
Abstract
The growing potential of low-dimensional metal-halide perovskites as conversion-type cathode materials is limited by electrochemically inert B-site cations, diminishing the battery capacity and energy density. Here, we design a benzyltriethylammonium tellurium iodide perovskite, (BzTEA)2TeI6, as the cathode material, enabling X- and B-site elements with highly reversible chalcogen- and halogen-related redox reactions, respectively. The engineered perovskite can confine active elements, alleviate the shuttle effect and promote the transfer of Cl- on its surface. This allows for the utilization of inert high-valent tellurium cations, eventually realizing a special eleven-electron transfer mode (Te6+/Te4+/Te2-, I+/I0/I-, and Cl0/Cl-) in suitable electrolytes. The Zn||(BzTEA)2TeI6 battery exhibited a high capacity of up to 473 mAh g-1Te/I and a large energy density of 577 Wh kg-1 Te/I at 0.5 A g-1, with capacity retention up to 82% after 500 cycles at 3 A g-1. The work sheds light on the design of high-energy batteries utilizing chalcogen-halide perovskite cathodes.
Similar content being viewed by others


Introduction
The need for efficient and affordable energy storage devices to manage intermittent renewable energy has kindled immense interest in functional perovskite materials, representing a distinctive class of ionic crystals comprising several oxide and halide structures with similar octahedral features1,2,3,4. ABO3 (A and B are divalent and tetravalent metallic cations, respectively) structured oxide perovskites are known as promising ferroelectric, dielectric, magnetic, and energy storage materials5,6,7. Owing to the abundance of oxygen vacancies, oxide perovskites have been successfully applied as catalytic electrode materials8,9,10,11. Instead, halide perovskites (ABX3, A is monovalent cation, B is divalent cation, and X is I, Br, or Cl) possess relatively narrower bandgaps due to the large difference in electronegativity between halogens and oxygen, thereby used for optoelectronic applications such as the light absorber and emissive layer in solar cells and light-emitting diodes, respectively12,13,14,15. Nevertheless, the study of halide perovskites in batteries has reached a rudimentary stage, though being contained to a subsidiary role in storage applications.
The research on molecular-level low-dimensional (LD) crystals such as two-dimensional (2D), one-dimensional (1D), and zero-dimensional (0D) perovskites are advanced in the meanwhile due to demand for structural stability16,17,18,19, some of which reflect deficient formation energy at ambient conditions being suitable for multifunctional purposes20,21,22. Among them, the LD organic-inorganic hybrid perovskites demonstrate a substantial quantum and dielectric confinement due to the breakdown of three-dimensional (3D) frameworks as the inclusion of insulating organic moieties23. This special lattice ordering promotes improved structural stability and expanded bandgaps that bring out an exciting intersection of the halide and oxide perovskites’ applications, including ferroelectricity and energy storage devices24,25,26,27,28,29,30,31,32.
Unlike ancillary oxide perovskites that provide oxygen vacancies for metal-air batteries33,34, halide perovskites perform as the halogen reservoir and restrain the shuttle effect by using Van Der Waals forces (hydrogen bonds and halogen bonds) and steric hindrance from A-site organics, eventually realizing a three-electron transfer process35. However, those encouraging attempts to develop halide perovskite cathodes failed to avail the full range of perovskite materials given that the redox reaction of the B-site cations sits electrochemical inert during the conversion process (of halogens). This undermines the whole discharge capacity. In this regard, replacing those noble B-site metal cations with tetravalent chalcogenide cations to construct chalcogen-halide octahedra is expected to offer full utilization of cathode materials and guarantee a reliable multiple electron transfer. Nevertheless, the high-valent hexavalent and tetravalent chalcogen cations are neither stable nor electrochemically active in aqueous electrolytes, causing irreversible redox processes in batteries36,37,38,39,40.
Herein, we designed a benzyltriethylammonium tellurium iodide perovskite, (BzTEA)2TeI6, as the cathode material for problem-solving and demonstrated its special chemical processes in aqueous zinc ion batteries. In our design, the high charge density surrounding each tetravalent Te4+ cation causes the formation of the tellurium-iodide octahedron unit, which is embedded in the A-site organic ligands (BzTEA) matrix and supplies high elemental iodine and tellurium content of over 71 wt.%. The robust Van Der Waals forces such as Te-I…Cl-Te and Te-I…I halogen bonds on the perovskite surface promote the localization of active elements and avert the undesired shuttling of oxidative polyiodide and tellurium polychloride ions, as supported by the density functional theory (DFT) calculations. Consequently, the Zn||(BzTEA)2TeI6 battery delivers a highly reversible eleven-electron transfer mode on account of the two-electron I+/I0/I- and eight-electron Te6+/Te4+/Te0/Te2- redox reactions, and one-electron Cl0/Cl- transfer gained from the chloride electrolyte, all of which favor a high discharge capacity up to 473 mAh g-1Te/I and a large energy density of 577 Wh kg-1Te/I at 0.5 A g-1 represented by five prominent voltage plateaus at 1.81 V, 1.64 V, 1.53 V, 1.26 V, and 0.51 V. The feasibility of the (BzTEA)2TeI6 perovskite cathode is further verified by the zinc pouch cells.
Results
(BzTEA)2TeI6 perovskite cathode
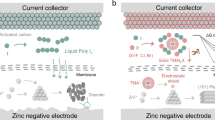
Chalcogens, such as sulfur, selenium, and tellurium, display a broad spectrum of valence states (−2, 0, +2, +4, +6) and exhibit significant redox potentials (Fig. 1a). This characteristic theoretically renders them suitable for energy storage, while the stability of high-valent chalcogen cations remains a crucial consideration. Halogens, on the other hand, are proven effective in giving high redox potentials in aqueous zinc ion batteries41,42. A proper integration of chalcogen and halogen chemistry should promote problem-solving and actualize high-energy zinc ion batteries. Implementing halogen redox in halide perovskites brings our attention to potential structural design and prompts the exploration of chalcogen halide perovskites as cathode materials. Actually, the typical ABX3 perovskites consist of corner-sharing [BX6]4- octahedra and offset of A-site atoms in octahedrons cavities throughout the whole 3D matrix43. In contrast, the molecular-level LD perovskite materials crystalize in a way where the [BX6]4- octahedron unit is separated by A-site cations in specific directions and upholds a high structural tunability (Supplementary Fig. 1)44,45. As such, we have now aim to replace conventional electrochemical inert B-site cations with tetravalent chalcogenide cations while retaining the octahedral BX6 motif to enable the formation of A2BIVX6 vacancy-ordered perovskites46. This change is expected to maximize the utilization of perovskite materials by enabling multivalent reactions (B2-/B0/B4+/B6+) of both chalcogen and halogen elements, as depicted in Fig. 1a. The special lattice arrangement ensures the confinement of chalcogen and halogen elements in the same perovskite structure, which creates the platform for chalcogen- and halogen-related redox reactions for high-energy batteries. As summarized in Supplementary Table 1, the cogitation theoretically upholds high redox potential and multiple electron transfer associated with B-site chalcogen and X-site halogens of the perovskite cathode materials, attempting to overcome the deficiency of conventional chalcogen cathodes and enable the electrochemically inert high-valent chalcogen redox47,48. As a proof-of-concept, (BzTEA)2TeI6 perovskite, which possesses a high proportion of active elements (>71 wt.%), is thus proposed as conversion-type cathodes for aqueous zinc ion batteries with a special eleven-electron transfer process. (Fig. 1b).

a Redox potential of chalcogens and halogens. b Structural arrangement of low-dimensional (BzTEA)2TeI6 perovskite where tellurium and iodine elements sit as the B- and X-site components, respectively. The CV curves of the perovskite cathode in c 2 M ZnSO4 (first two cycles), d 30 M ZnCl2, and e Ch0.4Zn0.6Cl1.6·1.5H2O.
Regardless of the lattice arrangement at molecular levels, (BzTEA)2TeI6 microcrystals prepared by a modified saturation recrystallization method crystallized into bulk rod shape with an average length of less than 50 µm, as illustrated in the scanning electron microscope (SEM) image in Supplementary Fig. 219. The perovskite microcrystals delivered identical XRD diffractions with the theoretical simulation in Supplementary Fig. 3 and appeared black due to its narrow optical bandgap, aligning with the ultraviolet absorption spectra in Supplementary Fig. 4. SEM-mapping in Supplementary Fig. 5 further revealed the even elemental distribution of (BzTEA)2TeI6 microcrystals associated with 14.5 at.% of Te and 85.5 at.% of I according to the energy-dispersive X-ray spectroscopy (EDS) test. Fourier Transform Infrared (FTIR) spectra in Supplementary Fig. 6 indicated the protonation of A-site N-H bond in (BzTEA)2TeI6. Thermogravimetric analysis (TGA) and derivative thermogravimetry (DTG) curves in Supplementary Fig. 7 confirmed the structural stability and hydrophobic feature of the tellurium iodide perovskite according to the negligible weight loss below 211 °C. Raman spectra in Supplementary Fig. 8 reveal symmetric A1g and asymmetric Eg stretching of Te-I bond at approximately 153 cm-1 and 107 cm-1 in (BzTEA)2TeI6, resembling the Te-O bond observed in TeO249.
Tellurium and halogen redox
The halide perovskite cathode was first examined in Zn||2 M ZnSO4||(BzTEA)2TeI6 Swagelok cells and achieved with two redox peaks, corresponding to I0/I- and Te4+/Te0, the latter of which experienced fast decay of current according to the cyclic voltammetry (CV) curves in Fig. 1c. In comparison, 2 M Zn(OTF)2, 2 M Zn(OAc)2, and 15 M ZnCl2 electrolytes all failed to activate the redox reaction of high-valent tellurium cations (Supplementary Figs. 9, 10). 30 M ZnCl2, containing reduced amount of free water, was then used as the electrolyte with an attempt to stabilize the tetravalent tellurium cations50. Surprisingly, it enabled the perovskite cathode with an additional Te6+/Te4+ redox at around 1.5 V. It should be noted that the Te6+/Te4+ redox was utterly absent in the “TeO2 + ZnI2” and “Te + I2” cathodes (Fig. 1d and Supplementary Fig. 11), which reveals the special structure of (BzTEA)2TeI6 for activation of the new redox. The observation also suggested that the activity of water and chloride anions may play an essential role in stabilizing high-valent tellurium cations. Based on that, we introduced choline chloride (ChCl) into the concentrated ZnCl2 electrolyte to devise the Ch0.4Zn0.6Cl1.6 · 1.5H2O electrolyte, which was found to have limited water activity and enough mobility of chloride ions to coordinate with high-valent tellurium cations during cycling. As shown in Fig. 1e, the activity endowed Zn||Ch0.4Zn0.6Cl1.6 · 1.5H2O||(BzTEA)2TeI6 prominent discharge peaks, including Te6+/Te4+ and Te4+/Te0 redox pairs at 1 mV. In sharp contrast, both “TeO2” and “Te + I2” cathodes failed to enforce the Te6+/Te4+ redox even when coupled with the same modified electrolyte, again highlighting the special chemical design of (BzTEA)2TeI6 (Supplementary Fig. 12).
In order to figure out the critical role of (BzTEA)2TeI6 perovskite structure for the rich chemistry, we subsequently conducted detailed electrochemical studies of these batteries. As shown in Fig. 2a, the “TeO2 + ZnI2” cathode presented two types of redox, including I0/I+/I- and Cl0/Cl-, which experienced an unusual attenuation as the scan rate increased, together with a mild Te6+/Te4+ redox reaction probably due to the catalytic role of iodine atom (Supplementary Fig. 13). The (BzTEA)2TeI6 cathode, on the contrary, featured four sharp discharge peaks from the Cl0/Cl-, I+/I0, Te6+/Te4+, and Te0/Te2- redox pairs and one broad peak due to the overlay of Te4+/Te0 and I0/I- as will be discussed later (Fig. 2b and Supplementary Fig. 14). Further, as illustrated in Fig. 2c, the surface-controlled process of the battery based on perovskite cathodes gradually grew from 21.5% at 0.5 mV s-1 to 40.2% at 3 mV s-1. In comparison, that for the “TeO2 + ZnI2” cathode started from 11.1% at 0.5 mV s-1 to 23.5% at 3 mV s-1 due to weak adsorption of active elements at a high scan rate (Supplementary Fig. 15). The derived b value of each cathodic peak generally fell between 0.5 and 1.0 and exhibited a combined action of the faradic and capacitive processes during the conversion reactions (Fig. 2d)51. However, the b values of the control sample were far below 0.5 and turned negative for Cl0/Cl-, suggesting the poor cycling stability of the “TeO2 + ZnI2” cathode and uncontrolled loss of chlorine elements (Supplementary Figs. 15, 16). The summary in Fig. 2e demonstrated their difference in b values. It highlighted the importance of (BzTEA)2TeI6 and ChCl for properly operating batteries, aside from taking full advantage of the B-site tellurium elements. As a comparison, the Zn||(BzTEA)2TeI6 battery in 30 M ZnCl2 experienced severe degradation under identical test conditions, even though it delivered a Te0/Te2- redox pair with a larger b value (0.6) and a mild Te6+/Te4+ redox pair (Supplementary Fig. 17). Figure 2f presents a typical galvanostatic discharge curve of Zn||(BzTEA)2TeI6 battery at 0.5 A g-1, which brought out an eleven-electron transfer process, including three-electron transfer from the iodine element, eight-electron transfer from tellurium element and one from chlorine element, following the result from the differential capacity (dQ/dV) plot in Supplementary Fig. 18.

CV curves of batteries coupled with a “TeO2 + ZnI2” cathode and b “(BzTEA)2TeI6” cathode in Ch0.4Zn0.6Cl1.6·1.5H2O electrolyte. c Diffusion-/surface-controlled contribution at different scan rates and d the fitting plots between log(i) and log(v) of the cathodic peaks, and e the derived b value a summary diagram. f Discharge curve at a current density of 0.5 A g−1 demonstrates the redox reactions of tellurium and halogen elements.
Mechanism of the eleven-electron transfer
The Raman spectra recorded during the charge and discharge process indicated the presence of I5- at 169 cm-1, consistent with theoretical calculations, while signals from high-valence tellurium cations were indistinct (Supplementary Fig. 19). This may be due to the overlap of Te6+ signal with the [ZnCl2+x(H2O)y] cluster at around 300 cm-1 and the Te4+ signal below 100 cm-1 being affected by intense Rayleigh scattered laser light (noise)33,40,52,53. The XRD and its enlarged patterns in Supplementary Fig. 20 reveal a broad peak around 10°, close to the main diffraction peaks of (BzTEA)2TeI6 perovskites, whereas the blank electrode consisting solely of Ketjen black and PVDF binder exhibits no characteristic peak at that position. X-ray photoelectron spectroscopy (XPS) measurements were further performed to evaluate the perovskite cathode’s conversion reactions. As demonstrated in Fig. 3a, Te0 and Te2- dominated at discharge states; the signal of Te4+ first appeared at 576 eV when charged to 1.2 V and shifted to 576.9 eV, verifying the formation of Te6+. The presence of Te-O at 579.2 eV is attributed to the oxidation of high-valent tellurium ions in damp air. The Cl 2p3/2 core level spectra in Fig. 3b exhibit the signals from lattice Cl- changing from 198.9 eV at 0.5 eV, 199.4 eV at 1.7 V, to 199.6 eV at 1.9 V, contributing to an energy span of over 0.7 eV. Instead, the adsorbed Cl- changed from 198.2 eV at 0.5 V to 198.7 eV at 1.7 V and slightly increased to 198.73 eV at 1.9 V, corresponding to an energy change of 0.5 eV. The energy evolution of the two types of Cl- revealed that a certain degree of (dynamic) halide exchange between iodide and chloride ions could promote a reliable Cl0/Cl- redox reaction, consistent with the CV performance discussed above. The I 3d3/2 core level spectra in Fig. 3c demonstrate the conversion from I+ at 1.7 V, to I0 at 1.3 V and I- at 0.5 V, verifying the successful operation of the I+/I0/I- redox pairs54.

XPS spectra of a Te 3d, b Cl 2p, and c I 3d3/2 core levels.
Molecular dynamics (MD) simulations and DFT calculations were subsequently conducted to understand the working principle of the chalcogen halide perovskite cathode. As depicted in Fig. 4a, the ZnCl2 · 1.4H2O electrolyte was dominated by tetrahedral [ZnCl4]2- and octahedral [ZnCl4(H2O)2]2- clusters. Notably, as the inclusion of ChCl, the coordinated water molecules surrounding Zn2+ increased in the form of [ZnCl3(H2O)3]- and octahedral [ZnCl2(H2O)4] cluster, giving rise to the free Cl- radicals that sat in the electrolyte voids (Supplementary Fig. 21). The mean squared displacement (MSD) study in Fig. 4b proved a much higher mobility of Cl- ion in Ch0.4Zn0.6Cl1.6 · 1.5H2O (4.30 ×10-8 cm2 s-1) while that for ZnCl2 · 1.4H2O was 8.64 ×10-9 cm2 s-1. The radial distribution function (RDF) result in Fig. 4c confirmed that the coordination number of Cl- decreased from 3.6 to 3.3 after introducing ChCl. Moreover, the amorphous features of Ch0.4Zn0.6Cl1.6 · 1.5H2O and the absence of undissolved solutes emphasized that the designed electrolyte was a homogeneous solution (Supplementary Figs. 22, 23), which offered high Cl- mobility to compensate and stabilize high-valent tellurium ions.

a Visualization of ZnCl2·1.4H2O (left) and Ch0.4Zn0.6Cl1.6·1.5H2O (right) electrolytes. b MSD and c RDF spectra derived from MD simulations. d Migration energy of chloride ions on TeO2 and perovskite surface. e Formation energy of associated redox pairs in vacuum and on perovskite surface. f Surface adsorption energy of (BzTEA)2TeI6 toward intermediate tellurium and halogen elements. Inset in f denotes the coordination between perovskite and TeCl5+ ions.
Of note, the (BzTEA)2TeI6 perovskite surface synergistically provided a fast channel for transporting Cl- ions, favoring the stabilization of high-valent tellurium ions. As shown in Fig. 4d, the migration energy of Cl- along the (1 0 −1) lattice plane of perovskite oscillated from −9.5 to 9.6 eV. In contrast, the amplitude of the migration barrier on TeO2 soared to over 156 eV. In addition to kinetic influences, the conversion reactions on the (BzTEA)2TeI6 surface were thermodynamically preferred. Specifically, as demonstrated in Fig. 4e, the formation energy of critical chalcogen- and halogen-related redox reactions all decreased, especially for TeCl3+ and TeCl5+, which reduced from 7.7 eV to 5.8 eV and 3.0 eV to 0.8 eV, respectively. Besides, the presence of perovskite surface ended up enhancing the confinement ability toward chalcogen and halogen elements by virtue of halogen bonds (such as Te-I…Cl-Te and Te-I…I) and positive dangling bonds, as summarized in Fig. 4f. The effective coordination among perovskite surface, high-valent cations, and passivating Cl- ligand jointly favored their stabilization and associated redox reactions (Supplementary Figs. 24, 25). The more significant adsorption energy of I5- (−1.97 eV) than I3- (−1.30 eV) helped explain the proliferation of I5- during the charging process, pointing out improved reaction kinetics and suppressed shuttle effects (Supplementary Fig. 26).
Electrochemical performance of Zn||(BzTEA)2TeI6 battery
We subsequently assembled batteries to examine the practical performance of the perovskite cathode in the Ch0.4Zn0.6Cl1.6 · 1.5H2O electrolyte. As presented in Fig. 5a, the Zn||(BzTEA)2TeI6 battery delivered a steady capacity of 473 mAh g-1Te/I at 0.5 A g-1 and 258 mAh g-1Te/I at 3 A g-1, which reverted to 411 mAh g-1Te/I after current reset, showing a high capacity retention of 87%. The relatively lower coulombic efficiency (CE) at lower C-rates could be attributed to the loss of chlorine gas during the slow charge/discharge process (Supplementary Fig. 27). This loss was suppressed at higher C-rates due to the shorter cycle time and saturated Cl₂ dissolution in the electrolytes. The corresponding galvanostatic charge-discharge (GCD) curves in Fig. 5b show five stable discharge plateaus at 1.81 V (for Cl0/Cl-), 1.64 V (for I+/I0), 1.53 V (for Te6+/Te4+), 1.26 V (for overlayed Te4+/Te0 and I0/I-), and 0.51 V (for Te0/Te2-), outperforming the Zn||Ch0.4Zn0.6Cl1.6·1.5H2O||“TeO2 + ZnI2” and Zn||30 M ZnCl2||(BzTEA)2TeI6 batteries (Supplementary Fig. 28) The highly reversible chalcogen- and halogen-related redox reactions jointly contributed to an eleven-electron transfer mode and verified the feasibility of the organic-inorganic hybrid tellurium iodide perovskite cathode. Figure 5c highlights the advantages of the (BzTEA)2TeI6 perovskite cathode that offered a record capacity compared with related references55,56,57,58,59,60,61,62,63. Long-term cycling performance was provided in Fig. 5d, e and Supplementary Fig. 29. The Zn||(BzTEA)2TeI6 battery successfully cycled 500 times at 1 A g-1 and 3 A g-1 with a high CE of approaching 98% and capacity retention of over 77% and 82%, respectively, superior to the reported counterparts (Supplementary Fig. 30). Moreover, the Zn||(BzTEA)2TeI6 battery retained 79.4% of its initial capacity after a storage period of 5 h due to the suppressed shuttle effect, while that for the Zn||“TeO2 + I2” battery was down to 16.7% (Fig. 5f and Supplementary Fig. 31). The viability of the Zn||(BzTEA)2TeI6 battery was additionally confirmed using a DC-DC converter, which maintained a consistent voltage output of 3.29 V for more than 250 min (Supplementary Fig. 32). As a summary, Fig. 5g emphasizes the importance of the special eleven-electron transfer mode actualized by the perovskite cathode and proper electrolyte design39,61,62,63,64,65,66,67,68. The structural advance endowed the zinc ion batteries with a high average voltage of 1.3 V and a large energy density of over 577 Wh kg-1Te/I. The pouch cell based on the (BzTEA)2TeI6 perovskite cathode further gave a high capacity of 113 mAh (at 10 mA cm-2) with a capacity retention of over 66% after 100 cycles (Fig. 5h). The pouch cell also presented good storage stability with a capacity loss of less than 24% after a resting time of 12 h (Supplementary Fig. 33).

a Rate performance, b the corresponding GCD curves, and c comparison with typical cathode of zinc ion batteries. d Long-term cycling property of zinc battery at 1 A g−1, e selected GCD curves, and f the self-discharge profile. g comparison of relative aqueous zinc ion batteries regarding average voltage and energy density. h Cycling performance of the pouch cell with a high loading mass of 12 mg cm−2. The sources of referential samples in c and g are cited as ref. 55 for I2, ref. 56 for MnO2, ref. 58 for Te, ref. 59 for KMO (K0.27MnO2·0.54H2O), ref. 60 for AVO (Ag0.3V2O5), ref. 61 for IBr, ref. 63 for V2O5, ref. 62 for Co3O4, ref. 64 for PTZAN [4,4’-(10H-phenothiazine-3,7-diyl)bis(N,N-diphenylaniline)], ref. 65 for PANI (Polyaniline), ref. 66 for MnO2, ref. 67 for I2, ref. 61 for IBr, ref. 39 for Te, ref. 68 for PPy (Polypyrrole).
Discussion
In conclusion, the designed (BzTEA)2TeI6 perovskite, as a proven methodology to actualize both B-site chalcogen and X-site halogen redox reactions, were examined upfront as the conversion-type cathode materials. The (BzTEA)2TeI6 cathode effectively confined active chalcogen and halogen elements and allowed fast transfer of chloride ions, stabilizing high-valent tellurium cations in the form of tellurium chloride ions. After coupling with the adaptive Ch0.4Zn0.6Cl1.6 · 1.5H2O electrolyte, an eleven-electron transfer was successfully realized in the Zn||(BzTEA)2TeI6 batteries as the emerging redox pairs including Cl0/Cl-1 (1.81 V), I+/I0/I- (1.64 and 1.26 V), Te6+/Te4+ (1.53 V), Te4+/Te0 (1.26 V), and Te0/Te2- (0.51 V), all of which benefited a high energy density of over 577 Wh kg-1Te/I. As a result, the Zn||(BzTEA)2TeI6 battery demonstrated a high capacity of 473 mAh g-1Te/I at 0.5 A g-1 and experienced 500 cycles at 1 A g-1 with a capacity of over 77%. The corresponding pouch cell delivered a high capacity of 113 mAh, showcasing promising storage stability.
Methods
Chemicals and reagents
Tellurium oxide (≥99%), benzyltriethylammonium chloride (BzTEACl or TEBAC, 98%), hydrogen iodide (HI, 48%), isopropanol (99%), zinc chloride (98%), choline chloride (ChCl, 98%), zinc sulfate (ZnSO4, AR), zinc acetate (Zn(OAc)2, 99%), zinc trifluoromethanesulfonate (Zn(OTF)2, 98%) and 1-methyl-2-pyrrolidinone (NMP, 98%) were purchased from Aladdin. Polyvinylidene difluoride (PVDF) was purchased from SOLVAY (Solef 1008). The conductive agent, Ketjenblack EC-300J, was purchased from Nouryon. All chemicals were used as received without further treatment.
Preparation of (BzTEA)2TeI6 microcrystal cathode and battery assembly
The perovskite microcrystals were obtained by a saturated recrystallization method. Specifically, 0.1 mmol of TeO2 and 0.2 mmol of BzTEACl were added into 1 ml HI solution. The mixture was heated to 110 °C and held for over 1 h with vigorous stirring, subsequently cooling to room temperature. The obtained black residue was washed with isopropanol and dried under a vacuum. The (BzTEA)2TeI6 perovskite microcrystals were mixed and ground with Ketjenblack and PVDF blinder in NMP with a mass ratio of 7:2:1 for 1 h. The obtained liquid slurry was evenly covered on the carbon cloth substrate, followed by a vacuum bakeout process at around 80 °C overnight. The mass loading of the perovskite cathode was estimated to be 1 ~ 1.2 mg cm-2. 39 mmol ZnCl2 was dissolved in 1 ml H2O at 100 °C and slowly cooling to 30 °C, and the clear supernatant was used as the ZnCl2 · 1.4H2O electrolyte. 0.4 mol ChCl and 0.6 mol ZnCl2 were dissolved in 1.5 mol H2O to form the Ch0.4Zn0.6Cl1.6 · 1.5H2O electrolyte. Specifically, 1 g of Ch0.4Zn0.6Cl1.6 · 1.5H2O contains 0.16 g H2O, 0.51 g ChCl, and 0.33 g ZnCl2. The cathode, zinc metal anode (100 µm, 1 cm2), and a glass fiber (420 µm, 1.1 cm2) sat in between were packed in a Swagelok cell for further evaluation (at room temperature without the need for a chamber).
Material and electrochemical characterizations
Absorption spectra were collected using a Shimadzu UV 3600 UV/visible/IR spectrophotometer, while Fourier-transform infrared (FTIR) measurements were conducted with a Perkin Elmer FT-IR spectrophotometer. Raman measurements were performed on a WITec Alpha300 R confocal Raman imaging system with a 532 nm laser. Powder X-ray diffraction (XRD) patterns were obtained using a Rigaku Smartlab X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å). X-ray photoelectron spectroscopy (XPS) was carried out on a PHI model 5802, with the carbon spectrum serving as a calibration reference. The FEI Quanta 250 scanning electron microscope (SEM) and energy dispersive spectroscopy (EDS) were utilized to examine the morphology and elemental composition of the samples. Cyclic voltammograms (CVs) and related electrochemical data were collected on a CHI 660E electrochemical workstation. The long-term stability and rate performance of the batteries were evaluated using the LAND battery testing system at room temperature (with a cut-off potential of 2.04 V).
Calculation and theoretical simulations
The b value was derived from the equation \(i=a{v}^{b}\) where \(i\) is the response current, \(v\) is the sweep rate. The b value of 0.5 signifies a purely faradaic (diffusion-controlled) process while a b value of 1 indicates a purely capacitive (surface-controlled) process, as outlined in the initial report by Lindquist et al. in 199769. The quantitative contribution of the charge storage process was evaluated by the equation \(i={k}_{1}v+{k}_{2}{v}^{0.5}\) where \({k}_{1}v\) is the surface-controlled process and \({k}_{2}{v}^{0.5}\) is the diffusion-controlled part33. The energy density was calculated by integrating the battery voltage over the specific capacity which was determined based on the mass of active elements (tellurium and iodine). The density functional theory (DFT) was employed for first-principles calculations using the Dmol3 mode within a numerical atom-centered basis function framework. The Perdew-Burke-Ernzerhof (PBE) method was used for electronic exchange-correlation interactions, while the Generalized Gradient Approximation (GGA) method with PBE formulation was applied for structural optimization70. DFT semi-core pseudopotentials were chosen for the core treatment of relativistic effects, replacing core electrons with a single effective potential71. The adsorption energy, \({E}_{{ads}}\), was calculated using the formula \({E}_{{ads}}={E}_{{ensemble}}-{E}_{{absorbent}}-{E}_{{adsorbate}}\), where the subscript represents the energy of the absorbent, adsorbate, or the entire system post adsorption72. The Raman spectrum of I5- was simulated under the excitation of 532 nm laser at 273 K, with Lorentzian smearing of 20 cm-1.
Data availability
All data that support the findings of this study are presented in the manuscript and Supplementary Information or are available from the corresponding author upon request. Source data are provided with this paper.
References
Ricciardulli, A. G., Yang, S., Smet, J. H. & Saliba, M. Emerging Perovskite Monolayers. Nat. Mater. 20, 1325–1336 (2021).
Morteza Najarian, A. et al. Homomeric Chains of Intermolecular Bonds Scaffold Octahedral Germanium Perovskites. Nature 620, 328–335 (2023).
Wang, J. et al. Templated Growth of Oriented Layered Hybrid Perovskites on 3D-Like Perovskites. Nat. Commun. 11, 582 (2020).
Li, J. et al. Grain-Orientation-Engineered Multilayer Ceramic Capacitors for Energy Storage Applications. Nat. Mater. 19, 999–1005 (2020).
Yang, B. et al. Engineering Relaxors by Entropy for High Energy Storage Performance. Nat. Energy 8, 956–964 (2023).
Yang, L. et al. Perovskite Lead-Free Dielectrics for Energy Storage Applications. Prog. Mater. Sci. 102, 72–108 (2019).
Sun, C., Alonso, J. A. & Bian, J. J. A. E. M. Recent Advances in Perovskite‐Type Oxides for Energy Conversion and Storage Applications. ACS Energy Lett. 11, 2000459 (2021).
Tan, P., Liu, M., Shao, Z. & Ni, M. Recent Advances in Perovskite Oxides as Electrode Materials for Nonaqueous Lithium–Oxygen Batteries. Adv. Energy Mater. 7, 1602674 (2017).
Zhang, L. et al. Lithium Lanthanum Titanate Perovskite as An Anode for Lithium Ion Batteries. Nat. Commun. 11, 3490 (2020).
Deng, R. et al. An Aqueous Electrolyte Densified by Perovskite SrTiO3 Enabling High-Voltage Zinc-Ion Batteries. Nat. Commun. 14, 4981 (2023).
Bai, Z. et al. Engineering Triple-Phase Interfaces Enabled by Layered Double Perovskite Oxide for Boosting Polysulfide Redox Conversion. Nano Lett. 23, 4908–4915 (2023).
Wang, S. et al. CsPbI3/PbSe Heterostructured Nanocrystals for High-Efficiency Solar Cells. ACS Energy Lett. 5, 2401–2410 (2020).
Liu, H. et al. Pure-Red Electroluminescence of Quantum-Confined CsPbI3 Perovskite Nanocrystals Obtained by the Gradient Purification Method. Mater. Today Energy 41, 101533 (2024).
Fakharuddin, A. et al. Perovskite Light-Emitting Diodes. Nat. Electron. 5, 203–216 (2022).
Liu, J. et al. Electron Injection and Defect Passivation for High-Efficiency Mesoporous Perovskite Solar Cells. Science 383, 1198–1204 (2024).
Hong, Y. et al. Local-Electrostatics-Induced Oxygen Octahedral Distortion in Perovskite Oxides and Insight into the Structure of Ruddlesden–Popper Phases. Nat. Commun. 12, 5527 (2021).
Zhu, T. & Gong, X. Low-Dimensional Perovskite Materials and Their Optoelectronics. Nat. Commun. 3, 1039–1069 (2021).
Hoye, R. L. Z. et al. The Role of Dimensionality on the Optoelectronic Properties of Oxide and Halide Perovskites, and their Halide Derivatives. Adv. Energy Mater. 12, 2100499 (2022).
Biswas, A. et al. Lead-Free Zero Dimensional Tellurium (IV) Chloride-Organic Hybrid with Strong Room Temperature Emission as A Luminescent. Mater. J. Mater. Chem. C. 9, 4351–4358 (2021).
Wang, S. et al. Strongly Luminescent Dion–Jacobson Tin Bromide Perovskite Microcrystals Induced by Molecular Proton Donors Chloroform and Dichloromethane. Adv. Funct. Mater. 31, 2102182 (2021).
Wang, S., Kershaw, S. V. & Rogach, A. L. Bright and Stable Dion-Jacobson Tin Bromide Perovskite Microcrystals Realized by Primary Alcohol Dopants. Chem. Mater. 33, 5413–5421 (2021).
Qi, J., Wang, S., Portniagin, A., Kershaw, S. V. & Rogach, A. L. Room Temperature Fabrication of Stable, Strongly Luminescent Dion–Jacobson Tin Bromide Perovskite Microcrystals Achsieved through Use of Primary Alcohols. Nanomater 11, 2738 (2021).
Na Quan, L. et al. Edge Stabilization in Reduced-Dimensional Perovskites. Nat. Commun. 11, 170 (2020).
Li, L. et al. 2D Antiferroelectric Hybrid Perovskite with a Large Breakdown Electric Field and Energy Storage Density. Adv. Funct. Mater. 33, 2305524 (2023).
Manzi, M. et al. Ferroelectricity in Hybrid Perovskites. J. Phys. Chem. Lett. 14, 3535–3552 (2023).
Deswal, S. et al. Neutral 1D Perovskite-Type ABX3 Ferroelectrics with High Mechanical Energy Harvesting Performance. Chem. Mater. 32, 8333–8341 (2020).
Li, D. et al. A Novel Zero-Dimensional Organic–Inorganic Hybrid Ferroelectric Material. J. Mater. Chem. C. 12, 4267–4272 (2024).
Ali, M. S. et al. Ultrahigh Energy Density Solid State Supercapacitor Based on Metal Halide Perovskite Nanocrystal Electrodes: Real-Life Applications. J. Energy Storage 65, 107215 (2023).
Jung, Y.-K., Han, I. T., Kim, Y. C. & Walsh, A. Prediction of High Thermoelectric Performance in the Low-Dimensional Metal Halide Cs3Cu2I5. Npj Comput. Mater. 7, 51 (2021).
Li, N. et al. Shedding Light on the Energy Applications of Emerging 2D Hybrid Organic-Inorganic Halide Perovskites. iScience 25, 103753 (2022).
Tewari, N., Shivarudraiah, S. B. & Halpert, J. E. Photorechargeable Lead-Free Perovskite Lithium-Ion Batteries Using Hexagonal Cs3Bi2I9 Nanosheets. Nano Lett. 21, 5578–5585 (2021).
Zhou, Y. et al. Cesium Lead Bromide Perovskite-Based Lithium–Oxygen Batteries. Nano Lett. 21, 4861–4867 (2021).
Wang, S. et al. Conversion‐Type Organic‐Inorganic Tin‐Based Perovskite Cathodes for Durable Aqueous Zinc‐Iodine Batteries. Adv. Energy Mater. 13, 2300922 (2023).
Li, X. et al. Perovskite Cathodes for Aqueous and Organic Iodine Batteries Operating Under One and Two Electrons Redox Modes. Adv. Mater. 36, e2304557 (2023).
Wang, S. et al. Halide Exchange in Perovskites Enables Bromine/Iodine Hybrid Cathodes for Highly Durable Zinc Ion Batteries. Adv. Mater. 36, e2401924 (2024).
Chen, Z. et al. Aqueous Zinc-Tellurium Batteries with Ultraflat Discharge Plateau and High Volumetric Capacity. Adv. Mater. 32, e2001469 (2020).
Wu, S. C. et al. High-Performance Rechargeable Aluminum-Selenium Battery with a New Deep Eutectic Solvent Electrolyte: Thiourea-AlCl3. ACS Appl. Mater. Interfaces 12, 27064–27073 (2020).
Chen, Z. et al. Zinc/Selenium Conversion Battery: a System Highly Compatible with Both Organic and Aqueous Electrolytes. Energy Environ. Sci. 14, 2441–2450 (2021).
Wang, H. et al. MoO2 Nanoclusters Embedded in Hierarchical Nitrogen Doped Carbon Nanoflower as Electrocatalytic Mediators in Aqueous Zinc-Tellurium Batteries: Enhancing Electrochemical Kinetics of Tellurium Redox Reaction. Small 19, e2304504 (2023).
Chen, Z. et al. Tellurium with Reversible Six-Electron Transfer Chemistry for High-Performance Zinc Batteries. J. Am. Chem. Soc. 145, 20521–20529 (2023).
Li, G. et al. Developing Cathode Materials for Aqueous Zinc Ion Batteries: Challenges and Practical Prospects. Adv. Funct. Mater. 34, 2301291 (2023).
She, L. et al. Rechargeable Aqueous Zinc-Halogen Batteries: Fundamental Mechanisms, Research Issues, and Future Perspectives. Adv. Sci. 11, e2305061 (2024).
Sun, J.-K. et al. Polar Solvent Induced Lattice Distortion of Cubic CsPbI3 Nanocubes and Hierarchical Self-Assembly into Orthorhombic Single-Crystalline Nanowires. J. Am. Chem. Soc. 140, 11705–11715 (2018).
Zhu, P. & Zhu, J. Low‐Dimensional Metal Halide Perovskites and Related Optoelectronic Applications. InfoMat 2, 341–378 (2020).
Zhou, C. et al. Low Dimensional Metal Halide Perovskites and Hybrids. Mater. Sci. Eng. R. Rep. 137, 38–65 (2019).
Kavanagh, S. R. et al. Frenkel Excitons in Vacancy-Ordered Titanium Halide Perovskites. (Cs2TiX6). J. Phys. Chem. Lett. 13, 10965–10975 (2022).
Zhang, L. & Hou, Y. The Rise and Development of MOF-Based Materials for Metal-Chalcogen Batteries: Current Status, Challenges, and Prospects. Adv. Energy Mater. 13, 2204378 (2023).
Zhang, L. & Liu, Y. Aqueous Zinc–Chalcogen Batteries: Emerging Conversion-Type Energy Storage Systems. Batteries 9, 62 (2023).
Vazquez-Fernandez, I. et al. Vacancy-Ordered Double Perovskite Cs2TeI6 Thin Films for Optoelectronics. Chem. Mater. 32, 6676–6684 (2020).
Du, J. et al. A High-Energy Tellurium Redox-Amphoteric Conversion Cathode Chemistry for Aqueous Zinc Batteries. Adv. Mater. 36, e2313621 (2024).
Li, X. et al. Bis-Ammonium Salts with Strong Chemisorption to Halide Ions for Fast and Durable Aqueous Redox Zn Ion Batteries. Nano Energy 98, 107278 (2022).
Smirnov, M. et al. Raman Spectra and Structural Peculiarities of TeO2-TeO3 Mixed Oxides. J. Phys. Condens. Matter 30, 475403 (2018).
Wilcox, R. J. et al. Crystalline and Liquid Structure of Zinc Chloride Trihydrate: A Unique Ionic Liquid. Inorg. Chem. 54, 1109–1119 (2015).
Zhang, X. P., Li, Y. N., Sun, Y. Y. & Zhang, T. Inverting the Triiodide Formation Reaction by the Synergy between Strong Electrolyte Solvation and Cathode Adsorption for Lithium-Oxygen Batteries. Angew. Chem. Int. Ed. 58, 18394–18398 (2019).
Wang, F. et al. Fully Conjugated Phthalocyanine Copper Metal-Organic Frameworks for Sodium-Iodine Batteries with Long-Time-Cycling Durability. Adv. Mater. 32, e1905361 (2020).
Sun, P. et al. Simultaneous Regulation on Solvation Shell and Electrode Interface for Dendrite-Free Zn Ion Batteries Achieved by a Low-Cost Glucose Additive. Angew. Chem. Int. Ed. 60, 18247–18255 (2021).
Li, S. et al. Design and Synthesis of a pi-Conjugated N-Heteroaromatic Material for Aqueous Zinc-Organic Batteries with Ultrahigh Rate and Extremely Long Life. Adv. Mater. 35, e2207115 (2023).
Chen, Z. et al. Conversion-Type Nonmetal Elemental Tellurium Anode with High Utilization for Mild/Alkaline Zinc Batteries. Adv. Mater. 33, e2105426 (2021).
Zheng, Z., Guo, S., Yan, M., Luo, Y. & Cao, F. A Functional Janus Ag Nanowires/Bacterial Cellulose Separator for High-Performance Dendrite-Free Zinc Anode Under Harsh Conditions. Adv. Mater. 35, e2304667 (2023).
Sun, Q. et al. Silver Ion in Combination Intercalation/Deintercalation Reaction of Aqueous Zinc-Ion Batteries. J. Mater. Sci. 58, 12008–12019 (2023).
Chen, S. et al. Solid Interhalogen Compounds with Effective Br0 Fixing for Stable High-energy Zinc Batteries. Angew. Chem. Int. Ed. 62, e202301467 (2023).
Wang, X. et al. An Aqueous Rechargeable Zn//Co3O4 Battery with High Energy Density and Good Cycling Behavior. Adv. Mater. 28, 4904–4911 (2016).
Zhang, N. et al. Rechargeable Aqueous Zn–V2O5 Battery with High Energy Density and Long Cycle Life. ACS Energy Lett. 3, 1366–1372 (2018).
Qiu, X. et al. Molecular Tailoring of p-type Organics for Zinc Batteries with High Energy Density. Angew. Chem. Int. Ed. 62, e202304036 (2023).
Shi, H. Y., Ye, Y. J., Liu, K., Song, Y. & Sun, X. A Long-Cycle-Life Self-Doped Polyaniline Cathode for Rechargeable Aqueous Zinc Batteries. Angew. Chem. Int. Ed. 57, 16359–16363 (2018).
Gao, S. et al. High-Energy and Stable Subfreezing Aqueous Zn-MnO2 Batteries with Selective and Pseudocapacitive Zn-Ion Insertion in MnO2. Adv. Mater. 34, e2201510 (2022).
Ma, L. et al. Electrocatalytic Iodine Reduction Reaction Enabled by Aqueous Zinc-Iodine Battery with Improved Power and Energy Densities. Angew Chem. Int. Ed. 60, 3791–3798 (2021).
Wang, N. et al. Tridentate Citrate Chelation towards Stable Fiber Zinc-Polypyrrole Battery with Hybrid Mechanism. Energy Storage Mater. 43, 585–594 (2021).
Lindstrom, H. et al. Li+ Ion Insertion in TiO2 (Anatase). 2. Voltammetry on Nanoporous Films. J. Phys. Chem. B 101, 7717–7722 (1997).
Setyawan, W. & Curtarolo, S. High-Throughput Electronic Band Structure Calculations: Challenges and Tools. Comput. Mater. Sci. 49, 299–312 (2010).
Zhu, H. & Kee, R. J. Computational Modeling of Sodium-Iodine Secondary Batteries. Electrochim. Acta 219, 70–81 (2016).
Wang, S. et al. Co-Doping of Tellurium with Bismuth Enhances Stability and Photoluminescence Quantum Yield of Cs2AgInCl6 Double Perovskite Nanocrystals. Nanoscale 14, 15691–15700 (2022).
Acknowledgements
C.Z. acknowledges the National Key R&D Program of China under Project 2019YFA0705104, the Research Grants Council of the Hong Kong Special Administrative Region, China (Project No. R5019-22, Project No. CityU PDFS2122−1S05, and Project No. CityU 11214023), and the Talent Recruitment Project of Guangdong Province (No. 2019QN01C883). X. Y. acknowledges the National Natural Science Foundation of China (62174104, 12304032), the Shanghai Science and Technology Committee (22YF1413500), and the Program of Shanghai Academic/Technology Research Leader (22XD1421200).
Author information
Authors and Affiliations
Contributions
S.W., X.Y. and C.Z. designed the study. C.Z. supervised the experiments. S.W., Z.W., H.H., X.G., Y.W., Z.C., D.Z. and X.Z. conducted structural, electrochemical, and spectroscopic characterizations and analyzed the data. S.W. performed the theoretical calculations. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hannes Radinger, Agnes Nkele and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, S., Wei, Z., Hong, H. et al. A tellurium iodide perovskite structure enabling eleven-electron transfer in zinc ion batteries. Nat Commun 16, 511 (2025). https://doi.org/10.1038/s41467-024-55385-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55385-6
