
Abstract
Cortical layer 5 (L5) intratelencephalic (IT) and pyramidal tract (PT) neurons are embedded in distinct information processing pathways. Their morphology, connectivity, electrophysiological properties, and role in behavior have been extensively analyzed. However, the molecular composition of their synapses remains largely uncharacterized. Here, we dissect the protein composition of the excitatory postsynaptic compartment of mouse L5 neurons in intact somatosensory circuits, using an optimized proximity biotinylation workflow with high spatial accuracy. We find distinct synaptic signatures of L5 IT and PT neurons that are defined by proteins regulating synaptic organization and transmission, including cell-surface proteins (CSPs), neurotransmitter receptors and ion channels. In addition, we find a differential vulnerability to disease, with a marked enrichment of autism risk genes in the synaptic signature of L5 IT neurons compared to PT neurons. These results align with human studies and suggest that the excitatory postsynaptic compartment of L5 IT neurons is susceptible in autism. Our approach is versatile and can be broadly applied to other neuron types to create a protein-based, synaptic atlas of cortical circuits.
Similar content being viewed by others

Introduction
How different types of neurons become embedded in neural circuits, the basis of the brain’s capacity for information processing, is a major question in neuroscience. Cortical L5 pyramidal neurons (PNs) play a crucial role in information processing1,2,3. These neurons integrate feedforward sensory information converging on their basal dendritic compartment and feedback contextual information converging on their apical dendrites4,5. Their axons project to cortical and subcortical regions6,7. L5 PNs are thus at the heart of cortical input/output processing.
Two main populations of L5 PNs can broadly be distinguished: intratelencephalic (IT) and pyramidal tract (PT) L5 PNs, which differ in their morphology, electrophysiological properties and projection targets6,8. IT neurons are thin-tufted, regular spiking and intracortically projecting PNs mostly located in superficial L5 (L5a), whereas PT neurons are thick-tufted, bursting and subcortically projecting PNs mostly found in deeper L5 (L5b)9,10. Both L5 neuron types project to the striatum, however, they carry different types of information7,11. The two L5 PN types differ in input connectivity, which is best characterized for excitatory input from the thalamus. L5 IT neurons primarily receive input from higher-order thalamic nuclei, whereas PT neurons preferentially receive input from the primary thalamus12,13,14. Functionally, L5 IT and PT neurons are part of different cortical information processing pathways. Perception of tactile stimuli depends on activation of apical dendrites of L5 PT, but not IT, neurons in the somatosensory cortex5. In the motor cortex, L5 IT neurons are involved in preparation of movement, whereas PT neurons are important for initiation and execution10,15. In the visual cortex, PT neurons display higher direction selectivity index and prefer higher temporal frequencies than IT neurons, indicating that they integrate different visual information16. Differences in connectivity and function of the two L5 PN types may also relate to their differential vulnerability in neurodevelopmental and movement disorders6.
Despite the wealth of information on morphological and functional properties, the molecular composition of L5 PN synapses has not been characterized. Elucidating cell type-specific molecular synaptic signatures could contribute to our understanding of how L5 PN types are connected into circuits and how perturbations in this process might contribute to diseases, particularly those characterized by a strong synaptic component, such as neurodevelopmental disorders17. Proximity biotinylation uses a promiscuous biotin ligase to covalently attach a biotin molecule to nearby proteins within a short radius18, and offers an attractive approach to profile synaptic proteomes. This approach has been used to characterize the molecular composition of postsynaptic compartments from whole cortex and hippocampus19, and the subcellular proteome of astrocytes in the striatum20, as well as to profile the neuronal proteomes of major neuronal classes by expressing a cytosolic biotin ligase21,22,23. However, profiling synaptic protein composition of genetically defined populations of cortical neurons is challenging, due to the small number of starting cells and correspondingly low amounts of input material.
To pave the way for such a systematic description of the synaptic protein composition of cortical neurons, we optimize a cell type-specific proximity biotinylation-based workflow using the engineered biotin ligase TurboID24. We fuse TurboID to two different excitatory postsynaptic scaffold proteins, PSD95 and Homer1, and demonstrate the high spatial accuracy of this approach in L5 neurons in vivo. We then analyze the protein composition of the excitatory postsynaptic compartment of L5 IT and PT neurons in the somatosensory cortex. We find that the synaptic signatures of L5 IT and PT neurons are characterized by differential expression of CSPs, neurotransmitter receptors and ion channels. To assess disease vulnerability of the L5 IT and PT neuron synaptic signatures, we analyze the identified proteomes for disease association. We find a prominent enrichment of ASD risk genes in the excitatory postsynaptic proteome of L5 IT neurons compared to PT neurons, suggesting that the excitatory postsynaptic compartment of L5 IT neurons is susceptible in ASD. Cross-referencing our synaptic proteome results with human transcriptomics data reinforces this notion. Our results fit in a framework of L5 IT neuron vulnerability in ASD6 and align with RNA sequencing studies of individuals with autism that highlight vulnerability of IT neurons25,26,27,28. Taken together, our analysis sheds light on the proteins that regulate synaptic organization and function of L5 PN types and contribute to their vulnerability in disease.
Results
Efficient, compartment-specific protein biotinylation in genetically identified neurons
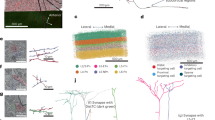
To compare the molecular synaptic signatures of L5 IT and PT neurons in the somatosensory cortex, we first optimized a workflow to identify postsynaptic proteomes in a cell-type specific manner, with optimal capture of biotinylated proteins from a small population of neurons in a specific brain region. This optimized workflow is based on the high-efficiency biotin ligase TurboID (Fig. 1a), which has a reported ~10-fold increase in in vitro activity compared to previously published biotin ligases24. To restrict the activity of TurboID to the excitatory postsynaptic density (PSD), we fused TurboID to the scaffold proteins PSD95 (PSD95.turboID) and Homer129 (Homer1.turboID) (Fig. 1b). As a control, we generated a TurboID construct without any localization sequence (cytosolic.turboID) (Fig. 1b). To target TurboID to L5 PNs in a cell-type and region-specific manner, we packaged Cre-dependent TurboID expression constructs into adeno-associated viral vectors (AAV) and injected them in the primary somatosensory (S1) cortex of postnatal day (P)28 mouse lines expressing Cre recombinase in L5 PNs. For this optimization stage, we used two different L5-targeting mouse lines based on animal availability. A week after stereotactic injection, we subcutaneously injected the TurboID substrate biotin and collected brains for immunofluorescence or biochemical analysis. Tissue sections stained for HA-tagged TurboID and for biotinylated proteins using fluorescent streptavidin showed colocalizing signal restricted to S1 and concentrated in L5 (Fig. 1c). A control sample in which only biotin but no virus was injected (no.turboID) showed that background biotinylation was below detection levels compared to the other conditions (Fig. 1c). High-resolution confocal imaging showed a diffuse pattern of biotinylated proteins throughout the neuronal cytoplasm in cytosolic.turboID samples (Fig. 1d). In PSD95.turboID and Homer1.turboID samples, streptavidin-labeled biotinylated proteins displayed a punctate pattern that colocalized with the excitatory postsynaptic markers Homer1 and PSD95, respectively (Fig. 1d, quantified in Supplementary Fig. 1a, b), confirming TurboID activity in the excitatory postsynaptic compartment.

a TurboID covalently binds a biotin molecule to nearby proteins. b A cytosolic.turboID construct labels cytosolic proteins. Fusion of TurboID to PSD95 (PSD95.turboID) or Homer1 (Homer1.turboID) directs its enzymatic activity to distinct postsynaptic compartments. c Whole-slice imaging of Tlx3-Cre mice injected with Cre-dependent TurboID constructs labelled for HA-tagged TurboID enzyme and biotinylated proteins. Scalebar is 200 μm. These experiments were independently repeated more than 3 times with similar results. d High resolution imaging shows biotinylated protein patterns from the TurboID constructs. Homer1 and PSD95 immunostaining labels postsynapses. Scalebar is 10 μm and 1 μm in the cropped region. These experiments were independently repeated more than 3 times with similar results. e Western blot quantification shows high levels of biotinylated proteins in dissected somatosensory cortices after 3 h of biotin injection. Tubulin is used as loading control. Molecular weight markers in kDa are indicated on the left (N = 2 mice per time point). Source data are provided as a Source Data file. f Western blot for TurboID (HA) confirms expression of constructs at the expected size. Both TurboID constructs produce strong biotinylated protein signal across molecular weights compared to the no.turboID sample, which shows only bands for endogenously biotinylated proteins. These experiments were independently repeated more than 3 times with similar results. g After streptavidin pulldown, postsynaptic marker proteins Gria1 and Homer1 are only detected in PSD95.turboID and in Homer1.turboID samples. Presynaptic marker Synaptophysin is not detected in any pulldown sample. These experiments were independently repeated more than 3 times with similar results.
To control for potential artefacts resulting from fusion protein expression, we analyzed spine density and morphology in TurboID-expressing samples using the Supernova labelling system, which allows for sparse yet bright GFP expression in genetically identified neurons30 (Supplementary Fig. 1c–f). In initial experiments, we noticed that higher PSD95.turboID viral titers resulted in an increase in mushroom-like spines (Supplementary Fig. 1c). We therefore adjusted viral titers and performed dendritic spine reconstruction and classification, and found no difference in spine density or morphology across conditions at lower titers (Supplementary Fig. 1d–f).
To optimize in vivo biotinylation, we expressed PSD95.turboID in L5 PNs in somatosensory cortex and provided biotin for different durations before collecting tissue for biochemical analysis. Western blot quantification showed a maximum, 4-fold increase in biotinylation levels compared to control conditions at 3 h following biotin injection (Fig. 1e). We, therefore, selected 3 h post-biotin injection for subsequent experiments to achieve maximum biotinylation while limiting off-target labelling31.
We then compared in vivo expression and activity of the different TurboID constructs. HA immunoblot showed that the TurboID constructs run at the expected size of 35 kDa for cytosolic.turboID, ~135 kDa for PSD95.turboID, and ~100 kDa for Homer1.turboID (Fig. 1f). Immunoblotting for biotinylated proteins showed two bands in the no.turboID condition, likely corresponding to endogenously biotinylated proteins32, while cytosolic, PSD95.turboID and Homer1.turboID produced biotinylation across all molecular weights (Fig. 1f). Western blot analysis showed that expression levels of PSD95.turboID and Homer1.turboID were low compared to endogenous PSD95 and Homer1 levels, yet produced high levels of biotinylated proteins (Supplementary Fig. 1g), adding confidence that our strategy does not interfere with synaptic protein homeostasis. Streptavidin pulldown from the same lysate detected signal for postsynaptic proteins Gria1 and Homer1 in the PSD95.turboID and Homer1.turboID samples, while the presynaptic protein Synaptophysin was not detected in any pulldown condition (Fig. 1g). Together, these results show that our proximity biotinylation strategy reliably targets the excitatory postsynaptic compartment of genetically identified L5 PNs in a specific cortical region and efficiently yields biotinylated proteins in a short timeframe.
Spatially accurate identification of excitatory postsynaptic proteins in cortical L5 neurons
The PSD95 and Homer1 scaffold proteins are core components of the PSD but reside in different locations29 (Fig. 2a). To determine the extent of coverage of PSD proteins and analyze potential differences in proteins identified by the two fusion proteins, we performed a comparison of PSD95.turboID and Homer1.turboID in L5 neurons. To this end, we injected L5 PN-specific Tlx3_PL56-Cre mice with TurboID-expressing AAVs at P28 (Fig. 2b). One week later, mice were injected with biotin for 3 h followed by tissue dissection and collection of the somatosensory cortex. Negative control samples (no.turboID) were prepared with mice injected only with biotin (Fig. 2a), to exclude endogenously biotinylated proteins and contaminants from the analysis. Four mice were used per replicate. Biotinylated proteins were purified from tissue lysates using streptavidin-coupled beads, eluted, and digested to peptides by trypsin and identified by label-free liquid chromatography-tandem mass spectrometry (LC-MS/MS) run in data-independent acquisition (DIA) mode (Fig. 2b) (see “Methods”). Protein identification and quantification performed with DIA-NN software33 yielded more than 4000 different proteins among all samples, which showed consistent intensity distribution at the whole sample level (Supplementary Fig. 2a).

a Representation of the different conditions tested and the expected pattern of biotinylated proteins at the synapse. b Mice are injected with AAVs in the somatosensory cortex at P28. At P35, biotin is provided 3 h before collection of somatodendritic material from somatosensory cortices. Biotinylated proteins are enriched by streptavidin pulldown and prepared for MS analysis. c PCA analysis shows replicates clustering by TurboID construct. d Relative protein enrichment for PSD95.turboID and cytosolic.turboID. Significant proteins (two-sided, unpaired t-test with permutation-based FDR correction at 10%, s0 > 0.1) are labelled, core excitatory postsynaptic proteins are highlighted. e Relative protein enrichment for Homer1.turboID and cytosolic.turboID. Significant proteins (two-sided, unpaired t-test with permutation-based FDR correction at 10%, s0 > 0.1) are labelled, core excitatory postsynaptic proteins are highlighted. f Gene ontology analysis of synaptic proteins in PSD95.turboID and Homer1.turboID samples. Gene ontology analysis was performed using Fisher’s one-tailed test with FDR multiple-comparison correction. g Relative protein enrichment by PSD95.turboID and Homer1.turboID. Significant proteins (two-sided, unpaired t-test with permutation-based FDR correction at 5%, s0 > 0.1) are labelled based on TurboID construct. h Gene ontology of differentially expressed proteins in PSD95.turboID vs. Homer1.turboID samples. Gene ontology analysis was performed using Fisher’s one-tailed test with FDR multiple-comparison correction. PCA principal component analysis, IT intratelencephalic, PN pyramidal neuron.
As expected, we detected high levels of endogenously biotinylated proteins in all samples (Supplementary Fig. 2b). Excitatory postsynaptic proteins on the other hand showed selective enrichment in the PSD95.turboID and Homer1.turboID conditions and depletion in the no.turboID samples (Supplementary Fig. 2c). Cytoskeletal proteins showed enrichment in the cytosolic.turboID condition, while inhibitory postsynaptic proteins showed no specific enrichment in any condition (Supplementary Fig. 2d). We used an approach similar to a previously published ratiometric analysis24 to create a list of proteins detected in the no.turboID condition and selected contaminants to be excluded from the analysis (Supplementary Fig. 2e) (see “Methods”). PCA analysis showed replicates clustering together by expressed TurboID construct (Fig. 2c).
To determine the extent of PSD protein coverage of the two constructs, we compared PSD95.turboID and Homer1.turboID with the cytosolic.turboID samples and selected proteins with a false discovery rate (FDR) < 0.1 and s0 > 0.1 (Fig. 2d, e), yielding 1359 synaptic proteins enriched in PSD95.turboID, Homer1.turboID or both. Both PSD95.turboID and Homer1.turboID fusion proteins identified PSD constituents such as Shank1 and Dlgap1, although the number of synaptic proteins enriched in Homer1.turboID was lower compared to PSD95.turboID (Fig. 2d, e). Gene Ontology (GO) analysis (cellular compartment) confirmed PSD localization of synaptically enriched proteins for PSD95.turboID and Homer1.turboID (Fig. 2f), and nuclear and organellar localization for cytosolic.turboID enriched proteins (Supplementary Fig. 2f). To further characterize the synaptic proteins identified by PSD95.turboID and Homer1.turboID, we queried the synaptic protein database published in Sorokina, Mclean, Croning et al. 34 (from here on referred to as the ‘synapse proteome’ database). This database lists the synaptic proteins detected in proteomic studies performed using various approaches in the last 20 years and collects and integrates information with several external databases. More than 80% of the proteins identified in the PSD95.turboID and Homer1.turboID samples were present in this database (Supplementary Fig. 2g and Supplementary Data 1). Cross-referencing with the curated synaptic protein database SYNGO35 showed an overlap of 21% of identified proteins in PSD95.turboID and 29% in Homer1.turboID, due to its relatively limited coverage (Supplementary Fig. 2g). Enrichment analysis on genes annotated in SYNGO for both PSD95.turboID and Homer1.turboID showed a strong bias towards the postsynaptic compartment (Supplementary Fig. 2h).
To analyze potential differences in the synaptic proteins detected in the PSD95.turboID and Homer1.turboID samples, we performed a differential expression analysis (Fig. 2g). PSD95 interactors, including neurotransmitter receptors such as the NMDA receptor subunit Grin2b36 and CSPs such as Lrrc4c37, were enriched in the PSD95.turboID sample (Fig. 2g). Homer1 interactors, such as the postsynaptic metabotropic glutamate receptors Grm1 and Grm538, were enriched in the Homer1.turboID sample (Fig. 2g). The analysis workflow to obtain this data is summarized in Supplementary Fig. 2i; synaptic proteins are annotated in Supplementary Data 1. GO analysis (biological process) showed more synaptic terms for PSD95.turboID compared to Homer1.turboID (Fig. 2h). Together, these results show that our optimized proximity biotinylation strategy combined with DIA-MS reliably detects postsynaptic proteins in genetically identified cortical neurons. The differential enrichment of proteins matching the localization of the specific bait protein fused to TurboID shows the spatial accuracy of this approach. Based on its broader synaptic protein coverage and stronger enrichment of neurotransmitter receptors and CSPs (Fig. 2d–h) that are important for synaptic function and organization, we chose to use PSD95.turboID for subsequent experiments.
Identification of excitatory postsynaptic proteins in cortical L5 IT and PT neurons
We next applied our approach to characterize the postsynaptic protein composition of L5 IT and PT neurons. To obtain genetic access to these neurons, we used the Tlx3_PL56-Cre and Sim1_KJ18-Cre mouse lines5,16,39,40. The projection patterns, as well as morphological and physiological properties of Tlx3-Cre+ and Sim1-Cre+ neurons in the cortex have been found to represent thin-tufted L5a IT and thick-tufted L5b PT neurons, respectively5,7. Imaging of tissue sections from Tlx3-Cre and Sim1-Cre mice injected with an AAV expressing Cre-dependent GFP confirmed the segregation of labeled Tlx3-Cre+ and Sim1-Cre+ cell bodies to superficial and deep L5, respectively (Fig. 3a, b). We injected P28 Tlx3-Cre or Sim1-Cre mice with cytosolic.turboID or PSD95.turboID AAVs and collected somatosensory cortex at P35 following 3 h of biotin injection (Fig. 3c, d). After further optimization, we pooled tissue from two mice per replicate instead of four, obtaining around 1 mg of total dissected tissue. Before biotinylated protein purification, TurboID expression and successful biotinylation were confirmed by western blot (Supplementary Fig. 3a). After LC-MS/MS, protein identification and quantification yielded more than 4500 different proteins among all samples, which showed consistent intensity distribution at the whole sample level (Supplementary Fig. 3b). Consistent with our previous experiments, endogenously biotinylated proteins were detected in all samples, while postsynaptic proteins showed highest enrichment in the PSD95.turboID samples (Supplementary Fig. 3c, d). We again created a list of contaminants to be excluded from the analysis based on the no.turboID samples (Supplementary Fig. 3e, f) (see “Methods”). PCA analysis showed replicates clustering together by condition with little separation relative to the Cre line used (Fig. 3e).

a Representation of L5 IT and PT neuron position and morphology in the somatosensory cortex with overview of input localization from other local and long-range excitatory neurons (adapted from Radnikow and Feldmeyer, 2018). b AAV-mediated Cre-dependent GFP expression in Tlx3-Cre+ and Sim1-Cre+ mouse lines recapitulates soma position for L5 IT and PT neurons, respectively. Layer 5 borders were identified by density of DAPI-positive nuclei. Scalebar is 200 μm. These experiments were independently repeated more than 3 times with similar results. c Workflow: mice are injected with AAVs in the somatosensory cortex at P28. At P35, biotin is provided 3 h before collection of somatodendritic material from somatosensory cortices. Biotinylated proteins are enriched by streptavidin pulldown and prepared for MS analysis. d MS replicates were collected from Tlx3-Cre+ and Sim1-Cre+ mice for no.turboID, cytosolic.turboID or PSD95.turboID conditions. e PCA analysis shows replicates clustering by condition but not by Cre line. f Statistical analysis of cytosolic.turboID and PSD95.turboID samples for each Cre line to determine synaptic protein enrichment. Core constituents of the excitatory postsynapse are annotated. Numbers of identified cytosolic and synaptic proteins for Tlx3-Cre+ and Sim1-Cre+ lines are indicated in bottom left and right corners of each graph. Two-sided, unpaired t-test with permutation-based FDR correction at 10%, s0 > 0.1. g Gene ontology analysis of the cytosolic and synaptic proteins from (f) showing cellular localization of identified proteins. VPM ventral posteromedial thalamic nucleus, POm posteromedial thalamic nucleus, IT intratelencephalic, PT pyramidal tract, PCA principal component analysis.
Comparison of the cytosolic.turboID and PSD95.turboID conditions for each Cre line showed core constituents of the excitatory postsynapse, such as glutamate receptors and scaffolding proteins, to be synaptically enriched in both L5 Tlx3-Cre + /IT and Sim1-Cre + /PT samples (Fig. 3f). Postsynaptic localization was confirmed by GO analysis (cellular compartment) (Fig. 3g), showing that we can reliably identify synaptic proteins even when halving the number of animals used.
Synaptic signatures of cortical L5 IT and PT neurons
To map the synaptic signatures of L5 Tlx3-Cre + /IT and Sim1-Cre + /PT neurons and analyze potential differences between IT and PT neurons, we next performed a differential expression analysis of the synaptically enriched proteins of the two cell types. As expected, most synaptic proteins did not differ significantly between L5 IT and PT neurons, but 135 proteins were differentially expressed between Tlx3-Cre + /IT (93 proteins) and Sim1-Cre + /PT (42 proteins) neurons (Fig. 4a and Supplementary Data 2). We first analyzed the function of these proteins using several GO databases41. Differentially expressed proteins showed strong postsynaptic localization (Supplementary Fig. 4a). ‘Synapse organization’ and ‘regulation of postsynaptic neurotransmitter receptor activity’ were among the most represented GO terms in the Biological Process category (Fig. 4b). The Molecular Function category showed enrichment mostly in terms related to PSD interactions (Supplementary Fig. 4b). Of the identified synaptic proteins in L5 Tlx3-Cre + /IT and Sim1-Cre + /PT neurons, 84% were represented in the synapse proteome database (Supplementary Fig. 4c) and 44% in SYNGO (Supplementary Fig. 4d).

a Relative protein enrichment in L5 IT and PT neurons. Significant proteins (two-sided, unpaired t-test with permutation-based FDR correction at 5%, s0 > 0.1) are labelled according to Cre line (93 proteins in Tlx3-Cre + /IT and 42 proteins in Sim1-Cre + /PT neurons). b Biological Process gene ontology analysis of differentially expressed proteins. Gene ontology analysis was performed using Fisher’s one-tailed test with FDR multiple-comparison correction. c CSPs are annotated to represent the ‘synapse organization’ module on the volcano plot from panel 4a (two-sided, unpaired t-test with permutation-based FDR correction at 5%, s0 > 0.1). d Ion channels and neurotransmitter receptors are annotated to represent the ‘postsynaptic neurotransmitter receptor activity’ module on the volcano plot from panel 4a (two-sided, unpaired t-test with permutation-based FDR correction at 5%, s0 > 0.1). e, g smFISH analysis of candidate genes in L5 IT and PT neurons labelled by Cre-dependent GFP expression in Tlx3-Cre and Sim1-Cre mice, respectively. Cells that have been quantified are indicated by dotted circles. Scale bars 10 μm. f, h Quantification of RNA expression (average number of RNA puncta per cell). N = 5 mice for Flrt3, Kcna1, Kcna6; n = 6 mice for every other mRNA per Cre line. For each mouse, 8 cells are quantified and averaged as one replicate. Boxplots show distribution of single cells, empty dots display the average of each biological replicate (N). The line across the box describes the mean. The lower and upper hinges correspond to the first and third quartiles. The upper whisker extends from the hinge to the largest value no further than 1.5 * inter-quartile range. The lower whisker extends from the hinge to the smallest value at most 1.5 * inter-quartile range of the hinge. Data beyond the end of the whiskers (outliers) are plotted individually as a small, solid dot. Statistical analysis is performed as an unpaired t-test on the replicates, alpha = 0.05, p-value is reported under transcript name. Source data are provided as a Source Data file. IT intratelencephalic, PT pyramidal tract, PN pyramidal neuron, L5 layer 5.
We next explored the proteins in the most represented GO modules ‘synapse organization’ and ‘regulation of postsynaptic neurotransmitter receptor activity’ (Fig. 4b and Supplementary Fig. 4e), as these are likely to contribute to connectivity and physiological properties of Tlx3-Cre + /L5 IT and Sim1-Cre + /PT neurons. To do so, we started from the proteins annotated in each term of the Biological Process category (Fig. 4b) and integrated these results with PANTHER42 and Uniprot43 information to assign a function to the differentially expressed proteins (see Supplementary Data 2). In the ‘synapse organization module’, CSPs were the most abundant protein class (Fig. 4c and Supplementary Fig. 4f). Extracellular leucine-rich repeat (LRR) proteins (Elfn2, Flrt2, Flrt3, Lrrtm3, Lrrtm4, Lrrc4c/Ngl-1, Ntrk3/Trkc, Lrfn2/Salm1, Slitrk2) were the most represented protein family among the differentially expressed CSPs in L5 IT and PT neurons (Fig. 4c). Interestingly, Flrt2 and Flrt3 belong to the same LRR subfamily but showed differential enrichment in IT and PT neuron synaptic signatures (Fig. 4c). LRR proteins control precise wiring and confer synaptic properties across species44,45,46, but their role in cortical circuit connectivity and function remains largely unexplored.
In the ‘postsynaptic neurotransmitter receptor activity’ module, neurotransmitter receptors and channels were the most enriched protein classes (Fig. 4d). Among differentially expressed receptors, kainate receptors (Grik2, Grik3) showed enrichment in the L5 PT neuron synaptic signature (Fig. 4d and Supplementary Fig. 4f). Kainate receptors have been observed to be enriched in deep cortical layers of the mouse brain using immunohistochemistry47 and receptor autoradiography48,49, but an enrichment in L5 PT neurons has not been reported to our knowledge. The NMDA receptor subunit Grin2b was enriched in IT neurons, whereas potassium and calcium ion channels showed different subunits specifically enriched in one of the two L5 neuron types (Fig. 4d and Supplementary Fig. 4f).
Due to limitations in availability of validated neuroscience antibodies50 and challenges in localizing synaptic proteins to specific neuron types in cortex, we decided to assess our proteomics results using an orthogonal approach measuring cell-type specific gene expression. While it has been reported that mRNA and protein rarely match at absolute levels, it has also been shown that mRNA can predict proteomic differences at a global level51,52,53. We selected four differentially expressed proteins from the ‘synapse organization’ and from the ‘regulation of postsynaptic neurotransmitter receptor activity’ modules (Fig. 4e–h). We injected AAVs expressing Cre-dependent GFP in Tlx3-Cre or Sim1-Cre mice and performed single-molecule Fluorescence In Situ Hybridization (smFISH) experiments (Fig. 4e, g). Quantification of mRNA puncta in GFP+ cells showed significant agreement between cell type enrichment of the selected synaptic proteins and their corresponding transcripts (Fig. 4f, h), except for Grik3, which showed a bimodal distribution in Tlx3-Cre + /IT neurons with cells expressing high levels of mRNA and cells expressing none (Supplementary Fig. 4g). These results show that for these selected proteins at least, cell type-specific differences in synaptic protein enrichment correlate with differences at the transcript level. In conclusion, these results show that the synaptic signatures of L5 IT and PT neurons are defined by differential expression of CSPs, neurotransmitter receptors and ion channels.
Differential vulnerability of L5 IT and PT synaptic signatures in neurodevelopmental disorders
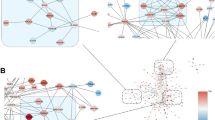
Neurodevelopmental disorders are associated with altered synaptic function and development17,54,55,56. We next tested whether the synaptic signatures of Tlx3-Cre + /IT and Sim1-Cre + /PT neurons might provide insight in disease vulnerability of these two L5 neuron types. We first ran the differentially expressed synaptic proteins in Tlx3-Cre + /IT and Sim1-Cre + /PT neurons (Fig. 4a) through the GeneTrek database57, which analyzes association of genes with neurodevelopmental disorders (Fig. 5a). Based on several disease association databases, genes are given a ‘high confidence’ or ‘candidate’ score. The synaptic signatures of both L5 IT and PT neurons showed a high proportion of genes in each of these two categories (Fig. 5a). Next, we queried the synapse proteome database for disease association and graphed the results for L5 IT and PT neurons, normalized to the total number of synaptic proteins identified in each neuron type (Fig. 5b and Supplementary Data 2). Overall, neurodevelopmental disorders showed a higher percentage of enriched genes compared to neurodegenerative diseases such as Parkinson’s or Alzheimer’s in IT and PT neurons (Fig. 5b and Supplementary Data 2). The synaptic signature of PT neurons showed a larger fraction of genes associated with schizophrenia and psychotic disorder, whereas that of IT neurons showed a larger fraction of genes associated with ASD and intellectual disability (Fig. 5b and Supplementary Data 2).

a Percentage of synaptic genes per cell type present in the ‘high confidence’ or ‘candidate’ neurodevelopmental disorder lists based on the Genetrek database published in Leblond et al. 202132. b Percentage of synaptic genes per cell type present in each disorder term based on the synapse proteome database6. c Synaptic proteins in L5 IT and PT neurons with an ASD risk score annotation in the SFARI gene database33 are indicated on the volcano plot from Fig. 4a (two-sided, unpaired t-test with permutation-based FDR correction at 5%, s0 > 0.1). d ASD proteins identified in the SFARI database33 listed per cell type and ranked by protein log fold change. e L5 IT and PT neurons were extracted from published human motor cortex snRNA-Seq data61 and re-clustered. f Differentially expressed genes (DEGs) between human IT and PT neurons (adjusted p-value < 0.01 and log fold change > 0.25) were plotted on top of mouse synaptic proteins enriched in IT and PT neurons from Fig. 4a (two-sided, unpaired t-test with permutation-based FDR correction at 5%, s0 > 0.1) to evaluate their overlap. 12/15 DEGs matched PT neuron-enriched synaptic proteins, 15/31 DEGs matched IT neuron-enriched synaptic proteins. g Synaptic proteins from mouse L5 IT and PT neurons that show matching differential gene expression in human L5 IT and PT neurons are shown. SFARI scores are annotated, IT intratelencephalic, PT pyramidal tract.
The SFARI gene database58 has a curated, up to date annotation of ASD genes scored by clinical and scientific evidence. Based on the larger fraction of genes associated with ASD and intellectual disability in IT neurons (Fig. 5b), we therefore cross-referenced the synaptic signatures of Tlx3-Cre + /IT and Sim1-Cre + /PT neurons with the SFARI gene database. This analysis revealed a marked enrichment of ASD risk genes in the synaptic signature of L5 IT neurons compared to that of PT neurons (Fig. 5c). ASD genes with the highest risk score annotation (‘syndromic’ and ‘high confidence’) in the SFARI database, such as Frmpd4, Cdkl5, Shank3, Cnksr2 and Slitrk2, were all found in the L5 IT synaptic signature (Fig. 5c, d), suggesting that the excitatory postsynaptic compartment of L5 IT neurons is susceptible in ASD.
Finally, to translate the significance of our findings to human neurons, we extracted and re-clustered L5 IT and PT neuron transcriptomes from a publicly available human motor cortex single-nucleus RNA sequencing dataset59 and calculated differential gene expression between IT and PT neurons (Fig. 5e) (See “Methods”). We next tested whether differentially expressed synaptic proteins in mouse L5 IT or PT neurons were also differentially expressed at the RNA level in human L5 IT and PT neurons. Of 93 differentially expressed synaptic proteins in mouse IT neurons, we found 31 to be differentially expressed at the RNA level in human neurons; 48% of which showed matching enrichment in human L5 IT neurons (Fig. 5f). Of 42 differentially expressed synaptic proteins in mouse PT neurons, 15 were differentially expressed at the RNA level in human neurons; 80% of which showed matching enrichment in human L5 PT neurons (Fig. 5f). Moreover, 6 out of 15 IT genes enriched both at the mouse protein and human RNA level were annotated in the SFARI database (Fig. 5g).
Previous analysis of differentially expressed genes (DEGs) in human, macaque, marmoset and mouse motor cortex showed fewer DEGs shared with greater evolutionary distance59. To evaluate whether we could find a greater match between mouse proteins and mouse transcripts, we tested the overlap between differentially expressed synaptic proteins from our proteomic experiment and mouse DEGs in L5 IT and PT neurons. We extracted and re-clustered L5 IT and PT neuron transcriptomes from a publicly available mouse motor cortex single-nucleus RNA sequencing dataset59 and calculated differential gene expression between IT and PT neurons as we did for the human data (Supplementary Fig. 5a). We then calculated the overlap between differentially expressed synaptic proteins from our proteomic dataset and DEGs from the single-nucleus RNA sequencing dataset. Of 93 differentially expressed synaptic proteins in mouse IT neurons, we found 36 to be differentially expressed at the RNA level in mouse neurons, 94% of which showed matching enrichment in mouse L5 IT neurons (Supplementary Fig. 5b, c). Of 42 differentially expressed synaptic proteins in mouse PT neurons, 22 were differentially expressed at the RNA level in mouse neurons, 91% of which showed matching enrichment in mouse L5 PT neurons (Supplementary Fig. 5b, c). The better match between mouse synaptic proteome and mouse transcriptome data supports our smFISH experiments (Fig. 4e–h) and highlights conserved synaptic genes between mouse and human that are associated with disease in a population-specific manner. Together, these results show that L5 IT and PT synaptic signatures display differential vulnerability to neurodevelopmental disorders such as ASD, and that these results can be translated to human neurons.
Discussion
Here, we characterize the synaptic signatures of two closely related L5 cortical neuron types from a specific brain region, using an optimized proximity biotinylation workflow with postsynaptic resolution. CSPs, neurotransmitter receptors and ion channels with a role in synapse organization and synaptic transmission define the synaptic signatures of L5 IT and PT neurons. The synaptic signatures of L5 neuron types are differentially vulnerable in disease, with the L5 PT synaptic signature showing a larger fraction of genes associated with schizophrenia and psychotic disorder, and the IT synaptic signature displaying a marked enrichment of ASD risk genes.
We find that our approach is sufficiently sensitive to enrich bait-specific proteins at the post-synapse from small populations of genetically identified L5 cortical neurons in vivo. PSD95.turboID and Homer1.turboID both identified PSD proteins, but interactors of each fusion protein showed differential enrichment in the corresponding condition. CSPs and neurotransmitter receptors were enriched in the PSD95.turboID samples, whereas the postsynaptic metabotropic glutamate receptors Grm1 and Grm5 were enriched in the Homer1.turboID samples. We focused on PSD95.turboID for our experiments because of the relevance of the identified proteins for synapse function and organization, but depending on the application, other parts of the synapse can be targeted using this approach.
The cell type-specific synaptic signatures we identify represent promising targets for future investigation into regulators of synaptic properties, physiology and connectivity of L5 IT and PT neurons. Especially evident in this context are the LRR family proteins, which dominate the CSPs in the L5 Tlx3-Cre + /IT and Sim1-Cre + /PT neuron synapse organization modules. LRR proteins are important regulators of connectivity and synaptic properties60,61,62. Input-specific combinations of postsynaptic LRR proteins define synaptic structure and function in hippocampal PNs63. Analogous to hippocampal PNs63,64,65, LRR proteins may differentially distribute across the dendritic tree of cortical PNs to regulate connectivity, structure and function of specific inputs. While our approach does not provide spatial information about synaptic protein distribution in L5 PN dendrites, this remains an important point that will need to be explored in further studies.
L5 Tlx3-Cre + /IT neurons displayed a higher number of differentially expressed synaptic proteins than Sim1-Cre + /PT neurons. PCA and sample level intensity analysis exclude differences in labelling efficiency between the two cell types. The higher number of differentially expressed synaptic proteins in L5 IT neurons compared to PT neurons could reflect differences in synapse number between the two neuron types or differences in type of excitatory input. Compared to L5 PT neurons, L5 IT neurons have a less extensive dendritic arborization, which would argue against an increased number of synapses in IT neurons compared to PT neurons. The two L5 PN neuron types have excitatory input in common but also show cell type-specific differences in input connectivity patterns12. First-order input from the ventral posteromedial (VPM) thalamic nucleus preferentially, though not exclusively, targets L5 PT neurons, whereas higher-order thalamic input from the posteromedial (POm) nucleus is concentrated on L5 IT neurons12,13,14,66,67. It will be of interest to determine whether the synaptic signatures of L5 IT and PT neurons contribute to differences in thalamic input connectivity.
We find that the synaptic signatures of L5 Tlx3-Cre + /IT and Sim1-Cre + /PT neurons display differential vulnerability to neurodevelopmental disorders. L5 PT neurons showed a larger fraction of synaptic proteins associated with schizophrenia, whereas IT neurons showed a larger fraction of synaptic proteins associated with ASD. Cross-referencing with the SFARI gene database revealed a strong enrichment of syndromic and high confidence ASD risk genes in the synaptic signature of L5 IT neurons. These findings are in line with RNA sequencing studies that find IT neurons to be the most affected cell types in ASD cortex25,26,68 and suggest altered intracortical processing in ASD27,69. Our synaptic compartment-specific proteome data indicates that compared to Sim1-Cre + /PT neurons, excitatory input of Tlx3-Cre + /IT neurons is particularly vulnerable in ASD. A comparison of the differentially expressed proteins with single-nuclei RNA sequencing data from human motor cortex samples shows good agreement between mouse proteins and human genes in IT and PT neurons, considering the differences (protein vs. transcript; somatosensory vs. motor cortex) between the datasets. Based on the differences in excitatory input connectivity between L5 IT and PT neurons, it is tempting to speculate that higher-order thalamic input on IT neurons is affected in ASD. Brain imaging studies have observed altered thalamocortical functional connectivity in individuals with autism70,71,72,73, with some studies suggesting altered connectivity between higher-order thalamic nuclei and cortex.
In addition to formulating hypotheses on L5 IT and PT connectivity and function, the synaptic signatures we identify here can also be used to explore specific hypotheses on protein function. An interesting candidate from our analysis is Tbr1, which is enriched in the Tlx3-Cre + /IT synaptic signature and a high confidence ASD risk gene. Tbr1 is a transcription factor but shows a dendritic distribution in cortical pyramidal neurons and localizes to synaptic fractions in biochemical analysis74. Tbr1 binds the synaptic scaffold protein CASK, which translocates to the nucleus75. Possibly, PSD-associated Tbr1 shuttles between synapse and nucleus, where it can regulate expression levels of Grin2b76, which we also find to be enriched in the L5 IT synaptic signature. Another interesting candidate we identified is Pelo, which is highly enriched in the synaptic signature of Tlx3-Cre + /IT neurons. Pelo rescues stalled ribosomes and triggers degradation of damaged mRNA in a process termed No-Go Decay77,78 but its role at synapses is unknown.
To obtain these results, we optimized every step of the proximity biotinylation workflow, resulting in the identification of thousands of proteins starting from single, genetically identified L5 neuron populations in the somatosensory cortex. We found that a single subcutaneous biotin injection 3 h before sample collection is sufficient to produce strong protein biotinylation in the brain. Previous in vivo studies have relied on multiple injections of biotin over several days19,22,23,79. As prolonged labeling with biotin increases the number of proteins identified at the expense of specificity24, minimizing the biotinylation window is important for the analysis of compartment-specific protein composition. Short biotinylation windows also open new possibilities, for example to study how the composition of synaptic proteomes changes upon neuronal activity. For similar reasons, we limited the duration of expression of TurboID constructs to 1 week (compared to multiple weeks or months in previous studies19,23,79,80,81), as low levels of protein biotinylation using endogenous biotin can occur while TurboID is expressed.
A possible limitation of our approach is the use of Cre driver lines to target IT and PT neurons. Although we selected the Tlx3-Cre and Sim1-Cre lines based on their thorough characterization for L5 IT and PT neurons in somatosensory cortex5, this approach is inherently limited to the selectivity of the Cre line used. An interesting alternative approach to explore would be to rely on connectivity patterns to target IT and PT neurons, using retrograde AAV. Another possible limitation of our approach is the use of PSD95 to target TurboID to the postsynaptic compartment. We carefully titrated our viral vectors to control PSD95.turboID expression levels and analyzed spine density and morphology in vivo to verify the absence of overexpression artefacts. This strategy is suitable for mature neurons but carries a risk of driving precocious postsynaptic maturation in developing neurons. Since we are targeting a small and defined population of cortical neurons in a specific brain region, efficient transduction is a requirement for obtaining sufficient amounts of protein for analysis. CRISPR/Cas9-mediated knock-in80,82 might be an alternative. This approach avoids the overexpression of exogenous proteins, however, the low percentage of successful recombination makes it challenging to target single neuronal populations in specific cortical regions80,82. A possible way to avoid modifying expression of synaptic proteins, while retaining short duration of TurboID expression and the high efficiency of labeling, might be the use of recently published PSD95 binders83, which are reported not to alter PSD95 interactions.
In conclusion, our analysis of cell type- and compartment-specific protein composition sets a foundation for the systematical description of cortical circuit connectivity at the molecular synaptic level. As our approach enables the comparison of the synaptic proteome of single neuronal populations, and mouse Cre driver lines for cortical neuronal cell types are increasingly available40,84, systematic application of this workflow could create a protein-based, synaptic atlas of the cortex. Integration of synaptic signature information with RNA sequencing and electrophysiology data will be useful for computational modeling efforts and contribute to our understanding of circuit connectivity and function.
Materials and methods
Animals
All animal experiments were conducted according to the KU Leuven ethical guidelines and approved by the KU Leuven Ethical Committee for Animal Experimentation. Mice were maintained under standard housing conditions with continuous access to food and water. Mice used in the study were 4–5-weeks old and were maintained on 14 h light, 10 h dark light cycle from 7 to 21 h. Wild-type (WT) C57BL/6 J mice were obtained from JAX. Rpb4.Cre and Tlx3_P56-Cre lines were generated by GENSAT. Sim1_KJ18-Cre was obtained from Dr. Charles Gerfen (National Institute of Mental Health, USA). Genotypes were regularly checked by PCR analysis. Heterozygous Cre-positive males were bred to Bl6 females. For euthanasia, animals were either anesthetized with isoflurane and decapitated, or injected with an irreversible dose of ketamine–xylazine.
Cell lines
HEK293T-17 human embryonic kidney cells were obtained from American Type Culture Collection (ATCC) cat# CRL-11268. HEK293T-17 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and penicillin/streptomycin (Invitrogen).
Plasmids
TurboID sequence was a gift from Alice Ting (Addgene plasmid #107169) and synthesized for Gibson assembly as a gBlock by IDT. Mouse PSD95 sequence was a gift from Gary Bassell (Addgene plasmid #102949). Homer1 sequence was cloned from mouse cDNA. TurboID was fused at the C-terminus of PSD95 or Homer1 and inserted into a Cre-dependent AAV expression vector under the synapsin promoter by Gibson assembly (NEB). Plasmids will be deposited at plasmids.eu or made available upon request.
AAV purification
High titer AAV production and purification was carried out as previously reported85. Briefly, 6 plates of 70% confluent HEK cells were transfected with 20 μg of pDelta F6 plasmid, 10 μg of RepCap 2/9 plasmid and 10 μg AAV genome plasmid per plate using PEI transfection. 3 days later cells were collected by scraping and centrifugation. AAV particles in the supernatant were precipitated by PEG and added to cell lysates. Lysates were treated by Benzonase nuclease to remove cellular DNA. Lysates were loaded onto a iodixanol gradient (60%, 40%, 25%, 15%) and ultracentrifuged 1:40 h at 50.000 RPM at 12 °C. The 40% fraction containing the purified AAV was carefully collected and desalted and concentrated on a 100 kDa MWCO 4 ml Amicon column (Sigma). AAV purity was tested by silver staining using the ProteoSilver™ Silver Stain Kit (Sigma PROTSIL1-1KT). AAV titer was determined by qPCR to normalize titers between different viral vector batches using LightCycler 480 SYBR Green I Master (Roche 04707516001). Primers for qPCR targeting the synapsin promoter were as follows: FWD tgataggggatgcgcaatttgg REV gtgcaagtgggttttaggacca.
Stereotactic injections
P28 mice were anesthetized with 5% isoflurane. Duratear artificial tears was applied to the eyes to prevent them from drying out. Mice were placed in a mouse stereotact (KOPF) on a hot plate kept at 37 °C. During the rest of the procedure 2.5% isoflurane was constantly administered. After shaving and disinfecting the mouse’s head, local anesthesia was administered by a subcutaneous injection with 100 μl lidocaine (xylocain 1%). An incision was made on the skin to reveal the skull. A hole was drilled at coordinates for somatosensory cortex and 200 nl of AAV mix injected using a Nanoject III (Drummond) through a beveled capillary at 2 nl/s. After a 5 min recovery, the capillary was pulled out at ~0.1 mm/5 s. The incision was stitched with surgical glue (Millpledge Veterinary). After 6 h, their health was examined and mice were injected with 0.1 mg/kg buprenorphine. Two injections per hemisphere were performed for biochemistry experiments to cover a larger area of the somatosensory cortex and recover more material (x: 3.2, y: 0.3 and 1.5 from bregma, z: 0.8 from the surface of the brain). One injection per hemisphere was performed for every other experiment (x: 3.2, y: 0.9 from bregma, z: 0.8 from the surface of the brain). Cytosolic.turboID was injected at ~1.6 * 10 ^ 12 GC/mL, PSD95.turboID was injected at ~3 * 10 ^ 11 GC/mL, Homer1.turboID was injected at ~5 * 10 ^ 11 GC/mL, Cre-dependent GFP was injected at ~10 ^ 12 GC/mL.
Biotinylated protein collection
One week after AAV injection, mice were injected subcutaneously between 9 and 10 AM with 1 ml of 2 mg/mL biotin in 1X PBS, adjusting the pH to 7.4 to increase biotin solubility. 3 h later mice were anaesthetized by isoflurane and brains collected in ice cold HBSS. 500 μm coronal brain sections were produced using an ice-cold brain matrix (AgnThos 69-2165-1), and somatosensory cortices dissected under a fluorescence stereomicroscope using Cre-dependent GFP signal expressed by infected cells and morphological landmarks to identify the somatosensory cortex. For experiments in Fig. 2, tissue from L4 and L5 around GFP was dissected. For experiments in Figs. 3 and 4, the whole cortical column around GFP was dissected. Tissue pieces from each mouse were collected in a separate tube, flash-frozen in liquid nitrogen and stored at −80 °C until needed. For protein extraction and pulldown, we extensively optimized published protocols86. Frozen tissue was slowly thawed on ice and tissue from 4 animals (Figs. 2) or 2 animals (Figs. 3, 4) was combined in a single tube as a replicate. Tissue was homogenized and lysed in 500 ul 1% SDS RIPA buffer (50 mM Tris pH 8, 150 mM NaCl, 1% SDS, 0.5% Sodium Deoxycholate, 1% Triton-X 100, 1X protease inhibitors) using a small glass Dounce homogenizer. Lysates were sonicated in a bath sonicator (124-9721 RS-online) at max power 3x for 30 s with 30 s rest on ice in between. Lysates were boiled at 95 °C for 5 min to dissociate PSD and trans-synaptic complexes and immediately diluted to 0.5% SDS with ice cold SDS-free RIPA buffer. This step will release all trans-synaptic complexes and increase the specificity of the pulldown, as reported in87. After 1 h incubation at 4 °C with rotation, lysates were cleared by ultracentrifugation at 100.000 G for 30 min at 4 °C and the supernatant was collected. Bicinchoninic acid assay (BCA) was used to quantify protein concentration. 10 μg protein input was used for western blot experiments in Fig. 1e and Fig. 1f. 200 μg protein input was used for pulldown for western blots in Fig. 1g. Around 1000 μg protein input was used for pulldown for MS. 5 μl of streptavidin magnetic beads (Pierce) per 100 μg of protein input was used for every condition. Beads were washed 3x in 0.5% SDS RIPA buffer using a magnetic rack and incubated overnight at 4 °C with rotation with protein input in 1 ml of 0.5% SDS RIPA buffer final volume. The following day, beads were washed 3x with 0.5% SDS RIPA buffer, 1x with 1 M KCl, 1x with 0.1 M Na2CO3, 1x with 2 M Urea in 50 mM Tris-HCl pH 8. MS sample preparation steps are described below, for western blot analysis beads were further washed 3x with 0.5% SDS RIPA buffer and proteins eluted in 1X protein loading buffer supplemented with 10 mM DTT and 2 mM biotin 5 mins at 95 °C.
Western blot
After boiling 5 min at 95 °C, samples were loaded on 4–20% or 7.5% polyacrylamide gels and run at 180 V. Proteins were transferred to 0.2 um nitrocellulose membrane using semi-dry transfer (Biorad) using the mixed-molecular weight program. Membranes were blocked in 5% milk in TBS-T buffer (25 mM Tris-base pH 7.5, 300 mM NaCl, 0.05% Tween-20) 1 h at RT. Primary and secondary antibodies were diluted in 5% milk in TBS-T. Primary antibodies were incubated O/N at 4 °C, secondary antibodies were incubated 1 h at RT. When blotting for biotinylated proteins, HRP-conjugated streptavidin was diluted 1:500 in 5% BSA to avoid binding to the biotin present in the milk. We obtain best results in streptavidin signal while blocking membranes in milk, however we noticed that some powdered milk formulations gave higher background than other. Blocking in 5% BSA is an alternative. After primary and secondary antibody incubation, membranes were washed 5 times with TBS-T and once with 1X PBS before development at ImageQuant 800 (Cytiva) using SuperSignal West Pico PLUS and Femto Chemiluminescent Substrate (Thermo Fisher 34577 and 34094). Western blot quantification in Fig. 1e was performed using the FIJI Gel tool normalizing first to Tubulin signal and then normalized to the 0 h timepoint signal.
MS sample preparation
Beads were washed 2 more times with 2 M Urea in 50 mM Tris-HCl pH 8 and moved to a new tube to avoid collecting contaminants stuck to the plastic. Proteins were eluted from the beads and digested to peptides using S-Trap kit (Protifi) following manufacturer instructions. Briefly, beads were resuspended in 92 μl of 5% SDS 50 mM TEAB pH 8, boiled 5 min at 95 °C and let cool down. Eluates were reduced with 4 μl of a 120 mM TCEP buffer at 55 °C for 15 mins. Eluates were alkylated with 4 μl of 500 mM MMTS in isopropanol at RT for 10 mins in the dark. Eluates were acidified with 10 μl of 27.5% phosphoric acid in dH2O. Using a magnetic rack to separate the beads, supernatants were collected in a different tube and diluted in 660 μl of binding/wash buffer. Supernatants were loaded onto an S-trap column, centrifuged at 4000 G 1 min. Columns were washed 10 times with binding/wash buffer. Proteins were digested to peptides applying 1 μg of trypsin diluted in 20 μl of digestion buffer directly to the column and incubated overnight at 37 °C in a humidified oven. Peptides were sequentially eluted from the column using 40 μl of elution buffer 1, 2 and 3 and dried using a SpeedVac before being shipped for LC-MS/MS.
Mass spectrometry
Peptides were analyzed by data-independent acquisition (DIA) mass spectrometry as described previously88,89. In summary, peptides were injected onto a nanoscale C18 reverse-phase chromatography system (UltiMate 3000 RSLC nano, Thermo Scientific) and electrosprayed into an Orbitrap Exploris 480 Mass Spectrometer (Thermo Fisher). For liquid chromatography the following buffers were used: buffer A (0.1% formic acid in Milli-Q water (v/v)) and buffer B (80% acetonitrile and 0.1% formic acid in Milli-Q water (v/v). Samples were loaded at 10 μl/min onto a trap column (100 μm × 2 cm, PepMap nanoViper C18 column, 5 μm, 100 Å, Thermo Scientific) equilibrated in 0.1% trifluoroacetic acid (TFA). The trap column was washed for 3 min at the same flow rate with 0.1% TFA then switched in-line with a Thermo Scientific, resolving C18 column (75 μm × 50 cm, PepMap RSLC C18 column, 2 μm, 100 Å). Peptides were eluted from the column at a constant flow rate of 300 nl/min with a linear gradient from 3% buffer B to 6% buffer B in 5 min, then from 6% buffer B to 35% buffer B in 115 min, and finally to 80% buffer B within 7 min. The column was then washed with 80% buffer B for 4 min and re-equilibrated in 3% buffer B for 15 min. Two blanks were run between each sample to reduce carry-over. The column was kept at a constant temperature of 50 °C.
The data was acquired using an easy spray source operated in positive mode with spray voltage at 2.445 kV, and the ion transfer tube temperature at 250 °C. The MS was operated in DIA mode. A scan cycle comprised a full MS scan (m/z range from 350-1650), with RF lens at 40%, AGC target set to custom, normalized AGC target at 300%, maximum injection time mode set to custom, maximum injection time at 20 ms, microscan set to 1 and source fragmentation disabled. MS survey scan was followed by MS/MS DIA scan events using the following parameters: multiplex ions set to false, collision energy mode set to stepped, collision energy type set to normalized, HCD collision energies set to 25.5, 27 and 30%, orbitrap resolution 30,000, first mass 200, RF lens 40%, AGC target set to custom, normalized AGC target 3000%, microscan set to 1 and maximum injection time 55 ms. Data for both MS scan and MS/MS DIA scan events were acquired in profile mode.
MS data analysis
RAW MS data were analysed by DIA-N-N version 1.8.133. Uniprot Mus musculus UP000000589 reference proteome was used for spectral library preparation as well as a custom FASTA file containing the TurboID sequence. Precursor m/z range was set at 350 to 1650. Neural network classifier was set at double-pass mode. Every other parameter was kept at default. The unique.gene result file was loaded onto Perseus version 1.6 for analysis90.
A list of contaminants was created from the no.turboID samples: first, only proteins identified in all no.turboID samples were kept and ranked by intensity. Then, a dataset specific threshold was set between known PSD proteins (DLG1, DLGAP1, GRIN2a, SHANK1) and known contaminants (endogenously biotinylated proteins ACACA, PCCA, MCCC1) to define true contaminants (Supplementary Figs. 2e, 3e). All keratins were considered contaminants and included in this list. Proteins in this list were removed from all other samples. Samples were log2 transformed and normalized by ‘substract median’ method. Only proteins present at least 3 times in at least one condition were kept (e.g. in all three Tlx3-Cre PSD95.turboID replicates) and missing values imputed with standard parameters. To enrich for synaptic proteins, we applied a two sample t-test with permutation-based FDR at 10% and s0 > 0.1 between the cytosolic.turboID and synaptic turboID. We then merged significant proteins from PSD95.turboID and Homer1.turboID (for experiments in Fig. 2) or PSD95.turboID in Tlx3-Cre and Sim1-Cre (for experiments in Fig. 3) to create the ‘synaptic protein list’ used for gene ontology and further comparison. To identify differentially expressed proteins between conditions (Figs. 2g, 4a), a t-test was performed on the synaptic proteins using an FDR < 0.05 and s0 > 0.1 to calculate significance. From Perseus, results tables were exported as “.tsv” for further analysis (described below) or data visualization using R.
PCA analysis
PCA analysis was performed after missing data imputation with standard parameters in Perseus and plotted using R (Figs. 2c, 3e).
Gene ontology analysis
Exported data from Perseus were imported to R and analyzed with the package gprofiler2 (0.2.2) based on gProfiler41. All proteins identified by MS were used as a reference background.
Protein annotation
Identified proteins were annotated using several published databases, complete annotation for the experiments presented in this work can be found in Supplementary Data 1 and 2. Data from Sorokina, Mclean, Croning34 were accessed with the R package RSQLite (2.3.1) or directly with MySQLlite software. SYNGO35 database (release 20210225) was imported to R from https://syngoportal.org/. Genetrek database was imported to R from https://genetrek.pasteur.fr/. SFARI database was imported to R from https://gene.sfari.org/. PantherDB functional classification was manually downloaded from https://pantherdb.org/. UNIPROT annotation was downloaded from the website and imported into R from https://www.uniprot.org.
Immunofluorescence
P35 mice were anesthetized by intraperitoneal injection with a lethal dose of 1 μl/g xylazine (VMB Xyl-M 2%), 2 μl/g ketamine (Eurovet Nimatek, 100 mg/ml), and 3 μl/g 0.9% saline. Next, mice were transcardially perfused with 4% PFA, in PBS. Brains were dissected, postfixed in 4% PFA in PBS at 4 °C overnight, washed in PBS and sliced to 50 μm thick by vibratome. Sections were blocked in 10% normal horse serum (NHS), 0.5 M Glycine, 0.5% Triton-X 100, 0.02% gelatin in 1X PBS for 2 h before incubation with antibodies in 5% NHS, 0.5% Triton-X 100, 0.02% gelatin in 1X PBS with extensive washing with 0.05% Triton-X 100 in 1X PBS between primary and secondary antibodies. Sections were counterstained with DAPI before being mounted on microscope slides with Mowiol-4-88 (Millipore). Imaging was performed on a Slide Scanner Axio Scan.Z1 (Zeiss) or LSM880 with Airyscan (Zeiss).
For spine density analysis, mice were perfused with 4% PFA (EM grade, Science Services), 2% Sucrose in 0.1 M PB buffer pH 7.4 and post-fixed 2 h at RT. Brains were sliced at 120 μm to increase the portion of dendritic tree for each positive neuron.
Mice injected with turboID AAVs were subcutaneously injected on the back of the animal with 2 mg/ml biotin in 1 mL PBS 3 h before perfusion.
Colocalization analysis of biotinylated proteins and postsynaptic markers
1–2 months old mice were injected with AAV expressing Homer1- or PSD95-turboID for 1 week in the somatosensory cortex as described. 3 h before perfusion biotin was administered subcutaneously. 60 μm coronal slices were stained for biotinylated proteins and for either PSD95 or Homer1. Image analysis was performed in FIJI: a mask for each image was created (using the Triangle threshold method) for Homer1, PSD95 and for biotinylated proteins produced by Homer1.turboID and PSD95.turboID. Biotinylated protein puncta signal was filtered for debris and out-of-focus signal removing particles <0.1 μm and >1 μm. Biotinylated protein puncta with postsynaptic marker signal were counted as positive.
RNAscope in situ hybridization
Mice injected with Cre-dependent GFP AAV were transcardially perfused at P35 as described. 20 um slices were produced on a Vibrating Microtome 7000 (Campden Instruments LTD), mounted on SuperFrost ultra Plus adhesion slides (Thermo-Fisher) and dried at RT. Sections were stained using the RNAscope™ Multiplex Fluorescent Detection Kit v2 (ACDbio, 323110) following manufacturer protocols. Briefly, sections were baked at 40 °C for 30 min to improve adhesion, treated for 10 min with hydrogen peroxide solution and 12 min with protease IV washing with 1X PBS in between. RNAscope probes were hybridized at 40 °C for 2 h and subsequently HRP signal developed using OPAL dyes. To counterstain the GFP, sections were blocked for 1 h at RT in 5% NHS, 0.3% TritonX-100, in PBS. GFP antibody was diluted in blocking solution 1:500 and added O/N at 4 °C (chicken anti-GFP). After 3 washes in 1X PBS, anti-chicken 488 secondary antibody was added 1:1000 for 1 h at RT. After 3 more washes in 1X PBS, sections were counterstained with DAPI for 5 min, and coverslipped with Mowiol. Images of GFP+ neurons were acquired on a Zeiss LSM900 using a 20X objective. Same laser power and gain settings were used to image the same probe in the two different mouse lines. Images were despeckled and single dots were counted manually in FIJI by a blinded observer. Statistical analysis was performed as an unpaired t-test with alpha = 0.05 after assessing normality of the data by Shapiro-Wilk test. Tests results are provided in Source Data files.
Dendritic spine imaging and analysis
Adult mice were injected in the barrel cortex with 200 nl of AAV mix consisting of pAAV-TRE-DIO-FLPo (Addgene #118027), pAAV-TRE-fDIO-GFP-IRES-tTA (Addgene #118026) and either cytosolic.turboID, PSD95.turboID or nothing. A week later, mice were injected with 1 ml of 2 mg/ml biotin in 1X PBS for 3 h before perfusion with 4% PFA, 2% sucrose in 0.1 M PB buffer pH 7.4 and post-fixed 2 h at RT. Brains were sliced at 120 μm to increase the portion of dendritic tree for each positive neuron. Brain slices were stained for GFP (antibody) and biotin (antibody). Dendritic spine images were acquired on a Zeiss LSM880 with airyscan detector using a 63X objective and 2.0 zoom. Optimal settings suggested by the software were used for pixel size and optical section size. Laser intensity and gain were adjusted for each image to obtain the best signal to noise ratio for GFP channel. Laser intensity and gain were kept the same for all conditions for the biotin channel. For each brain, 3 dendrites were imaged. Dendritic spines were reconstructed using Neurolucida 360 software and classified by morphology. Statistical analysis was performed with ANOVA test after assessing normality of the data by Shapiro-Wilk test. Tests results are provided in the Source Data files.
Human cortex data analysis
Processed SNARE-seq2 human cortex dataset was downloaded from Bakken et al.59. The gene expression data consists of 84178 cells selected by Bakken et al. Data analysis was performed using Seurat v4.391. Single-nucleus transcriptomics data was first normalized and highly variable genes were selected using SCTransform (variance stabilizing method) function from Seurat. Principal component analysis was then performed on normalized data. FindNeighbors function was subsequently used to compute the shared nearest neighbor graphs using 35 principal components (chosen using ElbowPlot function in Seurat). Finally, non-linear dimensionality reduction was performed using the uniform manifold approximation and projection (UMAP) method and clusters were obtained using Louvain clustering92 (resolution 0.5). Clusters were then manually annotated based on marker gene expressions.
Human layer 5 neuron cluster analysis
Cells belonging to layer 5 IT and layer 5 PT clusters were then selected for downstream analysis. Selected cells (5039 cells) gene expression matrix was normalized and scaled using SCTransform function from Seurat v4.391. Differentially expressed genes between cells from layer 5 IT and cells from layer 5 PT clusters were identified by performing Wilcoxon rank sum test using FindMarkers function from Seurat. Genes with adjusted p-value < 0.01 and log fold change > 0.25 were considered as differentially expressed.
Mouse cortex data analysis
Processed mouse cortex single-nucleus RNA-seq dataset was downloaded from Bakken et al.59. Low-quality nuclei were excluded if they did not meet all quality-control criteria: a minimum of 200 features per cell, a maximum of 9000 features per cell, and less than 5% mitochondrial content. In total 159719 nuclei passed quality control and were used for downstream analysis. First, gene expression data was normalized using SCTransform93 (variance stabilizing method) function from Seurat v4.3.091. Next, PCA was performed on the data. After inspecting the ElbowPlot, the first 35 dimensions were chosen to compute the shared nearest neighbor graphs using the FindNeighbors function in Seurat. Nuclei were then projected on non-linear dimensionality reduction space using UMAP method and clustering was performed using the Louvain clustering algorithm92 in Seurat (FindClusters function) with 0.5 resolution. The resulting clusters were then annotated based on marker genes expression.
Mouse layer 5 neuron cluster analysis
After performing quality control steps and cell type annotation, layer 5 IT and layer 5 PT nuclei (total of 33365 nuclei) were selected for further analysis. Variance stabilizing (SCTransform) normalization was applied on the selected nuclei raw expression data. Seurat FindMarkers function was subsequently applied using Wilcoxon rank sum test to identify differentially expressed genes between layer 5 IT and PT neurons. Genes with absolute log fold change value higher than 0.25 and adjusted pvalue lower than 0.01 were considered differentially expressed between layer 5 IT and PT neurons.
Antibodies
rat anti HA (Merck, 11867423001, WB: 1:1000, IHC: 1:1000), Alexa 647-conjugated streptavidin (Thermo Fisher Scientific, S32357, IHC: 1:500), Alexa 647-conjugated PSD95 sdAb FluoTag-X2 (Synaptic Systems N3702-AF647- IHC: 1:500), mouse anti-PSD-95 (NeuroMab clone K28/43, WB: 1:1000), rabbit Homer1 (Synaptic Systems, 160 003, WB: 1:5000, IHC: 1:1000), HRP conjugated streptavidin (Thermo Fisher Scientific, SA10001, WB: 1:5000), mouse anti Gria1 (Millipore, MAB2263, WB: 1:500), chicken anti-GFP (Aveslabs, GFP-1020, IHC: 1:500), rabbit anti Tubulin 3 (Abcam, ab18207, WB: 1:2500), mouse anti Synaptophysin (Sigma, S5768, WB 1:2000), Alexa fluor conjugated secondary antibodies from Invitrogen (1:1000).
Data availability
The RAW MS data generated in this study have been deposited in the ProteomeXchange via the PRIDE database: PXD046812 and PXD052081. The Human and Mouse transcriptomic data used in this study are available in the NeMo database under accession code nemo:dat-ek5dbmu [https://assets.nemoarchive.org/dat-ek5dbmu]. Source data are provided with this paper as a separate excel file. Source data are provided with this paper.
Code availability
Data analysis was performed using R version 4.3.2. Packages used for analysis are listed in the relevant paragraph in the method section. New code was not generated.
References
Harris, K. D. & Mrsic-Flogel, T. D. Cortical connectivity and sensory coding. Nature 503, 51–58 (2013).
Spruston, N. Pyramidal neurons: dendritic structure and synaptic integration. Nat. Rev. Neurosci. 9, 206–221 (2008).
Harris, K. D. & Shepherd, G. M. G. The neocortical circuit: themes and variations. Nat. Neurosci. 18, 170–181 (2015).
Suzuki, M. & Larkum, M. E. General anesthesia decouples cortical pyramidal neurons. Cell 180, 666–676.e13 (2020).
Takahashi, N. et al. Active dendritic currents gate descending cortical outputs in perception. Nat. Neurosci. 23, 1277–1285 (2020).
Shepherd, G. M. G. Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci. 14, 278–291 (2013).
Hooks, B. M. et al. Topographic precision in sensory and motor corticostriatal projections varies across cell type and cortical area. Nat. Commun. 9, 1–16 (2018).
Radnikow, G. & Feldmeyer, D. Layer- and cell type-specific modulation of excitatory neuronal activity in the neocortex. Front. Neuroanat. 12, 1 (2018).
Feldmeyer, D. Excitatory neuronal connectivity in the barrel cortex. Front. Neuroanat. 6, 24 (2012).
Baker, A. et al. Specialized subpopulations of deep-layer pyramidal neurons in the neocortex: bridging cellular properties to functional consequences. J. Neurosci. 38, 5441–5455 (2018).
Reiner, A., Hart, N., Lei, W. & Deng, Y. Corticostriatal projection neurons – dichotomous types and dichotomous functions. Front. Neuroanat. 4, 142 (2010).
Petreanu, L., Mao, T., Sternson, S. M. & Svoboda, K. The subcellular organization of neocortical excitatory connections. Nature 457, 1142–1145 (2009).
Sermet, B. S. et al. Pathway-, layer- and cell-type-specific thalamic input to mouse barrel cortex. eLife 8, e52665 (2019).
Audette, N. J., Urban-Ciecko, J., Matsushita, M. & Barth, A. L. POm thalamocortical input drives layer-specific microcircuits in somatosensory cortex. Cereb. Cortex 28, 1312–1328 (2018).
Li, N., Chen, T.-W., Guo, Z. V., Gerfen, C. R. & Svoboda, K. A motor cortex circuit for motor planning and movement. Nature 519, 51–56 (2015).
Kim, E. J., Juavinett, A. L., Kyubwa, E. M., Jacobs, M. W. & Callaway, E. M. Three types of cortical layer 5 neurons that differ in brain-wide connectivity and function. Neuron 88, 1253–1267 (2015).
Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 16, 551–563 (2015).
Roux, K. J., Kim, D. I., Raida, M. & Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 (2012).
Uezu, A. et al. Identification of an elaborate complex mediating postsynaptic inhibition. Science 353, 1123–1129 (2016).
Soto, J. S. et al. Astrocyte–neuron subproteomes and obsessive–compulsive disorder mechanisms. Nature 616, 764–773 (2023).
Hobson, B. D. et al. Subcellular proteomics of dopamine neurons in the mouse brain. eLife 11, e70921 (2022).
Sun, X. et al. Deep single-cell type proteome profiling of mouse brain by nonsurgical AAV-mediated proximity labeling. Anal. Chem. 94, 5325–5334 (2022).
Rayaprolu, S. et al. Cell type-specific biotin labeling in vivo resolves regional neuronal and astrocyte proteomic differences in mouse brain. Nat. Commun. 13, 2927 (2022).
Branon, T. C. et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887 (2018).
Velmeshev, D. et al. Single-cell genomics identifies cell type–specific molecular changes in Autism. Science 364, 685–689 (2019).
Willsey, A. J. et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of Autism. Cell 155, 997–1007 (2013).
Wamsley, B. et al. Molecular cascades and cell type–specific signatures in ASD revealed by single-cell genomics. Science 384, eadh2602 (2024).
Velmeshev, D. et al. Single-cell analysis of prenatal and postnatal human cortical development. Science 382, eadf0834 (2023).
Dani, A., Huang, B., Bergan, J., Dulac, C. & Zhuang, X. Superresolution imaging of chemical synapses in the brain. Neuron 68, 843–856 (2010).
Luo, W. et al. Supernova: a versatile vector system for single-cell labeling and gene function studies in vivo. Sci. Rep. 6, 35747 (2016).
May, D. G., Scott, K. L., Campos, A. R. & Roux, K. J. Comparative application of BioID and TurboID for protein-proximity biotinylation. Cells 9, 1070 (2020).
Kirkeby, S., Moe, D., Bøg-Hansen, T. C. & van Noorden, C. J. F. Biotin carboxylases in mitochondria and the cytosol from skeletal and cardiac muscle as detected by avidin binding. Histochemistry 100, 415–421 (1993).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Sorokina, O. et al. A unified resource and configurable model of the synapse proteome and its role in disease. Sci. Rep. 11, 9967 (2021).
Koopmans, F. et al. SynGO: an evidence-based, expert-curated knowledge base for the synapse. Neuron 103, 217–234.e4 (2019).
Kornau, H.-C., Schenker, L. T., Kennedy, M. B. & Seeburg, P. H. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Sci. N. Ser. 269, 1737–1740 (1995).
Kim, S. et al. NGL family PSD-95-interacting adhesion molecules regulate excitatory synapse formation. Nat. Neurosci. 9, 1294–1301 (2006).
Stillman, M., Lautz, J. D., Johnson, R. S., MacCoss, M. J. & Smith, S. E. P. Activity dependent dissociation of the Homer1 interactome. Sci. Rep. 12, 3207 (2022).
Gouwens, N. W. et al. Classification of electrophysiological and morphological neuron types in the mouse visual cortex. Nat. Neurosci. 22, 1182–1195 (2019).
Gerfen, C. R., Paletzki, R. & Heintz, N. GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and Basal Ganglia circuits. Neuron 80, 1368–1383 (2013).
Raudvere, U. et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Thomas, P. D. et al. PANTHER: making genome-scale phylogenetics accessible to all. Protein Sci. 31, 8–22 (2022).
The UniProt Consortium. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
de Wit, J. et al. Unbiased discovery of glypican as a receptor for LRRTM4 in regulating excitatory synapse development. Neuron 79, 696–711 (2013).
Cao, Y. et al. Interplay between cell-adhesion molecules governs synaptic wiring of cone photoreceptors. Proc. Natl Acad. Sci. USA 117, 23914–23924 (2020).
Choi, Y. et al. NGL-1/LRRC4C deletion moderately suppresses hippocampal excitatory synapse development and function in an input-independent manner. Front. Mol. Neurosci. 12, 119 (2019).
Kohama, S. G. & Urbanski, H. F. Distribution of glutamate receptor subunits in the primate temporal cortex and hippocampus. Brain Res. 769, 44–56 (1997).
Cremer, C. M., Lübke, J. H. R., Palomero-Gallagher, N. & Zilles, K. Laminar distribution of neurotransmitter receptors in different reeler mouse brain regions. Brain Struct. Funct. 216, 201–218 (2011).
Bailey, A. et al. Regional mapping of low-affinity kainate receptors in mouse brain using [3H](2S,4R)-4-methylglutamate autoradiography. Eur. J. Pharmacol. 431, 305–310 (2001).
Ayoubi, R. et al. Scaling of an antibody validation procedure enables quantification of antibody performance in major research applications. eLife 12, RP91645 (2023).
Schwanhäusser, B. et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011).
Carlyle, B. C. et al. A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci. 20, 1787–1795 (2017).
Li, J. et al. Cell-surface proteomic profiling in the fly brain uncovers wiring regulators. Cell 180, 373–386.e15 (2020).
Hall, J. & Bray, N. J. Schizophrenia genomics: convergence on synaptic development, adult synaptic plasticity, or both? Biol. Psychiatry 91, 709–717 (2022).
Trubetskoy, V. et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022).
de Wit, J. & Ghosh, A. Control of neural circuit formation by leucine-rich repeat proteins. Trends Neurosci. 37, 539–550 (2014).
Leblond, C. S. et al. Operative list of genes associated with autism and neurodevelopmental disorders based on database review. Mol. Cell. Neurosci. 113, 103623 (2021).
Abrahams, B. S. et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4, 36 (2013).
Bakken, T. E. et al. Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature 598, 111–119 (2021).
de Wit, J., Hong, W., Luo, L. & Ghosh, A. Role of Leucine-Rich repeat proteins in the development and function of neural circuits. Annu. Rev. Cell Dev. Biol. 27, 697–729 (2011).
Schroeder, A. & de Wit, J. Leucine-rich repeat-containing synaptic adhesion molecules as organizers of synaptic specificity and diversity. Exp. Mol. Med. 50, 10 (2018).
Ko, J. & Kim, E. Leucine-rich repeat proteins of synapses. J. Neurosci. Res. 85, 2824–2832 (2007).
Schroeder, A. et al. A modular organization of LRR protein-mediated synaptic adhesion defines synapse identity. Neuron 99, 329–344.e7 (2018).
DeNardo, L. A., de Wit, J., Otto-Hitt, S. & Ghosh, A. NGL-2 regulates input-specific synapse development in CA1 pyramidal neurons. Neuron 76, 762–775 (2012).
Nishimura-Akiyoshi, S., Niimi, K., Nakashiba, T. & Itohara, S. Axonal netrin-Gs transneuronally determine lamina-specific subdendritic segments. Proc. Natl Acad. Sci. 104, 14801–14806 (2007).
Bureau, I., von Saint Paul, F. & Svoboda, K. Interdigitated paralemniscal and lemniscal pathways in the mouse barrel cortex. PLoS Biol. 4, e382 (2006).
Young, H., Belbut, B., Baeta, M. & Petreanu, L. Laminar-specific cortico-cortical loops in mouse visual cortex. eLife 10, e59551 (2021).
Parikshak, N. N. et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in Autism. Cell 155, 1008–1021 (2013).
Gandal, M. J. et al. Broad transcriptomic dysregulation occurs across the cerebral cortex in ASD. Nature 611, 532–539 (2022).
Cerliani, L. et al. Increased functional connectivity between subcortical and cortical resting-state networks in Autism spectrum disorder. JAMA Psychiatry 72, 767–777 (2015).
Woodward, N. D., Giraldo-Chica, M., Rogers, B. & Cascio, C. J. Thalamocortical dysconnectivity in autism spectrum disorder: an analysis of the Autism brain imaging data exchange. Biol. Psychiatry Cogn. Neurosci. Neuroimag. 2, 76–84 (2017).
Iidaka, T., Kogata, T., Mano, Y. & Komeda, H. Thalamocortical hyperconnectivity and amygdala-cortical hypoconnectivity in male patients with Autism spectrum disorder. Front Psychiatry 10, 252 (2019).
Ayub, R. et al. Thalamocortical connectivity is associated with autism symptoms in high-functioning adults with autism and typically developing adults. Transl. Psychiatry 11, 93 (2021).
Hong, C.-J. & Hsueh, Y.-P. Cytoplasmic distribution of T-box transcription factor Tbr-1 in adult rodent brain. J. Chem. Neuroanat. 33, 124–130 (2007).
Hsueh, Y.-P., Wang, T.-F., Yang, F.-C. & Sheng, M. Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature 404, 298–302 (2000).
Chuang, H.-C., Huang, T.-N. & Hsueh, Y.-P. Neuronal excitation upregulates Tbr1, a high-confidence risk gene of autism, mediating Grin2b expression in the adult brain. Front. Cell Neurosci. 8, 280 (2014).
Doma, M. K. & Parker, R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440, 561–564 (2006).
Guydosh, N. R. & Green, R. Dom34 rescues Ribosomes in 3′ untranslated regions. Cell 156, 950–962 (2014).
Takano, T. et al. Chemico-genetic discovery of astrocytic control of inhibition in vivo. Nature 588, 296–302 (2020).
Gao, Y. et al. Proximity analysis of native proteomes reveals interactomes predictive of phenotypic modifiers of Autism and related neurodevelopmental conditions. Nat. Commun. 15, 6801 (2024).
Sharma, K. et al. Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 18, 1819–1831 (2015).
Willems, J. et al. ORANGE: a CRISPR/Cas9-based genome editing toolbox for epitope tagging of endogenous proteins in neurons. PLOS Biol. 18, e3000665 (2020).
Rimbault, C. et al. Engineering selective competitors for the discrimination of highly conserved protein-protein interaction modules. Nat. Commun. 10, 4521 (2019).
Daigle, T. L. et al. A suite of transgenic driver and reporter mouse lines with enhanced brain-cell-type targeting and functionality. Cell 174, 465–480.e22 (2018).
Challis, R. C. et al. Widespread and targeted gene expression by Systemic AAV Vectors: production, purification, and administration. https://doi.org/10.1101/246405 (2018).
Cho, K. F. et al. Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat. Protoc. 1–29 https://doi.org/10.1038/s41596-020-0399-0 (2020).
Loh, K. H. et al. Proteomic analysis of unbounded cellular compartments: synaptic clefts. Cell 166, 1295–1307.e21 (2016).
Walgrave, H. et al. microRNA-132 regulates gene expression programs involved in microglial homeostasis. iScience 26, 106829 (2023).
Molina-Gonzalez, I. et al. Astrocyte-oligodendrocyte interaction regulates central nervous system regeneration. Nat. Commun. 14, 3372 (2023).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, P10008 (2008).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Acknowledgements
We thank Esther Klingler, Patrik Verstreken, Pierre Vanderhaeghen and Anthony Holtmaat for critical reading of the manuscript, and De Wit lab members for helpful discussion and comments. We are grateful to Joris Vandenbempt for mouse colony maintenance and genotyping. We thank Keimpe Wierda for help with pilot experiments. We thank Charles Gerfen (National Institute of Mental Health, USA) and Nathaniel Heintz (The Rockefeller University, USA) for generously providing Sim1_KJ18-Cre mice. We thank Aya Takeoka (VIB-Imec-KU Leuven NeuroElectronics Research Flanders) for sharing Tlx3_P56-Cre and Rbp4-Cre mice. We thank Kristofer Davie (VIB Center for Brain & Disease Research Single-Cell Bioinformatics Expertise Unit) for bioinformatic support. G.M. is supported by Fonds Wetenschappelijk Onderzoek (FWO, Belgium) PhD fellowship 11F1219N and 11F1221N; Dan Dascenco is supported by FWO Postdoctoral fellowship 12W5218N, 12W5221N and FWO Research Grant 1513320 N; B.L-E. is supported by FWO PhD fellowship 1120821 N and 1120823 N; L.B. is supported by FWO Postdoctoral fellowship 12ZK221N. J.d.W. is supported by FWO Project Grants G0C4518N, G0A8720N and G0A8320N; FWO EOS Grant G0H2818N; ERANET-NEURON TAO2PATHY G0I3118N and a Methusalem Grant of KU Leuven/Flemish Government.
Author information
Authors and Affiliations
Contributions
G.M., D.Das., and J.d.W. conceived the study and designed experiments. G.M., D.Das., B.L.E., D.Daa., J.V., E.L., L.B. and A.J.M.H., performed experiments and analyzed data. A.J.M.H. provided guidance on proteomic sample preparation and analysis. G.M., D.Das., and J.d.W. wrote the paper with input from all authors. All authors contributed to and approved the final version.
Corresponding author
Ethics declarations
Competing interests
J.d.W. is scientific co-founder and served as scientific advisory board member of Augustine Tx. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Thomas Biederer, Tabrez Siddiqui and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Marcassa, G., Dascenco, D., Lorente-Echeverría, B. et al. Synaptic signatures and disease vulnerabilities of layer 5 pyramidal neurons. Nat Commun 16, 228 (2025). https://doi.org/10.1038/s41467-024-55470-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55470-w
This article is cited by
-
An integrated transcriptomic and proteomic map of the mouse hippocampus at synaptic resolution
Nature Communications (2025)
