
Abstract
Electrochemical CO2 capture driven by renewable electricity holds significant potential for efficient decarbonization. However, the widespread adoption of this approach is currently limited by issues such as instability, discontinuity, high energy demand, and challenges in scaling up. In this study, we propose a scalable strategy that addresses these limitations by transforming the conventional single-step electrochemical redox reaction into a stepwise electrochemical-chemical redox process. Specifically, the hydrogen evolution reaction (HER) at the cathode and the oxidation of a redox carrier at the anode are employed to modulate the pH of the electrolyte, thereby facilitating effective CO2 capture. By decoupling the electrochemical swing for CO2 capture from redox carrier regeneration in both temporal and spatial domains, this approach mitigates unwanted side reactions and enhances system stability. Our results demonstrate a stable CO2 capture process sustained for over 200 h, with a electrical work of 49.16 kJe mol-1 CO2 at a current density of 10 mA cm-2. Furthermore, a scaled-up system capable of producing approximately 0.4 kg of pure CO2 per day maintained stable operation for 72 h, highlighting the potential feasibility of this method for large-scale decarbonization applications.
Similar content being viewed by others

Introduction
Direct carbon dioxide (CO2) capture from industrial sources or the atmosphere plays a crucial role in managing global CO2 emissions. However, it faces a significant challenge due to the high energy consumption associated with current methods1,2,3. Electrochemical CO2 capture has emerged as a promising alternative that efficiently utilizes intermittent renewable energy resources without relying on traditional thermal processes, such as chemical absorbent scrubbing or high-pressure-driven membrane separation. This approach offers the potential for low-power CO2 purification and improved energy efficiency4,5. One key advantage of electrochemical CO₂ capture is its ability to separate CO2 from gas streams through an electricity-controllable electro-swing system. Unlike conventional methods, this technique does not require heating, making it highly flexible for capturing CO2 at different times and locations6,7.
There are two main types of electrochemical CO2 capture methods that have been extensively studied. The direct method involves the electrochemical generation of nucleophilic agents in organic solvents via electro-reduction8. These nucleophilic agents react with CO2 to form adducts, capturing low concentrations of CO2. The CO2 is later released and purified through the electro-oxidation of these adducts. Despite its potential, this method faces challenges, such as high ohmic resistance and interference from water in organic solvents, which limit its efficiency9.
The indirect methods, including pH-swing10,11,12 and metal-amine complexing electro-swing13,14, use absorbents like alkali or amines to capture CO2. Desorption occurs when redox components are electrically regenerated, which displaces the CO2. These methods benefit from lower ohmic resistance and can operate in aqueous solutions. In addition, the use of organic redox mediators in these systems typically enhances electrode kinetics on graphite electrodes. This design avoids common issues like flooding in gas diffusion electrodes15 and dendrite formation or corrosion in copper electrodes16.
However, a critical limitation of electrochemical CO2 capture systems, particularly those using organic redox couples, is their instability during continuous CO2 absorption-desorption cycles. When exposed to real CO2 sources containing oxygen (O2), such as atmospheric air or flue gas, redox molecules can degrade, limiting the long-term application of these systems5. Addressing this instability is essential for advancing electrochemical CO2 capture technologies for practical use.
Recent advancements have introduced several innovative strategies to mitigate the effects of oxygen-side reactions in electrochemical carbon capture systems. For direct electrochemical carbon capture methods, it remains challenging to overcome the coupling between the oxygen tolerance of redox carriers and their binding constants with CO2. Some advanced studies have reported that hydrogen bonding can be employed to develop redox carriers that exhibit both oxygen tolerance and favorable CO2 binding constants17,18. For indirect electrochemical carbon capture methods, strategies like electrochemical rebalancing19 and solid electrolyte reactors20 have been proposed to develop oxygen-tolerant electrochemical carbon capture systems. In addition, extensive research has focused on creating O2-tolerant electrochemical redox molecules21,22 and absorbent systems23, which demonstrate good oxygen resistance. However, these technologies still face the complex challenge of balancing oxygen tolerance, operational continuity, and energy consumption for carbon capture.
In this paper, we present an approach to decoupled redox electrochemical CO2 capture, which enables continuous and stable CO2 capture in O2-containing flue gas. This method separates the conventional single-step electrochemical reaction into two processes: electrochemical and chemical redox reactions. Electrochemical reactions are used to modulate the pH at the anode and cathode, allowing for efficient CO2 capture and desorption. Specifically, at the cathode, the hydrogen evolution reaction (HER) produces an alkaline solution, while at the anode, the oxidation of an organic redox mediator (QH2) generates an acidic solution. The alkaline solution absorbs low concentrations of CO2, forming bicarbonate ions. These ions migrate through an anion exchange membrane, driven by an electric field, to the acidic region at the anode, where high-purity CO2 is desorbed. The redox mediator at the anode is then regenerated via spontaneous hydrogenation in a separate tower, a process that does not require electrical energy (Fig. 1). This design enables each module to operate independently, facilitating continuous CO2 capture.

In the electrolytic system, hydrogen evolution reactions occurring at the cathode increase the alkalinity of the solution, facilitating the absorption of low-concentration CO2. At the anode, the electrochemical oxidation of AQDSH2 acidifies the solution, promoting the desorption of high-purity CO2. Bicarbonate ions generated at the cathode migrate to the anode through the anion exchange membrane, maintaining charge balance and providing a carbon source for the CO2 desorption reaction at the anode. In the regeneration tower, the oxidized AQDS is regenerated through a spontaneous chemical hydrogenation reaction with H2 over a Pt/C catalyst.
By decoupling the electrochemical CO2 capture process from the regeneration of the redox mediator both spatially and temporally, this strategy effectively reduces the impact of oxygen on the regeneration process. In this study, we developed and validated a laboratory-scale decoupled redox electrochemical CO2 capture (DREC) system. We established a thermodynamic theoretical model for the DREC system and demonstrated a scaled-up electrochemical carbon capture system capable of continuous and stable CO2 capture in oxygen-containing environments.
Results and discussions
Concept and verification of DREC system
The decoupled redox electrochemical CO2 capture (DREC) system employs an asymmetric electrolysis design consisting of three modules: an electrolysis module, a CO2 capture module, and a redox regeneration module. In the electrolysis module, the hydrogen evolution reaction (HER) at the cathode increases the solution’s alkalinity, facilitating CO2 absorption in the capture module. Simultaneously, the anode utilizes an organic redox mediator (QH2) in its reduced state. At the anode, QH2 undergoes electrochemical oxidation, releasing H⁺ ions and acidifying the solution through a reversible proton-coupled electron transfer (PCET) reaction. Following this, the oxidized form, Q, is regenerated by H2 in the redox regeneration module, catalyzed by Pt/C, to restore QH2. This regeneration process occurs spontaneously without the need for electrical input. The hydrogen oxidation reaction, characterized by rapid kinetics, is mediated by anthraquinone disulfonate (2,7-AQDS). Meanwhile, bicarbonate ions (HCO3-) formed at the cathode migrate through an anion exchange membrane (AEM) to the anode, where they react with H+ ions to release pure CO2.
This asymmetric design decouples the sites of CO2 absorption-desorption from the redox regeneration process, eliminating the need for frequent electrode replacement as required in traditional electrochemical CO2 capture systems. In addition, it prevents direct contact between oxygen on the absorption side and the anodic redox mediator, which would otherwise reduce efficiency. It overcomes the slow kinetics of H2 oxidation at the gas diffusion anode.
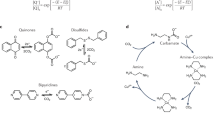
We began by establishing the selection criteria for the redox mediator in the DREC system (Fig. 2A). The organic redox mediator at the anode must satisfy thermodynamic requirements under CO2 dissolution equilibrium to maintain an optimal pH for efficient CO2 absorption and desorption. Thus, the mediator must be stable in an aqueous CO2 environment, possess a redox potential higher than H₂/H⁺, and enable reversible proton-coupled electron transfer (PCET) reactions. In addition, energy consumption for CO2 capture is primarily affected by the potential range, which varies with pH (∆EpH and ∆EpH’ in Fig. 2B), and the intrinsic potential difference (∆E₀ in Fig. 2B). To minimize energy consumption, we aim for a low intrinsic potential difference (∆E₀) while meeting the other necessary criteria. We selected 2,7-AQDS as the redox mediator due to its low reduction potential (− 0.1265 V vs. SHE at pH 6, Fig. 2B, red line), high solubility (1.34 mol L−1 in a phosphate-mixed solution; Supplementary Fig. 1), and excellent electrochemical stability and reaction kinetics (Supplementary Figs. 2, 3)24. It can be efficiently reduced by H2 using metal-based catalysts25,26, such as the Pt-based catalysts employed in this study. The 2e⁻/2H⁺ PCET reaction of 2,7-AQDS effectively drives CO2 desorption at pH 6. At pH values above 11, the reduced form of quinone, hydroquinone, deprotonates before electrochemical oxidation and exhibits a pH-independent reduction potential (Fig. 2C). This behavior is determined by the pKa of the redox-active center and can be fine-tuned through molecular design to accommodate various pH conditions.

A Selection criteria for active substances in anodic oxidation-reduction. B Cyclic voltammogram of AQDS (red line) and the HER (blue line) scanned at 50 mV s−1 on a glassy carbon electrode and a platinum-plated nickel mesh electrode. The difference between the two CV scans indicated by the red solid line and the red dashed line represents a pH difference of 0.5 in the anolyte. The difference between the two CV scans indicated by the blue solid line and the blue dashed line represents a pH difference of 1.49 in the catholyte. C Pourbaix diagram of 2,7-AQDS. Data are fitted to three solid lines with slopes of − 59.3 mV pH−1, − 28 mV pH−1, and 0 mV pH−1, corresponding to two-, one- and zero-proton processes, respectively. The data shown are an average from three experimental runs; the error bars indicate the standard deviation. D LSV curve of the HER cathode (the blue line is measured under the mixture gas of 15% CO2 and 85% N2, and the yellow line is measured under the mixture gas of 15% CO2, 80% N2, and 5% O2). The electrolyte (1 M KHCO3) is injected with a mixture of gas at 40 mL min−1 for 30 min before the test. Throughout the test, the same mixture of gas was maintained. The potential at the intersection point of the dot-dash line and the dashed line is − 0.545 V. E Stability test of the HER cathode under different O2 concentrations. The electrolyte is injected with an oxygen-containing gas mixture. The concentration of O2 was measured by the O2 concentration sensor. All figures have not been IR-corrected.
Since AQDS operates efficiently below pH 7 and offers proton buffering capabilities, we selected a K2HPO4/KH2PO4 buffer for the anode and a KHCO3 buffer for the cathode. These buffers maintain appropriate pH levels for CO2 desorption and absorption, enabling low-voltage operation with minimal pH fluctuation. The small pH variations observed between the outlet of catholyte and anolyte during electrolysis further demonstrate the strong proton buffering capacity of the electrolyte (Supplementary Fig. 4).
We then assessed the impact of oxygen on the cathode during CO2 absorption. A simulated gas mixture containing 15% CO2, 80% N2, and 5% O2, representative of typical flue gas composition from coal-fired power plants, was employed. Results from cathode linear sweep voltammetry (LSV) tests under both O2-free and O2-containing conditions showed nearly identical curves (Fig. 2D). No obvious effect of oxygen on our electrochemical process was observed. The stability of cathodic constant current electrolysis was further validated across a range of oxygen concentrations (0–20%, Fig. 2E), demonstrating the DREC system’s robust performance in O2-containing CO₂ environments.
While oxygen could be reduced at the potential of H2/H⁺, the low solubility of oxygen in water (~ 0.6 mg L−1 under atmospheric conditions or simulated flue gas with 5% O2) plays a critical role in maintaining electrolysis stability (Supplementary Fig. 5). On the anode side, where CO2 desorption occurs, AQDSH2 is exposed to a pure CO2 atmosphere. The anion exchange membrane (AEM) effectively acts as a barrier, preventing dissolved oxygen from the cathode from reaching the anode, with oxygen levels measured at 0 mg L−1 on the anode side (Supplementary Fig. 5). This design ensures that AQDSH2 is not oxidized by oxygen in the CO2 capture process, eliminating the degradation issues typically observed in prior electrochemical carbon capture systems using quinones as redox couples, which were susceptible to side reactions with O2. Constant current tests for both the anode and cathode demonstrated stability over a 72-hour period (Supplementary Fig. 6). In addition, the system showed stable operation even under high current density conditions, further confirming its durability in practical applications (Supplementary Fig. 7).
Matching of electrochemical oxidation and chemical reduction of redox mediator
The relative redox potentials of H2 and AQDS indicate that the hydrogenation of AQDS is thermodynamically favorable. We monitored the reduction of 2,7-AQDS by H₂ using in situ UV-vis spectroscopy (Fig. 3A, B). The presence of electron-donating phenolic hydroxyl groups facilitated electron transfer within the AQDS system through the p-π conjugated network, decreasing the transition energy and causing a redshift in the absorption band (from 328 nm to 390 nm). This redshift aligns with previous reports and confirms the formation of AQDSH2 following hydrogen reduction27.

A Schematic diagram of the in situ UV‒vis monitoring system. B In situ UV‒vis spectra obtained during AQDS hydrogenation. C Schematic showing the potential monitoring system for hydrogenation reduction. D OCV curve of the AQDS hydrogenation reduction process. The electrolyte was 100 mL of 0.1 M AQDS in phosphate buffer. The catalyst loading volume was 50 mL. The H2 flow rate was 30 mL min−1. The temperature was 50 °C. The electrolyte was injected with Ar during the tests. E The Q/QH2 ratio during the hydrogenation process was calculated with the Nernst equation. The dots are reaction data, and the line reflects a linear fit to the data. F Schematic of the full-cell electrolysis system. G, H Constant voltage electrolysis test of the full cell under different pump flow rates. The initial electrolyte was fully oxidized to 0.1 M AQDS in phosphate buffer and 1 M KHCO3. Hydrogen gas was introduced for hydrogenation reduction at 20 min.
Next, we tracked the hydrogenation reaction rate by monitoring the open circuit potential of the anode (Fig. 3C, D). Using the Nernst equation, we calculated the QH2/Q ratio based on the reaction potential (Fig. 3E and Supplementary Note 1). The initial reaction rate was relatively slow due to the adsorption of hydrogen gas on the catalyst surface. However, as the surface became saturated with hydrogen, the reaction rate stabilized. We modeled the reaction process to estimate the rate of AQDSH2 production, which was found to be 0.0627 M h−1. This corresponds to a maximum current of 1.41 A for electrochemical oxidation. It is important to note that this observed rate was influenced by both internal and external diffusion limitations, meaning it reflected only the rate of AQDSH2 production within the decoupled capture system and not the intrinsic hydrogenation rate of AQDS.
In addition, we verified the compatibility of the hydrogenation and electrochemical oxidation reactions within the full cell. By applying a constant cell voltage of 1.5 V, within the water stability window, we determined the maximum achievable current (Fig. 3F–H). As the liquid flow rate increased from 200 to 1000 mL min−¹, the cell current gradually rose and then plateaued. This initial increase was attributed to improved mass transfer on the electrode surface as flow rates increased, which enhanced the overall mass transfer and boosted cell current. However, as the flow rate continued to rise, the current increase diminished, indicating that the system was nearing the mass transfer limit under the test conditions.
Long-term operation at higher flow rates was restricted by the pump equipment’s limitations. At a flow rate of 1000 mL min−1, we achieved a maximum current density of 160 mA cm−2. However, higher flow rates led to greater parasitic energy consumption. We estimated that energy loss from the pump during fluid delivery was approximately 2.55 and 20.11 kJe mol−1 CO2 at a flow rate of 200 and 1000 mL min−1, respectively (Supplementary Note 2).
The stability of the anode was verified over 15 days of continuous electrolytic oxidation and hydrogen catalytic reduction (Fig. 4A). Proton nuclear magnetic resonance (¹HNMR) spectra showed no additional peak signals with no observed shifts (Fig. 4B), indicating the stability of 2,7-AQDS against chemical reduction by hydrogen in acidic aqueous solutions. These results were corroborated by carbon nuclear magnetic resonance (13CNMR) spectroscopy and liquid chromatography-mass spectrometry (LC-MS) (Fig. 4C and Supplementary Fig. 8). Through cyclic voltammetry (CV) tests using a glassy carbon electrode, no additional redox peaks were observed within the potential range of − 0.4 to 0.4 V versus the standard hydrogen electrode (SHE) (Fig. 4D). This confirms that 2,7-AQDS exhibits both electrochemical and chemical stability during prolonged electrolysis. Furthermore, scanning electron microscopy (SEM) images (Supplementary Fig. 9) confirm that the catalyst’s morphology remains unchanged after extended reduction, indicating the robustness of the catalyst under prolonged operation. Compared to direct electrochemical hydrogen oxidation using a gas diffusion electrode (GDE), our method effectively mitigates the problematic “flooding” process commonly observed at the gas-liquid-solid interface during the hydrogen oxidation reaction (HOR)15,28.

A Single-cell potential stability test in phosphate buffer solution at a current density of 10 mA cm−2 for 15 days. B 1HNMR spectra of AQDS recorded in D2O, showing AQDS after different reaction times of 48, 96, 144, 192, 240, 288, and 336 h. C 13CNMR spectra of AQDS recorded in D2O, showing AQDS after different reaction times of 48, 96, 144, 192, 240, 288, and 336 h. D CV curve of 0.1 M AQDS scanned at 50 mV s−1 on a glassy carbon electrode.
The hydrogen used in the redox regeneration module theoretically originates from the electrolysis cathode. To validate the hydrogen cycle concept, we designed a laboratory-scale testing device (Supplementary Fig. 10). During 24 h of constant current electrolysis, the volume of hydrogen gas in the system remained unchanged (Supplementary Fig. 10). This demonstrates that the hydrogen produced at the cathode is fully recycled within the redox regeneration module, with no detectable loss of hydrogen throughout the system, (Supplementary Fig. 10), demonstrating that the hydrogen generated at the cathode can be fully recycled in the redox regeneration module without any system-wide hydrogen loss. In addition, we measured the amount of hydrogen produced and consumed during the cathodic and anodic reactions (Supplementary Fig. 11A–C). The hydrogen evolution reaction (HER) efficiency at the cathode was 99.5%, while the hydrogen consumption efficiency in the redox regeneration module was 99.7%. The ratio of hydrogen production to consumption was ~ 99.8%, confirming the feasibility of the hydrogen cycle concept in the DREC system.
Demonstration and validation of a laboratory DREC system
The laboratory DREC system demonstrated stable electrolysis voltage over a 200 h period in 15% CO2, 80% N2, 5% O2 environment, maintaining an average voltage of 0.507 V at a current density of 10 mA cm−2 (Fig. 5A). To ensure the purity of the desorbed CO2, we release CO2 at a partial pressure of 1 bar. CO2 absorption and desorption efficiencies were determined to be 99.6% and 99.5% (Fig. 5B, Cand Supplementary Figs. 12–14), respectively. Carbon removal efficiency is 21.76% (Fig. 5D), corresponding to energy consumption for CO2 electrolysis was approximately 49.16 kJe per mole of CO2 (Fig. 5E). Compared to other advanced electrochemical carbon capture techniques19,20,21,23,29,30,31,32, our system exhibited lower energy consumption (Supplementary Figs. 15, 16). Carbon removal efficiency is defined as the ratio of the CO2 concentration at the system’s inlet to its outlet at low concentrations (Eq. 5).

A Electrolysis durability test at a constant current density of 10 mA cm−2 for simulated flue gas capture for a total period of 200 h. B Absorption efficiencies at different current densities. C Desorption efficiencies at different current densities. D Carbon removal efficiency of DREC system at different current densities. The carbon removal efficiency is calculated using Eq. 5. The error bars indicate that each group was subjected to three repeated experiments. E Energy consumption of DREC system at different current densities. The energy consumption is calculated using Eq. 8.
Subsequently, we evaluated the electrochemical CO2 capture performance of the full cell under higher current densities. With increasing current density, we observed a rise in the pH of the catholyte outlet, corresponding to a higher CO2 absorption rate and improved carbon removal efficiency (Supplementary Fig. 17). At 50 mA cm−2, absorption and desorption efficiencies decreased slightly to 95.8% and 94.7%, respectively, while the carbon removal efficiency is 63.23% with an average voltage of 0.814 V. This corresponded to an energy consumption of 82.93 kJe mol−1 for CO2 capture (Fig. 5B–Dand Supplementary Figs. 18, 19). At a higher current density of 120 mA cm−2, the absorption and desorption efficiencies further declined to 89.09% and 84.49%, respectively, with an average voltage of 1.25 V, resulting in an energy consumption of 142.7 kJe mol−1 for CO2 capture. The carbon removal efficiency can reach 92.78% (Fig. 5B–Dand Supplementary Figs. 20, 21).
The reduction in electrolysis efficiency at higher current densities was primarily attributed to the competitive transport of anions such as CO32− and OH− in the catholyte, which competed with HCO3− for transport towards the anode. Although an anion exchange membrane selective for HCO3− was employed, this competition persisted, especially as cathode alkalinity and operation time increased, leading to a drop in efficiency. Nevertheless, the system maintained relatively high efficiency (greater than 84%) due to the membrane’s strong selectivity for HCO3−. Higher current densities enhance carbon removal efficiency and absorption rates; however, this comes at the cost of increased energy consumption. In addition, we evaluated the membrane’s permeability to AQDS, finding that over a 21-day period, the AQDS permeance was 5.16 × 10−3 mol m−2 h−1 (Supplementary Fig. 22). The presence of SOx and NOx in real flue gases will also impact the system (Supplementary Fig. 23). Therefore, the development of catalysts that are tolerant to NOx and SOx will be crucial for advancing the industrial application of the DREC system in the future. In future applications, these factors will need to be carefully balanced.
The cathode in the DREC system facilitates the transfer of bicarbonate ions (HCO3-) to the anode, where they react with protons (H+) to form water, establishing a water balance within the system. Overextended operational periods, this balance may shift, resulting in water loss at the cathode and accumulation at the anode. However, water vapor is typically carried out by gases into the atmosphere, which mitigates water accumulation. Experimental results indicated that during a 15-day CO2 capture process with a gas flow rate of 20 ml min−1, water loss at the anode averaged approximately 1.93 ml per day. In industrial applications, flue gas absorbed at the cathode contains water vapor (H2O), which theoretically increases water content at the cathode. Thus, maintaining water equilibrium in the system is a complex challenge. Established methods, such as membrane separation33,34, offer potential solutions for managing water balance in future industrial-scale applications.
Thermodynamic model and energy consumption evaluation
The theoretical model analysis covered the entire CO2 capture process to evaluate energy consumption (refer to Supplementary Note 3). The analysis primarily focused on the equilibrium relationships of carbonates and phosphates (Supplementary Table 2), excluding the effects of anthraquinone. Unlike previous models11,14,35, this model introduces a theoretical framework for continuous CO2 capture without the need for electrode exchange. The model assumes that electrochemical acidification/alkalization and desorption/absorption processes occur independently.
Here, we quantify the amount of CO2 absorbed or desorbed by measuring the change in dissolved inorganic carbon (∆DIC). Total alkalinity (TA) is defined based on charge balance. In our system, we assume that bicarbonate ions transfer through the anion exchange membrane from the cathode to the anode, accompanied by an equal molar transfer of electrons. As a result, TA remains constant in our model, and ∆DIC is equal to the concentration of electrons transferred. Our model confirms that the DREC system can capture CO2 from sources with varying concentrations, such as 10–15% in flue gas or approximately 400 ppm in ambient air (Supplementary Fig. 24), and desorb CO2 at 1 bar. The ∆DIC can be calculated based on the number of electrons transferred, which allows us to determine the pH of the anolyte and catholyte. Using the Nernst equation, we then calculate the system’s theoretical minimum energy consumption. The minimum theoretical electrical energy required by the system is 12.28 kJe mol−¹ of CO2 for a 15% CO2 concentration and 23.35 kJe mol−¹ for 400 ppm CO2 (Supplementary Figs. 25–27). Because of the asymmetric structure of the DREC system, we conducted further analysis, which demonstrates that the membrane potential also affects the system’s energy consumption (Supplementary Fig. 28).
Performance of a scaled-up DREC system
To demonstrate the practical applicability of our approach, we developed a demonstration system for CO2 capture using a stacking strategy, capable of generating approximately 0.4 kg of pure CO2 per day (Fig. 6A). The electrolysis system was compact and modular, consisting of three stacked cells with a total effective geometric surface area of 1008 cm², compared to 4 cm² for the lab-scale system. The sizes of the modules can be adjusted to accommodate different capture quantities. During a 72 h test period, the system consistently captured CO2, operating stably at an average voltage of 0.95 V and a current density of 10 mA cm−2 (Fig. 6B), with an energy consumption of approximately 92.93 kJe mol−¹ CO2. Furthermore, CO2 absorption and desorption tests revealed that simulated flue gas with a CO2 concentration of 15% was absorbed in the cathode, while 100% CO2 was desorbed from the anode. The CO2 absorption and desorption efficiencies were approximately 100% and 98.5%, respectively, with a capture-removal efficiency of around 34% (Fig. 6C–F, Eq. 5).

A Optical image and schematic of a demonstration system. B Electrolytic durability test for the demonstration system operated at a constant current density of 10 mA cm−2. C Flow rate of the CO2 absorption tower inlet and outlet. The dashed line represents the fitted curve of the flow rate at the outlet of the CO2 absorption tower. D Concentrations of CO2 at the inlet and outlet of the absorption tower. E Flow rates of the CO2 desorption tower inlet and outlet. The dashed line represents the fitted curve of the flow rate at the outlet of the CO2 desorption tower. F Concentrations of CO2 at the outlet of the desorption tower.
Despite the successful scale-up, a disparity in energy consumption between the scaled-up system and the laboratory-scale system was observed, due to amplification effects. We anticipate that further reductions in energy consumption and improvements in capture efficiency can be achieved through optimized reactor design, implementation of comprehensive heat exchange networks, and fine-tuning of operational parameters. Overall, the preliminary validation of the decoupled redox electrochemical carbon capture strategy has been achieved.
The most established method for CO2 capture from flue gas is amine scrubbing, where organic amines absorb low concentrations of CO2, followed by desorption at temperatures between 120–140 °C. The regeneration heat requirement ranges from approximately 132–150 kJth mol−1 CO236. Heat exchange optimization can somewhat reduce this energy demand. Other carbon capture technologies, such as solid adsorption and membrane separation, aim to lower overall energy consumption by either using solid adsorbents or leveraging membrane selectivity, thereby avoiding the high thermal regeneration burden associated with solvents. Our system introduces an electrochemical decoupled strategy, utilizing a pH-swing mechanism to bypass the high energy costs of thermal regeneration. Given its potential integration with renewable energy sources in the future, our system offers a promising solution for carbon capture.
It is worth noting that thermal-driven technologies, such as amine scrubbing use thermal work for desorption. In contrast, membrane separation and electrochemical carbon capture technologies rely on electrical work to capture CO2. A direct comparison between electrical work and thermal work across different techniques is challenging. Therefore, we have indicated the different types of separation work in the table (Supplementary Table 3) to facilitate a more equitable assessment of each technology.
In summary, we have developed an electrochemical CO2 capture strategy that decouples the traditional single-step electrochemical redox process into a stepwise electrochemical-chemical redox mechanism. This approach enables continuous and stable CO2 capture via pH swings at the electrode, effectively avoiding undesired O2 side reactions. We demonstrated the efficacy of this system in a 200 h lab-scale experiment, achieving an energy consumption of approximately 49.16 kJe mol−1 CO2, and in a scaled-up 72 h experiment, achieving 92.93 kJe mol−1 CO2. Further optimization of performance is possible by designing molecular structures with reduction potentials closer to the hydrogen evolution reaction (HER), employing more efficient anion exchange membranes, and dynamically optimizing process parameters. This work presents a perspective on electrochemical CO2 capture and offers potential solutions to long-standing challenges of scalability and high energy consumption in this field.
Methods
Materials
Chemicals such as potassium bicarbonate (KHCO3), potassium dihydrogen phosphate (KH2PO4), and dipotassium hydrogen phosphate (K2HPO4), all of analytical grade, were procured from Shanghai Aladdin Bio-Chem Technology Co., Ltd. 9,10-Anthraquinone-2,7-disulfonic acid (AQDS, 97%) was purchased from HuaXia Technology Co., Ltd. The Pt/C granular catalyst (1 wt% loading, diameter 0.7 mm) was supplied by Shanxi Rock New Materials Co., Ltd. All chemicals were used without further purification. The anion exchange membrane (FAA-3-PK-75) with a thickness of 75 ± 5 μm was purchased from SCI Materials Hub. It was soaked in a 1 M KHCO3 solution for 24 h and then washed with deionized water three to four times before use. Deionized water with an electrical resistivity of 18.25 MΩ·cm at 25 °C was used as the solvent to prepare the electrolyte solutions. CO2, N2, and a mixed gas (80% N2, 15% CO2, 5% O2), all with 99.999% purity, were sourced from the Chengdu Heping Gas Plant.
Structure of the cell
The electrolytic cell was designed as a sandwich-type reactor, comprising two stainless steel plates, two polytetrafluoroethylene (PTFE) plates, and two graphite plates. The anode and cathode chambers were separated by an anion exchange membrane (FAA-3-PK-75). Graphite plates with serpent-like flow channels were employed to regulate the flow of reactants and products. Platinum-plated nickel mesh, with a geometric surface area of 4 cm2, served as the cathode for the hydrogen evolution reaction. The anode electrode was composed of pretreated graphite felt, also with an area of 4 cm2. The graphite felts were pretreated by soaking in 98% H2SO4 at 60 °C for 6 h, followed by soaking in deionized water for 4 h, and then vacuum-dried for 3 days.
Characterization
1HNMR and 13CNMR spectra were obtained using Bruker AV II-600 MHz. The NMR spectra were recorded in D2O (99.9%) to suppress the water signal. The half-cell stability test was conducted in an oxygen-free glovebox. Daily quantitative sampling was performed, and after vacuum drying in the glovebox, a measured amount of D₂O was added to the samples, which were then sealed in NMR tubes. The tubes were further sealed with parafilm and sent for testing.
Scanning electron microscopy (SEM) was performed using a Thermo Scientific HITACHI REGULUS 8230 at an accelerating voltage of 5.0 kV. A polaron sputtering gun was employed to coat the samples with a 25 nm gold film to enhance their electrical conductivity.
In situ UV-Vis spectroscopy was conducted using a PerkinElmer Lambda 750S spectrophotometer (USA), covering the wavenumber range of 300–600 nm. The reduction tower was loaded with 5 mL of catalyst, and the temperature was maintained at 50 °C. During the experiment, 1 mM AQDS in phosphate buffer was pumped from the tank into both the reduction tower and the flow-through cuvette at a flow rate of 100 mL min−¹. Simultaneously, hydrogen gas was introduced into the reduction tower at a flow rate of 30 mL min−¹.
Solubility measurement
A phosphate mixture solution (0.8 M KH2PO4 and 0.2 M K2HPO4) was used as the test environment for the solubility test. Different amounts of AQDS were added to 10 mL of the mixed solution and fully dissolved until there was no precipitate; then, standard solutions of 5 × 10−5 M, 1 × 10−4 M, 1.2 × 10−4 M, 1.4 × 10−4 M, 1.6 × 10−4 M and 1.8 × 10−4 M concentrations after dilution were prepared. The absorbance within the wavelength range of 300–600 nm was measured by means of an ultraviolet spectrophotometer (PerkinElmer Lambda 750S, USA) with a wavelength interval of 0.5 nm and an optical path length of the quartz cuvette of 10 mm. The peak absorbance values of different concentrations of AQDS at 328 nm were selected for fitting to plot a standard regression curve27. The sample consisted of 5 mL of a mixed solution with AQDS, reaching a saturation state after 24 h of equilibrium. There was still a small amount of solid insoluble matter. After resting, the supernatant was filtered and diluted to a constant volume, and the absorbance was measured with the same method. The peak absorbance at 328 nm was used to calculate the maximum solubility of AQDS in the mixed solution by means of the standard regression curve.
Dissolved oxygen test
To evaluate the effectiveness of the anion exchange membrane in isolating oxygen, we measured the dissolved oxygen concentration in an H-cell. The H-cell was divided into two chambers, separated by the anion membrane. One chamber was filled with 1 M KHCO₃ solution and continuously supplied with a mixed gas (15% CO2, 80% N2, 5% O2) at a flow rate of 40 mL min−1. The other chamber contained 0.1 M AQDS in a phosphate buffer solution (0.8 M KH2PO4 and 0.2 M K2HPO4). Initially, the solution was exposed to atmospheric conditions for 3 h. Subsequently, the mixed gas was bubbled into the AQDS chamber for an additional 3 h. To simulate practical application conditions, the solution was then bubbled with pure CO2 to remove any remaining oxygen and achieve degassing. The dissolved oxygen concentration in the solution decreased to nearly 0 mg L−1. To maintain a stable CO2 environment, CO2 gas was continuously bubbled into the upper portion of the solution. Dissolved oxygen concentrations were monitored using a dissolved oxygen detector (INESA Scientific Instrument Co., Ltd., JPB607A).
The permeance of AQDS test
The AQDS permeation experiment was conducted in an H-cell. One compartment contained a 0.1 M AQDS phosphate solution, while the other contained a 1 M KHCO3 solution. The area of the anion exchange membrane (AEM) was 4.9 × 10−4 m2. After sampling from the KHCO3 solution side, the concentration of AQDS was measured using UV-vis spectroscopy to calculate the permeance of AQDS.
Hydrogen evolution and consumption efficiency
For the hydrogen evolution efficiency test, N2 gas is used as a carrier gas and introduced into the cathode to sweep out the H2. The flow rate of the mixed gas and the concentration of H2 are then measured using a flow meter and gas chromatograph (GC). For the hydrogen consumption efficiency test, H2 is introduced into the reduction tower, where it mixes with N2 upon exiting. The flow rate of the mixed gas and the concentration of H2 are similarly measured using a flow meter and GC.
CO2 absorption and desorption
For CO2 absorption measurements, the catholyte was purged with 15% CO2, 5% O2 and 80% N2. The exit gas was passed sequentially through a drying tube, a gas flow sensor, and a CO2 sensor to measure the total gas flow rate and CO2 concentration. Two magnetic pumps were used to circulate both the catholyte and anolyte solutions.
For CO2 desorption measurements, pure CO2 gas was introduced into the anolyte chamber, and the flow rate was measured using a gas flow sensor. The exit gas was then passed through a drying tube, a gas flow sensor, and a CO2 sensor to determine the total gas flow rate and CO2 concentration. The amount of desorbed CO2 was calculated by multiplying the difference in flow rates between the inlet and outlet by the CO2 concentration.
Electrochemical characterization
Cyclic voltammetry (CV) measurements were performed at room temperature using a CHI660E electrochemical workstation (CH Instruments). All electrochemical data were exported in TXT or CSV format. No iR compensation was applied to any of the electrochemical tests. A glassy carbon electrode with a 3.0 mm diameter was employed as the working electrode in all three-electrode CV tests. The reference electrode was an Ag/AgCl electrode, presoaked in a 3.5 M KCl solution, while a platinum mesh (1 × 1 cm²) served as the counter electrode. The reference electrode was calibrated prior to the electrochemical measurements. The reference electrode was immersed in a 3.5 M potassium chloride standard solution, and its potential was calibrated by comparing the measured potential with the theoretical potential at room temperature. Prior to testing, the solution was degassed with argon for 30 min, and argon was continuously purged throughout the experiment to maintain an inert atmosphere. The glassy carbon electrode was polished sequentially with 0.5 μm and 0.03 μm alumina powders to ensure a smooth surface. The electrolyte volume is 100 mL. For the stability test, the electrolyte consisted of 1 mM AQDS in a phosphate buffer (0.8 M KH2PO4 and 0.2 M K2HPO4). In the potential comparison experiment (Fig. 2B), the electrolyte was 0.1 M AQDS in the same phosphate buffer.
To construct the Pourbaix diagram, 1 mM AQDS solutions (100 mL) were prepared in various pH buffers, using sulfuric acid (1 M, pH 0), HSO4−/SO42− (0.1 M, pH 1–2), AcOH/AcO− (0.1 M, pH 2.65–5), H2PO4−/HPO42− (0.1 M, pH 5.3–8), HPO42−/PO43− (0.1 M, pH 9.28–11.52), and KOH (0.1 M, pH 13). The pH of each solution was adjusted with 1 M H₂SO₄ or 0.1 M KOH and measured using a pHS-3E pH meter (INESA Scientific Instrument Co., Ltd.), which was calibrated using buffer solutions with pH values of 9.18, 6.86, and 4.00. The slope of the linear fit of the plot of E0 versus pH was used to determine the ratio of the proton number to the electron number (m/n), with the slope ratio of 0.059 approximating this value.
RDE measurements were conducted at room temperature using an AutoLab PGSTAT302N electrochemical workstation. A glassy carbon electrode with a 5.0 mm diameter was used as the working electrode, along with an Ag/AgCl reference electrode and a platinum mesh (1 × 1 cm²) as the counter electrode. The reference electrode was presoaked in a 3.5 M KCl solution. Argon gas was introduced into the system to degas the solution for 30 min and was maintained throughout the tests. The glassy carbon electrode was polished sequentially with 0.5 μm and 0.03 μm alumina powders and rinsed with deionized water before use. The electrolyte volume is 100 mL. The electrode was rotated at speeds of 2800, 2400, 2000, 1600, 1200, 800, 600, and 400 r.p.m., while the potential was swept linearly from 0 V to − 0.8 V (vs. Ag/AgCl) at a scan rate of 5 mV s−¹. The diffusion-limited current densities at -0.8 V were plotted against the square root of the rotation rate (ω) and fitted using a straight line. The slope of the fit, defined by the Levich equation, was used to determine the diffusion coefficient (D) of AQDS, yielding a value of 4.09 × 10−6 cm2 s−1.
The inverse of the current at overpotentials of 32, 42, 52, 61, 71, and 81 mV was plotted against the reciprocal of the square root of the rotation rate, and the data were fitted with a straight line. The intercept of the fit gave the inverse of the kinetic current (iK). A plot of log(iK) versus overpotential was linearly fitted, with a slope of 50 mV, consistent with the Butler–Volmer equation. The x-intercept of this fit provided the exchange current (i0), which was used to calculate the standard rate constant (k0) for AQDS, giving a value of 3.2 × 10−3 cm s−1.
Linear sweep voltammetry (LSV) curves were obtained at room temperature using a CHI660E in a single cell. The platinum-plated nickel mesh (1 × 1 cm2) was used as the working electrode for all three-electrode LSV tests. An Ag/AgCl reference electrode (presoaked in 3.5 M KCl solution), and a platinum net counter electrode (1 × 1 cm2) were used for the LSV tests. The scan rate was 1 mV s−1. The electrolyte (100 mL) was 1 M KHCO3.
Hydrogen circulation verification
The electrolysis module, along with all pipeline interfaces, was sealed with epoxy resin to prevent hydrogen leakage. Prior to electrolysis, AQDS was pre-reduced to AQDSH2 using external hydrogen, and the system was subsequently purged with hydrogen. Hydrogen gas produced at the cathode was stored in a buffer tank and circulated to the redox regeneration module via an H2 gas circulation pump, where it was utilized for AQDS hydrogenation. During 0.04 A constant current electrolysis, the pressure in the hydrogen buffer tank was monitored using the drainage method. If hydrogen was consumed, the liquid level in the detection water column would rise as the buffer tank pressure dropped, enabling quantification of net hydrogen consumption by measuring the change in liquid column volume.
Hydrogen reaction rate test
In the lab-scale system, the reduction tower was filled with 50 mL of Pt/C particle catalyst, having a loading density of 0.45 g mL−1. The anolyte comprised 100 mL of a 0.1 M AQDS phosphate buffer, which circulated between the reduction tower and a reservoir. The H2 flow rate was set at 30 mL min−1, and the system operated at 50 °C. The potential of the solution was monitored in real time using an electrochemical workstation. A glassy carbon electrode (3.0 mm diameter) was employed as the working electrode, while an Ag/AgCl reference electrode (pre-soaked in a 3.5 M KCl solution) and a platinum mesh counter electrode (1 × 1 cm2) were used for the test.
Half-cell measurement
Half-cell measurements were conducted at room temperature using a CHI660E electrochemical workstation (CH Instruments) in an H-cell. A platinum-plated nickel mesh (1 × 1 cm2) was used as the working electrode for the cathode test, and a carbon felt electrode (1 × 1 cm2) was used for the anode test. An Ag/AgCl reference electrode (pre-soaked in a 3.5 M KCl solution) and a platinum mesh counter electrode (1 × 1 cm2) were employed. The catholyte (250 mL) was 1 M KHCO3, bubbled with a gas mixture (80% N2, 15% CO2, 5% O2). The anolyte (250 mL) was a 0.1 M AQDS/phosphate buffer (0.8 M KH2PO4 and 0.2 M K2HPO4). An anion membrane (FAA-PK-3-75) with a thickness of 75 ± 5 μm separates two chambers. It was soaked in a 1 M KHCO3 solution for 24 h and then washed with deionized water three to four times before use.
Full-cell measurement
Measurements for the full cell were conducted using a direct current power supply (ITECH 6911, ITECH Electronic Co., Ltd.). An anion exchange membrane (FAA-3-PK-75) with a thickness of 75 ± 5 μm was selected. It was soaked in a 1 M KHCO3 solution for 24 h and then washed with deionized water three to four times before use. The effective area of the electrode is 4 cm2. The anolyte consisted of 100 mL of a 0.1 M AQDS/phosphate buffer solution, composed of 0.8 M KH2PO4 and 0.2 M K2HPO4, while the catholyte was 100 mL of 1 M KHCO3. Both the anolyte and catholyte were introduced into the cell through 3 mm diameter PFET tubing, controlled by two magnetic pump systems (Lead Fluid, CT3001F). The reduction tower was loaded with 50 mL of catalyst and maintained at 50 °C. The anolyte was pumped into the reduction tower, and the electrolyte, along with hydrogen, flowed from the top of the tower back to the anode chamber. The hydrogen flow rate was set at 30 mL min−1. Unless otherwise specified, the electrolyte flow rate is 200 mL min−1.
Calculation of CO2 absorption or desorption, carbon-removal, hydrogen evolution and consumption efficiency
The CO2 absorption and desorption efficiency is defined as the ratio of the actual CO2 absorbed or desorbed to the theoretical CO2 absorption or desorption under the corresponding current. The CO2 absorption or desorption efficiency was determined based on Faraday’s law and thermodynamic principles (Eqs. 1–4).
where QCO2 is the amount of absorbed or desorbed CO2, tf is the final time, ti is the start time, \({{{\rm{V}}}}_{{{{\rm{CO}}}}_{2}}^{{{\rm{base}}}}\) is the flow rate of inlet CO2, \({{{\rm{V}}}}_{{{{\rm{CO}}}}_{2}}^{{{\rm{n}}}}\) is the instantaneous volumetric flow rate of outlet CO2 at nth data recording time tn, n0 is the theoretical number of moles of absorbed or desorbed CO2 calculated by means of the Faraday formula, z is the amount of charge transferred by the molecule of the substance, which is 1 in this system, P is the gas pressure, R is the ideal gas constant, T is the temperature in Kelvin, and Qthe is the theoretical amount of absorbed or desorbed CO2.
The carbon removal efficiency is defined as the ratio between the CO2 outlet concentration and the CO2 inlet concentration. The carbon-removal efficiency was calculated by means of Eq. 5.
where Qin and Qout are the CO2 flow rates at the inlet and outlet of the absorption tower, respectively. Cin and Cout are the concentrations of CO2 in the inlet and outlet concentrations of the CO2 absorption tower, respectively.
The hydrogen evolution and consumption efficiency are defined as the ratio of the hydrogen generated at the cathode and consumed at the anode to their theoretical values under the corresponding current. These efficiencies are calculated using Faraday’s law and the laws of thermodynamics (Eqs. 6–7).
where QH2 is the actual amount of hydrogen measured by gas chromatography, tf is the final time, ti is the start time, \({V}_{{H}_{2}}^{{base}}\) is the flow rate of inlet H2, \({V}_{{H}_{2}}^{n}\) is the instantaneous volumetric flow rate of outlet H2 at nth data recording time tn, and Qthe is the theoretical hydrogen volume. The hydrogen evolution efficiency and hydrogen reduction efficiency were calculated to be 99.5% and 99.7%, respectively.
Calculation of energy consumption
The electrolysis energy consumption of the cell was estimated by means of Eq. 8.
where P is the electrolytic power and t is the time. U is the average cell voltage of the electrolytic process under a specified current density, F is the Faraday constant (96485 C mol−1), η is the average current efficiency, and n0 is the number of electrons transferred.
Data availability
Source data are provided in this paper.
References
Haszeldine, R. S. Carbon capture and storage: how green can black be? Science 325, 1647–1652 (2009).
Brethomé, F. M., Williams, N. J., Seipp, C. A., Kidder, M. K. & Custelcean, R. Direct air capture of CO2 via aqueous-phase absorption and crystalline-phase release using concentrated solar power. Nat. Energy 3, 553–559 (2018).
Hammond, G. P., Akwe, S. S. O. & Williams, S. Techno-economic appraisal of fossil-fuelled power generation systems with carbon dioxide capture and storage. Energy 36, 975–984 (2011).
Rochelle, G. T. Amine scrubbing for CO2 capture. Science 325, 1652–1654 (2009).
Renfrew, S. E., Starr, D. E. & Strasser, P. Electrochemical approaches toward CO2 capture and concentration. ACS Catal. 10, 13058–13074 (2020).
Diederichsen, K. M. et al. Electrochemical methods for carbon dioxide separations. Nat. Rev. Methods Prim. 2, 68 (2022).
Eisaman, M. D. et al. CO2 separation using bipolar membrane electrodialysis. Energy Environ. Sci. 4, 1319–1328 (2011).
Gurkan, B., Simeon, F. & Hatton, T. A. Quinone reduction in ionic liquids for electrochemical CO2 separation. ACS Sustain. Chem. Eng. 3, 1394–1405 (2015).
Barlow, J. M. et al. Molecular design of redox carriers for electrochemical CO2 capture and concentration. Chem. Soc. Rev. 51, 8415–8433 (2022).
Xie, H. et al. Low-energy-consumption electrochemical CO2 capture driven by biomimetic phenazine derivatives redox medium. Appl. Energy 259, https://doi.org/10.1016/j.apenergy.2019.114119 (2020).
Jin, S., Wu, M., Gordon, R. G., Aziz, M. J. & Kwabi, D. G. pH swing cycle for CO2 capture electrochemically driven through proton-coupled electron transfer. Energy Environ. Sci. 13, 3706–3722 (2020).
Rahimi, M. et al. Carbon dioxide capture using an electrochemically driven proton concentration process. Cell Rep. Phys. Sci. 1, https://doi.org/10.1016/j.xcrp.2020.100033 (2020).
Stern, M. C., Simeon, F., Herzog, H. & Hatton, T. A. Post-combustion carbon dioxide capture using electrochemically mediated amine regeneration. Energy Environ. Sci. 6, 2505–2517 (2013).
Wang, M. & Hatton, T. A. Flue gas CO2 capture via electrochemically mediated amine regeneration: desorption unit design and analysis. Ind. Eng. Chem. Res. 59, 10120–10129 (2020).
Moss, A. B. et al. In operando investigations of oscillatory water and carbonate effects in MEA-based CO2 electrolysis devices. Joule 7, 350–365 (2023).
Liu, W., Liu, P. & Mitlin, D. Tutorial review on structure–dendrite growth relations in metal battery anode supports. Chem. Soc. Rev. 49, 7284–7300 (2020).
Li, X. et al. Redox-tunable isoindigos for electrochemically mediated carbon capture. Nat. Commun. 15, 1175 (2024).
Barlow, J. M. & Yang, J. Y. Oxygen-stable electrochemical CO2 capture and concentration with quinones using alcohol additives. J. Am. Chem. Soc. 144, 14161–14169 (2022).
Jin, S., Wu, M., Jing, Y., Gordon, R. G. & Aziz, M. J. Low energy carbon capture via electrochemically induced pH swing with electrochemical rebalancing. Nat. Commun. 13, 2140 (2022).
Zhu, P. et al. Continuous carbon capture in an electrochemical solid-electrolyte reactor. Nature 618, 959–966 (2023).
Pang, S. et al. A phenazine-based high-capacity and high-stability electrochemical CO2 capture cell with coupled electricity storage. Nat. Energy 8, 1126–1136 (2023).
Seo, H. & Hatton, T. A. Electrochemical direct air capture of CO2 using neutral red as reversible redox-active material. Nature. Communications 14, 313 (2023).
Liu, A., Musgrave III, C. B., Li, X., Goddard III, W. A. & Liu, Y. Non-aqueous alkoxide-mediated electrochemical carbon capture. Nat. Energy 9, 1415–1426 (2024).
Huskinson, B. et al. A metal-free organic-inorganic aqueous flow battery. Nature 505, 195–198 (2014).
Miyamura, H., Tobita, F., Suzuki, A. & Kobayashi, S. Direct synthesis of hydroquinones from quinones through sequential and continuous-flow hydrogenation-derivatization using heterogeneous Au-Pt nanoparticles as catalysts. Angew. Chem. Int. Ed. Engl. 58, 9220–9224 (2019).
Miao, C., Zhang, R., Hui, T., Zang, G. & Wang, J. The metal-based catalysts for selective hydrogenation of anthraquinone to produce hydrogen peroxide. Catal. Surv. Asia 27, 115–131 (2022).
Preger, Y. et al. Anthraquinone-mediated fuel cell anode with an off-electrode heterogeneous catalyst accessing high power density when paired with a mediated cathode. ACS Energy Lett. 5, 1407–1412 (2020).
Leonard, M. E. et al. Editors’ choice—flooded by success: On the role of electrode wettability in CO2 electrolyzers that generate liquid products. J. Electrochem. Soc. 167, https://doi.org/10.1149/1945-7111/abaa1a (2020).
Rheinhardt, J. H., Singh, P., Tarakeshwar, P. & Buttry, D. A. Electrochemical capture and release of crbon dioxide. ACS Energy Lett. 2, 454–461 (2017).
Voskian, S. & Hatton, T. A. Faradaic electro-swing reactive adsorption for CO2 capture. Energy Environ. Sci. 12, 3530–3547 (2019).
Liu, Y., Ye, H.-Z., Diederichsen, K. M., Van Voorhis, T. & Hatton, T. A. Electrochemically mediated carbon dioxide separation with quinone chemistry in salt-concentrated aqueous media. Nat. Commun. 11, 1–11 (2020).
Wang, M., Herzog, H. J. & Hatton, T. A. CO2 Capture using electrochemically mediated amine regeneration. Ind. Eng. Chem. Res. 59, 7087–7096 (2020).
Liu, Q., Noble, R., Falconer, J. L. & Funke, H. Organics/water separation by pervaporation with a zeolite membrane. J. Membr. Sci. 117, 163–174 (1996).
Pal, P. Industrial Water Treatment Process Technology. (Butterworth-Heinemann, 2017).
Jiang, W. et al. Electrochemically regenerated amine for CO2 capture driven by a proton-coupled electron transfer reaction. Ind. Eng. Chem. Res. 61, 13578–13588 (2022).
Goto, K., Kodama, S., Higashii, T. & Kitamura, H. Evaluation of amine-based solvent for post-combustion capture of carbon dioxide. J. Chem. Eng. Jpn. 47, 663–665 (2014).
Acknowledgements
This work is supported by Program for National Natural Science Foundation of China (Grant No.52104400 to Y.Wu, No.51827901 to H.X., No. 52374133 to Y.Wu), National Natural Science 504 Foundation of China (Grant No.U2013603 to T.L.), National Key R&D Program of China (Grant No. 2022YFB4102100 to T.L.), Guangdong Introducing Innovative and Entrepreneurial Teams (Grant No. 2019ZT08G315 to H.X.) and Tianfu Yongxing Laboratory Organized Research Project Funding (Grant No. 2023KJGG01 to H.X., Grant No. 2023KJGG02 to T.L.). We are grateful to Yiri Li and Xinchen Song for their help.
Author information
Authors and Affiliations
Contributions
H.X., T.L., and Y.Wu conceived and designed the idea. T.L., Y.Wang, Y.Wu, W.J., and Q.L. performed the characterizations and experiments. Y.Wang, Y.Wu, Y.D., C.L., and Z.Z. analyzed the data. T.L., W.J., L.Z., and D.Y. designed the devices. H.X., T.L., Y.Wu, Y.Wang, W.J., and T.N. drafted the article and revised it critically. All the authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hussain Almajed, Carl Koval, and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, T., Wang, Y., Wu, Y. et al. Continuous decoupled redox electrochemical CO2 capture. Nat Commun 15, 10920 (2024). https://doi.org/10.1038/s41467-024-55334-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55334-3
