
Abstract
Iridium has the exclusive chemistry guaranteeing both high catalytic activity and sufficient corrosion resistance in a strong acidic environment under anodic potential. Complex iridates thus attract considerable attention as high-activity electrocatalysts with less iridium utilization for the oxygen evolution reaction (OER) in water electrolyzers using a proton-exchange membrane. Here we demonstrate the effect of chemical doping on the durability of hexagonal-perovskite Bax(M,Ir)yOz-type iridates in strong acid (pH ~ 0). Some aliovalent cations are directly visualized to periodically locate at the octahedral sites bridging the two face-sharing [Ir2O9] dimer or [Ir3O12] trimers in hexagonal-perovskite polytypes. In particular, highly ionic bonding of the d0 Nb5+ and Ta5+ cations with oxygen anions results in notable suppression of lattice oxygen participation during the OER and thus effectively preserves the connectivity between the [Ir3O12] trimers without lattice collapse. Providing an in-depth understanding of the correlation between the electronic structure and bonding nature in crystals, our work suggests that proper control of chemical doping in complex oxides promises a simple but efficient tool to realize OER electrocatalysts with markedly improved durability.
Similar content being viewed by others


Introduction
Water electrolysis by renewable electricity is a key pathway for hydrogen production without serious carbon emissions1. As the conventional alkaline electrolyzers utilize a large volumetric electrolyte solution and a thick diaphragm, it is fairly difficult to achieve >2 A/cm2 nominal current density and >70% voltage efficiency2,3,4. To overcome these intrinsic confinements in alkaline water electrolysis, many research endeavors have been focused on electrolyzers based on a polymer-type proton exchange membrane (PEM). Although PEM electrolysis offers many advantages in terms of compact size and efficiency of the device system, the highly acidic and corrosive characteristics of the sulfonated fluoropolymer membrane prevent the simple use of most first-row transition metals as electrocatalysts to facilitate the oxygen evolution reaction (OER) at the anode side5. Even though many noteworthy compositional combinations have been proposed for OER electrocatalysts with high stability6,7,8,9,10,11,12,13,14,15,16,17,18,19,20, Ir appears to be an indispensable element offering both high catalytic activity and substantial longevity under strong acidic conditions21,22,23,24,25,26,27,28,29,30,31,32,33,34,35.
Ir-based complex oxides thus have recently garnered significant attention, as they can provide notably high catalytic activity along with much less Ir utilization per unit catalyst mass36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54. In particular, the basic framework of AIrO3-type (A = Ca, Sr, and Ba) hexagonal-perovskite iridates consists of face-shared [IrO6] octahedra, in contrast to the corner-sharing configuration in cubic-perovskite iridates. Providing the highest degree of connectivity among the [IrO6] octahedra, the face-sharing structural basis in hexagonal perovskites strongly correlates with catalyst durability43. It is noted that this face-sharing connectivity has an exceptional advantage against lattice instability induced by lattice oxygen oxidation55,56,57,58 during the OER. Because Ir is highly resistive to electrochemical dissolution5, one of the major origins of the structural instability59,60,61 in many Ir-based oxides stems from the lattice oxygen oxidation and subsequent disassembly of the connected [IrOx] clusters22,29,62,63,64. Unless three oxygen anions between two adjacent face-shared octahedra in hexagonal perovskites are simultaneously oxidized as an O2 gas phase, the face-shared [IrO6] octahedral framework is very difficult to collapse. In this respect, hexagonal-perovskite iridates are exclusive candidates offering remarkable structural stability under a significantly acidic environment.
Using hexagonal-perovskite BaIrO3 as a starting material in this work, we systematically examine the effect of (post)transition-metal (Mn, Co, Ni, In, Nb, and Ta) solid solutions on the lattice stability in 1 M HClO4 (pH ≈ 0.1). One of the notable features at an atomic scale in these solid solutions is that the added transition metals induce structural transformation into other polytypes and subsequently occupy specific positions in a very ordered manner when they are aliovalent with Ir4+. In particular, it was identified that Nb and Ta ordering enhances structural durability during the OER, while the addition of Mn, Co, Ni, and In distinctively showed a detrimental effect. A combination of atomic-column-resolved scanning transmission electron microscopy (STEM), time-of-flight secondary ion mass spectroscopy (TOF-SIMS)65, and X-ray absorption spectroscopy (XAS) demonstrated that Nb5+ and Ta5+ play important roles in substantially suppressing the lattice oxygen oxidation, thereby preserving the strong connectivity among the [IrO6] octahedra. Density functional theory (DFT) calculations also consistently support that highly ionic-bonding characteristics between the Nb5+ 4d0 states and the O 2p states make a critical contribution to inhibiting lattice oxygen participation during the OER. Providing direct evidence for peculiar atomic-scale ordering in the solid solutions, our study suggests that control of chemical ordering and subsequent bonding nature by doping with d0 transition-metal cations will be an efficient approach to suppress the lattice oxygen oxidation for notable durability in many iridate-based high-activity catalysts.
Results
Structure transformation and chemical ordering
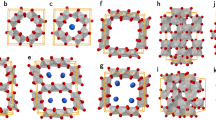
Figure 1 schematically illustrates the crystal structures of the hexagonal perovskites observed in this work. First, BaIrO3 has a 9R polytype in the space group of monoclinic C2/m. This consists of trimers of robust face-sharing Ir–O octahedra along the c axis and each trimer is connected by corner-sharing oxygens (gray spheres), as denoted in Fig. 1a. The 9R structure does not vary with Mn4+ addition, indicating a random distribution of Mn at the Ir sites. In contrast, as will be clarified below, structural transformation and chemical ordering at an atomic level are readily observed when aliovalent transition-metal cations are added in 9R BaIrO3. The 6H polytype with P63/mmc commonly appears in the cases of Co2+, Ni2+, and In3+ doping. It is noted that each dopant exclusively occupies the bridging site between two dimers of face-sharing octahedra in 6H Ba3(M’Ir2)O9, (M’ = Co, Ni, and In), as indicated by different colors in Fig. 1b. Ordered occupancy of pentavalent Nb5+ and Ta5+ is also identified at the bridging position to connect the face-sharing [Ir3O12] trimers in 12R-type Ba4(M”Ir3)O12, (M” = Nb and Ta) with the space group of R\(\bar{3}\)m (Fig. 1c). Three distinct types of octahedral connectivity (corner-, edge-, and face-sharing configurations) are illustrated in Supplementary Fig. 1.

Three polytypes, a 9R, b 6H, and c 12R, are presented. As indicated by black arrows in the illustrations in the left-hand column, each [IrO6] octahedron is face-sharing in the [Ir2O9] dimers of the 6H polytype and in the [Ir3O12] trimer in the 9R and 12R polytypes. Co, Ni, In, Nb, and Ta occupy the specific sites bridging the two adjacent dimers or trimers, whereas Mn distributes randomly in the lattice.
All the powder samples in this work were synthesized by a solid-state reaction (see scanning electron microscopy (SEM) images in Supplementary Fig. 2 for the size and morphology of each powder). Before direct visualization of atomic-scale chemical ordering, we carried out powder X-ray diffraction (XRD) for a macroscopic phase analysis. Figure 2a–c shows a series of XRD patterns representing the three hexagonal-perovskite polytypes, 9R, 6H, and 12R, together with their Bragg peak simulations. The whole series of XRD results are provided in Supplementary Fig. 3. As easily recognized in Fig. 2a, XRD data acquired from pristine BaIrO3 and Ba2(MnIr)O6 present a nearly identical pattern. This indicates that the 9R polytype structure does not change by adding a substantial amount of Mn. Consequently, Mn and Ir appear to be distributed in Ba2(MnIr)O6 in a random manner (see Supplementary Fig. 4 for details).

a Experimentally obtained XRD patterns are presented for BaIrO3 and Ba2(MnIr)O6. b, c In addition to the experimental patterns, XRD simulations are provided to support chemical ordering in the 6H and 12R polytypes. d–f Atomic-scale HAADF images are demonstrated for all seven iridates, showing the Z-contrast features. Yellow circles in each image denote the Ir columns. White arrows indicate the ordered configurations by dopants.
In contrast, notable ordering was found in the 6H- and 12R-type solid solutions, as exemplified in Fig. 2b, c. Because a periodic configuration of foreign dopants constructs a superlattice in the crystal, either new additional Bragg reflections or intensity inversion of existing peaks should be observed in the diffraction data. Indeed, a comparison of simulated XRD patterns between the ordered and disordered states in Fig. 2b, c directly indicates the appearance of a new reflection (the 1\(\bar{1}\)0 peak) in Ba3CoIr2O9 and enhanced intensity of the 004 peak in Ba3CoIr2O9 and the \(\bar{1}\)11 peak in Ba4NbIr3O12 in the ordered states. Agreeing well with the simulated diffractions, the experimentally obtained XRD results in Fig. 2b, c consistently support the presence of Co and Nb ordering in Ba3CoIr2O9 Ba4NbIr3O12, respectively. The detailed XRD results, diffraction simulations, and a further discussion of all the solid solutions containing Co, Ni, In, Nb, and Ta are provided in Supplementary Figs. 5–7. The valence oxidation state of Ir should change to satisfy the overall charge neutrality condition when aliovalent dopants are added into BaIrO3. To examine the oxidation state of Ir in Co2+, In3+, and Nb5+-doped samples, we carried out X-ray absorption near edge structure66 analyses to compare the positions of the white lines of the Ir L3 edge. As shown in Supplementary Fig. 8, each of the Ir oxidation states agrees well with the value expected from the charge neutrality condition, without additional vacancy formation.
Atomic-column-resolved STEM was utilized to directly verify the site-specific chemical ordering, as the image contrast in high-angle annular dark-field (HAADF) mode is sensitively proportional to the atomic number (Z)67,68,69,70,71,72,73,74,75,76,77,78. While Ir has the highest atomic number, ZIr = 77, among the compositions of the lattice, most of the transition-metal dopants in our study have a much lower atomic number (ZMn = 25; ZCo = 27; ZNi = 28; ZIn = 49; ZNb = 41). Consequently, if low-Z dopants selectively occupy some specific sites, their low intensity will be readily detected. Figure 2d–f presents atomic-scale HAADF–STEM images of all the solid solutions. As denoted by three yellow circles in the HAADF image for pristine BaIrO3 in Fig. 2d, the Ir trimers are clearly observed as the brightest atomic columns, in good agreement with the image simulations shown in Supplementary Fig. 9. In contrast, such a distinct column contrast of the trimers disappears in the image of Ba2(MnIr)O6 (Fig. 2d). As low-Z Mn is substituted for Ir, a remarkable reduction of the column intensity for the Ir trimers is easily identifiable, providing consistent support for the random distribution of Mn.
We captured distinct column contrast of the low-Z dopants in the 6H- and 12R-polytype solid solutions during the STEM analysis. As indicated by arrows in Fig. 2e, f, significantly low column intensity is observable specifically at the bridging sites in Ba3CoIr2O9, Ba3NiIr2O9, Ba3InIr2O9, and Ba4NbIr3O12, where Z values of the five dopants are much lower than ZIr = 77, in good accordance with the image simulations (see Supplementary Fig. 9). This series of STEM images thus directly verifies that the bridging positions are occupied by the aliovalent dopants, Co2+, Ni2+, In3+, and Nb5+, in the 6H and 12R polytypes. We note that the atomic number of Ta is very similar to that of Ir (ZTa = 73; ZIr = 77). Therefore, it is nearly impossible to distinguish whether the bridging sites are occupied by Ta or Ir in Ba4TaIr3O12. To overcome this ambiguity, we performed atomic-level energy-dispersive X-ray spectroscopy (EDS)73,79,80. As shown in Supplementary Fig. 10, Ta atoms were identified to locate at the bridging sites, demonstrating a high degree of Ta ordering as well.
Dissolution behavior of solid solutions
Electrochemical anodic cycling tests of the pristine and solid-solution iridates were carried out to compare their catalytic properties. Figure 3 shows a series of cycling measurements in 1 M HClO4 (pH ≈ 0.1) in a range of 1.10–1.63 V vs. a reversible hydrogen electrode (RHE) with a cycling scan rate of 10 mV/s (see Supplementary Fig. 11 for the plots of the OER current densities vs. potentials). Two discriminated features are easily identified from this set of measurements. First, the addition of Mn, Co, Ni, and In rather seriously deteriorates the OER activity with cycling, whereas pristine BaIrO3 shows strong retention of its initial activity even after 800 anodic cycles. In stark contrast, the Nb- and Ta-added 12R-type iridates preserve high OER current densities without significant degradation. We note that Ba4NbIr3O12 in particular presents remarkable long-term stability of high catalytic activity among all the iridates including pristine BaIrO3.

All the measurements were carried out in 1 M HClO4 in a range of 1.10–1.63 V vs. RHE with a scan rate of 10 mV/s and 0.5 mg/cm2 mass loading. All the current-density values were read at 1.53 V vs. RHE. While the samples doped with Mn, Co, Ni, and In show a substantial decrease of the OER current with cycling, no degradation of activity is observed in the Nb- and Ta-doped 12R-polytype samples.
As shown in Supplementary Fig. 12, the Pourbaix diagrams of Mn, Co, Ni, and In indicate that they are highly corrosive in acidic conditions (pH < 3), and thereby their dissolution into an electrolyte is difficult to avoid. We examined the dissolution behavior of cycled samples by using EDS in STEM. Figure 4a, b provides two sets of EDS maps of the seven iridates before and after 200 anodic cycles in 1 M HClO4 for comparison. As expected, substantial dissolution of Mn, Co, Ni, and In is directly visualized in the series of composition maps in Fig. 4b. Because alkaline-earth elements, including Ba, dissolve into electrolytes36,39,40,43,47,48 (see Supplementary Fig. 13), Ba dissolution is simultaneously observable in the maps as well. Additional EDS maps for these four doped iridates and pristine BaIrO3 are provided in Supplementary Figs. 14–18 for verification.

a, b EDS maps for the seven iridates particles are compared before and after 200 anodic cycles. No dissolution of Nb and Ta is identified, whereas Mn, Co, Ni, and In significantly dissolve, as expected from their Pourbaix diagrams. c Even after 800 cycles, Nb is observed to be stable without detectable dissolution. d As demonstrated in the EDS maps in (c), line-by-line EELS analyses consistently show a deficiency of alkaline-earth Ba in the surface region of Ba4NbIr3O12. This dissolution accompanies amorphization, as shown in the HAADF image.
In contrast, detectable dissolution of Nb and Ta is not observed in the EDS maps in Fig. 4b, consistent with the high stability of their pentoxides in acid, as can be verified in the Pourbaix diagrams in Supplementary Fig. 11. When we scrutinized the surface of Ba4NbIr3O12 and Ba4TaIr3O12 particles after 800 cycles, indeed the absence of Nb deficiency was confirmed by EDS (see the maps in Fig. 4c and Supplementary Figs. 19 and 20), verifying the remarkable corrosion resistance. The surface analysis based on X-ray photoelectron spectroscopy (XPS) also consistently supported the corrosion resistance of Nb and Ta (see Supplementary Fig. 21 for the XPS results). Instead, Ba dissolution and subsequent amorphization in the surface regions of ~20 nm thickness were unavoidable in most Ba4NbIr3O12 particles after long-term anodic cycling, as shown in Fig. 4c, d by STEM imaging, EDS, and electron energy-loss spectroscopy (EELS). In particular, the EELS analysis results in Fig. 4d systematically demonstrate the reduced intensity of the Ba–M4,5 peaks and finally their disappearance (line 8) near the particle surface (see Supplementary Fig. 22 for an additional set of EELS results). As shown in previous reports on iridate catalysts, the alkaline-earth dissolution induces significant surface roughening and thereby results in a considerable increase of the surface area of particles during initial cycling39,40,43 (see Supplementary Figs. 23 and 24 for double-layer (DL) capacitance measurements). Consequently, surface roughening induced by Ba dissolution makes a beneficial contribution to considerably enhancing the apparent OER activity, unless the robust connectivity among the [IrO6] octahedra is affected by the dissolution, as in Ba4NbIr3O12.
Suppression of lattice oxygen oxidation
Even though Ir-based oxides have a high degree of resistance against electrochemical corrosion under anodic conditions, structural instability can be induced by lattice oxygen oxidation62,63,64, which is one of the major origins of degradation in many oxide catalysts, irrespective of whether they are metals, charge-transfer insulators, or Mott–Hubbard insulators81 (see Supplementary Fig. 25 for details). The set of atomic-scale STEM images in Fig. 5a directly shows that the surfaces of a Ba4NbIr3O12 particle are terminated with Ir for the basal (001) plane and with Nb–Ir mix for the other planes. Unless three face-shared lattice oxygens simultaneously evolve as a gas phase, the connection between two neighboring [IrO6] octahedra in the trimer is very difficult to break (see Supplementary Fig. 26 for the illustration). The [IrOx] unit on the (001) surface termination is thus anticipated to be fairly stable against lattice oxygen evolution. In contrast, the bridging [NbO6] units on the other surfaces are corner-shared with neighboring [IrO6] octahedra, as shown in the illustrations in Figs. 1 and 5c. This weak binding is more vulnerable to breaking by lattice oxygen activation during the OER. However, a comparison of the density of states (DOS) in Fig. 5b directly indicates significantly suppressed O 2p states (red arrow) of the corner-shared [NbO6] beneath the Fermi level (EF), whereas the large O 2p states (black arrow) overlap with the Ir 5d states in the face-shared [IrO6].

a A representative morphology of Ba4NbIr3O12 particles is shown in this STEM image. The Ir-termination at the (001) surface and the Ir-Nb mixed termination at the (101) surface are observed in the magnified images. b, c DOS plots are presented for the [IrO6] octahedron in the trimer and the [NbO6] octahedron at the bridging site, as denoted in the illustration in (c). Arrows in the plots indicate the variation of O 2p states near EF. d An isosurface map is presented for the electron density difference field around each atom. No density distribution around Nb is noted. e Schematic energy band diagrams for [IrO6] and [NbO6] are illustrated. The O 2p states at a lower level and the empty Nb 4d states at an upper level are dominant in the [NbO6] case, indicating ionic Nb–O bonding. f, g DOS plots are provided for the [IrO6] [NbO6] units in the oxyhydroxide paracrystalline supercell without Ba, as illustrated in (g).
The electronic structure of Nb5+ consists of fully filled 4p6 and empty 4d0 states. As a consequence, most of the empty Nb 4d orbitals contribute to the upper electronic band above +2 eV, as plotted in the DOS for the [NbO6], while the Ir 5d5 states largely place in the lower band below EF and thereby form covalent bonding with the O 2p states, as shown in Fig. 5b (see Supplementary Fig. 27 for the spin-polarized DOS plots and Supplementary Data 1 for the atomic coordinates of the optimized unit cell). The isosurface contour acquired from the DFT calculation for the electron density distribution near EF at each atom is visualized in Fig. 5d, consistently verifying the absence of visible electron density around Nb. Although Nb5+ is not an OER active site, this cation provides a crucial contribution to suppressing the lattice oxygen association and preventing structural collapse at the bridging sites in addition to providing important corrosion resistance in acid.
The electron energy diagrams in Fig. 5e schematically depict the effect of Nb on suppressing lattice oxygen oxidation. As already shown in the DOS plot in Fig. 5b, the partially filled Ir 5d5 orbitals construct high-degree hybridization with the O 2p orbitals. Furthermore, the large DOS of both Ir 5d5 and O 2p orbitals near EF between −2 and 0 eV consistently support that sufficient hole states82,83,84,85 from Ir and the lattice oxygen in the trimers as active sites can be provided to facilitate the OER under an anodic potential. As the Nb 4d orbitals are empty, highly ionic-bonding characteristics of Nb–O are noted for the [NbO6] units. Therefore, while the upper band (pink) above EF in Fig. 5e is dominated by the empty 4d states of Nb, the lower band (gray) largely consists of a stabilized O 2p band, the center of which thus locates at a lower level than that of the [IrO6] units. In this regard, Nb plays multiple noteworthy roles in both offering high corrosion resistance and simultaneously inhibiting the lattice oxygen evolution for robust bridging sites in 12R-Ba4NbIr3O12.
Ba dissolution inevitably induces the charge imbalance in the lattice. To examine how the charge neutrality was satisfied after Ba leaching, we carried out additional XPS analyses. As shown in Supplementary Fig. 28a, the position of the Ir 4f peaks does not vary with anodic cycling, indicating that the valence state of Ir hardly changes. In contrast, the O 1s peak for OH becomes dominant, while the peak for lattice O diminishes (Supplementary Fig. 28a). This directly demonstrates that the charge neutrality without Ba is achieved through protonation from the electrolyte, resulting in the formation of oxyhydroxide.
We carried out ab initio molecular dynamics (AIMD) simulations to support the robust framework of face-sharing [Ir3O12] trimers in the 12R structure. The simulation was performed by using an oxyhydroxide supercell without Ba at 300 K to emulate the Ba leaching condition (see Supplementary Fig. 28b). As shown in Fig. 5g and Supplementary Fig. 28b, despite substantial perturbations from the original positions of atoms, the overall medium-range-ordered configurations of the [Ir3O12] trimers and Nb are clearly discernible (see Supplementary Data 1 for the initial and final atomic configurations from the AIMD simulations). This result demonstrates that the surface region, after electrochemical Ba leaching at room temperature, is not entirely amorphous but rather paracrystalline with medium- and short-range orders (see Supplementary Movie 1 for a video clip). Figure 5f illustrates a set of DOS plots for the [IrO6] and [NbO6] units in the paracrystalline oxyhydroxide supercell prepared by the AIMD simulation. The relatively lower DOS of O 2p near the Fermi level in Nb–O can be verified, indicating that the role of Nb in suppressing lattice oxygen contribution remains unaffected in the paracrystalline state.
To quantitatively compare the extent of the lattice oxygen contribution between pristine BaIrO3 and Ba4NbIr3O12, we carried out TOF-SIMS65, using samples anodically cycled in a 1 M HClO4 electrolyte prepared with water containing 18O. As this type of SIMS using a tine-of-flight analyzer is a surface-sensitive analytical tool capable of detecting isotopes, the relative concentration of 18O out of the total amount of 18O and 16O at the particle surfaces could be traced with cycling in each sample. Figure 6a presents a bar graph showing the 18O (m = 18) isotope concentrations at the surface of BaIrO3 and Ba4NbIr3O12 (see Supplementary Fig. 29 for the ion mass counts of m = 18). We note that the detection of ~0.5% 18O in both iridates before anodic cycling stems from the naturally present oxygen isotope. This SIMS result directly demonstrates substantially lower 18O concentration, especially after 100 cycles, in Ba4NbIr3O12, agreeing well with the DOS plots in Fig. 5b and the description in Fig. 5e. Much lower mass counts of 18O in Ba4TaIr3O12 were consistently verified as well (see Supplementary Fig. 30).

a TOF-SIMS analyses directly reveal less detection of 18O at the surface of Ba4NbIr3O12 particles. Each error bar represents the range from the maximum to the minimum value of three data points. b A series of O K-edges obtained from XAS are shown. The absorption energy difference (Δ), which is proportional to the ligand field splitting, between the first and second peaks is observed to increase after 600 cycles (Δ1 < Δ2).
As in many other Ba-based complex oxides, the outmost 5p electron states of Ba2+ in Ba4NbIr3O12 are far below the O2− 2p states (see the DOS plot in Supplementary Fig. 31), representing the absence of covalent bonding characteristics of Ba–O86. Therefore, even if Ba dissolves out of the lattice during the OER, the basic framework of [Ir3O12] trimers bridged by [NbO6] would not be perturbed. A series of O K-edges obtained from XAS for anodically cycled samples in Fig. 6b reveals that the first peak (~530 eV), associated with the Ir 5d t2g states87,88, shifts to lower energy, whereas the second peak (~532 eV), hybridized with the eg states87,88, shifts to higher energy. This indicates that the ligand field splitting (Δ) of the Ir 5d t2g and eg levels does not diminish but rather slightly increases with cycling (Δ1 < Δ2), as schematically depicted in Fig. 6b (see Supplementary Fig. 32 for the O K-edges in a wider range of energy). The strong ligand field of six oxygens in each [IrO6] unit thus does not appear to change with cycling, supporting that the [Ir3O12]-trimer-based robust framework is preserved despite the amorphization induced by Ba dissolution in Ba4NbIr3O12.
Comparison of catalytic longevity
Since a report benchmarking many OER catalysts in 201389, chronopotentiometry (CP) has been frequently utilized as a representative measurement technique for evaluating catalyst stability. However, the durability results from this method vary considerably, strongly depending on the particle size of the catalysts. We performed CP tests with two IrO2 specimens having different particle sizes (see the SEM images in Supplementary Fig. 33): a coarse-particle IrO2 specimen purchased from Sigma-Aldrich (a surface area of 1.35 m2/g) and a specimen with a much smaller particle size (11.0 m2/g) prepared by ball-milling the coarse-particle specimen. The CP results in Fig. 7a show that the OER activity of the fine-particle specimen is preserved for a much longer time, even though the two specimens are of identical rutile IrO2. As observed in this plot, if the catalyst surface area is smaller, a higher potential (red arrow) is required to maintain a constant OER current (10 mA/cm2geo) and thereby more rapid degradation should take place. Consequently, a catalyst consisting of fine particles may show substantially better performance in the CP measurement, even if it is intrinsically less stable than the other catalyst. Indeed, it is noteworthy that recent studies, reporting exceptional catalytic longevity based on CP, utilized nanoscale particles (less than 20 nm in size)10,14,16,19,29,30. In this respect, electrochemical tests performed under the same overpotential condition should be offered as more reasonable protocols to evaluate the intrinsic durability of materials in a size-insensitive manner.

Three distinct electrochemical tests based on a CP, b CV, and c CA were carried out in 1 M HClO4 with 0.5 mg/cm2 mass loading to measure the catalytic longevity of rutile IrO2 samples with two different particle sizes. The CP test in (a) shows significant size-dependent variation, revealing that much lower overpotential is required to achieve 10 mA/cm2 for the fine-particle sample and thereby far longer lifetime is attained. In contrast, analogous behavior of the current variation insensitive to the particle size is observed in the CV and CA measurements in (b, c). d, e Three materials, IrO2, 9R-BaIrO3, and 12R-Ba4NbIr3O12, were measured by the CV and CA methods, respectively. Ba4NbIr3O12 demonstrates notable durability.
We provide other test results of the two IrO2 specimens measured by anodic cyclic voltammetry (CV) and chronoamperometry (CA) methods in Fig. 7b and Fig. 7c, respectively. The same overpotential range of 1.10–1.63 V vs. RHE was applied for the CV and a constant overpotential, 1.58 V vs. RHE, was adopted to measure the specific current variation during the CA. A common feature of these two methods is that the degradation behavior of OER activity appears to be nearly identical between the two IrO2 specimens. In particular, when we plotted the normalized values of the specific current, as shown in the third panels in Fig. 7b, c, it is easily recognizable that the degradation proceeds in an almost identical manner in each of the tests, irrespective of the particle size of the specimens. The CV and CA methods, both of which are performed under the same overpotential conditions, are thus believed to be more suitable test protocols than CP if the relative durability among catalyst materials should be demonstrated.
Figure 7d, e presents further anodic CV and CA results measured from three different samples of IrO2 (Sigma-Aldrich), 9R-BaIrO3, and 12R-Ba4NbIr3O12 in 1 M HClO4. The fourth panel in each of the figures shows plots normalized by the initial current value so that the relative longevity of the catalytic activity is easily identifiable. As consistently demonstrated in previous reports, these two sets of CV and CA measurements verify that BaIrO3 based on the face-shared octahedra shows notable durability of the high OER activity comparable with that of rutile IrO2 having the corner- and edge-shared geometry despite Ba dissolution. More importantly, the CA measurements in Fig. 7e provide straightforward evidence indicating that Ba4NbIr3O12 is prominent among the three high-stability catalysts in terms of preserving the OER activity. We note that a combination of the significant suppression of the lattice oxygen oxidation by Nb ordering and the face-shared robust trimers is the key advantage of Ba4NbIr3O12 to attain exceptional durability.
Discussion
The findings in this work offer several implications regarding the durability of oxide catalysts and their test protocols. First, although we focus on the addition of pentavalent Nb5+ in the present study, the addition of other d0 metal cations having highly ionic bonding with oxygen90 is anticipated to be beneficial to suppress the lattice oxygen evolution in the same manner by lowering the O 2p states near EF. Second, the face-sharing covalent [Ir3O12] trimers in 12R-Ba4(M”Ir3)O12 appear to be fairly stable even after Ba dissolution. Although Pauling’s third rule suggests better stability of the corner-sharing configuration in ionic compounds, the highly covalent characteristics of Ir–O in Ba4NbIr3O12 stabilize the face-sharing trimers while fulfilling the third rule for the corner-sharing ionic Nb–O. Finally, our work clarifies that even a small difference in the particle size of catalysts would cause a significant variation in CP results. Although the CP provides useful information regarding the catalyst performance from the viewpoint of practical devices in operation, CV and CA tests under identical overpotentials appear to be alternative protocols to perform a relative comparison of the material durability independent of the particle size.
As demonstrated in a previous study on the phase relations of TiO2–IrO2 and SnO2–IrO291, it is noted that solubility limits of insulating TiO2 and SnO2 into metallic IrO2 are very low (less than 5% at 900 °C), even though the valence state of Ti4+, Sn4+, and Ir4+ is identical and even the crystal structure of TiO2, SnO2, and IrO2 is the same rutile structure. Because the crystal structure of Nb2O5 and the chemical bonding nature of Nb–O completely differ from the crystal structure of IrO2 and the bonding characteristics of Ir–O, the solubility of Nb is thus difficult to exceed 10%. To directly prove this considerably low solubility of Nb in rutile IrO2, we carried out an additional XRD analysis using a powder mixture of 75IrO2–25NbO2.5 annealed at 800 °C for a sufficiently long period of time, 100 h, in air. As directly shown in Supplementary Fig. 34, the diffraction result verifies the appearance of the high-intensity Braggs reflections for the secondary Nb2O5 phase, demonstrating that the solubility of Nb in rutile IrO2 is considerably low (evidently less than 10% based on the diffraction pattern). In this respect, attaining the 25% solubility of Nb and Ta in the 12R-type iridate and directly visualizing its ordered configuration are other significant findings in this work.
In conclusion, we have demonstrated that structural stability and OER catalytic longevity under a strong acidic environment can be significantly enhanced by atomic-scale chemical ordering in hexagonal-perovskite barium iridates. When aliovalent transition metals are added to BaIrO3, the 9R perovskite polytype transforms into either 6H or 12R type. Furthermore, it is directly identified that the added transition metals exclusively occupy specific sites bridging the face-sharing [Ir2O9] dimers in the 6H type and the [Ir3O12] trimers in the 12R type. While most of the transition-metal cations (Mn, Co, Ni, and In) dissolve into an acidic electrolyte, pentavalent Nb5+ and Ta5+ in 12R-Ba4(M”Ir3)O12 show notable corrosion resistance. In addition, their electronic configuration with the empty d orbitals (d0) is found to critically contribute to suppressing lattice oxygen evolution during the OER. As oxygen anion oxidation and subsequent evolution under a large anodic potential are difficult to avoid in most oxides, our study suggests that solid solutions, including atomic-scale ordering, with d0 cations having ionic-bonding characteristics can provide an important route to prevent structural degradation and achieve high-durability oxide catalysts.
Methods
Powder synthesis
All of the seven iridates were synthesized by a conventional solid-state reaction through a mixed oxide technique. BaCO3 (99.999%, Sigma-Aldrich), IrO2 (99.99%, Alfa Aesar), MnO2 (99.996%, Alfa Aesar), Co3O4 (99.5%, Sigma-Aldrich), NiO (99.8%, Sigma-Aldrich), In2O3 (99.9%, Sigma-Aldrich), Nb2O5 (99.99%, Sigma-Aldrich), and Ta2O5 (99.99%, Sigma-Aldrich) were used as starting materials for particle synthesis. Powder mixtures of the starting materials with their own stoichiometry were ball-milled by using a zirconia jar and balls in high-purity ethanol for 24 h. Each of the dried slurries after evaporation of ethanol was calcined in a box-type furnace at 1000 °C for 3 h in air for BaIrO3 and at 1200 °C for 3 h in air for the other six iridates, 6H-Ba3(M’Ir2)O9, (M’ = Co, Ni, In) and 12R-Ba4(M”Ir3)O12, (M” = Nb, Ta). Commercially available rutile-type IrO2 (99.8%, Sigma-Aldrich) was also utilized as a reference material for electrochemical tests.
XRD and Bragg peaks simulations
The phase identification of all the synthesized powders was carried out by X‐ray diffractometry (SmartLab, Rigaku) with Cu‐Kα radiation. To examine the presence of chemical ordering in each of the doped iridates for Co, Ni, In, Nb, and Ta, XRD pattern simulations were performed by using CrystalDiffract (CrystalMaker Software Ltd) along with CrystalMaker (CrystalMaker Software Ltd). Two distinct supercells were used for simulations: one is based on a unit cell containing the dopant exclusively at the specific bridging sites in the 6H and 12R polytypes. The other contains each dopant and Ir evenly at all the cation sites with a stoichiometric ratio. When chemical ordering is present, the appearance of new Bragg reflections and the intensity inversion of existing peaks were identified in the simulated patterns and thus the presence of ordering could be easily recognized.
SEM, STEM, EELS, and EDS
The overall polycrystalline morphologies and particle sizes of synthesized powders were observed in scanning electron microscopes (Magellan 400, Thermo Fisher Scientific; SU8230, Hitachi) at 5–15 kV. Each of the powder samples for STEM observation was ultrasonically dispersed in high-purity ethanol for 15 min and several drops were taken on a lacey-carbon-film Cu or Ni grid and subsequently dried. Atomic-column-resolved STEM images were acquired in a transmission electron microscope (Titan cubed G2 60‐300, Thermo Fisher Scientific) in HAADF and BF modes at 300 kV with spherical aberration correction of the electron probe. The collection semiangles of the STEM detectors were set to 79.5–200 mrad for HAADF imaging and 0–43.3 mrad for BF imaging. An EELS analysis was conducted using a Gatan Image Filter (GIF Quantum 965, Gatan Inc.) within the Titan cubed G2. Electron energy-loss spectra for the O–K and Ba–M4,5 edges were acquired for spectrum imaging with a dispersion of 0.25 eV per channel and a collection aperture of 5 mm diameter. EDS mapping was performed in another transmission electron microscope (Talos F200X, Thermo Fisher Scientific) at 200 kV along with four integrated silicon-drift EDS detectors at a high beam-current rate of >600 pA/cm2, to visualize the overall compositional variations before and after electrochemical tests. Atomic-level EDS was also carried out in the Titan cubed G2 60‐300 at 300 kV at 100 pA/cm2. The Ba-Lα (4.5 keV), Ir-Lα (9.2 keV), Nb-Kα (16.6 keV), Ta-Lα (8.1 keV), Mn-Kα (5.9 keV), Co-Kα (6.9 keV), Ni-Kα (7.5 keV), and In-Lα (3.3 keV) lines were selected for elemental mapping and subsequent quantification. The EDS maps were low-pass filtered using Bruker ESPRIT software to reduce background noise.
STEM image simulations
HAADF–STEM image simulations were carried out by using Dr. Probe software92 based on the multislice algorithm. The specific parameters for simulations include a beam energy of 300 kV and spherical aberration coefficients of Cs = 0 mm, C5 = 0 mm, and C7 = 0 mm without coma or astigmatism. An electron probe size of 1 Å at the full width of half maximum and a slice thickness of 2 Å were set during the simulations. The real collection semiangle of the HAADF–STEM detector (79.5–200 mrad) was also utilized for precise comparison with experimentally obtained images.
SIMS, XPS, and XAS
Samples for TOF-SIMS and XAS were fabricated on PTFE-treated carbon papers (Alfa Aesar). The ink was prepared in the same manner, as will be described below, and drop-casted onto the carbon papers. To track the variation of the extent of lattice oxygen exchange by SIMS, an 18O-containing 1 M HClO4 electrolyte solution was used. The solution was prepared with 0.862 mL of 70% HClO4 (99.999%, Sigma-Aldrich), 4.138 mL of deionized water (18.2 MΩ·cm), and 5 mL of water-18O (97 at% of 18O, Sigma-Aldrich) such that approximately a half of the aqueous solution consists of H218O. The amounts of both 16O and 18O at the particle surface were measured in a secondary ion mass spectrometer with a time-of-flight analyzer (TOF.SIMS 5, IONTOF GmbH) during the count of negative ions generated by Bi+-ion bombardment at 30 kV in 100 × 100 μm2 areas. The 18O exchange concentrations after electrochemical cycling reported in this work were calibrated values, as not all of the oxygen elements in the electrolyte solution are isotope 18O. To verify the corrosion-resistant behavior of Nb and Ta, the composition at the particle surfaces in Ba4NbIr3O12 and Ba4TaIr3O12 was analyzed using an X-ray photoelectron spectroscope (K-Alpha XPS, Thermo Scientific) with monochromatic Al-Kα radiation (1486.7 eV) and flood gun emission of 150 μA. X-ray absorption spectra for the O K-edge were acquired at the 6 A beamline of Pohang Light Source II (PLS-II) at the Pohang Accelerating Laboratory. To confine the XAS analysis to the surface region of particles within <10 nm depth, a soft X-ray source in total electron yield mode was utilized. The measured spectra were normalized by setting the baseline intensity before the absorption edge to zero. X-ray absorption spectra for the Ir L3 edge were also obtained at the 7D and 8C beamlines of PLS-II to examine the position of the white lines of the samples.
Electrochemical measurements
All the electrochemical tests were performed using a potentiostat (SP-300, Biologic) in 1 M HClO4. A 1 M HClO4 solution (pH ~ 0.3) was prepared by mixing milli-Q water (18.2 MΩ·cm) and 70% HClO4 (99.999%, Sigma-Aldrich). A Pt wire and Ag/AgCl (3 M NaCl) were used as the counter electrode and the reference electrode, respectively. After the electrochemical experiments, the measured potentials were converted to the RHE scale at 25 °C according to the following equation: ERHE = Emeasure + 0.059·pH + E°ref, where ERHE is the converted potential vs. RHE, Emeasure is the measured potential with respect to the reference electrode, and E°ref is the standard potential of Ag/AgCl at 25 °C, i.e., 0.21 V. For the electrochemical experiments, the ink was prepared by using catalyst particles, a K+-exchanged 5-wt% Nafion solution (Nafion 117, Sigma-Aldrich) as a binder, and acetylene black (Alfa Aesar) along with high-purity ethyl alcohol. The mass ratios were adjusted to be 10:1 for the catalyst to acetylene black and 3:1 for the catalyst to Nafion. The mixture was stirred and sonicated for 1 h to ensure thorough mixing. The ink was drop-casted onto a gold rotating ring disk electrode (3 mm in diameter, RRDE-3A, ALS) such that the amount of loaded catalysts loaded on the disk electrode was adjusted to be 0.5 mg/cm2. The electrode was then dried at 60 °C in a drying oven for 2 h. The rotation speed of the electrode was set to 2400 rpm and was intermittently raised to 5000 rpm for 5 s every 5 min during CA measurements and every five cycles during CV measurements for the removal of O2 bubbles on catalyst particles.
CV tests were carried out with a scan rate of 10 mV/s in a potential range of 1.1–1.63 V vs. RHE. The DL capacitance was measured in a non-faradaic potential range (0.95–1.20 V vs. RHE) by recording the current values as a function of scan rates every five cycles to the 20th cycle; every 20 cycles to the 100th cycle; and every 50 cycles to the 800th cycle to monitor the change in surface area of catalysts. CP and CA tests were also performed at a current density of 10 mA/cm2geo and at a potential of 1.53 (or 1.58) V vs. RHE, respectively. The DL capacitance was measured in the same range (0.95–1.20 V vs. RHE) every 1 h in both tests. Because the intermittent capacitance measurement was carried out in a non-faradaic potential range, this was beneficial for the diffusion of O2 gas bubbles under the condition of no further OER and thereby their efficient removal from the catalyst surfaces. Indeed, the recovery of a high OER current density was observed after each of the capacitance measurements during the CA tests. Electrochemical impedance spectroscopy was also carried out in the same potentiostat in a frequency range from 0.1 Hz to 1 MHz with an amplitude of 10 mV for iR correction of the uncompensated series resistance (R). All electrochemical results were verified by performing additional measurements.
The DL capacitance was also used to estimate the electrochemically active surface area (ECSA), as demonstrated in a previous study89. The ECSA was acquired from the following equation:
where CDL is the measured DL capacitance of a catalyst material and Cs is the specific capacitance of the sample per unit area under a certain identical electrolyte condition. Following the suggestion of the previous study, we took Cs = 0.035 mF/cm2 as the general specific capacitance in an acidic condition for a reasonable general value. To precisely evaluate the ECSA of active materials, we excluded the ECSA of acetylene black and the Nafion binder from the total ECSA.
DFT calculations and AIMD simulations
Ab initio DFT calculations were performed to acquire the DOS and the isosurface of electron density difference of Ba4NbIr3O12, employing the spin-polarized generalized-gradient approximation (GGA) along with the PBEsol functional optimized for exchange-correlation of densely packed solids. The ultrasoft pseudopotentials were utilized for ionic cores, as implemented in the CASTEP code (Biovia Inc.). Low-spin (t2g5)(eg0) for the Ir4+ 5d5 states and (t2g6)(eg0) for the Ir3+ 5d6 states were assumed, as the intensity of the peak, corresponding to the hybridization with the Ir 5d eg orbitals, in the O K-edge is much higher. The plane-wave basis set for the kinetic energy cutoff was set to 500 eV. Relaxation of the internal coordinates in the unit cell performed using the Broyden–Fletcher–Goldfarb–Shanno algorithm with convergence tolerances of 0.1 eV Å−1 for the maximum ionic force, 5 × 10−5 eV/atom for the total energy, and 0.005 Å for the maximum ionic displacement. AIMD simulations were utilized to construct supercells representing the paracrystalline state without Ba in agreement with the STEM observation. To accelerate the atomic displacement, the dynamic relaxation was conducted at 1200 K in the isothermal–isovolumetric (NVT) canonical ensemble with the Nose thermostat in a time step of 1.0 fs to induce displacement of each atom by thermal agitation. The GGA-PBEsol functional for exchange-correlation and the ultrasoft pseudopotentials for ionic cores were consistently employed in the CASTEP code. The plane-wave basis set for the kinetic energy cutoff was 260 eV with k-space integration of a Γ-point (1 × 1 × 1). Optimal MD steps were carried out at 300 K until the overall lattice energy was stabilized. The cell was subsequently quenched to 10 K, and further DFT calculations for DOS were performed to examine the variation of the electronic states of Ir and O in the paracrystalline supercell.
Data availability
The data that support the findings of this study are shown in the main text figures, the Supplementary Information, and the Supplementary Data. The data generated in this study are provided in the Source Data file. Source data are provided with this paper.
References
Glenk, G. & Reichelstein, S. Economics of converting renewable power to hydrogen. Nat. Energy 4, 216–222 (2019).
Kraglund, M. R. et al. Ion-solvating membranes as a new approach towards high rate alkaline electrolyzers. Energy Environ. Sci. 12, 3313–3318 (2019).
Ehlers, J. C., Feidenhans’l, A. A., Therkildsen, K. T. & Larrazábal, G. O. Affordable green hydrogen from alkaline water electrolysis: key research needs from an industrial perspective. ACS Energy Lett. 8, 1502–1509 (2023).
Feng, Q. et al. A review of proton exchange membrane water electrolysis on degradation mechanisms and mitigation strategies. J. Power Sources 366, 33–55 (2017).
Wang, Z., Guo, X., Montoya, J. & Nørskov, J. K. Predicting aqueous stability of solid with computed Pourbaix diagrams using SCAN functional. NPJ Comput. Mater. 6, 160 (2020).
Frydendal, R., Paoli, E. A., Chorkendorf, I., Rossmeisl, J. & Stephens, I. E. L. Toward an active and stable catalyst for oxygen evolution in acidic media: Ti-stabilized MnO2. Adv. Energy Mater. 5, 1500991 (2015).
Moreno-Hernandez, I. A. et al. Crystalline nickel manganese antimonate as a stable water-oxidation catalyst in aqueous 1.0 M H2SO4. Energy Environ. Sci. 10, 2103–2108 (2017).
Zhou, L. et al. Rutile alloys in the Mn–Sb–O system stabilize Mn3+ to enable oxygen evolution in strong acid. ACS Catal. 8, 10938–10948 (2018).
Wang, Z., Zheng, Y.-R., Chorkendorff, I. & Nørskov, J. K. Acid-stable oxides for oxygen electrocatalysis. ACS Energy Lett. 5, 2905–2908 (2020).
Hao, S. et al. Dopants fixation of ruthenium for boosting acidic oxygen evolution stability and activity. Nat. Commun. 11, 5368 (2020).
Lin, C. et al. In-situ reconstructed Ru atom array on α-MnO2 with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012–1023 (2021).
Huang, J. et al. Modifying redox properties and local bonding of Co3O4 by CeO2 enhances oxygen evolution catalysis in acid. Nat. Commun. 12, 3036 (2021).
Li, A. et al. Enhancing the stability of cobalt spinel oxide towards sustainable oxygen evolution in acid. Nat. Catal. 5, 109–118 (2022).
Wen, Y. et al. Introducing Brønsted acid sites to accelerate the bridging-oxygen-assisted deprotonation in acidic water oxidation. Nat. Commun. 13, 4871 (2022).
Pan, S. et al. Efficient and stable noble-metal-free catalyst for acidic water oxidation. Nat. Commun. 13, 2294 (2022).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2023).
Shi, Z. et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 14, 843 (2023).
Zhang, D. et al. Construction of Zn-doped RuO2 nanowires for efficient and stable water oxidation in acidic media. Nat. Commun. 14, 2517 (2023).
Chong, L. et al. La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science 380, 609–616 (2023).
Liu, H. et al. Eliminating over-oxidation of ruthenium oxides by niobium for highly stable electrocatalytic oxygen evolution in acidic media. Joule 7, 558–573 (2023).
Kim, Y.-T. et al. Balancing activity, stability and conductivity of nanoporous core-shell iridium/iridium oxide oxygen evolution catalysts. Nat. Commun. 8, 1449 (2017).
Geiger, S. et al. The stability number as a metric for electrocatalyst stability benchmarking. Nat. Catal. 1, 508–515 (2018).
Nong, H. N. et al. A unique oxygen ligand environment facilitates water oxidation in hole-doped IrNiOx core-shell electrocatalysts. Nat. Catal. 1, 841–851 (2018).
Gao, J. et al. Breaking long-range order in iridium oxide by alkali ion for efficient water oxidation. J. Am. Chem. Soc. 141, 3014–3023 (2019).
Hao, S. et al. Torsion strained iridium oxide for efficient acidic water oxidation in proton exchange membrane electrolyzers. Nat. Nanotechnol. 16, 1371–1377 (2021).
Li, R. et al. IrW nanochannel support enabling ultrastable electrocatalytic oxygen evolution at 2 A cm−2 in acidic media. Nat. Commun. 12, 3540 (2021).
Shi, Z. et al. Confined Ir single sites with triggered lattice oxygen redox: toward boosted and sustained water oxidation catalysis. Joule 5, 2164–2176 (2021).
Fan, Z. et al. Extraordinary acidic oxygen evolution on new phase 3R-iridium oxide. Joule 5, 3221–3234 (2021).
Wen, Y. et al. Stabilizing highly active Ru sites by suppressing lattice oxygen participation in acidic water oxidation. J. Am. Chem. Soc. 143, 6482–6490 (2021).
Zheng, Y.-R. et al. Monitoring oxygen production on mass-selected iridium-tantalum oxide electrocatalysts. Nat. Energy 7, 55–64 (2022).
Shi, X. et al. Efficient and stable acidic water oxidation enabled by low-concentration, high-valence iridium sites. ACS Energy Lett. 7, 2228–2235 (2022).
Ouimet, R. J. et al. The role of electrocatalysts in the development of gigawatt-scale PEM electrolyzers. ACS Catal. 12, 6159–6171 (2022).
Liu, S. et al. Structurally-distorted RuIr-based nanoframes for long-duration oxygen evolution catalysis. Adv. Mater. 35, 2305659 (2023).
Ge, S. et al. A robust chromium–iridium oxide catalyst for high-current–density acidic oxygen evolution in proton exchange membrane electrolyzers. Energy Environ. Sci. 16, 3734–3742 (2023).
Wang, S., Shen, T., Yang, C., Luo, G. & Wang, D. Engineering iridium-based oxygen evolution reaction electrocatalysts for proton exchange membrane water electrolyzers. ACS Catal. 13, 8670–8691 (2023).
Seitz, L. C. et al. A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction. Science 353, 1011–1014 (2016).
Diaz-Morales, O. et al. Iridium-based double perovskites for efficient water oxidation in acid media. Nat. Commun. 7, 12363 (2016).
Yang, L. et al. Efficient oxygen evolution electrocatalysis in acid by a perovskite with face-sharing IrO6 octahedral dimers. Nat. Commun. 9, 5236 (2018).
Song, C. W., Suh, H., Bak, J., Bae, H. B. & Chung, S.-Y. Dissolution-induced surface roughening and oxygen evolution electrocatalysis of alkaline-earth iridates in acid. Chem 5, 3243–3259 (2019).
Lee, K., Osada, M., Hwang, H. Y. & Hikita, Y. Oxygen evolution reaction activity in IrOx/SrIrO3 catalysts: correlations between structural parameters and the catalytic activity. J. Phys. Chem. Lett. 10, 1516–1522 (2019).
Chen, Y. et al. Exceptionally active iridium evolved from a pseudo-cubic perovskite for oxygen evolution in acid. Nat. Commun. 10, 572 (2019).
Strickler, A. L., Higgins, D. & Jaramillo, T. F. Crystalline strontium iridate particle catalysts for enhanced oxygen evolution in acid. ACS Appl. Energy Mater. 2, 5490–5498 (2019).
Song, C. W., Lim, J., Bae, H. B. & Chung, S.-Y. Discovery of crystal structure-stability correlation in iridates for oxygen evolution electrocatalysis in acid. Energy Environ. Sci. 13, 4178–4188 (2020).
Zhang, R. et al. First example of protonation of Ruddlesden–Popper Sr2IrO4: a route to enhanced water oxidation catalysts. Chem. Mater. 32, 3499–3509 (2020).
Liang, X. et al. Perovskite-type solid solution nano-electrocatalysts enable simultaneously enhanced activity and stability for oxygen evolution. Adv. Mater. 32, 2001430 (2020).
Yang, C. et al. Cation insertion to break the activity/stability relationship for highly active oxygen evolution reaction catalyst. Nat. Commun. 11, 1378 (2020).
Edington, J., Schweitzer, N., Alayoglu, S. & Seitz, L. C. Constant change: exploring dynamic oxygen evolution reaction catalysis and materials transformations in strontium zinc iridate perovskite in acid. J. Am. Chem. Soc. 143, 9961–9971 (2021).
Li, N. et al. Identification of the active-layer structures for acidic oxygen evolution from 9R-BaIrO3 electrocatalyst with enhanced iridium mass activity. J. Am. Chem. Soc. 143, 18001–18009 (2021).
Wan, G. et al. Amorphization mechanisms of SrIrO3 electrocatalyst: how oxygen redox initiates ionic diffusion and structural reorganization. Sci. Adv. 7, eabc7323 (2021).
Li, N. et al. Origin of surface amorphization and catalytic stability of Ca2−xIrO4 nanocrystals for acidic oxygen evolution: critical roles of acid anions. ACS Catal. 12, 13475–13481 (2022).
Retuerto, M. et al. Highly active and stable OER electrocatalysts derived from Sr2MIrO6 for proton exchange membrane water electrolyzers. Nat. Commun. 13, 7935 (2022).
Liu, Y. et al. Operando identification of dual active sites in Ca2IrO4 nanocrystals with yttrium substitutions boosting acidic oxygen evolution reaction. ACS Energy Lett. 7, 3798–3806 (2022).
Yang, L. et al. A highly active, long-lived oxygen evolution electrocatalyst derived from open-framework iridates. Adv. Mater. 35, 2208539 (2023).
Wang, L. et al. Structurally robust honeycomb layered strontium iridate as an oxygen evolution electrocatalyst in acid. ACS Catal. 13, 7322–7330 (2023).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457–465 (2017).
Grimaud, A. et al. Activation of surface oxygen sites on an iridium-based model catalyst for the oxygen evolution reaction. Nat. Energy 2, 16189 (2017).
Zhang, N. & Chai, Y. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647–4671 (2021).
Wang, X., Zhong, H., Xi, S., Lee, W. S. V. & Xue, J. Understanding of oxygen redox in the oxygen evolution reaction. Adv. Mater. 34, 217956 (2022).
Chen, F.-Y., Wu, Z.-Y., Adler, Z. & Wang, H. Stability challenges of electrocatalytic oxygen evolution reaction: from mechanistic understanding to reactor design. Joule 5, 1704–1731 (2021).
Yun, T. G. et al. Surface dissolution and amorphization of electrocatalysts during oxygen evolution reaction: atomistic features and viewpoints. Mater. Today 58, 221–237 (2022).
Wang, Q. et al. Long-term stability challenges and opportunities in acidic oxygen evolution electrocatalysis. Angew. Chem. Int. Ed. 62, e202216645 (2023).
Kasian, O. et al. Degradation of iridium oxides via oxygen evolution from the lattice: correlating atomic scale structure with reaction mechanisms. Energy Environ. Sci. 12, 3548–3555 (2019).
Schweinar, K., Gault, B., Mouton, I. & Kasian, O. Lattice oxygen exchange in rutile IrO2 during the oxygen evolution reaction. J. Phys. Chem. Lett. 11, 5008–5014 (2020).
Zagalskaya, A., Evazzade, I. & Alexandrov, V. Ab initio thermodynamics and kinetics of the lattice oxygen evolution reaction in iridium oxides. ACS Energy Lett. 6, 1124–1133 (2021).
Park, C. H. et al. Atomic-level observation of potential-dependent variations at the surface of an oxide catalyst during oxygen evolution reaction. Adv. Mater. 36, 2403392 (2024).
Risch, M., Morales, D. M., Villalobos, J. & Antipin, D. What X-ray absorption spectroscopy can tell us about the active state of earth-abundant electrocatalysts for the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202211949 (2022).
Choi, S.-Y., Chung, S.-Y., Yamamoto, T. & Ikuhara, Y. Direct determination of dopant site selectivity in ordered perovskite CaCu3Ti4O12 polycrystals by aberration-corrected STEM. Adv. Mater. 21, 885–889 (2009).
Ryoo, H. et al. Frenkel-defect-mediated chemical ordering transition in a Li–Mn–Ni spinel oxide. Angew. Chem. Int. Ed. 54, 7963–7967 (2015).
Chung, S.-Y., Choi, S.-Y., Yoon, H.-I., Kim, H.-S. & Bae, H. B. Subsurface space-charge dopant segregation to compensate surface excess charge in a perovskite oxide. Angew. Chem. Int. Ed. 55, 9680–9684 (2016).
Yoon, H.-I. et al. Probing dopant segregation in distinct cation sites at perovskite oxide polycrystal interface. Nat. Commun. 8, 1417 (2017).
Heo, Y. et al. Symmetry-broken atom configurations at grain boundaries and oxygen evolution electrocatalysis in perovskite oxides. Adv. Energy Mater. 8, 1802481 (2018).
Bak, J., Bae, H. B. & Chung, S.-Y. Atomic-scale perturbation of oxygen octahedra via surface ion exchange in perovskite nickelates boosts water oxidation. Nat. Commun. 10, 2713 (2019).
Jung, H. J. et al. Direct identification of antisite cation intermixing and correlation with electronic conduction in CuBi2O4 for photocathodes. ACS Appl. Mater. Interfaces 11, 43720–43727 (2020).
Bak, J., Bae, H. B., Oh, C., Son, J. & Chung, S.-Y. Effect of lattice strain on the formation of Ruddlesden–Popper faults in heteroepitaxial LaNiO3 for oxygen evolution electrocatalysis. J. Phys. Chem. Lett. 11, 7253–7260 (2020).
Bak, J., Heo, Y., Yun, T. G. & Chung, S.-Y. Atomic-level manipulations in oxides and alloys for electrocatalysis of oxygen evolution and reduction. ACS Nano 14, 14323–14354 (2020).
Hong, Y. et al. Local-electrostatics-induced oxygen octahedral distortion in perovskite oxides and insight into the structure of Ruddlesden–Popper phases. Nat. Commun. 12, 5527 (2021).
Kim, D. et al. Direct probing of lattice-strain-induced oxygen release in LiCoO2 and Li2MnO3 without electrochemical cycling. Adv. Mater. 35, 2212098 (2023).
Yoo, S. J. et al. Comparing the impacts of strain types on oxygen-vacancy formation in a perovskite oxide via nanometer-scale strain fields. ACS Nano 18, 18465–18476 (2024).
Kim, H.-S., Bae, H. B., Jung, W. & Chung, S.-Y. Manipulation of nanoscale intergranular phases for high proton conduction and decomposition tolerance in BaCeO3 polycrystals. Nano Lett. 18, 1110–1117 (2018).
An, J.-S. et al. Unveiling of interstice-occupying dopant segregation at grain boundaries in perovskite oxide dielectrics for a new class of ceramic capacitors. Energy Environ. Sci. 16, 1992–2002 (2023).
Zaanen, J., Sawatzky, G. A. & Allen, J. W. Band gaps and electronic structure of transition-metal compounds. Phys. Rev. Lett. 55, 418–421 (1985).
Nong, H. N. et al. Key role of chemistry versus bias in electrocatalytic oxygen evolution. Nature 587, 408–413 (2020).
Yun, T. G., Heo, Y., Bae, H. B. & Chung, S.-Y. Elucidating intrinsic contribution of d-orbital states to oxygen evolution electrocatalysis in oxides. Nat. Commun. 12, 824 (2021).
Bak, J., Yun, T. G., An, J.-S., Bae, H. B. & Chung, S.-Y. Comparison of Fe-enhanced oxygen evolution electrocatalysis in amorphous and crystalline nickel oxides to evaluate the structural contribution. Energy Environ. Sci. 15, 610–620 (2022).
Alkhalifah, M. et al. Correlating orbital composition and activity of LaMnxNi1−xO3 nanostructures toward oxygen electrocatalysis. J. Am. Chem. Soc. 144, 4439–4447 (2022).
Kim, H.-S., An, J.-S., Bae, H. B. & Chung, S.-Y. Atomic-scale observation of premelting at 2D lattice defects inside oxide crystals. Nat. Commun. 14, 2255 (2023).
Tsai, H. M. et al. Comparison of electronic structures of RuO2 and IrO2 nanorods investigated by X-ray absorption and scanning photoelectron microscopy. Appl. Phys. Lett. 90, 042108 (2007).
Frati, F., Hunault, M. O. J. Y. & de Groot, F. M. F. Oxygen K-edge X-ray absorption spectra. Chem. Rev. 120, 4056–4110 (2020).
McCrory, C. C. L., Jung, S., Peters, J. C. & Jaramillo, T. F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977–16987 (2013).
Ryoo, H., Lee, S.-G., Kim, J.-G. & Chung, S.-Y. Effect of chemical bonding characteristics on ordering structure in Li spinel oxides. Adv. Funct. Mater. 29, 1805972 (2019).
McDaniel, C. L. & Schneider, S. J. Phase relations in the systems TiO2–IrO2 and SnO2–IrO2 in air. J. Res. Natl. Bur. Stand. A. Phys. Chem. 71A, 119–123 (1967).
Barthel, J. Dr. Probe: a software for high-resolution STEM image simulation. Ultramicroscopy 193, 1–11 (2018).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF), grant numbers 2022M3H4A1A01008918, RS−2024-00347287, RS−2023-00222411, and RS-2024−00435493. S.-Y.C. was also supported by Samsung Research Funding & Incubation Center of Samsung Electronics (project number SRFC-MA2401-04).
Author information
Authors and Affiliations
Contributions
S.-Y.C. conceived and designed the project, carried out the DFT calculations, STEM image simulations, and AIMD simulations, and wrote the paper. Y.S., T.G.Y., and K.H.P. carried out powder synthesis, XRD, and electrochemical CP and CV measurements. They also conducted TOF-SIMS and XAS. D.K. performed EDS compositional analyses and data processing. H.B.B. performed atomic-scale STEM, EDS, and EELS analyses, and contributed to the acquisition of image data. All the authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sim, Y., Yun, T.G., Park, K.H. et al. Effect of ionic-bonding d0 cations on structural durability in barium iridates for oxygen evolution electrocatalysis. Nat Commun 16, 4152 (2025). https://doi.org/10.1038/s41467-024-55290-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55290-y
