
Abstract
Energy deprivation and metabolic rewiring of cardiomyocytes are widely recognized hallmarks of heart failure. Here, we report that HEY2 (a Hairy/Enhancer-of-split-related transcriptional repressor) is upregulated in hearts of patients with dilated cardiomyopathy. Induced Hey2 expression in zebrafish hearts or mammalian cardiomyocytes impairs mitochondrial respiration, accompanied by elevated ROS, resulting in cardiomyocyte apoptosis and heart failure. Conversely, Hey2 depletion in adult mouse hearts and zebrafish enhances the expression of mitochondrial oxidation genes and cardiac function. Multifaceted genome-wide analyses reveal that HEY2 enriches at the promoters of genes known to regulate metabolism (including Ppargc1, Esrra and Cpt1) and colocalizes with HDAC1 to effectuate histone deacetylation and transcriptional repression. Consequently, restoration of PPARGC1A/ESRRA in Hey2- overexpressing zebrafish hearts or human cardiomyocyte-like cells rescues deficits in mitochondrial bioenergetics. Knockdown of Hey2 in adult mouse hearts protects against doxorubicin-induced cardiac dysfunction. These studies reveal an evolutionarily conserved HEY2/HDAC1-Ppargc1/Cpt transcriptional module that controls energy metabolism to preserve cardiac function.
Similar content being viewed by others


Introduction
Heart failure (HF) is a clinical syndrome of cardiovascular diseases characterized by impaired contractile function of the heart and its reduced capacity to pump blood, a leading cause of morbidity and mortality with rising prevalence worldwide1. Although HF pathophysiologies are complex and include numerous etiologies2,3, mitochondria dysregulation causing insufficient energy supply is considered a shared pathology4. Mitochondrial abnormalities include impaired mitochondrial electron transport chain (ETC) activity, increased formation of reactive oxygen species (ROS), shifted metabolic substrate utilization and aberrant mitochondrial dynamics4. In the adult heart, cardiomyocytes (CMs) adopt fatty acid oxidation (FAO) as the primary energy source for mitochondrial ATP generation4,5. During HF, CMs shift from FAO to glycolysis to generate ATP4,6. Peroxisome proliferator-activated receptor gamma coactivator 1 (PPARGC1, also as PGC-1) is known as a central transcriptional coactivator complex that modulates the expression of mitochondrial oxidative phosphorylation genes in diverse organs7. This transcriptional complex comprises PPARGC1A (PGC-1α) and PPARGC1B (PGC-1β) that regulate genes for mitochondrial biogenesis and oxidative metabolism in interaction with multiple nuclear receptors including estrogen-related receptor (ESRR), peroxisome proliferator-activated receptor (PPAR), retinoid X receptor (RXR), and nuclear respiratory factor (NRF)8. Loss of function of these complex components in animal hearts causes defects in mitochondrial biogenesis and oxidative respiration, concurrent with increased ROS, resulting in cardiomyopathy and HF4,9,10. While temporal induction of Ppargc1a can improve mitochondrial oxidation and cardiac function, prolonged Ppargc1a overexpression deteriorates mitochondrial structure, dynamics, and increases ROS, causing adverse effects on the aged heart11. Whether and how excessive PPARGC1A activity is dampened and controlled to sustain cardiac homeostasis remains incompletely understood.
The family of HEY (Hairy/Enhancer-of-split related with YRPW motif, also known as HRT or HESR) transcriptional repressors, comprising HEY1, HEY2/GRIDLOCK (GRL) and HEYL, play important roles in development of cardiac, vascular, neuronal and other diverse tissues12,13,14,15,16,17. During embryogenesis, hey2 (grl) is initially expressed in arterial endothelial cells, then restricted to ventricular CMs and the postnatal myocardium17,18,19. We and others previously reported that Hey2 (Grl) interacts with Hey1 to regulate arterial-venous differentiation in response to Notch signaling12,15, and can limit expansion of cardiac progenitor cells and embryonic CMs in zebrafish embryos18,20. During mouse heart development, HEY1 and HEY2 participate in atrioventricular boundary patterning13,21,22, ventricular maturation16,23, and formation of the compact ventricular wall24,25. In humans, mutations or duplications of HEY2 contribute to congenital heart defects, thoracic aortic aneurysms26 and cognitive impairment27. Molecularly, HEY1, and HEY2 mediate transcriptional repression by directly binding E-box motif or suppressing GATA4-mediated transcription via interacting GATA factors18,28,29. Hey2 (Grl) is also demonstrated to impede CM renewal and zebrafish heart regeneration19. While HEY/HRT family members play prominent roles in cardiovascular development and regeneration, little is known about their impact on mitochondrial respiration and cardiac homeostasis.
Here, we describe that HEY2 is upregulated in patients with DCM and DOX-treated CMs. Using modified fish, mouse, and cell culture models and unbiased genome-wide analyses entailing transcriptomic, epigenomic, chromatin occupancy, and accessibility assays, we investigated the roles and mechanisms of Hey2 in cardiac pathophysiology. Our findings uncover HEY2 (GRL) as a bona fide modulator of mitochondrial oxidative metabolism in the adult heart. We find that HEY2 interacts with HDAC1 to impede the expression of genes encoding the master energy regulator PPARGC1A, its coactivators ESRRA, NRF1, and FAO enzyme CPT1A/CPT2 through histone deacetylation and chromatin condensation. These findings uncover a HEY2/HDAC1-Ppargc1/Cpt transcriptional mechanism coordinating mitochondrial oxidation and cardiac homeostasis in health and disease, which provides potential therapeutic strategies whereby silencing HEY2-mediated repression at local regions may empower failing heart.
Results
Hey2 upregulation in adult CMs results in contractile dysfunction and lethality
We analyzed human heart samples from cardiomyopathy patients (Supplementary Data 1). We detected the increased HEY2 expression in dilated cardiomyopathy (DCM) hearts (normal hearts vs DCM hearts: 0.9 vs 1.6, P < 0.05) (Fig. 1a, b). HEY2 upregulation has been also reported in human HF patients30. Adult zebrafish is an emerging vertebrate model to study cardiomyopathy and HF31,32,33,34. To further explore functional consequences of Hey2 induction in the adult heart, we generated an inducible hey2 overexpression zebrafish transgenic line Tg(cmlc2: loxP-NLSmCherry-Stop-loxP-Hey2-EGFP) [referred as Tg(cmlc2:nRSGG)]. Zebrafish harboring cmlc2:nRSGG were crossed with a CM-specific Cre driver line [Tg(cmlc2:CreER)]35, permitting hey2 induction specifically in the adult myocardium after 4-hydroxytamoxifen (4-HT) treatment (referred as hey2-mOE) (Fig. 1c)19. We subjected Tg(cmlc2:CreER;cmlc2:nRSGG) animals and Cre-negative littermates [Tg(cmlc2:nRSGG)] (Ctrl) to 4-HT treatment at 1 month old and analyzed at 2, 3, 4 and 5 months of age (Fig. 1d). The hey2-mOE hearts were conspicuously enlarged compared to Tg(cmlc2:nRSGG) control hearts (Supplementary Fig. 1a, b), exhibiting gradually sparse myofibers, thinner myocardial wall and expanded cardiac chambers from the age of 2 to 3 months (Fig. 1e–h). Accordingly, hey2-mOE hearts displayed increased CM size compared to control hearts (Supplementary Fig. 1c, d). The total CM number quantified by FACS analysis in hey2-mOE hearts was gradually reduced from 1-week post hey2 induction (1 wpi) to 1-month post induction (1 mpi) (Fig. 1i). We also assessed CM proliferation in hey2-mOE hearts at 2 mpi (3 months of age) and found a moderate increase in CM proliferation index (Supplementary Fig. 1e, f), suggesting a compensatory proliferation mechanism. We next performed in situ hybridization (ISH) on mOE hearts and observed the increased expression of cardiac fetal and stress markers myh7 and nppa (Fig. 1j). Notably, hey2-mOE hearts showed elevated expression of extracellular matrix collagen I and fibronectin compared with controls (Fig. 1k), which coincided with augmented collagen fiber depositions, revealed by Masson’s trichrome and Sirius red staining (Fig. 1l–n). RT-qPCR validated the upregulation of cardiac fetal and stress markers nppa, nppb, myh7, and fibrosis markers col1a2, fn1b in hey2-mOE hearts (Fig. 1o). By contrast, the expression of ventricle myosin heavy chain-like (vmhcl), a predominant myosin isoform of adult zebrafish heart, was declined (Fig. 1o), leading to an increased ratio of myh7/vmhcl in hey2-mOE hearts (Fig. 1p), indicating cardiac stress resurgence and pathologic remodeling.

a, b Western blots analysis and quantification of HEY2 in DCM patients and normal human hearts. 3 biological replicates. c Diagram of transgenic zebrafish used for inducible expression of Hey2 in CMs. d Schematic of 4-HT treatment and cardiac analysis for hey2-mOE and Ctrl zebrafish. e, f Section images stained with anti-MHC antibody (red) and quantification of ventricular area of hey2-mOE and Ctrl ventricles at 2 and 3 months. n = 5 (Ctrl), n = 7 (OE). Scale bar: 500 µm. g Immunofluorescence images of anti-MHC antibody in hey2-mOE and Ctrl fish at 3 months old. The dotted line depicts the ventricular wall. Scale bar: 100 µm. h Quantification of ventricular wall thickness of hey2-mOE and Ctrl fish. 2 months: n = 6 (Ctrl), n = 7 (OE). 3 months: n = 6 (Ctrl, OE). i Quantification of CM number of hey2-mOE and Ctrl fish at 1 wpi and 1 mpi by FACS analysis. 1 wpi (4 biological replicates); 1 mpi (3 biological replicates). j ISH analyses of myh7 and nppa expression in hey2-mOE and control ventricles. Scale bar: 100 µm. k Section images of ventricles from hey2-mOE and Ctrl fish. Co-staining: anti-fibronectin (green) and anti-MHC antibodies (red); anti-collagen I (green) and anti-MHC antibodies (red). n = 6 hearts. Scale bar: 50 µm. l, m Masson’s trichrome and Sirius red staining of hey2-mOE and control hearts. Higher-magnification of the dashed boxes (right). Scale bar: 50 µm. n Fibrosis area quantification in hey2-mOE and control hearts. n = 5. o, p RT-qPCR analysis of myh7, vmhcl, nppa, nppb, col1a, fn1b and myh7/vmhcl ratio in hey2-mOE and control hearts. 3 biological replicates. q Representative M-mode echocardiography images of hey2-mOE and control hearts at 3 months old. r–t Analysis of EF, VIDs and VIDd of hey2-mOE and control hearts. 3 months: n = 9 (Ctrl), n = 10 (OE). 4 months: n = 4 (Ctrl, OE). 5 months: n = 4 (Ctrl), n = 3 (OE). Data presents as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Student’s t-test (unpaired, two-tailed) (b, p) or Multiple t tests (two-tailed) (f, h, i, n, o, r–t). 2 months old (j, k, l, m). Source data are provided as a Source Data file.
We next assessed cardiac function by performing echocardiography on hey2-mOE hearts at 3-, 4-, and 5-month-old animals. The systolic function of hey2-mOE hearts was significantly impaired at 3 months of age (mOE versus control: ejection fraction 31% versus 52%, P < 0.0001; fractional shortening 13% versus 24%, P < 0.0001) (Fig. 1q, r and Supplementary Fig. 1g). The ventricular internal dimensions in both systole and diastole were profoundly increased (mOE versus control: VID at end-systole 1.5 mm versus 0.8 mm, P < 0.0001; VID at end-diastole 1.7 mm versus 1 mm, P < 0.0001), disclosing enlarged chamber of mOE heart (Fig. 1e, f, s, t). From 4 to 5 months of age, hey2-mOE animals exhibited a gradual reduction of ejection fraction and a progressive increase of chamber dimension (Fig. 1r–t and Supplementary Fig. 1g), suggesting progression into DCM and HF. Survival analysis of 4-HT treated cmlc2:nRSGG;cmlc2:CreER (n = 34) animals showed that 83% died after 12-month 4-HT induction, with 100% mortality by 16 months (Supplementary Fig. 1h).
CM-specific Hey2 expression impairs mitochondrial biogenesis and oxidative respiration
To investigate the mechanism underlying the development of HF in hey2-mOE hearts, we assessed cardiac ultrastructure and mitochondrial morphology. Transmission electron microscopy (TEM) revealed that hey2-mOE hearts possess fewer mitochondria than control hearts (Fig. 2a–c) and display profound morphological ultrastructure changes, which manifests irregular matrix and less cristae with empty vacuoles and broken membrane (Fig. 2a–I, a–II; b–I, b–II; d, e). However, mitochondrion size and sarcomere structure and organization were unaltered (Fig. 2f, Supplementary Fig. 1i). These results suggest that increased Hey2 expression in CMs disrupts mitochondrial biogenesis and morphogenesis.

a, b Representative TEM images of 2-month-old hey2-mOE and control hearts. Red arrow: degraded mitochondria. Higher-magnification images of the dashed boxes are shown on the right. Scale bar: 2 µm. c–f Quantifications of mitochondria density, damaged mitochondria density, cristae number and mitochondria (mito) area in hey2-mOE and control hearts. density (mito number per 25 µm2): 20 fields. cristae: 28 mito. area: 50 mito. ****P < 0.0001, Student’s t-test (unpaired, two-tailed). g Diagram of experimental design for OCR, MMP, ROS or TUNEL staining using adenovirus-infected or siRNAs-transfected NRVMs. h Western blots analysis of Flag-HEY2 using anti-Flag antibody in Adv-Hey2- and Adv-Ctrl-infected NRVMs. i Western blots analysis of HEY2 in Ctrl- and Hey2-siRNA-transfected NRVMs. j–m OCR analyses and quantification of basal respiration, maximal respiration, spare respiratory capacity and ATP production in Adv-Hey2- and Adv-Ctrl-infected NRVMs (j, k), as well as in Ctrl- and Hey2-siRNA-transfected NRVMs (l, m). n = 3 (Adv-Ctrl), n = 4 (Adv-Hey2), n = 6 (Ctrl-siRNA), n = 5 (Hey2-siRNA). n, p TMRM (red) staining (n) and JC-1 staining (p) in Adv-Hey2- and Adv-Ctrl-infected NRVMs. JC-1 aggregates (red, polarization), JC-1 monomers (green, depolarization). Scale bar: 50 µm. o Quantification of TMRM fluorescent intensity. Adv-Ctrl: n = 44, Adv-Hey2: n = 18. q Quantification of JC-1 aggregates/monomers ratio. n = 49 (Adv-Ctrl), n = 57 (Adv-Hey2). r TUNEL (red) staining of Adv-Hey2-Flag- and Adv-Flag- (Ctrl) infected NRVMs. α-actinin antibody: NRVMs (green). Flag antibody: infected-adenovirus (yellow). Scale bar: 50 µm. s Apoptosis percentage of Adv-transduced NRVMs. n = 8 fields. t Quantification of CM apoptosis in hey2-mOE and control hearts at 2 months old by FACS analysis. 3 biological replicates. u ATP level in hey2-mOE and control hearts at 2 months old. Ctrl: 6 biological replicates, hey2-mOE: 4 biological replicates. Box plots displayed median ± interquartile range (IQR), with whiskers extending to the minimum and maximum values. ****P < 0.0001, Student’s t-test (unpaired, two-tailed) (o, q). Data presents as mean ± SEM, *P < 0.05, **P < 0.01, ****P < 0.0001, Student’s t-test (unpaired, two-tailed) (k, m, s, t, u). Data presents as mean ± SD (j, l). Source data are provided as a Source Data file.
To further delineate the effects of HEY2 on mitochondrial biogenesis and respiratory function in mammalian CMs, Hey2 was infected using adenoviruses (Adv-Hey2) in isolated neonatal rat ventricular myocytes (NRVMs) and lentiviruses in human AC16 ventricular CM-like cells36 (Fig. 2g, h). We measured oxygen consumption rates (OCRs) in Adv-Hey2-transduced NRVMs and lentiviral Hey2-transduced AC16 cells. A significant OCR reduction was observed in basal and maximal respiration, and ATP production in NRVMs or AC16 cells transduced with Hey2 compared with respective controls (Fig. 2j, k and Supplementary Fig. 2a, b). In contrast, siRNA-mediated Hey2 reduction resulted in elevated OCR levels in maximal respiration, spare capacity and ATP production (Fig. 2i, l, m), suggesting that HEY2 negatively regulates mitochondrial oxidative respiration. We next measured mitochondrial membrane potential (MMP) for ATP sysnthesis37 using TMRM38, a cell-permeant dye that accumulates in polarized mitochondria. We found that mitochondria were depolarized in NRVMs or human AC16 cells transduced with HEY2, resulting in lower MMPs in comparison with control cells (Fig. 2n, o and Supplementary Fig. 2c, d). To substantiate the findings, JC-1, a fluorescent cationic probe that enters into polarized mitochondria, was employed to assess mitochondrial respiratory function39. Hey2-transduced NRVMs or AC16 CM-like cells exhibited a decreased ratio of mitochondrial polarization (red fluorescent aggregates/green fluorescent monomers) compared with control cells (Fig. 2p, q and Supplementary Fig. 2e, f). Accordingly, electron transport chain activity in complex I (NADH dehydrogenase complex) decreased in Adv-Hey2-infected NRVMs compared to control NRVMs (Supplementary Fig. 2g). Derangement of ETC complex and damaged mitochondrial ultrastructure also contribute to elevated ROS production40,41. We detected the increased ROS in NRVMs or AC16 cells ectopically expressing HEY2, as revealed by MitoSOX staining (Supplementary Fig. 2h–k). Enhanced ROS was associated with increased apoptosis in Hey2-overexpressing NRVMs or zebrafish CMs isolated from hey2-mOE hearts at 1 mpi (Fig. 2r–t), accompanied by the reduced ATP production (Fig. 2u). Collectively, these findings indicate that HEY2 upregulation disrupts mitochondrial biogenesis, reduces ETC activity and impairs oxidative respiration.
Hey2 repressor negatively regulates mitochondrial bioenergetics pathways and maintains cardiac homeostasis
To decipher the molecular underpinnings of Hey2-induced mitochondrial and cardiac dysfunction, we assessed early transcriptomic profiling changes in hey2-mOE hearts. Young adult animals at 1-week post hey2 induction by 4-HT were subjected to RNA-seq analyses before the emergence of an overt cardiac phenotype to minimize the secondary effect of cardiac remodeling and dysfunction. Pearson correlation coefficient analysis indicated high reproducibility of RNA-seq data after normalization (Fig. 3a). RNA-seq analyses identified an increase in expression of 1573 genes and a reduction in expression of 1065 genes in hey2-mOE-1 wpi hearts (Fig. 3b). Gene Ontology analysis revealed the downregulated genes that were enriched in the categories of mitochondrion, mitochondrial respiratory chain complex, and ATP synthase complex (Fig. 3c). ETC complex I, II, III, IV, V genes were systematically reduced (Fig. 3d). These findings were further supported by transcriptome analyses of hey2-mOE hearts 1-month post hey2 induction by 4-HT (Supplementary Fig. 3a–f). We found that hey2 mRNAs levels were 1.8-fold increase in hey2-mOE hearts compared to endogenous hey2 mRNA levels of control hearts (Supplementary Fig. 3g). Hey2 protein was increased in hey2-mOE hearts (Supplementary Fig. 3h, i). Furthermore, the protein levels of selected ETC genes were evaluated by immunoblot analysis that confirmed a reduction of Ndufb8 (NADH:ubiquinone oxidoreductase subunit b8), Sdhb (succinate dehydrogenase complex, subunit B, iron sulfur), Cox1(cytochrome c oxidase subunit I) and Atp5a1 (ATP synthase, H+ transporting, mitochondrial F1 complex, subunit a1) in hey2-mOE hearts (Fig. 3e, f). We then interrogated whether key transcriptional regulators of mitochondrial metabolic gene program were downregulated in hey2-mOE hearts at early stages. Analyses of 1 wpi-mOE transcriptome dataset revealed a decrease in the expression of master energy regulator genes ppargc1a and ppargc1b, along with transcriptional coactivator genes esrra, pparaa, and rxraa42,43 (Fig. 3g). Furthermore, carnitine palmitoyltransferase 1ab (cpt1ab), a gene encoding enzyme essential for the transport of long-chain fatty acids into mitochondria for β-oxidation44 was also reduced (Fig. 3g). Together, these findings suggest that the increased Hey2 level in CMs causes a reduction in mitochondrial biogenesis and oxidative respiration. This was further supported by the reduced PPARGC1A, ESRRA and CPT1 proteins (Fig. 3h, i), the declined mitochondrial mass (Fig. 3j, k and Supplementary Fig. 3j, k) as well as the decreased FAO activity (Fig. 3l), in Hey2 overexpressing NRVMs or AC16 CM-like cells. In human DCM hearts, levels of PPARGC1A/B, ESRRA and NRF1 mRNA were also found to be reduced (Supplementary Fig. 3l). Furthermore, we observed a decrease of metabolite intermediates such as pyruvate and α-ketoglutarate (α-KG), but an increased glycogen level in zebrafish hey2-mOE hearts (Supplementary Fig. 3m). Genes regulating glycolysis, TCA cycle and glycogen synthesis were accordingly altered in hey2-mOE hearts (Supplementary Fig. 3n). Finally, a concordant upregulation of gene groups pertaining to apoptotic cell signaling, cardiac development and extracellular matrix was observed in hey2-mOE hearts at 1 mpi (Supplementary Fig. 3o), suggesting that many of the increased genes are a response to Hey2-induced mitochondrial and cardiac dysfunction, reminiscent to DCM and HF pathogenesis (Fig. 1e–t).

a Pearson correlation coefficients analysis showing the correlation between biological replicates of aligned RNA-seq data form hey2-mOE and control hearts. Correlation coefficient closer to 1 indicates better agreement. 3 biological replicates. b Volcano plot of expressed genes in hey2-mOE-1 wpi and control hearts. The colored dots were the differentially expressed genes (DEGs). Red: upregulated DEGs, green: downregulated DEGs. The horizontal dot line indicated Q < 0.05. The vertical line indicated fold change of 2. c GO analysis based on cellular component of downregulated genes in the hey2-mOE-1 wpi profile. d Heatmap of expression profile of affected mitochondrial ETC complex I, II, III, IV and V genes in hey2-mOE-1 wpi and control heart (FC > 1.5 and Q < 0.05). e, f Western blots analysis and quantification of ETC complexes in hey2-mOE and control heart at the age of 2 months. 3 biological replicates. g Heatmap of expression profile of master regulators of mitochondria biogenesis in hey2-mOE-1 wpi and control heart (FC > 1.4 and Q < 0.05). h, i Western blots analysis and quantification of PGC1A, ESRRA, CPT1A and Flag-HEY2 protein in Adv-Hey2- and Adv-Ctrl-infected NRVMs. 3 biological replicates. j MitoTracker Red CMXRos (red) staining measuring the mitochondrial mass of Adv-Hey2- and Adv-Ctrl-infected NRVMs. The NRVMs was marked by CM marker α-actinin (green). The anti-Flag antibody showed the nuclear-located HEY2-Flag protein (yellow). Scale bar: 50 µm. k Quantification of MitoTracker Red fluorescent intensity in Adv-Hey2- and Adv-Ctrl-infected NRVMs. Adv-Ctrl: n = 69, Adv-Hey2: n = 38. l Measurement of FAO activities in Adv-Hey2- and Adv-Ctrl-infected NRVMs. 3 biological replicates. m GO analysis based on cellular component of upregulated genes in the hey25nt-/- profile. n Representative long-axis M-mode echocardiography images of hey2+/+ and hey25nt-/- hearts at the age of 3 months. o Echocardiography analysis of EF and VIDd of hey2+/+ and hey25nt-/- hearts. hey2+/+: n = 9, hey25nt-/-: n = 12. Data presents as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Student’s t-test (unpaired, two-tailed) (k, l, o) or Multiple t tests (two-tailed) (f, i). Source data are provided as a Source Data file.
To assess the effects of Hey2 deficiency, as opposed to Hey2 increase, on mitochondrial bioenergetics and cardiac function, we performed transcriptome analyses in hey25nt-/- mutant zebrafish at 3-month-old. hey25nt-/- hypomorph mutants can survive to young adulthood19 and exhibited a reduction in myh7, col1a1a, and fibronectin, compared to the age-matched WT hearts (Supplementary Fig. 4a, b). GO analysis identified upregulated genes in hey25nt-/- mutant hearts were enriched in cellular component ‘mitochondria matrix’, ‘ATP synthase complex’ and ‘membrane-bound organelle’ (Fig. 3m). Genes encoding Ppargc1 energy regulator complex (ppargc1a, esrra, pparab, and rxrga), as well as genes of ETC complex I-V were upregulated in hey25nt-/- hearts (Supplementary Fig. 4c). Furthermore, hey2 mutant hearts contained increased numbers of mitochondria with regular shape and ordered cristae (Supplementary Fig. 4d–i). Echocardiography revealed enhanced contractile function with increased EF and approximately unaltered VIDd in hey25nt-/- hearts (mutant vs control: EF, 56% vs 44%, P < 0.01; VIDd, 1.14 mm vs 1.12 mm, P = 0.79) (Fig. 3n, o). At the age of 12 months, hey25nt-/- hearts even exhibited thicker myocardial walls (Supplementary Fig. 4j, k). Together, these findings indicate that reduced Hey2 activity in adult zebrafish heart stimulates mitochondrial biogenesis and elevates cardiac function.
Hey2 directly impedes the transcription of Ppargc1 energy coactivator complex genes in the setting of cardiac dysfunction
Reduced expressions of Ppargc1 energy coactivators and ETC complex subunits in hey2-mOE hearts suggested that increased Hey2 represses the transcription of energy regulatory genes. To test this hypothesis, we performed cleavage under targets and tagmentation-sequencing (CUT&Tag-seq) and transposase-accessible chromatin sequencing (ATAC-seq) assays in isolated hey2-mOE and control sibling hearts at the age of 2 months. Spearman correlation analysis indicated high reproducibility of CUT&Tag-seq and ATAC-seq data after normalization (Fig. 4a and Supplementary Fig. 5a). About 29% of detected Hey2 binding sites were located in promoter and 5’ UTR regions, 26% distributed over gene body regions and 31% to intergenic regions (Fig. 4b), suggesting that Hey2 binding is more concentrated in promoter regions compared to intergenic regions. We found that hey2 overexpression caused a significant increase in detectable Hey2 bindings to promoter regions (<=2 kb from TSS) compared to control hearts (74%; 12213 increased peaks/16411 total differential peaks) (Fig. 4c). Regions with reduced Hey2 bindings (26%; 4198 decreased peaks/16411 total differential peaks) were also detectable (Fig. 4c), suggesting indirect mechanisms preventing Hey2 bindings to those regions. By profiling chromatin accessibility, we observed that the majority of differential transposase-accessible regions in the promoter areas exhibited the reduced chromatin accessibility (94%; 11791/12522 total differentially marks, FC > 1.5) in hey2-mOE hearts compared to control hearts (Fig. 4d), consistent with the analysis of genome-wide ATAC-seq signals (Supplementary Fig. 5b). Furthermore, the decrease differences in ATAC signals between hey2-mOE and control hearts were significantly larger in Hey2-occupied promoter regions than in Hey2-bound intergenic regions, whereas ATAC peaks in non-Hey2-bound regions were slightly increased (Fig. 4e), suggesting that Hey2 binding primarily facilitates chromatin closure at its occupancy sites in the promoter regions. Motif analysis of Hey2-occupied genome regions identified Hey2 DNA-binding site28 (E box) as one of top-enriched motives (Fig. 4f). Lastly, 4324 genes with increased Hey2 occupancy and reduced chromatin accessibility in the promoter regions were identified, which were related to functional categories in mitochondrial morphogenesis, energy homeostasis, chromosome organization and histone modification (Fig. 4g). Among these genes were the key energy regulatory genes ppargc1a, esrra, nrf1 and FAO enzyme cpt1ab (Fig. 4h). Using Integrative Genomics Viewer (IGV), a marked increase in Hey2 occupancy was observed at the promoter regions of ppargc1a, esrra, or cpt1ab, which was associated with reduced chromatin accessibility in hey2-mOE hearts compared to control hearts (Fig. 4i), revealing these key metabolic regulatory genes as direct targets of Hey2-induced transcriptional repression.

a Spearman correlation coefficients analysis showing the correlation between biological replicates of ATAC-seq samples from hey2-mOE and control hearts. Correlation coefficient closer to 1 indicates better agreement. b Pie chart of Hey2 peak distribution determined from whole-genome CUT&Tag sequencing data. Promoter was defined as ± 500 bp from the transcription start site (TSS). c, d Heatmaps of Hey2 CUT&Tag-seq and ATAC-seq signals in hey2-mOE hearts compared to control hearts, using a 2 kb window centered on promoter regions and sorted in descending order by signal intensity. e Boxplot showing ATAC-seq mark signals in the Hey2-occupied promoter regions (n = 6752 regions each group), Hey2-bound intergenic regions (n = 20826 regions each group), and non-Hey2-occupied regions (n = 6752 regions each group) in hey2-mOE hearts and control hearts. Signal intensity is represented by the log10 (RPKM + 1) value of ATAC-seq data. Box plots displayed median ± interquartile range (IQR), with whiskers extending to 1.5*IQR. P value by paired Wilcoxon test. f Top enriched motifs of SP2, HEY2 (E box) and NFYA at Hey2-occupied genome regions. Hey2-occupied areas were defined by peaks called from Hey2 CUT&Tag-seq. Binomial test by “CentriMo” algorithm of software package “MEME Suite”. P value was adjusted by Bonferroni correction. g Bar graph showing -log10 P values for GO categories significantly represented in the overlapped genes with increased Hey2 binding peaks and reduced chromatin accessibility on the promoter regions in hey2-mOE hearts. Hypergeometric test by “enrichGO” function of R package “clusterProfiler”. P value was adjusted by Benjamini-Hochberg correction. h Venn diagram outlining 4324 overlapped genes between genes with increased Hey2 binding (CUT&Tag-seq up) and reduced chromatin accessibility (ATAC-seq down) in hey2-mOE hearts (including ppargc1a, ppargc1b, esrra, esrrga, nrf1 and cpt1ab). i The Genome browser views at loci of ppargc1a, esrra and cpt1ab showing Hey2 CUT&Tag-seq, ATAC-seq, and RNA-seq signals from hey2-mOE and control hearts. The signal values are indicated on the left. 2 biological replicates. Source data are provided as a Source Data file.
HEY2 interacts with HDAC1 to mediate transcriptional repression and chromatin remodeling during cardiac energy homeostasis
To further evaluate the conserved effects of HEY2-mediated transcriptional modulation in mammalian CMs, we performed HEY2 CUT&Tag-seq and RNA-seq analyses of NRVMs transduced with Adv-Hey2 (Fig. 5a and Supplementary Fig. 5c). In addition, H3K9ac and H3K27ac CUT&Tag-seq analyses of the Adv-Hey2-transduced NRVMs were performed (Fig. 5a–c), given that histone deacetylation was implicated in HEY-induced transcriptional repression45. Spearman correlation coefficient analysis indicated high reproducibility of CUT&Tag-seq data after normalization (Supplementary Fig. 5d). Notably, we observed a genome-wide reduction of H3K9ac (93.9%; 6234 reduced peaks/6638 total altered peaks) and H3K27ac depositions (94.7%; 3295 reduced peaks/3480 total altered peaks) in Hey2-transduced NRVMs compared to controls (Fig. 5b, c). Furthermore, the H3K9ac/H3K27ac signals were significantly decreased genome-wide in HEY2-occupied regions compared to non-HEY2-bound regions in Adv-Hey2-transduced NRVMs (Supplementary Fig. 5e, f), suggesting that HEY2 facilitates histone deacetylation at its occupancy sites. We next performed the overlap analysis between HEY2 CUT&Tag-seq and H3K9ac/H3K27ac CUT&Tag-seq data and observed that a reduction of H3K9ac and/or H3K27ac depositions was associated with a gained HEY2 occupancy at downregulated genes encoding PPARGC1 energy coactivator complex regulators (Ppargc1a/Ppargc1b, Esrra/Esrrg, Nrf1, Rxra/Rxrg, Ppara/Pparg), as well as fatty acid oxidation enzymes (Cpt1a, Cpt2) (Fig. 5d). Specifically, we observed that decreased H3K27ac/H3K9ac marks were associated with increased HEY2 binding peaks at the promoter regions of Ppargc1b, Esrra or Cpt2, which was accompanied by the declined gene transcription (Fig. 5e, f and Supplementary Fig. 5i). Furthermore, basal HEY2 binding signals were detectable at those promoter regions in control NRVMs, suggesting its roles in limiting the excessive energy metabolism gene expression during cardiac homeostasis. Given that class I histone deacetylases are involved in cardiac homeostasis22,46, we postulated that HEY2 recruits HDAC1, an abundantly expressed HDAC in adult hearts47,48, to mediate the transcriptional repression. To test the idea, we performed HDAC1 CUT&Tag-seq, ATAC-seq in Adv-Hey2-transduced NRVMs and siRNA-mediated Hey2 knockdown NRVMs. Spearman correlation analysis indicated high reproducibility of HDAC1 CUT&Tag-seq and ATAC-seq data after normalization (Supplementary Fig. 5g, h). We observed a reduction of HDAC1 marks at the promoter regions of Ppargc1b, Esrra or Cpt2, in Hey2-knockdown NRVMs, which was associated with the increased chromatin accessibility (Fig. 5e, f and Supplementary Fig. 5i). In contrast, Hey2 overexpression resulted in an elevation of HDAC1 occupancy in the promoters of these energy regulatory genes, accompanied by the reduced chromatin accessibility (Fig. 5e, f and Supplementary Fig. 5i). Accordingly, an increased expression of energy metabolism genes and ETC complex genes was detectable in Hey2-reduced NRVMs (Supplementary Fig. 5j). Furthermore, the enrichment of E box was significantly detectable not only in the reduced chromatin accessibility regions in Adv-Hey2-transduced NRVMs, but also in the increased ATAC signal areas in Hey2 knockdown NRVMs (Supplementary Fig. 5k), suggesting E box as a primary binding site of HEY2. We next performed the analysis of the relationship between genome-wide HEY2 occupancy, HDAC1 association, histone acetylation and chromatin accessibility. The results showed that at the HEY2 occupancy regions in both promoter and intergenic regions, HDAC1 binding signals were significantly increased in Hey2-OE NRVMs compared to control cells, whereas H3K27ac and ATAC signals were reduced (Supplementary Fig. 5l). On the contrary, in Hey2-KD NRVMs, the HDAC1 signals were decreased in HEY2-occupied promoter and intergenic regions, whereas H3K27ac and ATAC signals were increased. Notably, the differences in signals between Hey2-OE/KD NRVMs and control cells were larger in promoter regions than in intergenic regions (Supplementary Fig. 5l), suggesting that the promoter regions are the major sites of HEY2-mediated HDAC1 recruitment, histone deacetylation and changes in chromatin accessibility changes.

a Schematic diagram for CUT&Tag-seq and ATAC-seq analysis using Adv-Hey2-transduced or siRNAs-transfected NRVMs. b, c Heatmaps of H3K9ac and H3K27ac CUT&Tag-seq signals in Adv-Hey2-infected NRVMs compared to controls, using a 3 kb window centered on peak regions and sorted in descending order by signal intensity. d Venn diagram outlining 1484 overlapped genes between genes with HEY2 binding peaks and reduced H3K9 acetylation and/or H3K27 acetylation in the promoter regions. e, f The Genome browser views at locus of Ppargc1b and Esrra showing marks of Flag-HEY2, H3K9ac, H3K27ac CUT&Tag-seq and RNA-seq from Adv-Ctrl- and Adv-Hey2-infected NRVMs; and also showing marks of HDAC1 CUT&Tag, ATAC-seq from Ctrl-siRNA- and Hey2-siRNA (knockdown, KD)-transfected NRVMs or Adv-Ctrl- and Adv-Hey2-infected NRVMs. 2 biological replicates. g, h Hey2 or Hdac1 immunoprecipitation with anti-Flag antibody or anti-HA antibody, respectively. Hey2 or Hdac1 was detected using anti-Flag or anti-HA antibody. 3 independent experiments. i Co-immunoprecipitation analysis of Flag-HEY2 and endogenous HDAC1 in Adv-Ctrl- and Adv-Hey2-infected NRVMs. 3 independent experiments. j Luciferase activities in 293 T cells that were transfected with pGL3-PGC1A-Fluc, pGL3-ESRRA-Fluc or pGL3-CPT2-Fluc with control pGL3, pGL3-HEY2, pGL3-HDAC1, and pGL3-HEY2 and pGL3-HDAC1 together. The relative flyfire luciferase activity was normalized by Renilla luciferase and calculated as the ratio of each experimental group to the control group. 3 biological replicates. Data presents as mean ± SD, *P < 0.05, **P < 0.01, ****P < 0.0001, Student’s t-test (unpaired, two-tailed). Source data are provided as a Source Data file.
To further assess whether HEY2 physically interacts with HDAC1, we performed immunoprecipitation experiments and detected the forward and reciprocal interactions between HA-HDAC1 and Flag-HEY2 in 293 T cells (Fig. 5g, h). The interaction was also observed between endogenous HDAC1 and Flag-HEY2 in NRVMs (Fig. 5i). We next performed luciferase reporter assays to verify the extent to which HDAC1 mediates HEY2-induced transcriptional suppression. We generated luciferase reporter vectors in which the promoter regions of PPARGC1A, ESRRA or CPT2 drive firefly luciferase (Fluc). These Fluc expression plasmids were co-transfected with pcDNA3-HEY2 and/or pcDNA3-HDAC1. We observed that HEY2 transfections reduced PPARGC1A-, ESRRA-, or CPT2-mediated luciferase activities (Fig. 5j), whereas HDAC1 transfections alone did not reduce the luciferase activities (Fig. 5j). Notably, HDAC1 together with HEY2 further reduced luciferase activities mediated by these gene promoters compared to HEY2 alone (Fig. 5j). Altogether, these findings demonstrate that HEY2 recruits HDAC1 to mediate transcriptional repression and chromatin remodeling during cardiac energy homeostasis.
Increased PPARGC1A and ESRRA expression ameliorates the HEY2-induced mitochondrial bioenergetics deficit
Giving that Hey2 negatively regulates mitochondrial bioenergetics by suppressing Ppargc1 energy regulatory complex genes, we reasoned that increased Ppargc1a and Esrra expression may rescue Hey2-induced mitochondrial biogenesis and function defects. To test the idea, we introduced human PPARGC1A and/or ESRRA in lentiviral HEY2-transduced human AC16 CM-like cells. We observed a moderate elevation in mitochondrial mass, ATP production or ETC complex gene expression in PPARGC1A and/or ESRRA introduced-AC16 CM-like cells (Fig. 6a–d). Notably, the HEY2-mediated reduction of mitochondrial volume, ATP levels or ETC gene expression was substantially recovered by transfection of both PPARGC1A and ESRRA (Fig. 6a–d). Accordingly, HEY2-mediated reduction of ETC complex I activity was also greatly restored by transfections of PPARGC1A and ESRRA in comparison with controls (Fig. 6e). Furthermore, transfections of PPARGC1A and/or ESRRA substantially reduced ROS levels in HEY2-transduced AC16 cells, but failed to decrease ROS in control cells (Supplementary Fig. 6a, b). These findings indicate that HEY2 regulates mitochondrial energy metabolism by specifically targeting PPARGC1A and ESRRA.

a MitoTracker Red CMXRos staining measuring the mitochondrial mass of control AC16 or HEY2-OE AC16 cells transfected with control vectors, PPARGC1A or ESRRA vectors, as well as PPARGC1A and ESRRA plasmids together. Scale bar: 50 µm. b Quantification of MitoTracker Red fluorescent intensity of transfected HEY2-OE AC16 cells and control AC16 cells. Data were relative to fluorescent intensity in control group. n = 38 (ctrl+vector, OE + PPARGC1A + ESRRA), n = 41 (ctrl+PPARGC1A), n = 39 (ctrl+ESRRA), n = 43 (the rest groups). Box plots displayed median ± interquartile range (IQR), with whiskers extending to the minimum and maximum values. *P < 0.05, **P < 0.01, ****P < 0.0001, Student’s t-test (unpaired, two-tailed). c ATP levels of control AC16 and HEY2-OE AC16 cells transfected with control vectors, PPARGC1A or ESRRA vectors, and PPARGC1A and ESRRA plasmids together. Data were relative to ATP level in control group. 3 biological replicates. d RT-qPCR analysis showing ETC complex genes that expressed in control AC16 and HEY2-OE AC16 cells transfected with control vectors, PPARGC1A or ESRRA vectors, and PPARGC1A and ESRRA plasmids together. 3 biological replicates. e Measurement of mitochondrial ETC complex I activity in control AC16 and HEY2-OE AC16 cells transfected with control vectors, and PPARGC1A and ESRRA plasmids together. 3 biological replicates. f Schematic synthesis diagram of Flag-ppargc1a and HA-esrra mRNA with N1mΨ and Cap analogs modifications. g Experimental diagram for 4-HT treatment, modRNAs injection and cardiac analysis. h Echocardiography analyses depicting EF and FS in hey2-mOE and control hearts injected with vehicle, ppargc1a modRNA, esrra modRNA, or ppargc1a and esrra modRNAs together. n = 10 (hey2-mOE+ppargc1a+esrra), n = 11 (the rest groups). i RT-qPCR analysis showing ETC complex genes that differentially expressed in hey2-mOE and control hearts injected with vehicle, ppargc1a modRNA, esrra modRNA, or ppargc1a and esrra modRNAs together. 3 biological replicates. Data presents as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Student’s t-test (unpaired, two-tailed) (c, e, h) or Multiple t tests (two-tailed) (d, i). Source data are provided as a Source Data file.
To further test whether the increased expression of ppargc1a and esrra recovers the depressed cardiac function of hey2-mOE hearts, we performed intracardiac injections of ppargc1a and esrra mRNAs with N1-methylpseudouridine (N1mΨ) modifications (modRNAs)49,50,51. ppargc1a and esrra modRNAs were first synthesized using N1mΨ and Cap analogs to increase their stabilities (Fig. 6f). We next performed a single injection of N1mΨ-modified ppargc1a and/or esrra modRNAs (1.5 µg) into the 4-HT-induced hey2-mOE heart and control heart (Fig. 6g). Western blot analyses revealed the high expression levels of Flag-tagged Ppargc1a and HA-tagged Esrra proteins at 1 day post-injections (dpi) and 3 dpi, which was reduced at 7 dpi (Supplementary Fig. 6c–f). The localization of Flag-Ppargc1a and HA-Esrra in the nuclei of CMs of modRNA-injected hearts was further verified by double immunostaining analyses using antibodies against Mef2 and Flag or HA. We observed that the proteins of Flag-Ppargc1a and HA-Esrra were localized in the nuclei of CMs throughout the entire ventricle at 1 dpi (Supplementary Fig. 6g, h). The CM transfection percentages of ppargc1a and esrra modRNAs were quantified to be approximately 80% and 60%, respectively (Supplementary Fig. 6i, j). As a result, the reduced expression of ETC complex genes was substantially restored in hey2-mOE hearts co-injected with ppargc1a and esrra modRNAs at 3 dpi in comparison with injections of either modRNAs (Fig. 6g, i). Importantly, injections of both ppargc1a and esrra modRNAs recovered the depressed EF and FS in hey2-mOE hearts at 7 dpi (Fig. 6g, h), whereas injections of either ppargc1a or esrra modRNAs failed to restore the compromised cardiac function (Fig. 6h). These results substantiate the findings that Hey2-induced mitochondrial and cardiac dysfunction is mediated by reduced Ppargc1a and Esrra.
Inducible Hey2 depletion protects mouse heart against doxorubicin-induced cardiac injury
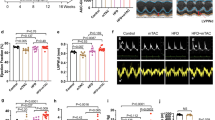
To further corroborate the functions of Hey2 in the adult mouse heart, we generated a Hey2 flox allele (designated Hey2flox) (Fig. 7a). These Hey2flox mice were crossed to mice heterozygous for the Myh6-Mer-Cre-Mer (Myh6-MCM) allele52 to generate Myh6-MCM; Hey2flox/flox inducible knockout mice (designed Hey2-iCKO) (Fig. 7a and Supplementary Fig. 7a). We next subjected 1-month-old Hey2-iCKO mice and control littermates (Myh6-MCM) to tamoxifen treatment for 5 days to inactivate Hey2 and analyzed cardiac morphology and function at 7-, 14-, 21- and 28-days post induction (dpi) (Supplementary Fig. 7b). Immunoblot and fluorescent analyses confirmed an efficient loss of HEY2 protein in isolated Hey2-iCKO hearts (Fig. 7b and Supplementary Fig. 7c). Histological analysis revealed comparable chamber size and morphology between Hey2-iCKO hearts and control hearts (Supplementary Fig. 7d). Notably, Hey2-iCKO heart exhibited persistently enhanced LVEF and reduced LVIDs from 7 days to 28 days post tamoxifen treatment (Fig. 7c–f), indicative of elevated systolic function. Furthermore, we observed the upregulation of Ppargc1a, Esrra, Nrf1, and ETC complex I-V genes in Hey2-iCKO heart (Fig. 7g), which also carried the increased number of mitochondria with regular shape and cristae (Supplementary Fig. 7e–j). Thus, inducible Hey2 ablation in adult mouse heart enhances mitochondrial biogenesis, oxidative phosphorylation and cardiac function.

a Design of induced Hey2 deletion by Myh6-MerCreMer. Tam, tamoxifen. b Western blots analysis of HEY2 in Hey2-iCKO and control hearts. 3 independent experiments. c Representative B-mode and M-mode echocardiography images of Hey2-iCKO and control hearts at 28 days post tamoxifen injection (dpi). d–f Analysis of LVEF, LVIDs and LVIDd of Hey2-iCKO and control hearts from 7 to 28 dpi. Control: n = 15 (7 dpi,14 dpi), n = 13 (21 dpi, 28 dpi). Hey2-iCKO: n = 14 (7 dpi,14 dpi), n = 12 (21 dpi, 28 dpi). Data presents as mean ± SD, ****P < 0.0001, Two-way ANOVA with Tukey multiple comparisons test. g RT-qPCR analysis of Ppargc1a, Esrra, Nrf1 and ETC complex genes in Hey2-iCKO and control hearts. 3 biological replicates. h RT-qPCR analysis of HEY2, PPARGC1 coactivator, ETC complex genes in DOX-treated and untreated AC16 cells. 3 biological replicates. i Diagram for tamoxifen and DOX injections, and hearts analysis in Hey2-iCKO and control mice. j, k Representative whole-mount hearts and quantification of heart weight (HW)/ body weight (BW) ratio from saline-injected control mice (Ctrl+saline, n = 5), DOX-injected control mice (Ctrl+DOX, n = 4), saline-injected Hey2-iCKO mice (iCKO+saline, n = 5) and DOX-injected Hey2-iCKO mice (iCKO+DOX, n = 3). Scale bar: 1000 µm. l, m Masson’s trichrome staining and fibrosis quantification of Ctrl+saline, Ctrl+DOX, iCKO+saline and iCKO+DOX hearts (n = 8 mice). Scale bar: 100 µm. n Quantification of TUNEL+ cells in Ctrl+saline, Ctrl+DOX, iCKO+saline and iCKO+DOX hearts (n = 6 mice). Box plots displayed median ± interquartile range (IQR), with whiskers extending to the minimum and maximum values. **P < 0.01, ****P < 0.0001, Student’s t-test (unpaired, two-tailed). o Analysis of LVEF, LVFS, LVIDs and LVIDd in hearts of Ctrl+saline (n = 5), Ctrl+DOX (n = 6), iCKO+saline (n = 5) and iCKO+DOX (n = 6) groups. p–r The serum levels of CK, CK-MB and LDH in Ctrl+saline (n = 7), Ctrl+DOX (n = 6), iCKO+saline (n = 3) and iCKO+DOX (n = 4) groups. Data presents as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Student’s t-test (unpaired, two-tailed) (k, m, o–r) or Multiple t tests (two-tailed) (g, h). Source data are provided as a Source Data file.
To assess the effect of Hey2 overexpression on adult mouse hearts, 2-month-old mice were injected with AAV9-cTNT-Hey2-GdGreen (AAV9-Hey2) and control AAV9, and analyzed 4 weeks later (Supplementary Fig. 7k). We found that AAV9-Hey2-injected mice exhibited a reduction in LVEF and LVFS compared to control animals (Supplementary Fig. 7n). Probably, because of heterogenous infections of AAV9-Hey2 in the adult heart (~26% CMs were infected) (Supplementary Fig. 7l, m), both LVIDs and LVIDd were only modestly increased (Supplementary Fig. 7o). Importantly, the expressions of PPARGC1 energy coactivator complex genes, ETC complex genes and Cpt1a/Cpt2 genes were detectably reduced in AAV9-Hey2-injected mice (Supplementary Fig. 7p), consistent with the response of energy metabolism regulatory genes in the hey2-mOE zebrafish heart.
Given the uplifted cardiac function caused by CM-specific Hey2 ablation, we tested the possibility that silencing Hey2 in adult CMs protects against doxorubicin (DOX)-induced mitochondrial and cardiac injury53. DOX has been shown to cause cardiotoxicity that impairs mitochondrial bioenergetics and leads to cardiomyopathy and HF53,54. We noticed that DOX treatment can substantially induce HEY2 expression in human AC16 CM-like cells, coincided with reduced expressions of PPARGC1A, ESRRA, NRF1, and ETC complex genes (Fig. 7h). Hey2 was also upregulated in DOX-treated mouse hearts and rodent H9C2 CM-like cell line (Supplementary Fig. 7q, r). We next subjected 2-month-old Hey2-iCKO mice and Myh6-MCM (control) mice to DOX or saline administration and analyzed treated hearts after 3 weeks (Fig. 7i). The control mice injected with DOX exhibited enlarged hearts, increased heart/body weight, enhanced cardiac fibrosis and CM apoptosis compared to saline-injected control mice (Fig. 7j–n). Induced-ablation of Hey2 in adult CMs alleviated the DOX-induced heart dilation, heart/body weight increase, cardiac fibrosis and CM apoptosis (Fig. 7j–n). The compromised cardiac function in DOX-injected control mice was also reversed in DOX-treated Hey2-iCKO mice (Fig. 7o). We observed an increase in the serum levels of HF biomarkers creatine kinase (CK), creatine kinase-MB (CK-MB), and lactic dehydrogenase (LDH) in DOX-treated control mice, but not in Hey2-iCKO mice (Fig. 7p–r). Furthermore, the saline-treated control and iCKO mice did not show significant differences in cardiac weight, fibrosis and HF biomarkers (Figs. 7j–n and 7p–r). These findings suggest that inducible Hey2 ablation in the adult mouse heart protects against DOX-induced cardiac injury.
Discussion
In this study, we provide evidence that HEY2 regulates mitochondrial respiration and cardiac energy homeostasis, and its increased expression in failing hearts mediates cardiac disease states. Specifically, HEY2 is upregulated in the hearts of patients with DCM and in DOX-treated CMs. Induction of Hey2 expression in adult zebrafish hearts or cultured NRVMs causes defective mitochondrial biogenesis and respiration, leading to cardiac energy deficit and an HF-like phenotype. In contrast, depletion of Hey2 in adult mouse hearts or adult zebrafish stimulates mitochondrial oxidation pathways and elevates cardiac function, suggesting HEY2 as a pivotal modulator in cardiac energy homeostasis. Furthermore, Hey2-induced mitochondrial and cardiac dysfunction in zebrafish heart or AC16 CM-like cells can be rescued by increased expression of PPARGC1A and ESRRA. Based on our observations in fish, mouse and cell culture models, we propose a mechanism (Fig. 8), in which HEY2 interacts with HDAC1 in CMs to repress the promoters of energy metabolism regulatory genes Ppargc1a, Esrra, Nrf1, and Cpt1a/Cpt2, through histone deacetylation and chromatin condensation. We propose that in normal conditions, the basal level of HEY2 along with HDAC1 limits the excessive expression of oxidative phosphorylation genes to preserve mitochondrial and cardiac homeostasis. Upon cardiac stress-mediated HEY2 induction, mitochondrial ETC activity declines owing to transcriptional repression of PPARGC1A coactivator complex genes and FAO regulatory genes, which in turn results in depressed energy production, cardiac dilation, and failure. These findings provide unique insights into the regulation of cardiac energy homeostasis in health and disease and lead to the development of potential strategies for intervention of cardiomyopathy and HF.

Hey2 is induced in adult CMs in response to pathological stress (i.e., DCM hearts or DOX treatment). Elevated HEY2 recruits HDAC1 to occupy the promoters of PPARGC1 coactivator complex genes and Cpt1a/Cpt2 regulatory genes to mediate histone deacetylation and transcriptional repression, which results in depressed energy production, ROS increases, cardiac dilation and failure. Under normal conditions, the basal level of HEY2 along with HDAC1 restrains the rampant energy metabolic gene expression to maintain cardiac homeostasis. FAO: fatty-acid oxidation; TCA: tricarboxylic acid cycle; MMP: mitochondrial membrane potential; ROS: reactive oxygen species; I-V: mitochondrial electron transport chain complex I-V; Cpt1, Cpt2: FAO enzyme.
Cardiac contractile function is dependent on the ability of CMs to sustain mitochondrial oxidation and proper energy production7. Cardiomyopathy caused by genetic variants in mitochondrial regulatory genes suggests a direct link between mitochondrial dysfunction and heart failure4,55. The role of HEY proteins in constraining mitochondrial energy metabolism during pathophysiological conditions appears to be conserved in different organ systems. An increase in HEY2 expression was observed in brain tissues in Alzheimer’s disease associated with dysfunctional neuronal cell mitochondria56. HEY1 was also found to suppress Ppargc1a transcription for mitochondrial energy transfer that inhibits glucose metabolism and adipose browning57. We detected the increased mitochondrial oxidation gene expression and cardiac function in young adult mice in which Hey2 was specifically ablated in CMs and in hearts of zebrafish hey25nt-/- mutants. These findings support view that, in the adult heart, HEY2 plays a role in dampening mitochondrial respiration in the context of maintaining cardiac homeostasis. This function is distinct from its roles during embryonic heart development. When Hey2 was deleted under the control of Nkx2.5-Cre16, atrial gene expression is ectopically upregulated in the ventricle at E16.5, highlighting its functions in cardiac chamber patterning and differentiation. Notably, inducible ablation of Hey2 in the adult mouse heart protects against DOX-induced mitochondrial and cardiac dysfunction, suggesting that HEY2 mediates DOX-induced mitochondrial damage. We speculate that the HEY2-mediated reduction of energy metabolism upon cardiac stress may initially protect compromised cardiomyocytes from damage and death, but that a prolonged reduced energy state ultimately contributes to cardiac remodeling and dysfunction.
It has been shown that damage of mitochondrial structure, dynamics and function contributes to the pathogenesis of cardiomyopathy and HF2. Our study links HEY2-mediated transcriptional suppression with mitochondrial respiration, cardiac energy deficits, and dysfunction. Multifaceted genome-wide analyses revealed that alterations in Hey2 occupancy and chromatin state are associated with altered gene expression in mitochondrial biogenesis and oxidation. HEY2 binding has been shown to decline the expression of genes encoding cardiac transcriptional factors and atrial/trabecular differentiation genes28,45,58. However, much of the underlying epigenetic regulatory mechanism of HEY2-mediated repression has remained unexposed. Our findings establish that H3K9ac/H3K27ac deacetylation mediates the state of chromatin accessibility for HEY2-controlled transcriptional repression. Consistent with the roles of class I histone deacetylases during cardiac homeostasis59, we find that HEY2 interacts with HDAC1 to regulate histone deacetylation and chromatin remodeling. In addition to Ppargc1 transcriptional coactivator genes, Cpt1a and Cpt2 are also repressed by HEY2, suggesting the existence of a hierarchy in this regulatory network, as Cpt1a/Cpt2 transcription is also regulated by PPARGC1a60. These findings suggest that the rate-limiting step for the transportation of long-chain fatty acids into mitochondria is likely to be governed by HEY2-mediated transcriptional repression. Although our integrative analyses of bulk RNA-seq data with genome-wide CUT&Tag-seq and ATAC-seq data provide valuable insights into the HEY2-dependent transcriptional regulation on cardiac energy metabolism, it still has limitations in uncovering the distinct roles of CM subsets or cellular cross-talks between cardiac heterogenous cells. It is important to conduct single-nucleus RNA-seq analysis to elaborate further the HEY2-mediated transcriptional regulation during energy homeostasis in future studies. Because prolonged Ppargc1a upregulation deteriorates mitochondrial structure, dynamics, and cardiac function11, future experiments will also be focused on the dosage effects of the HEY2/HDAC1-Ppargc1/Cpt transcriptional module on cardiac energy homeostasis and function, as well as their activities in cardiomyopathy and HF, which may be relevant in developing viable concepts and therapies for cardiac protection.
Methods
Zebrafish and mouse lines
In this study, we used wild-type (WT) and genetically modified zebrafish of the AB strain. The published transgenic and mutant lines employed are as follows: Tg(cmlc2:CreER)35, Tg(cmlc2:loxP-nlsmCherry-STOP-loxP-Hey2-EGFP)(also known as cmlc2:nRSGG)19, hey25nt-/- mutants(also known as grl5nt-/-)19. Hey2flox/+ mouse was purchased from GemPharmatech company (Nanjing, China). Myh6-MerCreMer mouse was reported previously61. Myh6-MerCreMer; Hey2flox/+ mouse were crossed with Hey2flox/flox to generate Myh6-MerCreMer; Hey2flox/ flox (referred as Hey2-iCKO) mice. All mice were housed at 20-25 °C with humidity of 45-65% and a light-dark cycle of 12 h. All animal experiments were conducted in accordance with the animal use guidelines at East China Normal University and received approval from the Institutional Animal Care and Use Committee of East China Normal University (Institutional Review Board No. mf20210201).
Human samples
Human control and dilated cardiomyopathy (DCM) hearts were obtained from Guangdong Provincial People’s Hospital (Guangdong, China), following the principles outlined in the Declaration of Helsinki and with approval by the Institutional Review Board of Guangdong Provincial People’s Hospital (Institutional Review Board No. GDREC2016255H). Informed consents were signed by the patients or their medical proxies (Informed consent form No. 20160628).
Histology, Immunostaining and in situ hybridization
Hearts were fixed in 4% paraformaldehyde (PFA), dehydration, embedded in paraffin, and sectioned frontally at 5-μm thickness. HE and Masson’s Trichrome/ Sirius Red staining were performed for detecting cardiac structure and fibrosis, according to the manufacturer’s protocol of HE, Masson’s Trichrome or Sirius Red Kit (Servicebio) respectively. For immunostaining, hearts were fixed in 4% PFA at room temperature. The fixed hearts were washed in PBS and immersed in 30% sucrose for 12 h. All primary antibodies used in this study included: anti-α-actinin (Sigma, A7732), anti-Fibronectin (Sigma, F3648), anti-Collagen I (Abcam, ab34710), anti-MHC (DSHB, MF20), anti-HEY2 (Sigma, HPA030205), FITC-conjugate WGA (Sigma, L4895), anti-Mef2 (Santa Cruz, sc-313), anti-PCNA (Sigma, P8825), anti-Flag (Proteintech, CL594-66008), anti-HA (Proteintech, CL594-66006). Secondary antibodies in this study were as follows: Alexa Fluor 594 goat anti-rabbit IgG (H + L) (Invitrogen, A11012), Alexa Fluor 488 goat anti-mouse IgG (H + L) (Invitrogen, A11001), Alexa Fluor 594 goat anti-mouse IgG (H + L) (Invitrogen, A11005), Alexa Fluor 488 goat anti-rabbit IgG (H + L) (Invitrogen, A11008). DAPI was used for nuclear staining. For TUNEL assay, sections were incubated in TUNEL reaction mixture for 1 h at 37 °C according to In Situ Cell Death Detection Kit (Sigma, 12156792910). ISH was performed using digoxigenin-labeled RNA antisense probes as described19. The hybridization signal was detected by anti-digoxigenin-AP (Roche) and developed by the NBT/BCIP (Roche) substrate.
Zebrafish and mouse hearts were Imaged by Leica M205 FA dissecting scope. HE, Sirius Red, Masson’s Trichrome and ISH images were taken by Olympus BX53 microscope with the DP74 digital color camera. Fluorescence immunostaining imaging was performed using Zeiss Axio Observer Z1 microscope and Andor Dragonfly 500 High Speed confocal microscope.
Annexin V-APC/Cyanine7 staining assay and flow cytometry
Primary cardiomyocytes from adult zebrafish hearts were isolated as described62. The isolated cardiomyocytes were incubated in Annexin V-APC/Cyanine7 and DAPI reagent for 20 min in the dark at 28°C according to the Annexin V-APC/Cyanine7/DAPI Apoptosis Kit (Elabscience Biotechnology). The apoptosis rate of the cardiomyocytes was detected by Flow cytometers.
Transmission electron microscopy
Transmission electron microscopy (TEM) for morphological analysis was performed according to standard operating procedures (Servicebio Technology). The Ultrathin sections were obtained using Leica UC7 ultramicrotome and imaged with transmission electron microscopy. The mitochondrial density, size, and cristae were assessed by analyzing TEM images using ImageJ.
Immunoprecipitation and western blot analysis
For immunoprecipitation analysis, the HEK293T cells were transfected with Flag tagged Hey2 and/or HA tagged Hdac1 plasmids using PEI 40 K transfection reagent (Servicebio, G1802). The NRVMs were infected by Adv-Hey2 or Adv-Ctrl. After 48 h, HEK293T cells or NRVMs were scraped and lysed in lysis buffer (P0013, Beyotime) supplemented with 1 mM PMSF (ST507, Beyotime) for 30 min at 4°C. Lysates were centrifuged at 13,000 g for 20 min at 4°C, and supernatants were incubated with 25 μl anti-Flag nanobody agarose beads (Alpalifebio, KTSM1308) or anti-HA Agarose (Thermo Fisher Scientific, 26181) for 2 h at 4°C. Beads were washed 5 times with washing buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 10% Glycerol, 5 mM EDTA, 0.1% TritonX-100) and boiled in 5 x SDS-loading buffer. Proteins were analyzed by western blot.
For western blot analysis, proteins were extracted on ice using a RIPA lysis buffer containing protease inhibitors (APExBIO). The lysate was centrifuged and the concentration of the supernatant was measured using the Pierce BCA Protein Assay Kit (Invitrogen, Cat. 23225). Antibodies used for western blot in this study were as follows: anti-Hey2 (Proteintech, 10597-1-AP), total OXPHOS rodent WB antibody cocktail (Abcam, ab110413), anti-Flag (MBL, M185-3L), anti-HA (Proteintech, 51064-2-AP), anti-HDAC1 (Abcam, ab280198), anti-PGC1A (Santa Cruz, sc-517380), anti-ESRRA (Abcam, ab76228), anti-CPT1A (Abcam, ab128568), HRP-conjugated Flag antibody (Proteintech, HRP-66008), anti-HA (Santa Cruz, sc-7392), anti-GAPDH (Abcam, ab181602), anti-Tubulin (Proteintech, 66031-1-Ig), anti-HSP90 (Cell Signaling Technology, 4877).
RNA isolation, RNA-seq and real-time PCR
The total RNAs of the ventricles were isolated using TRIzol Reagents (Life Technologies). Whole mRNA Library and RNA-seq were performed as described previously19. Briefly, the library was quality-checked and quantified using the Agilent 2100 Bioanalyzer. Amplification of the final library was amplified with phi29 to produce DNA nanoballs (DNBs). These DNBs were loaded into a patterned nanoarray, and paired-end 150-base reads were generated using the BGIseq500 platform (BGI-Shenzhen, China). We sequenced 3 replicates each group with more than 4xRNA-seq read depth and an average generation of about 6.7 G bases per sample.
1ug of RNA was extracted and reverse transcribed to cDNA with Prime-Script II 1st Strand cDNA Synthesis Kit (TaKaRa) for gene expression analysis. The Real-time PCR reactions were carried with SYBR Premix Ex Taq II (Takara) using Roche LightCycler 480 II system. Primers are listed in the Supplementary Data 2–5.
Echocardiography
Echocardiography was performed on all fish using Vevo 3100 with a MX550s linear array transducer (Visual Sonics, FUJIFILM). Fish was anesthetized by tricaine for 1 min and placed ventral side up in a small sponge holder submerged in a plastic plate filled with aquaria water. All research processes were performed by blind-operators at 28°C for no more than 4 min. Transthoracic echocardiography was performed on mice using a Vevo 3100 system with a MX400 transducer (Visual Sonics, FUJIFILM) as described63. Parasternal long-axis views were used for M-mode analysis. Data were analyzed using Vevo LAB software (FUJIFIL). End diastolic and end systolic left ventricular inner diameters (LVIDd, LVIDs) were measured and fractional shortening (FS) and ejection fraction (EF) were calculated using the following equations: FS (%) = (LVIDd − LVIDs) / LVIDd × 100%, EF (%) = [(LVIDd)3 − (LVIDs)3] (LVIDd)3 × 100%.
NRVMs isolation, siRNA transfection, adenovirus and lentivirus infections
Neonatal rat ventricular myocytes (NRVMs) were isolated from 1 ~ 3-day-old Sprague-Dawley rats as described64. Briefly, the ventricles were minced with scissors into 1 mm3 fragments and digested by trypsin. Cells were then plated in dish for 1 hour to remove fibroblasts. After isolation, NRVMs were maintained in DMEM medium containing 10% FBS and 1% antibiotics. siRNAs were purchased from GenePharma (Shanghai, China). The target sequence for the Hey2 sense strand was 5’- GCCGCUGCAACAGCAAUCATT- 3’ and 5’-UGAUUGCUGUUGCA GCGGCTT- 3′. A nonspecific siRNA sequence was used as a negative control. NRVMs (2 × 106) were transfected with siRNA (150 nM) using Lipo2000 transfection reagents (Invitrogen) in Opti-MEM medium overnight. The media were replaced with fresh DMEM for another 12 h. The rat Hey2 coding sequence was cloned into the adenovirus vector (pADV-mCMV-MCS-3xFlag) and adenovirus packaging (Adv-Hey2) was produced by OBiO Technology (Shanghai, China). The vector adenovirus was used as the control (Adv-Ctrl). The monolayer NRVMs were cultured for 24 h, and infected with the adenoviruses (Adv-Ctrl or Adv-Hey2) at the multiplicity of infection (MOI) of 50. The AC16 cell with lentiviral HEY2 overexpression (HEY2-OE) was constructed as following: HEY2 cDNA was cloned into the pCDH-CMV-MCS-EF1-puro vector (the empty vector was used as a negative control). pCDH-CMV-MCS-EF1-puro vector, pMD2.G, and psPAX2 were co-transfected into HEK293T cells by LentiFitTM (Hanbio Biotechnology), and viral supernatant was harvested after transfection. The AC16 cells were infected with the viruses at 37 °C for 24 h. The positive cells were selected by incubating with 1 ug/mL puromycin.
TMRM, JC-1, Mito-Tracker CMXRos, MitoSOX Staining and ATP measurement
Mitochondrial membrane potentials (ΔΨm, MMP) were measured using TMRM (Thermo Fischer I34361) and JC-1 (Beyotime, C2003S) according to the manufacturer’s protocols. Cells were imaged by Andor Dragonfly 500 High-Speed confocal microscope. ImageJ software was used to analyze red fluoresce intensity. Mitochondrial mass was estimated using Mito-Tracker Red CMXRos dye (Beyotime, C1049) according to the manufacturer’s instructions. Andor Dragonfly 500 High-Speed confocal microscope was used for observation. ImageJ software was used to analyze red fluoresce intensity. The MitoSOX Red (Invitrogen, M36008) was used to visualize mitochondrially derived superoxide in cells according to the manufacturer’s instructions. Adv-Hey2- and Adv-Ctrl- infected live NRVMs were first stained with MitoSOX Red or Mito-Tracker Red CMXRos, then fixed and immunostained with α-actinin and Flag antibodies. Cells were imaged by Andor Dragonfly 500 High-Speed confocal microscope. ImageJ software was used to analyze red fluoresce intensity. The myocardial ATP content was determined using an Enhanced ATP Assay Kit (Beyotime, S0027) according to the manufacturer’s instructions. ATP levels were measured using a multi-mode microplate reader (FLUOstar Omega).
Oxygen consumption rate measurement
The oxygen consumption rate (OCR) was measured using Seahorse XF Pro analyzer (Agilent, USA) following the manufacturer’s instruction. NRVMs or AC16 cells were seeded onto a Seahorse XF96 Cell Culture Microplate (Agilent) at a density of 4 × 104. After 24 h, cells were prepared by adding 1 mM pyruvate, 2 mM glutamine and 10 mM glucose to Base medium. NRVMs/AC16 were then cultured in a 37 °C and non-CO2 incubator prior for 1 h. The mitochondrial stress test kit (Agilent) was used to monitor OCR after the addition of the following drugs: 1 μM oligomycin, 2 μM fluoro-carbonyl cyanide phenylhydrazone (FCCP), 100 nM rotenone, and 1 μM antimycin A (Agilent). Data were analyzed using Seahorse Wave Pro software (Agilent). The OCR measurements were normalized to the protein levels in each microplate well.
ETC complex I activity and FAO activity assays
The colorimetric enzyme activity assay kits (Abcam, ab109721) were used to assess the activity of Complex I (NADH dehydrogenase) enzyme. The NRVMs were homogenized in PBS, and the BCA was used to extract protein. 500 µg protein was mixed with 5 volumes of detergent and incubated on ice for 30 minutes, and then added to the plate. 200ul assay solution (NADH and dye in 1x Dilution Buffer) was loaded to the plate. The BioTek Synergy NEO2 microplate reader was used to detect the ETC Complex I enzyme activity at OD 450 for 30 min. The fatty acid β-oxidation activity assay is based on the oxidation of octanoyl-CoA, which is coupled to NADH dependent reduction of INT to INT-formazan. According to the assay kit (Assay Genie, BR00001), 1 × 105 NRVMs were lysate on ice with Lysis Solution. The Supernatant exhibits an absorption maximum at 492 nm. The BioTek Synergy NEO2 microplate reader was used to detect the FAO enzyme activity.
Detection of pyruvate, α-ketoglutaric acid and glycogen
Total heart tissues from zebrafish (n = 10) were homogenized and centrifuged at 12,000 g for 10 min at 4°C. The amounts of pyruvate, α-ketoglutaric acid and glycogen were measured in lysed heart tissue using Pyruvate Content Assay Kit (Solarbio, BC5260, China), α-Ketoglutaric Acid Content Assay kit (Solarbio, BC5420, China) and Glycogen Content Assay Kit (Solarbio, BC0340, China) according to the instructions of the manufactures.
Luciferase assay
HEK293T cells were cultured at 8 × 104 cells per well in 24-well plates. Firefly luciferase pGL3-PPARGC1A-Fluc, pGL3-ESRRA-Fluc or pGL3-CPT2-Fluc plasmid vector (0.5 µg) and Renilla luciferase pRL-TK plasmid vector (10 ng, as a normalization control) were co-transfected with HEY2 and/or HDAC1 expression plasmids (0.5 µg) using PEI 40 K transfection reagent (Servicebio, G1802) according to the manufacturer’s protocol. Cell lysates were collected and luciferase activities were measured using a Dual-Luciferase Reporter Assay System (Promega) after 48 h. The FLUOstar Omega multi-mode microplate reader was used to measure luminescence.
Cleavage under targets and tagmentation (CUT&Tag) assay
CUT&Tag assay was performed using NovoNGS CUT&Tag 3.0 High-Sensitivity Kit (Novoprotein, N259-YH01-01B). Briefly, isolated cardiomyocytes from zebrafish hearts (n = 6) or NRVMs (2 × 106) were enriched with ConA beads and incubated with primary antibody buffer of anti-Hey2 antibody (Proteintech, 10597-1-AP), anti-Flag antibody (MBL, M185-3L), anti-H3K9ac antibody (Abcam, ab32129), anti-H3K27ac antibody (Abcam, ab4729) or anti-HDAC1 antibody (Abcam, ab280198). Then incubated with the secondary antibody buffer of anti-mouse IgG antibody (Abcam, ab6708) or anti-rabbit IgG antibody (Abcam, ab6701). The cells were then incubated with protein A/G-Tn5 transposome and the tagmentation buffer (10 mM MgCl2 in ChiTaq Buffer). The DNA fragments were extracted by Tagment DNA extract beads and the libraries were amplified using 5x AmpliMix. The DNA libraries were extracted by DNA clean beads for sequencing.
ATAC-seq assay
ATAC-seq assay was performed using the Chromatin Profile Kit for Illumina following the manufacturer’s instructions (Novoprotein, N248-01A). Briefly, isolated cardiomyocytes from zebrafish hearts (n = 6) or NRVMs (2 × 106) were resuspended in ATAC lysis buffer (10 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl2, 0.1% NP40, 0.1% Tween 20, 0.01% Digitonin) and centrifuged for 5 min at 500 g at 4°C. Nuclei were incubated in tagmentation buffer for 30 min at 37 °C. Tagmented DNA was collected using Tagment DNA extract beads and then amplified by PCR. The DNA libraries were purified using DNA clean beads and then sequenced.
RNA-seq, ATAC-seq and CUT&Tag-seq data processing
The sequencing data files in FastQ format were preprocessed using TrimGalore65 (v0.6.7) to exclude low-quality reads and trim adapter sequences (options: -q 20 --length 25 -e 0.1 --phred33 --gzip --stringency 3). Paired-end reads were mapped to the zebrafish genome (danRer11) or rat genome (rn7) using HISAT266 (v2.2.1, options: ‘--no-spliced-alignment’ for ATAC-seq and CUT&Tag-seq data, default for RNA-seq). Sambamba67 was employed to filter out duplicated reads and produce BAM format results. Normalized BigWig files were generated using Deeptools68 (v3.5.3, bamCoverage module, options: --binSize 10 --normalizeUsing RPKM –ignoreDuplicates). Peaks were identified from replicates using MACS3 (v3.0.1, callpeak module, options: ‘-g 2.63e9 -B -f BAM’ for rat, ‘-g 1.37e9 -B -f BAM’ for zebrafish). Differential peaks were determined using the diffpeak module, revealing changes in signal within peak regions between experiments and categorizing peaks into up, down, and common groups.
Gene expression levels were quantified by FPKM of RNA-seq data. Differentially expressed genes calculated by DESeq269 R packages (v1.38) were defined as those with a Q-value < 0.05 and an absolute log2 fold change > log2 (1.5). Chromatin accessibility and Hey2 binding intensity of genes were quantified as the RPKM in promoter regions of ATAC-seq and CUT&Tag-seq data, respectively. Differentially regulated genes were defined as those with an absolute log2 fold change > log2(1.5), with a pseudo count of 1.
Integrative analyses for RNA-seq, ATAC-seq and CUT&Tag-seq data
To assess the reproducibility of CUT&Tag-seq and ATAC-seq experiments (Fig. 4a and Supplementary Fig. 5a, d, g, h), we first gathered all previously called peaks for each group of experiments. We then extracted the signal within these peaks for all samples using the multiBigwigSummary module of Deeptools and calculated the Spearman correlation coefficients to measure sample correlation. For RNA-seq experiments (Fig. 3a and Supplementary Fig. 3a), it was done by calculating Pearson correlation coefficients of the expression level of all genes between samples. Given the high correlation among replicates, averaged BigWig files for each condition were generated for visualization and cross-experiment comparative analysis.
For the genomic distribution of Hey2 binding sites (Fig. 4b), we calculated the base-pair overlaps between Hey2 Ctrl peaks and predefined regions, including promoters (±500 bp of TSS), 3’/5’-UTRs, gene bodies of RefSeq genes, and transposable elements defined by RepeatMasker70. Local motif enrichment analyses (Fig. 4f and Supplementary Fig. 5k) were performed using the CentriMo module of MEME Suite71 (v5.5.5), with genome sequences extracted by bedtools (v2.30.0, options: slop -i summits.bed -g chrom.size -l 500 -r 500 -s). The motif file in MEME format was downloaded from the JASPAR 2024 database. Comparative analyses of signal intensity of Non-Hey2 peak regions and Hey2 peak regions in Ctrl and Hey2-OE groups (Fig. 4e, Supplementary Fig. 5f, g) were performed to investigate the correlation between Hey2 binding and chromatin accessibility or histone acetylation using ATAC-seq and H3K9ac/H3K27ac CUT&Tag-seq data. For the genome-wide relationship between HEY2 occupancy, HDAC1 bindings, histone acetylation and chromatin accessibility, statistical analyses were conducted using HDAC1 and H3K27ac CUT&Tag-seq datasets, ATAC-seq datasets on signal intensity variations in HEY2-occupied promoter and intergenic regions, non-HEY2-occupied regions between HEY2-OE/KD NRVMs and control cells (Supplementary Fig. 5l). HEY2 peak regions were called from HEY2 CUT&Tag experiments in NRVMs and zebrafish hearts. Non-Hey2 peaks were generated by randomly sampling regions of equal length (average length of Hey2-peaks) from the whole genome excluding Hey2 peak regions. Genomic regions from TSS-2kb to TSS + 100 bp were defined as promoter regions, and regions from TES + 1 kb to TSS-2kb for intergenic regions. Paired Wilcoxon tests were conducted to calculate the significance level.
modRNA synthesis and zebrafish heart injection
The optimized DNA templates for HA-esrra and Flag-ppargc1a RNA were cloned into the plasmid with backbone elements (T7 promoter, 5’ and 3’ UTR, poly(A) tail). These two mRNAs were in vitro-transcribed by T7 RNA polymerase in the presence of the Cap1 analog (APExBIO, USA) and nucleotides with a global substitution of uridine with N (1)-methylpseudouridine (N1mΨ) (APExBIO, USA). Under the condition of appropriate loading and washing buffer, mRNAs were purified using a column packed with oligo (dT)25 affinity resin (Thermo Scientific, USA). The commercial double-antibody sandwich enzyme-linked immunoassay kit (Vazyme Medical Technology, China) was used to detect the content of dsRNA in the synthesized mRNA stock solution. RNA integrity was assessed by microfluidic capillary electrophoresis, and pH and the concentration were determined as well.
Flag-ppargc1a/HA-esrra modRNA and RNAiMAX transfection reagent (Invitrogen, USA) were dissolved separately in Opti-MEM (Invitrogen, USA), combined and incubated to generate the transfection mixture. 0.5 µl of RNAiMAX reagent was used for every microgram modRNA transfection. The 1.5 µg of modRNA mixture was injected intracardially into the hey2-mOE or control zebrafish hearts.
AAV9 injection, and DOX-induced cardiotoxicity
The myocardial specific Hey2 was overexpressed in vivo by using an adeno-associated virus type 9 (AAV9) vector (pAAV-cTNT-Hey2-3xFlag-P2A-GdGreen-WPRE, Obio Biotechnology, Shanghai, China). A total of 1 × 1011 viral particles (vp) of AAV9-CTNT-Hey2 or AAV9-EGFP vector virus (pAAV-cTNT-GdGreen-WPRE) (Obio Biotechnology, Shanghai, China) were injected into the tail vein of mice at 8 weeks of age.
Six-week-old Hey2-iCKO and male C57BL/6 mice were administered either normal saline (0.9% NaCl) or DOX (cumulative dose of 15 mg/kg) via intraperitoneal injection 4 times over 14 days period. All the mice were echocardiographic assessment, weighed and sacrificed to collect the hearts under anesthesia at 7 days after the final injection of doxorubicin.
Statistics and reproducibility
All experiments were conducted with at least three biological replicates or three independent experiments, and analyzed in double-blind and randomized manners. Results are presented as mean ± SEM or mean ± SD of bar plots. Box plots displayed median ± interquartile range (IQR), with whiskers extending to 1.5*IQR or the minimum and maximum values. Statistical analyses were performed using Student’s t-test (unpaired, two-tailed), Multiple t tests (unpaired, two-tailed) with Benjamini, Krieger, and Yekutieli’s correction and two-way ANOVA with Tukey’s multiple comparison test, as appropriate, utilizing GraphPad Prism version 8.0 software. The statistical analyses used for each figure are indicated in the corresponding figure legends. Differences with a P-value of less than 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The ATAC-seq and CUT&Tag-seq data of this study have been deposited in the NCBI Sequence Read Archive database with the accession codes PRJNA983929, PRJNA984420, PRJNA1148500, and PRJNA1143894. The RNA-seq data have been deposited in the NCBI Sequence Read Archive database with the accession codes PRJNA984676 and PRJNA1148340. Source data of Supplementary Fig 5l have been deposited in Figshare repository (https://doi.org/10.6084/m9.figshare.27936759). All other data generated in this study are provided in the Source data file. Source data are provided with this paper.
References
Ziaeian, B. & Fonarow, G. C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 13, 368–378 (2016).
McNally, E. M. & Mestroni, L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circulation Res. 121, 731–748 (2017).
Lee, J. et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 572, 335–340 (2019).
Brown, D. A. et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 14, 238–250 (2017).
Paredes, A. et al. γ-Linolenic acid in maternal milk drives cardiac metabolic maturation. Nature 618, 365–373 (2023).
Kumar, A. A., Kelly, D. P. & Chirinos, J. A. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation 139, 1435–1450 (2019).
Vega, R. B., Horton, J. L. & Kelly, D. P. Maintaining ancient organelles: mitochondrial biogenesis and maturation. Circulation Res. 116, 1820–1834 (2015).
Ventura-Clapier, R., Garnier, A. & Veksler, V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1. Cardiovascular Res. 79, 208–217 (2008).
Riehle, C. et al. PGC-1beta deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circulation Res. 109, 783–793 (2011).
Martin, O. J. et al. A role for peroxisome proliferator-activated receptor gamma coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circulation Res. 114, 626–636 (2014).
Zhu, X. et al. Fine-tuning of pgc1alpha expression regulates cardiac function and longevity. Circulation Res. 125, 707–719 (2019).
Zhong, T. P., Childs, S., Leu, J. P. & Fishman, M. C. Gridlock signalling pathway fashions the first embryonic artery. Nature 414, 216–220 (2001).
Kokubo, H., Tomita-Miyagawa, S., Hamada, Y. & Saga, Y. Hesr1 and Hesr2 regulate atrioventricular boundary formation in the developing heart through the repression of Tbx2. Dev. (Camb., Engl.) 134, 747–755 (2007).
Fujitani, M. et al. TAp73 acts via the bHLH Hey2 to promote long-term maintenance of neural precursors. Curr. Biol.: CB 20, 2058–2065 (2010).
Fischer, A., Schumacher, N., Maier, M., Sendtner, M. & Gessler, M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 18, 901–911 (2004).
Xin, M. et al. Essential roles of the bHLH transcription factor Hrt2 in repression of atrial gene expression and maintenance of postnatal cardiac function. Proc. Natl Acad. Sci. USA 104, 7975–7980 (2007).
Zhong, T. P., Rosenberg, M., Mohideen, M. A., Weinstein, B. & Fishman, M. C. gridlock, an HLH gene required for assembly of the aorta in zebrafish. Sci. (N.Y., N.Y.) 287, 1820–1824 (2000).
Jia, H. et al. Vertebrate heart growth is regulated by functional antagonism between Gridlock and Gata5. Proc. Natl Acad. Sci. USA 104, 14008–14013 (2007).
She, P. et al. The Gridlock transcriptional repressor impedes vertebrate heart regeneration by restricting expression of lysine methyltransferase. Dev. (Camb., Engl.) 147, dev190678 (2020).
Gibb, N. et al. Hey2 regulates the size of the cardiac progenitor pool during vertebrate heart development. Dev. (Camb., Engl.) 145, dev167510 (2018).
Rutenberg, J. B. et al. Developmental patterning of the cardiac atrioventricular canal by Notch and Hairy-related transcription factors. Dev. (Camb., Engl.) 133, 4381–4390 (2006).
Stefanovic, S. et al. GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development. Nat. Commun. 5, 3680 (2014).
Koibuchi, N. & Chin, M. T. CHF1/Hey2 plays a pivotal role in left ventricular maturation through suppression of ectopic atrial gene expression. Circulation Res. 100, 850–855 (2007).
Tian, X. et al. Identification of a hybrid myocardial zone in the mammalian heart after birth. Nat. Commun. 8, 87 (2017).
Miao, L. et al. Notch signaling regulates Hey2 expression in a spatiotemporal dependent manner during cardiac morphogenesis and trabecular specification. Sci. Rep. 8, 2678 (2018).
van Walree, E. S. et al. Germline variants in HEY2 functional domains lead to congenital heart defects and thoracic aortic aneurysms. Genet. Med. 23, 103–110 (2021).
Jordan, V. K., Rosenfeld, J. A., Lalani, S. R. & Scott, D. A. Duplication of HEY2 in cardiac and neurologic development. Am. J. Med Genet A 167A, 2145–2149 (2015).
Heisig, J. et al. Target gene analysis by microarrays and chromatin immunoprecipitation identifies HEY proteins as highly redundant bHLH repressors. PLoS Genet. 8, e1002728 (2012).
Kathiriya, I. S. et al. Hairy-related transcription factors inhibit GATA-dependent cardiac gene expression through a signal-responsive mechanism. J. Biol. Chem. 279, 54937–54943 (2004).
Das, S. et al. Transcriptomics of cardiac biopsies reveals differences in patients with or without diagnostic parameters for heart failure with preserved ejection fraction. Sci. Rep. 9, 3179 (2019).
Shih, Y.-H. et al. Cardiac transcriptome and dilated cardiomyopathy genes in zebrafish. Circulation: Cardiovascular Genet. 8, 261–269 (2015).
Wang, Y. et al. atg7-based autophagy activation reverses doxorubicin-induced cardiotoxicity. Circulation Res. 129, e166–e182 (2021).
Ma, X. A.-O. et al. Retinoid X receptor alpha is a spatiotemporally predominant therapeutic target for anthracycline-induced cardiotoxicity. Sci. Adv. 6, eaay2939 (2020).
Akerberg, A. A. et al. RBPMS2 is a myocardial-enriched splicing regulator required for cardiac function. Circulation Res. 131, 980–1000 (2022).
Kikuchi, K. et al. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 464, 601–605 (2010).
Davidson, M. M. et al. Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 39, 133–147 (2005).
Wang, T. et al. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 33, 531–546 e539 (2021).
Creed, S. & McKenzie, M. Measurement of mitochondrial membrane potential with the fluorescent dye tetramethylrhodamine methyl ester (TMRM). Methods Mol. Biol. 1928, 69–76 (2019).
Sivandzade, F., Bhalerao, A. & Cucullo, L. Analysis of the mitochondrial membrane potential using the cationic jc-1 dye as a sensitive fluorescent probe. Bio Protoc. 9, e3128 (2019).
Ide, T. et al. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circulation Res. 85, 357–363 (1999).
Balaban, R. S., Nemoto, S. & Finkel, T. Mitochondria, oxidants, and aging. Cell 120, 483–495 (2005).
Arany, Z. et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 1, 259–271 (2005).
Lehman, J. J. et al. The transcriptional coactivator PGC-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am. J. Physiol. Heart circulatory Physiol. 295, H185–196, (2008).
Wong, B. W. et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature 542, 49–54 (2017).
Weber, D. et al. Mechanisms of epigenetic and cell-type specific regulation of Hey target genes in ES cells and cardiomyocytes. J. Mol. Cell. Cardiol. 79, 79–88 (2015).
Trivedi, C. M. et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3β activity. Nat. Med. 13, 324–331 (2007).
Montgomery, R. L. et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 21, 1790–1802 (2007).
Conlon, F. L. et al. Histone deacetylase 1 controls cardiomyocyte proliferation during embryonic heart development and cardiac regeneration in zebrafish. PLoS Genet. 17, e1009890 (2021).
Zangi, L. et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat. Biotechnol. 31, 898–907 (2013).
Sun, J. et al. CCND2 Modified mRNA activates cell cycle of cardiomyocytes in hearts with myocardial infarction in mice and pigs. Circulation Res. 133, 484–504 (2023).
Carlsson, L. et al. Biocompatible, purified VEGF-A mRNA improves cardiac function after intracardiac injection 1 week post-myocardial infarction in swine. Mol. Ther. - Methods Clin. Dev. 9, 330–346 (2018).
Sohal, D. S. et al. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circulation Res. 89, 20–25 (2001).
Luo, W. et al. Critical role of the cGAS-STING pathway in doxorubicin-induced cardiotoxicity. Circulation Res. 132, e223–e242 (2023).
Wallace, K. B., Sardao, V. A. & Oliveira, P. J. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circulation Res. 126, 926–941 (2020).
Bates, M. G. et al. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur. Heart J. 33, 3023–3033 (2012).
Chen, F. Z., Zhao, Y. & Chen, H. Z. MicroRNA-98 reduces amyloid β-protein production and improves oxidative stress and mitochondrial dysfunction through the Notch signaling pathway via HEY2 in Alzheimer’s disease mice. Int. J. Mol. Med. 43, 91–102 (2019).
Bi, P. et al. Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nat. Med. 20, 911–918 (2014).
Churko, J. M. et al. Defining human cardiac transcription factor hierarchies using integrated single-cell heterogeneity analysis. Nat. Commun. 9, 4906 (2018).
Gillette, T. G. HDAC inhibition in the heart: erasing hidden fibrosis. Circulation 143, 1891–1893 (2021).
Kang, H. M. et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21, 37–46 (2015).
Yu, H. et al. LARP7 protects against heart failure by enhancing mitochondrial biogenesis. Circulation 143, 2007–2022 (2021).
Sander, V., Suñe, G., Jopling, C., Morera, C. & Belmonte, J. C. I. Isolation and in vitro culture of primary cardiomyocytes from adult zebrafish hearts. Nat. Protoc. 8, 800–809 (2013).
Xie, S. et al. Discovering small molecules as Wnt inhibitors that promote heart regeneration and injury repair. J. Mol. cell Biol. https://doi.org/10.1093/jmcb/mjz023 (2019).
Du, J. et al. A small-molecule cocktail promotes mammalian cardiomyocyte proliferation and heart regeneration. cell stem cell 29, 545–558.e513 (2022).
Krueger, F., James, F., Ewels, P., Afyounian, E. & Schuster-Boeckler, B. FelixKrueger/TrimGalore: v0.6.7 - DOI via Zenodo (0.6.7). Zenodo. https://doi.org/10.5281/zenodo.5127899 (2021).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907 (2019).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic acids Res. 44, W160–W165 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0 http://www.repeatmasker.org (2013–2015).
Bailey, T. L., Johnson, J., Grant, C. E. & Noble, W. S. The MEME Suite. Nucleic acids Res. 43, W39–W49 (2015).
Acknowledgements
We thank Dr. Mei Gao for helping with zebrafish echocardiography analysis, Shujun Zhai for zebrafish care, Jiwei Chen for bioinformatics data discussion, ECNU Multifunctional Platform for Innovation (004) for image acquisition and T.P.Z. laboratory members for comments on the manuscript. This work was supported by grants from the Ministry of Science and Technology of China (2018YFA0800103 to T.P.Z., 2018YFA0801004 to T.P.Z.), National Natural Science Foundation of China (NSFC31970780 to T.P.Z., NSFC82300275 to P.S., NSFC82170256 to P.Z., NSFC31530044 to T.P.Z.), Science and Technology Planning Project of Guangzhou (2024A04J4487 to P.S.), Guangdong Major Project of Basic and Applied Basic Research (2023B0303000005 to P.Z.), Guangdong Provincial Special Support Program for Prominent Talents (2021JC06Y656 to P.Z.), Science and Technology Planning Project of Guangdong Province (2022B1212010010 to P.Z.), and the Special Project of Dengfeng Program of Guangdong Provincial People’s Hospital (KJ012019119 to P.Z.).
Author information
Authors and Affiliations
Contributions
T.P.Z. and P.S. conceived and designed the project. T.P.Z., P.Z., and Y.W. supervised and managed the project. P.S. and B.G. performed most experiments in mitochondrial respiration and cardiac function using zebrafish, mouse and cell culture models. D.L., X. Zhu, Y.H., and F.M. contribute to these experiments. C.W. and P.X. performed bioinformatics analyses. Y.Q. and Y.C. performed Co-IP analyses. X.Zhao and J.Lin conducted modRNA synthesis. T.P.Z., V.M.C., C.L., P.X., B.Z., J. Li, H.H., D.J., P.S., and B.G. analyzed and interpreted data. T.P.Z. and P.S. wrote the manuscript. V.M.C. and B.Z. proofread the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ian Scott, Shinichi Oka and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
She, P., Gao, B., Li, D. et al. The transcriptional repressor HEY2 regulates mitochondrial oxidative respiration to maintain cardiac homeostasis. Nat Commun 16, 232 (2025). https://doi.org/10.1038/s41467-024-55557-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55557-4
This article is cited by
-
Mongolian medicine Eerdun-Wurile promotes myocardial regeneration by regulating MVDA in zebrafish
Cell Regeneration (2025)
-
Mitochondrial homeostasis restoring peptide-drug conjugates with ROS-responsive NO releasing ability for targeted therapy of myocardial infarction
Journal of Nanobiotechnology (2025)
