regeneration and transformation studies on tef. Tef Improvement. 67, 15–18 (2013).
Liu, Y. et al. Establishment of genome‐editing system and assembly of a near‐complete genome in broomcorn millet. J. Integr. Plant Biol. 66, 1688–1702 (2024).
Mamidi, S. et al. A genome resource for green millet Setaria viridis enables discovery of agronomically valuable loci. Nat. Biotechnol. 38, 1203–1210 (2020).
Lin, Z. J. D., Taylor, N. J. & Bart, R. Engineering Disease-Resistant Cassava. Cold Spring Harb. Perspect. Biol. 11, a034595 (2019).
Guo, L. et al. Echinochloa crus-galli genome analysis provides insight into its adaptation and invasiveness as a weed. Nat. Commun. 8, 1031 (2017).
Zhang, H. et al. Glutathione S-transferase activity facilitates rice tolerance to the barnyard grass root exudate DIMBOA. BMC Plant Biol. 24, 117 (2024).
Tay Fernandez, C. G. et al. Pangenomes as a resource to accelerate breeding of under-utilised crop species. Int. J. Mol. Sci. 23, 2671 (2022).
Chen, J. et al. Pangenome analysis reveals genomic variations associated with domestication traits in broomcorn millet. Nat. Genet 55, 2243–2254 (2023).
Zhao, J. et al. Trait associations in the pangenome of pigeon pea (Cajanus cajan). Plant Biotechnol. J. 18, 1946–1954 (2020).
Khan, A. W. et al. Cicer super-pangenome provides insights into species evolution and agronomic trait loci for crop improvement in chickpea. Nat. Genet. 56, 1225–1234 (2024).
Varshney, R. K. et al. A chickpea genetic variation map based on the sequencing of 3,366 genomes. Nature 599, 622–627 (2021).
Liu, C. et al. High-quality genome assembly and pan-genome studies facilitate genetic discovery in mung bean and its improvement. Plant Commun. 3, 100352 (2022).
Cortinovis, G. et al. Adaptive gene loss in the common bean pan-genome during range expansion and domestication. Nat. Commun. 15, 6698 (2024).
Ruperao, P. et al. Sorghum Pan-Genome Explores the Functional Utility for Genomic-Assisted Breeding to Accelerate the Genetic Gain. Front Plant Sci. 12, 666342 (2021).
Tao, Y. et al. Extensive variation within the pan-genome of cultivated and wild sorghum. Nat. Plants 7, 766–773 (2021).
Hufnagel, B. et al. Pangenome of white lupin provides insights into the diversity of the species. Plant Biotechnol. J. 19, 2532–2543 (2021).
Garg, G. et al. A pan-genome and chromosome-length reference genome of narrow-leafed lupin (Lupinus angustifolius) reveals genomic diversity and insights into key industry and biological traits. Plant J. 111, 1252–1266 (2022).
Xia, Z. et al. Pan-genome and Haplotype Map of Cultivars and Their Wild Ancestors Provides Insights into Adaptive Evolution of Cassava (Manihot esculenta Crantz). bioRxiv. 2023.07. 02.546475 (2023).
Garg, V. et al. Chromosome-length genome assemblies of six legume species provide insights into genome organization, evolution, and agronomic traits for crop improvement. J. Adv. Res. 42, 315–329 (2022).
Scheben, A., Wolter, F., Batley, J., Puchta, H. & Edwards, D. Towards CRISPR/Cas crops–bringing together genomics and genome editing. N. Phytologist 216, 682–698 (2017).
Hurgobin, B. & Edwards, D. SNP discovery using a pangenome: has the single reference approach become obsolete? Biology 6, 21 (2017).
Jensen, S. E. et al. A sorghum practical haplotype graph facilitates genome-wide imputation and cost-effective genomic prediction. Plant Genome 13, e20009 (2020).
Mascher, M., Jayakodi, M., Shim, H. & Stein, N. Promises and challenges of crop translational genomics. Nature https://doi.org/10.1038/s41586-024-07713-5 (2024).
Mascher, M. et al. Genebank genomics bridges the gap between the conservation of crop diversity and plant breeding. Nat. Genet 51, 1076–1081 (2019).
Bellucci, E. et al. The INCREASE project: Intelligent Collections of food-legume genetic resources for European agrofood systems. Plant J. 108, 646–660 (2021).
Jamnadass, R. et al. Enhancing African orphan crops with genomics. Nat. Genet. 52, 356–360 (2020).
Zhong, Y. et al. Pan-transcriptomic analysis reveals alternative splicing control of cold tolerance in rice. Plant Cell 36, 2117–2139 (2024).
Sibbesen, J. A. et al. Haplotype-aware pantranscriptome analyses using spliced pangenome graphs. Nat. Methods 20, 239–247 (2023).
Kawakatsu, T. et al. Epigenomic Diversity in a Global Collection of Arabidopsis thaliana Accessions. Cell 166, 492–505 (2016).
Dong, X. et al. Dynamic and Antagonistic Allele-Specific Epigenetic Modifications Controlling the Expression of Imprinted Genes in Maize Endosperm. Mol. Plant 10, 442–455 (2017).
Huang, Y., Liu, Y., Liu, C., Birchler, J. A. & Han, F. Prospects and challenges of epigenomics in crop improvement. Genes Genomics 44, 251–257 (2022).
Wang, P. et al. A large-scale proteogenomic atlas of pear. Mol. Plant 16, 599–615 (2023).
Lin, J. et al. SVision: a deep learning approach to resolve complex structural variants. Nat. Methods 19, 1230–1233 (2022).
Cai, L., Wu, Y. & Gao, J. DeepSV: accurate calling of genomic deletions from high-throughput sequencing data using deep convolutional neural network. BMC Bioinforma. 20, 665 (2019).
Ondov, B. D. et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 132 (2016).
Garrison, E. et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 36, 875–879 (2018).
Li, H., Feng, X. & Chu, C. The design and construction of reference pangenome graphs with minigraph. Genome Biol. 21, 265 (2020).
Hickey, G. et al. Pangenome graph construction from genome alignments with Minigraph-Cactus. Nat. Biotechnol. 42, 663–673 (2024).
Garrison, E. et al. Building pangenome graphs. Nat. Methods 21, 2008–2012 (2024).
Wick, R. R., Schultz, M. B., Zobel, J. & Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352 (2015).
Guarracino, A., Heumos, S., Nahnsen, S., Prins, P. & Garrison, E. ODGI: understanding pangenome graphs. Bioinformatics 38, 3319–3326 (2022).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (32400512). The GuangDong Basic and Applied Basic Research Foundation (2024A1515011981). The “YouGu” Plan and “Outstanding youth Researcher” of Rice Research Institute of Guangdong Academy of Agricultural Sciences (2023YG04&2024YG01), the Introduction of Young Key Talents of Guangdong Academy of Agricultural Sciences (R2023YJ-QC001) and Guangdong Key Laboratory of Rice Science and Technology (2023B1212060042). We gratefully acknowledge funding from the Australia Research Council (Projects LP230100351, DP210100296 and DP200100762).
Author information
Authors and Affiliations
Contributions
H.H. and D.E. wrote the manuscript. H.H., J.Z., W.J.W.T., J.B., and D.E. contributed to the editing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Linkai Huang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, H., Zhao, J., Thomas, W.J.W. et al. The role of pangenomics in orphan crop improvement. Nat Commun 16, 118 (2025). https://doi.org/10.1038/s41467-024-55260-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55260-4
This article is cited by
-
From the genome to super-pangenome: a new paradigm for accelerated crop improvement
npj Science of Plants (2026)
-
Revitalizing orphan crops to combat food insecurity
Nature Communications (2025)
-
Wheat genomics frontiers for gene discovery and breeding applications
WheatOmics (2025)
-
Harnessing genomics for sustainable food systems with orphan crops
Discover Agriculture (2025)
-
Integrating multi-omics and machine learning for disease resistance prediction in legumes
Theoretical and Applied Genetics (2025)
