
Abstract
Natural materials with highly oriented heterogeneous structures are often lightweight but strong, stiff but tough and durable. Such an integration of diverse incompatible mechanical properties is highly desired for man-made materials, especially weak hydrogels which are lack of high-precision structural design. Herein, we demonstrate the fabrication of hierarchically aligned heterogeneous hydrogels consisting of a compactly crosslinked sheath and an aligned porous core with alignments of nanofibrils at multi-scales by a sequential self-assembly assisted salting out method. The produced hydrogel offers ultrahigh mechanical properties among the reported hydrogels, elastomers and natural materials, including a toughness of 1031 MJ · m-3, strength of 55.3 MPa, strain of 3300%, stiffness of 6.8 MPa, fracture energy of 552.7 kJ · m-2 and fatigue threshold of 40.9 kJ · m-2. Furthermore, such a tough and strong hydrogel facilely achieves stable regeneration and rapid adhesion owing to the highly crystallized and aligned network structure. The regenerated specimen presents the reinforced strength, toughness and fatigue resistance over 10 regeneration cycles. This work provides a simple method to produce hydrogels with bioinspired heterostructures and combinational properties for real applications.
Similar content being viewed by others
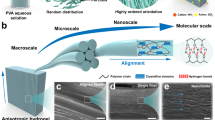

Introduction
Hydrogels possessing a combination of high strength, high stiffness, strong toughness, and fatigue resistance are highly attractive as load-bearing materials in the fields of tissue engineering1,2, soft robotics3,4,5, and artificial muscles6,7,8. However, these mechanical properties are hardly to be integrated into a single material due to their mutually exclusive characteristics, especially for synthetic hydrogels composed of polymer networks swollen with water, which limits their real application. Attributable to hierarchically structural design9, natural materials are generally porous but strong10, stiff but tough11,12, and water-containing but anti-fatigue enough13, providing a rich source of inspiration for the fabrication of revolutionary materials with high performance.
Typically, arising from hierarchically aligned fibrous micro/nanostructures, skeletal muscles can withstand a high stress of 1 MPa over 1 million cycles per year, exhibiting a fatigue threshold over 1000 J m-2 13. Fascinated by such combinational mechanical properties, various structural orientation strategies including ice-templating3,14,15, mechanical stretching16,17 and hot pressing18 have been developed to produce highly anisotropic hydrogels, achieving multiple orders of magnitude increases in stiffness and strength compared to the counterparts with homogeneous structures. A recent report showed that the hydrogels with a high alignment of hierarchical fibrous structure offered ultrahigh strength ranking with counterpart of natural ligaments by a facile drying in confined condition method, ascribed to strong inter-molecular hydrogen-bond interaction and formation of considerable stiff aligned fibrils17. However, due to the inherent conflict between stiffness and toughness according to the Lake-Tomas model19,20,21, uniformly oriented architecture provides considerable stiffness but suffers from a limited toughness, resulting in catastrophic failure when encountering flaws. More recently, He et al. have constructed tough hydrogels with hierarchical structure at multiple lengths comprising aligned pore walls at the microscale and interconnected nanofibrillar meshes by a combination of freezing and salting out22. Despite with these advancements23,24, the hierarchy and precision of the assembly structure in the man-made gels are lagging behind that of natural biological tissues. It is highly desired to create high-precision structures with the inspiration of natural materials, thereby coordinately addressing the inherent strength-toughness trade-off in the hydrogel.
Biological materials with hierarchical heterogeneous structures (i.e., teeth) arrange stiff constituents and structures at their surfaces to bear the wear and tear while tough ones in inner layers to adapt deformation and shield cracks25,26,27, which simultaneously combine high stiffness and toughness. Unfortunately, no stiff, tough and fatigue-resistant hydrogels based on engineering hierarchical heterogeneous structure have been reported till now due to the lack of a delicate assembly method involving simultaneous generation of density gradient and all-in-one configuration within preponderant polymer network. Herein, elaborately regulating gradient interfaces is highly essential to eliminate mismatched modulus between soft and stiff phases and avoid catastrophic destruction from stress concentration, yet remains challenging. Currently, biomimetic structural materials have witnessed the reliability of heterogeneous structural design strategy in combining mutually exclusive mechanical properties, achieving synergistically improved stiffness, toughness, and impact-resistance28. Therefore, we can expect the creation of hierarchically structured hydrogels with combinations of high strength, stiffness, stretchability, toughness, and fatigue resistance so long as to develop an effective method to engineer highly oriented heterogeneous architecture with seamless interfaces at multi-scales within anisotropic polymer network.
Herein, inspired by structural orientation and heterogeneous structure conception, a kind of strong and ultratough hydrogels with hierarchically anisotropic heterogeneous core-sheath structure has been fabricated by a directional freezing-induced assembly and prestretch-assisted salting out method. Owing to highly effective energy dissipation mechanism by dissociating and reforming the densified nanocrystalline domains within the compact shell and slipping and fracturing hierarchically aligned nanofibrils gradually, the hydrogels offered ultrahigh toughness (tensile toughness of 903~1031 MJ·m-3 and fracture toughness of 330~552.7 kJ·m-2), astonishing stiffness (5.7~9.5 MPa) and strength (41.9~64.6 MPa) while maintaining high elongation at break (2590~3850%). Besides, they also showed high energy dissipation capacity (92.4%) and fatigue resistance (fatigue threshold of 40.9 kJ·m-2). Such an integration of incompatible mechanical properties made these heterogeneous hydrogels highly competitive among the reported hydrogels, tough elastomers and natural materials. Furthermore, a facile hydrogen bonds competition-driven network reconfiguration strategy was developed to reborn the hydrogels with substantial residual strain after stretch by a swelling and resalting out treatment. The regenerated hydrogel presented pronouncedly improved strength of 80.2 MPa and ultrahigh toughness of 1898 MJ·m-3 which were 1.45 and 1.84 times higher than that of original specimen. This work provides a simple route to produce strong, tough and durable hydrogels with anisotropic hierarchical heterostructure which are comparable with natural materials and meanwhile, illuminates the critical role of elaborate architecture in comprehensively boosting diverse incompatible properties.
Results
Fabrication of hierarchically heterostructural hydrogels
Poly (vinyl alcohol) (PVA) and cellulose nanofibers (CNFs) produced by the aerosol-assisted biosynthesis (diameter: ~ 15 nm), which contain abundant hydroxyl groups were selected as the model constituents for the assembly of hierarchically oriented and heterogeneous architecture due to their tunable conformation and crystallinity via hydrogen bonding (Supplementary Fig. 1). As depicted in Fig. 1a, a delicate heterogeneous aligned structure design method combining directional freezing-induced self-assembly and prestretch-assisted salting out was proposed. Typically, a precursor hydrogel was first prepared by the directional freezing (DF) assembly of the mixture of PVA and CNFs, named as DF-PVA/CNF. With the growth of parallel ice crystals along the temperature gradient far below the freezing point at −20 °C, the polymer chains and CNFs were extruded from the solution and squeezed in gaps for the crystallization among the aligned ice fingers. After freezing for 12 h and thawing for additional 3 h, the honeycomb-like DF-PVA/CNF was produced by delivering a continuous, crosslinked 3D network structure composed of the interconnected 2D films in parallel arrangement (Supplementary Fig. 2a-2c)14,22. With the introduction of CNFs with a sharp stretching vibration of O-H at 3345 cm-1 in the FT-IR spectrum, the bands at 3277 cm-1 and 1086 cm-1 of the DF-PVA gel assigned to the υ(O-H) and υ(C-O), respectively, were blue-shifted to 3309 cm-1 and 1089 cm-1 for the DF-PVA/CNF (Supplementary Fig. 3a), suggesting that the additional physically-crosslinked network was formed via considerable hydrogen bonds between PVA and CNFs at the expense of free OH groups in PVA and CNFs, and self-bonding OH groups of PVA29,30,31. To obtain hierarchically oriented structure at multi-length scales mimicking natural muscles, the DF-PVA/CNF gel was subsequently stretched to further align the network of PVA and CNFs along the axis parallel to the micro-walls, leading to the formation of a temporarily oriented architecture with the elongated microchannels (Supplementary Fig. 2d-2f).

a Schematic illustrations of fabrication of HHPC hydrogel composed of a compact sheath and porous core by directional freezing assembly and stretch-assisted salting out method. Firstly, the DF-PVA/CNF gel with a honeycomb structure was obtained through aggregation of PVA chains and CNFs repelled by ice crystals. By stretching the gel along the channel direction, a temporarily elongated architecture was constructed with the aligned PVA and CNFs species on the cellular wall. Immersing in salt solution, the aligned structure was fixed and heterogeneous HHPC hydrogel was finally fabricated. The δ+ and δ- symbols around the water molecules indicated the polarization of the water molecules by adjacent anions during salting out. b Hierarchically aligned nanofibril structures by PVA crystallization and interactions with CNFs via hydrogen bonding on the internal cellular wall, that is, belt-like fibril bundles at microscale, fibril bundles at submicroscale and fibrils at nanoscale. c Evolution of heterogeneous structure. Upon the Hofmeister effect, the gel surfaces were rapidly crosslinked arising from strong self-coalescence and phase-separation of amorphous PVA chains and CNFs. The impeded inward permeation of ions by dense surfaces led to a gradient concentration of kosmotropic ions. The dashed blue arrow represented the trajectory of the ion. Aging with salting out, the gel was thinner, and heterogeneous core-sheath structured hydrogel was finally constructed with distinct density gradient.
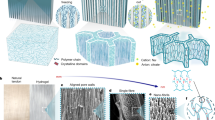
Upon immersing the prestretched hydrogel in a kosmotropic salt solution of sodium citrate, the temporarily aligned network was permanently fixed arising from strong self-coalescence and phase-separation of the preliminarily concentrated amorphous PVA chains and CNFs via hydrogen bonding under the Hofmeister effect that kosmotropic ions polarized water molecules22,32. With the generation of numerous crystalline domains, highly tough and hierarchically heterogeneous PVA/CNF hydrogels were finally fabricated, denoted as HHPC-x, where x is the prestretch. Typically, a 400 μm-thick HHPC-1.5 hydrogel exhibited a heterogeneous structure composed of a 3D-aligned porous inner layer with thickness of 220 μm coated with a 90 μm-thick compact sheath (Fig. 2a). It was found that the gel surface was rapidly crosslinked and densified at the initial salting out (Supplementary Fig. 4), which impeded ions to permeate inward and led to a gradient concentration of kosmotropic ions. Aging with the time, the gel was thinner with the internal polymer chains gradually self-aggregated and crystallized (Supplementary Fig. 5a). A 3D uniformly aligned porous network with the pore size of 4 μm was therefore formed due to dramatical aggregation and crosslinking of the microchannels in the inner layer (Fig. 2b). Furthermore, a transitional network region having a continuous gradient variation was generated to connect the densified and porous parts (Fig. 2c), which effectively eliminated stress concentration and mechanics mismatch, and was crucial to avoid catastrophic delamination and toughen the heterostructure28. Besides, a hierarchically aligned fibril network was presented on the elongated cellular wall, which comprised three levels, that was, belt-like fibril bundles at tens-of-micrometers, micro-fibril bundles at 1-2 μm, and nanofibrils at 60 nm (Figs. 2d–f).

a Side-view SEM image of HHPC−1.5 hydrogel. b SEM image of internal porous structure. c Magnified SEM image of transitional region in heterostructure. d–f Different magnifications of SEM images of the surface of cellular wall of HHPC−1.5. g XRD patterns of HHPC-1.5 and its sheath and core layer. h Master curves of frequency dependence of storage modulus G’ and loss modulus G” for surface and inner layer of HHPC-1.5. i Crystallinity of sheath and core layer in drying and swollen states. Data are presented as mean values ± SD (n = 3). Iq2-q SAXS profiles (j) and scattering intensity versus azimuthal angle plot (k) of HHPC hydrogels at different prestretches. The inserted image showing 2D SAXS pattern of HHPC-1.5. The colored arrows in (j) represent the qmax positions. l Variation of Herman’s factor at different prestretches. Data are presented as mean values ± SD (n = 3). m POM images of HHPC hydrogels at different prestretches. A: analyzer, P: polarizer.
According to the XRD patterns (Supplementary Fig. 6a), the PVA hydrogel alone presented a broad amorphous peak between 15° and 35° while the CNF hydrogel alone displayed a peak at 22.5° corresponding to the reflection plane (200) of cellulose crystalline structure33,34. As for the control hydrogel prepared without CNFs, denoted as HHP-1.5 hydrogel, an obvious peak centered at 19.6° corresponded to the typical reflection plane of \(\left(10\bar{1}\right)\) in semicrystalline PVA, while a shoulder peak at 22.5° belonged to (200) plane of PVA35,36. With the introduction of CNFs, the HHPC-1.5 hydrogel presented the stronger peaks at 19.6° and 22.5°, indicating the enhanced crystallinity induced by more hydrogen bonds among PVA chains and CNFs37. Furthermore, the individual sheath and core were obtained by a careful peeling-off method and carried out for the structural characterization (Supplementary Fig. 6c). A stronger diffraction peak at 19.6° was observed in XRD pattern of the sheath compared to that of the core layer, indicating the higher crystallinity of the sheath induced by the ion concentration gradient (Fig. 2g). The higher storage modulus G’ emerging in DMA curve of the surface was evidence of its higher crystallinity (Fig. 2h)38. Meanwhile, the sheath displayed a stronger endotherm peak at 230 °C in the DSC profiles assigning to the melting of PVA crystals by an increase of 10 °C compared with the inner layer (Supplementary Fig. 6b). Quantitatively, the surface crystallinity was estimated to be 23.2 wt% in a swollen state, far higher than 7.1 wt% of the inner part (Fig. 2i)14,39.
The mechanical stretch before salting out played an important role in controlling over core-sheath structure of the HHPC hydrogels. Without the prestretch, no nanofibril alignment occurred on the cellular wall of the HHPC-0, taking place of smooth thin walls in ~100 nm thickness, even though it maintained heterogeneous structure with a dense sheath and porous inner layer (Supplementary Fig. 7). Upon the prestretch, amorphous PVA chains and CNFs in the gel network got closer and aligned more orderly, which facilitated the self-coalescence and crystallinity under the salting out. As shown in low-field NMR spectra, the spin-spin relaxation rate in 10–200 ms belonging to immobilized water molecules40,41 was slower in the DF-PVA/CNF after the stretch (Supplementary Fig. 3b), indicating the weaker binding of the water to polymer chains and stronger interactions among the polymer chains in the stretched gel. Likewise, the greatly sluggish relaxation of immobilized water molecules in the HHPC-1.5 suggested that the interactions among the PVA and CNFs were further significantly enhanced. Thereby, the HHPC gel displayed 3.5 times decrease in thickness with increasing the prestretch from 0 to 2 (Supplementary Fig. 8). The decreased thickness was mainly arising from the porous core while the surface thickness showed negligible variation (Supplementary Fig. 5b). Meanwhile, the higher diffraction intensity was observed at the peak of 19.6° in XRD patterns of the HHPC hydrogels with the increase of prestretch, indicating the formation of more crystalline domains induced by the stronger aggregation of PVA chains after the salting out at the larger prestretch (Supplementary Fig. 9a). As calculated from DSC curves, the crystallinity in swollen state was greatly enhanced from 4.5 wt% to 8.9 wt% upon increasing the prestretch to 2, confirming mechanical stretch benefiting to the crystal growth (Supplementary Fig. 9b and c).
To trace the evolution of lamellar PVA crystalline domains in the HHPC hydrogels, the 1D SAXS profiles were plotted with Lorentz correction by multiplying q2, considering that the scattering signals of the randomly-distributed lamellar crystallization were dispersed in random directions rather than the ideally concentrated to the normal direction42,43,44. The PVA gel alone without crystalline domains only showed a low-q scattering (q < 0.03 Å-1) (Supplementary Fig. 10a), arising from the presence of heterogeneous structures constituted by polymer-rich and polymer-poor regions45,46. As for the HHPC-1.5 hydrogel, besides the low-q scattering, an obvious scattering peak was exhibited in middle-q region (0.03-0.15 Å-1), belonging to the long period of crystalline PVA in the polymer-rich phase, which was indicative of the interdomain spacing (Fig. 2j)47. Upon the prestretch increasing from 0 to 2, the scattering vector qmax corresponding to the maximum scattered intensity shifted to the large q direction, indicating the smaller interdomain spacing perpendicular to stretching direction owing to the stronger aggregation of polymer chains induced by the shearing effect at the higher prestretch48. Meanwhile, the clearer streak scattering was observed on 2D SAXS patterns of HHPC hydrogels with the higher prestretch, indicating the formation of microfibrils and enhanced orientation which can be verified by the stronger scattering intensity at θ = 90° in the azimuthal angle plot (Fig. 2k and Supplementary Fig. 10b). It was also quantified the Herman’s orientation factor increasing from 0.047 of the HHPC-0 to 0.182 of the HHPC-2 (Fig. 2l)49,50. The brighter POM image with the red-shifted interference color of the HHPC at a higher stretch confirmed the higher alignment of the nanofibrils with increasing elongation along the fiber axis (Fig. 2m)51,52.
Furthermore, to clarify the influence of the salt species on the formation of the heterogeneous structure, the HHPC-1.5 hydrogels were salted out in the salt solution at the top and middle of Hofmeister series, including Na2CO3, Na2SO4 and NaCl. The hydrogels salted out in two adjacent ions in the top Hofmeister series of Na2CO3 and Na2SO4 exhibited heterogeneous structure with the larger thickness composed of a thicker core layer of 500-600 μm coated with a thinner sheath layer of 40–50 μm compared with the hydrogel salted by sodium citrate (Supplementary Fig. 11a and b)22. Meanwhile, the aligned microfibrils with the diameter of ~1 μm were presented on the porous walls instead of hierarchical nanofibrils (Supplementary Fig. 11g and h), demonstrating the lower crystallinity resulting from the weaker aggregation of PVA chains. When salted out in NaCl solution at a relatively low Hofmeister series, a porous architecture rather than heterogeneous core-sheath structure was obtained (Supplementary Fig. 11c and f). Besides, no aligned hierarchical fibrils were observed on the pore walls (Supplementary Fig. 11i), indicating poor ability of Cl- to induce the crystallization of PVA chains. To be summarized, an integrated heterogeneous core-sheath structure with a distinct density gradient was formed in the HHPC hydrogel attributable to rapid crystallization of PVA chains by salting out in sodium citrate at the highest Hofmeister series, which consisted of tightly crosslinked high-density surface and aligned porous inner layer with hierarchically aligned fibrils at multi-scales by the directional freezing-induced self-assembly and prestretch-assisted salting out method.
Mechanical properties of heterostructural hydrogels
Owing to high alignment structure, the HHPC hydrogels exhibited prominent mechanical anisotropy with the stress in the orthogonal directions. For example, the extension parallel to the alignment direction yielded an astonishing tensile stress of 55.3 MPa at a stretchability of 3300% for the HHPC-1.5, which was 3.6 times the strength of 15.4 MPa and 3.0 times the elongation of 1080% under a perpendicular stretch, respectively (Supplementary Fig. 12). The importance of aligned structure on toughening the hydrogel was further investigated by performing tensile tests along the nanofibril axis on the HHPC hydrogels with different prestretches (Fig. 3a). Without the mechanical stretch, a strength of 20.2 MPa at the ultimate elongation of 2180% was measured for the HHPC-0. Once the salting out assisted by the stretch, both the strength and stretchability showed large enhancements. At a low prestretch of 0.5, the tensile strength was increased to 32.1 MPa at a maximum strain of 3520%, producing a toughness of 655 MJ·m-3. Upon further increasing the prestretch, the HHPC-1.5 hydrogel was toughened greatly by offering a high toughness of 1031 MJ·m-3, which was 3.8 times that of the HHPC-0 counterpart (Fig. 3b). At a high prestretch of 2, the tensile stress of the HHPC-2 reached as strong as 64.6 MPa, leading to a high toughness of 975 MJ·m-3 even with a decreased elongation of 2590%.

a Tensile stress-strain curves of HHPC hydrogels at the prestretch from 0 to 2 and HHP-1.5 hydrogel. b Variations in strength and toughness of HHPC hydrogels at different prestretches from 0 to 2. Data are presented as mean values ± SD (n = 3). c Tensile stress-strain curves of initial and notched HHPC-1.5. The fracture energy (Γ) can be calculated as the product of the integral area in the shading and the initial length (H) of the sample. The red “x” symbol represents critical strain for initiating crack propagation of core layer of pre-notched sample. d Variations in modulus and fracture energy of HHPC hydrogels. Data are presented as mean values ± SD (n = 3). e Photograph showing that a HHPC-1.5 hydrogel with a thickness of ~0.4 mm and a precut crack of 0.2 times its width lifted a 10 kg weight without crack propagation. f-h Ashby diagrams of toughness versus elongation at break (f), fracture energy versus fracture strength (g) and fracture energy versus modulus (h) of HHPC hydrogels (solid stars) and the reported tough hydrogels (hollow circles) (summarized in Supplementary Table 1), elastomers (hollow squares), synthetic fibrils (hollow diamonds) (summarized in Supplementary Table 2) and natural materials (hollow triangles) (summarized in Supplementary Table 3). The red dashed line in (h) indicates the trade-off between fracture toughness (Γ) and stiffness (E) (Γ = 234.234 kN3/2 m−2 × E-1/2) for the current tough hydrogels. i Cyclic loading-unloading curves of HHPC-1.5 hydrogel under different strains. j Calculated total energy, dissipated energy and dissipated ratio of HHPC-1.5 hydrogel under different strains. Data are presented as mean values ± SD (n = 3). k Crack extension per loading cycle dc/dN under increasing energy release rate G controlled by maximum elongation correspondingly.
Notably, the directional freezing of CNFs with PVA gave rise to a more highly stretchable hydrogel precursor, which was advantage to the subsequent prestretch-assisted salting out (Supplementary Fig. 13). Correspondingly, the HHP-1.5 hydrogel delivered a fracture stress of 36.2 MPa, weaker than the HHPC-1.5 counterpart (Fig. 3b). The PVA concentration also had a great influence on the mechanical behavior of the HHPC hydrogel. At a low PVA content of 5 wt%, the precursor hydrogel of DF-PVA/CNF was too mechanically weak to undergo the subsequent prestretch. The HHPC-1.5 hydrogel at a high content of PVA showed the decreased strength and toughness (Supplementary Fig. 14), for example, 34.4 MPa of tensile stress and 783 MJ·m-3 of toughness for 15 wt% PVA. Meanwhile, the heterostructural gel salted out in a 1.5 M sodium citrate for 3 h showed the optimal mechanical property (Supplementary Fig. 15). The water content was another important parameter to evaluate the mechanical properties of the gel. With the prestretch increasing from 0 to 2, the water content decreased from 46.5 wt% to 39.8 wt%, which was closed to that of the reported hydrogels toughened by salting-out method23,24. Comparatively, 5 ~ 20 times increase on toughness was presented for the HHPC hydrogel, indicating the superiority of the delicate heterogeneous structure. Furthermore, when the HHPC-1.5 hydrogel was dried in an ambient condition for 10 h (Supplementary Fig. 16), an obvious decline of 20 wt% after 2 h of drying was recorded. Afterwards, the trend slowed down due to strong interactions between the polymers and immobilized water molecules. In the drying process, the strength and strain at break remained to be 50 MPa and 3400% even after the weight decreased by ~30% during the drying process for 10 h, confirming the stable mechanical property of the HHPC hydrogels working in ambient conditions.
An unusual tendency appeared in the fracture energy-modulus correlation of the heterostructural hydrogels-their modulus and fracture energy increased simultaneously with the prestretch increased (Fig. 3c and Supplementary Fig. 17). With increasing the prestretch, the HHPC hydrogel stiffened drastically as proved by 3.7 times increase of Young’s modulus from 2.6 MPa for the HHPC-0 to 9.5 MPa for the HHPC-2 (Fig. 3d). It was noted that the fracture energy of the HHPC-1.5 hydrogel was determined to be a high value of 552.7 kJ·m-2, which was much higher than 351.0 kJ·m-2 of the HHPC-0 without the stretch. Specifically, a HHPC-1.5 hydrogel with a crack 0.4 times of its width even showed an ultrahigh strength of 13.6 MPa at elongation of 1500% and a pronounced fracture-blunting behavior was observed during further extension (Supplementary Fig. 18). This toughening was further confirmed by the crack blunting of a pre-notched HHPC-1.5 hydrogel of 0.2 g lifting a weight of 10 kg which was 50,000 times its own weight (Fig. 3e).
To highlight heterogeneous aligned structural hydrogels with high mechanical properties, Ashby charts in Fig. 3f–h comprehensively summarized and compared the toughness versus stretchability, fracture energy versus strength, and fracture energy versus modulus of various tough hydrogels, elastomers, and natural materials, respectively (Supplementary Tables 1-3). The HHPC hydrogels displayed tensile toughness an order of magnitude higher than reported tough hydrogels, elastomers, synthetic fibers and natural materials while maintaining super stretchability (Fig. 3f). Moreover, strength, stiffness and fracture energy, which were typically incompatible in a hydrogel according to Lake-Thomas model, were combined in HHPC hydrogels, fully illustrating these heterogeneous hydrogels as top performers among the reported hydrogels and even comparable with elastomers and natural materials (Fig. 3g and h). Overall, such an unusual integration of multiple mutually excluded mechanical properties achieved in HHPC hydrogel by alignment-assisted salting out confirmed the crucial role of the heterostructure with high orientations in simultaneous promotion of strength, stiffness and toughness.
The HHPC-1.5 hydrogel exhibited excellent energy dissipation capacity as indicated from apparent hysteresis loops in the tensile loading-unloading tests at the strains from 100% to 2000% (Fig. 3i). When the elongation was set as 100%, the work performed in the tensile and unloading process was 5.5 and 1.9 MJ·m-3, respectively, producing a dissipated energy per unit volume of 3.6 MJ·m-3 and dissipated ratio of 65.8% (Fig. 3j), implying that hydrogen bonds and crystalline domains were not completely dissociated at a low strain. Upon the strain exceeding 500%, the dissipated energy was increased sharply with a high and stable energy dissipation. At a large stretch of 2000%, the dissipated energy reached as high as 396.3 MJ·m-3, which was two orders of magnitude higher than that at 100%, yielding a high dissipated ratio of 92.4%. Such an energy dissipation at large tensile stains far exceeded the reported hydrogels, elastomers, and natural materials (i.e. spider silk) and comparable with synthetic fibers with exceptional damping capacity (Supplementary Fig. 19 and Supplementary Table 4), guaranteeing highly efficient energy dissipation mechanism to toughen the HHPC hydrogel.
To further evaluate the fatigue resistance, fatigue threshold Γ0, that was, the critical energy release rate above which fatigue fracture occurred, was quantified by the single-notch method (Supplementary Fig. 20)53. The HHPC-1.5 was determined to be a high fatigue threshold of 40.9 kJ·m-2 (Fig. 3k), larger than 33.4 kJ·m-2 of HHP-1.5 and 31.6 kJ·m-2 of HHPC-0 hydrogel (Supplementary Fig. 21). Specifically, no crack propagation was detected for the HHPC-1.5 after 1000 loading-unloading cycles at 1100% strain with a high energy release rate of 38.4 kJ·m-2 (Supplementary Figs. 22 and 23), validating their high durability. To sum up, the HHPC-1.5 with the optimized heterostructural design integrated multiple mutually exclusive properties together including high stiffness, strength, toughness and fatigue resistance.
Strengthening and toughening mechanism
To understand the toughening mechanism, the structural variation of HHPC-1.5 hydrogel across multiple length scales was demonstrated based on the stress-strain and its derivative curves (Supplementary Fig. 24). At the initial deformation (ε < 0.3), weak hydrogen bonds in network preferred to be dissociated to dissipate energy, resulting in the increased bond energy and frequency of O-H band, as confirmed by blue shift of υ(O-H) in FT-IR spectra from 3243 cm-1 to 3285 cm-1 with the ε increasing to 0.3 (Supplementary Fig. 25a)54. At this stage, crystalline domains were hardly dissolved as revealed from an upward trend in the derivative profile from the tensile stress-strain curve and smaller hysteresis on the cyclic tensile curve at small elongation (Supplementary Fig. 25b). Upon increasing the strain (0.3 < ε < 5), the modulus decreased as plotted by the sharply descended derivative, suggesting the melting of crystalline domains induced by stretch, but the polymer network still being locked by substantial rigid but dynamic crystalline domains47. Meanwhile, the declined diffraction intensity of crystalline peak at 19.6° in the XRD patterns demonstrated the melting of crystalline domains at a small elongation of ε = 5 (Fig. 4a), corresponding to the decreased crystallinity from 8.5 to 6.2 wt% as determined from the melting peak in DSC curves (Supplementary Fig. 26). In the subsequent stretching (5 < ε < 20), the crystallization was improved as indicated by the enhanced diffraction intensity, arising from tight aggregation of amorphous chains induced by tension which further facilitated the interactions between PVA chains and CNFs via hydrogen bonds. With the increase of ε, the interdomain spacing perpendicular to stretching direction was decreased, resulting from the stronger alignment and shearing effect during deformation (Fig. 4b)13. Furthermore, the continuous increase in crystallinity indicated that new crystalline domains were reconstructed induced by the dramatically aggregated polymer chains with the elongation increasing from 5 to 30 as estimated by DSC curves, for example, the crystallinity increased to 14.0 wt% at ε = 20 (Fig. 4c). These results illuminated the formation of the denser crystalline domains during the continuous stretch, which effectively dissipated energy for higher macroscopic ductility and toughness. Meanwhile, Herman’s factor as indicative of orientation degree along the stretching direction was increased from 0.146 to 0.241 with ε increasing from 0 to 30 (Figs. 4d and 4e). To this end, the densification of crystalline domains and increasing of orientation degree jointly accounted for continuous enhancement in strength and rise of derivative curve.

XRD patterns (a), Iq2-q SAXS profiles (b), variations of crystallinity in drying and swollen states (c), scattering intensity versus azimuthal angle plot (d), and Herman’s factor and corresponding 2D SAXS patterns (e) of HHPC-1.5 hydrogel stretched at different strains from 0 to 30. The colored arrows in (b) represent the qmax positions. Data in (c) and (e) are presented as mean values ± SD (n = 3). Schematic illustrations of coordinatively toughening mechanism, including the sheath-dominant energy dissipation mechanism by melting and reconstruction of crystalline domains (f, g), inner layer-dominant crack-blunting by crack bridge and crack deflection (g), and hierarchical fracture mechanism, that is, successive rupture of crystalline domains, nanofibrils and microfibrils for dissipating energy before macroscopic crack propagation (h).
When reaching a high strain (ε > 20), the significance of hierarchically aligned heterostructure to the toughness was highlighted. As a high energy was required to fracture tightly packed polymer chains in stiff surface, its network was generally extended by melting and recombination of crystalline domains under the stretch. Since the external loading surpassed the energy required to break the crystallized chains, the nanofibrils’ slippage and fracture preferentially occurred. Notably, the energy required to fracture a nanofibril packed by crystallization of considerable polymer chains was far higher than that broke the equivalent number of amorphous chains. Likewise, fracturing a microfibril composed of substantial nanofibrils required higher energy than a single nanofibril. Owing to effective crack blunting of hierarchically aligned structure, catastrophic damage to the bulk would not instantly occur, even though the microcracks appeared in inner network. It was illuminated at three length scales: microcracks tended to be deflected along the anisotropic micropores; aligned micro-/nanofibrils at the ahead of crack tips could bridge intact regions spanning the cracks to prevent their progress; and uncracked stiff crystalline domains would pin the crack (Supplementary Fig. 27). As a result, a gradual decrease was plotted on the derivative curve but stress was still raised with increasing the ε under the synergy of melting of crystalline domains and successive slippage and fracture of nano-/microfibrils. Importantly, no damage was observed on the transitional zone connecting the surface and inner layer even at an ultrahigh deformation of 3000% (Supplementary Fig. 28), demonstrating that continuous heterostructure effectively prevented the property mismatch and stress concentration.
Based on these analyses, our hierarchically aligned heterostructure strategy achieved the integration of ultrahigh strength, stiffness and toughness within a single hydrogel under the coordinative strengthening and toughening mechanism. The sheath provided hierarchical energy dissipation through the melting and recombination of crystalline domains during deformation (Figs. 4f and g), whereas the inner offered high anti-fracture performance by gradual slippage and fracture of hierarchically aligned nanofibrils and crack-blunting mechanism spanning multiple length scales (Fig. 4h). Consequently, the HHPC hydrogels presented strong strength approaching to stiff surface while kept a high notch-insensitivity as the tough inner behaved, affording high fracture toughness absent for the surface, thereby broke out the strength (stiffness)-toughness conflict what the reported hydrogels with homogeneous structures have suffered for a long time.
Regeneration and adhesion
A strong toughness with a large tensile strain was always accompanied with a plastic deformation, which was highly desired to be recovered for the real-world utilization, yet remained challenging due to their inherent contradictory mechanisms. For the HHPC hydrogels, kosmotropic ions polarized immobilized water molecules and enhanced the surface tension of the cavity surrounding the backbone and side chains which enabled strong hydrogen bonding among PVA chains and CNFs, resulting in the network crystallization and dehydration. Considering this conception and taking advantage of low activation barrier of hierarchically ordered HHPC-1.5 hydrogel, especially its highly densified sheath, we proposed a hydrogen bonds competition-driven network reconfiguration strategy to reborn the network structure and mechanical performance for boosting the sustainability and economy of tough hydrogels through a swelling and resalting out process (Fig. 5a). When swelling the plasticized HHPC hydrogel, water molecules without polarization plundered hydrogen bonds among intra- and inter-polymer chains, enabling crystalline domains to be dissociated and the plasticized network to be elastic. Further exerting a salting out treatment, phase separation reoccurred in the dissociated but still aggregated amorphous chains, and delicate network structure was reconstructed in the regenerated hydrogel.

a Schematic illustrations of the regeneration mechanism of HHPC hydrogel. It was realized by the dissociation and reformation of crystalline domains induced by the competition between water molecules and kosmotropic ions. b Adhesion mechanism of HHPC hydrogel by activating polymer chains at the highly densified surface via hydrogen bonds between water and polymer chains. c Photographs showing the recovery of the plasticized HHPC-1.5 with the initial clamp distance of 2 mm by a regeneration process. d Tensile stress-strain curves of the original and regenerated HHPC-1.5 hydrogels undergoing 1, 2, 5, and 10 regeneration cycles. e Strength and toughness of the HHPC-1.5 hydrogel over 10 regeneration cycles. Data are presented as mean values ± SD (n = 3). f Photograph showing that three pieces of HHPC-1.5 hydrogel with an adhesion area of 50 mm2 bearing a 5 kg weight. Adhesive strength-strain curves (g) and adhesive strength and energy (h) of the HHPC-0, HHPC-1.5 salted out in 1 M salt and HHPC-1.5 hydrogel. Data in (h) are presented as mean values ± SD (n = 3).
Typically, a HHPC-1.5 gel upon a 3000% strain with an irreversible deformation of 2000% recovered to its original length after a swelling-salting out regeneration treatment (Fig. 5c). The regenerated hydrogel showed greatly improved strength of 80.2 MPa at a stretchability of 4340% compared with initial 55.3 MPa at 3300% (Fig. 5d), while affording ultrahigh toughness of 1898 MJ·m-3 and fracture energy of 705.5 kJ·m-2, which were 1.84 times and 1.28 times higher than that of original counterpart, respectively (Fig. 5e and Supplementary Fig. 29). Notably, even after 10 regeneration cycles, the gel demonstrated much higher mechanical properties than the fresh by delivering a strength of 69.4 MPa, toughness of 1497 MJ·m-3 and fracture energy of 682.5 kJ·m-2. Furthermore, 92.4% of fatigue threshold was retained from 53.8 to 49.7 kJ·m-2 over 10 regeneration cycles, still exceeding the original 40.9 kJ·m-2, fully suggesting their stronger fatigue resistance (Supplementary Fig. 30). In contrast, the control samples of HHPC-0 and HHPC-1.5 salted out in low-concentration salt presented the smaller augmentation factor in toughness, defined as the ratio of regenerated toughness to original value, that was 0.98 and 0.79, respectively (Supplementary Fig. 31). These results validated the superiority of hierarchically aligned heterogeneous structure in toughening the regenerated hydrogels.
SEM images showed that anisotropic hierarchical heterostructure with highly aligned fibrils was well retained for the HHPC-1.5 even after 10 regeneration cycles (Supplementary Fig. 32). Furthermore, the regenerated HHPC-1.5 exhibited the enhanced crystallinity after the 1st regeneration, which kept constant in the subsequent 9 regeneration cycles (Supplementary Fig. 33a). Based on the 1D SAXS profiles, the interdomain spacing was obviously reduced in the 1st regenerated hydrogel revealing from the shift of qmax to the large q direction, and kept stable during the regeneration cycles (Supplementary Fig. 33b). The hierarchical heterogeneous structure with the denser crystalline domains after the regeneration contributed to the energy dissipation and fracture resistance for the dramatically enhanced toughness (Supplementary Fig. 34). We could safely conclude that continuous regeneration of the plasticized hydrogel was achieved with significantly enhanced mechanics (Supplementary Fig. 35).
Even possessing with ultrahigh stiffness and strength, the HHPC-1.5 demonstrated rapid and strong adhesion by a facile surface activation strategy in a few seconds. By a wetting and drying process, the HHPC-1.5 hydrogel could stick to each other with the tightly adhesive interfaces (Supplementary Fig. 36). Three pieces of HHPC-1.5 glued in pairs could lift a 5 kg weight without rupture of interface (Fig. 5f). Quantitatively, an adhesive strength of 21.0 MPa and adhesive energy as high as 622.8 kJ·m-2 were measured, two magnitude orders higher than 5.7 kJ·m-2 of HHPC-0 and 3.5 kJ·m-2 of HHPC-1.5 salted out by 1 M salt (Figs. 5g and h). This excellent adhesion capacity was owing to sensitive activation of aggregated polymer chains at the highly densified and aligned surface via the formation of hydrogen bonds between water molecules and polymer chains which favored releasing considerable amorphous chains (Fig. 5b). Thereafter, the released amorphous chains on the interphases were tightly packed and crystallized rapidly induced by kosmotropic ions during drying, enabling a strong interlocking interface and high adhesive energy. Based on the above demonstrations, regeneration and adhesion were realized for the tough hydrogels with excellent fatigue resistance, which were conflicted inherently.
With the advantages of ultrahigh modulus with increasing the prestretch, while being rapidly softened under the stimulation of water, the HHPC hydrogels presented the potentials in the field of flexible neural probes32,55. The high modulus could satisfy the implantation process of the neural probes. Meanwhile, a low modulus after stimulated by water could match the stiffness of soft brain tissue (0.4~15 kPa) after implantation. Furthermore, the aligned porous channels provided microfluidic channels for material delivery56. Therefore, the HHPC hydrogels can access a comprehensive neural probe system with an all-in-one configuration, featuring the adaptive modulus for both implantation and matching soft issues, as well as microfluidic channels for material delivery.
Discussion
In summary, we demonstrated a delicate hierarchically aligned heterogeneous structural design strategy to achieve an integration of multiple mutually exclusive mechanical properties within a single hydrogel by a facile sequential self-assembly-assisted salting out approach. The obtained HHPC hydrogel demonstrated the integrated heterogeneous core-sheath architecture with distinct density gradient, consisting of a compactly crosslinked sheath and an aligned porous inner layer with hierarchically aligned fibrils at multiple length scales. Attributed to melting and recombination of crystalline domains at the densified surface and gradual slippage and fracture of hierarchically aligned fibrils as well as strong crack blunting, the heterogeneous hydrogel presented high toughness (tensile toughness of 1031 MJ·m-3 and fracture toughness of 552.7 kJ·m-2) and large stretchability (3300%), while showing ultrastrong strength (55.3 MPa), high stiffness (6.8 MPa) and fatigue resistance (40.9 kJ·m-2). These integral performances enabled it to surpass the reported hydrogels, tough elastomers and natural materials. Furthermore, regeneration and robust adhesion that were conflicted to toughness and strength were realized with tough HHPC hydrogel owing to its densified and aligned microstructure. Furthermore, the regenerated hydrogels were much stronger, tougher and higher resistance to fatigue even over 10 regeneration cycles. This work paved a way to engineer hierarchical alignment of nanofibrils and gradient densification of nanocrystalline domains for the fabrication of heterogeneous hydrogels with combinational mechanical properties, exhibiting significant potentials for sustainable uses in soft robotics and biomedical applications.
Methods
Preparation of CNFs
The aerosol-assisted biosynthesis process of CNFs started with inoculation of Gluconacetobacter xylinus 1.1812 (China General Microbiological Culture Collection Center, CGMCC) onto a solid substrate in a bioreactor at an ambient temperature of 28 °C57. The composition of the solid substrate consisted of glucose (50 g L-1), yeast extract (5 g L-1), CaCO3 (10 g L-1), and agar (20 g L-1) in deionized water (DIW). The solid substrate was heated and stirred to dissolve all components thoroughly and then sterilized in an autoclave at 121°C for 30 min. In the later continuous fermentation process, liquid nutrient was introduced into the aerosol and gradually settled to the surface of the solid medium. Subsequently, after a period of continuous fermentation, the bacterial cellulose was obtained. The bacterial cellulose was mechanically treated using the blender and fully dispersed in DIW. This dispersion was further processed in a high-pressure homogenizer at 1000 bar at least 3 times to generate the final CNF suspension.
Fabrication of HHPC hydrogels
Typically, 10 wt% of PVA (Mw: 89000-98000) and 0.1 wt% of CNFs were dissolved in deionized water at 95 °C under stirring. After cooling, the mixture was poured into molds placing above a thermal conductive copper plate and partially submerged in ethanol at -90 °C. With the directionally frozen sample placing at −20 °C for 12 h, thawing and aging for additional 3 h, the DF-PVA/CNF hydrogel was firstly fabricated. Then, the obtained hydrogel was clamped and uniaxially stretched along the growth direction of ice crystals by using a fixture with sliding rails. The stretch was controlled by regulating the distance between two clamps. By immersing the prestretched hydrogel into 1.5 M sodium citrate solution for 3 h, the temporary orientation was permanently fixed and the HHPC hydrogel was finally fabricated.
Preparation of individual sheath and core layer
To make structural analyses of the individual sheath and core layer, a physical peeling-off method was applied to separate the semitransparent compact sheath and white porous core layer along the direction of nanofibril’s alignments. By sanding the sheath layer with 200 mesh-sandpaper to remove the adhered core residual, the individual parts of the sheath and core layer were obtained and carried out for characterizations.
Regeneration and adhesion procedures
For the regeneration process, the stretched HHPC hydrogels with residual strain were soaked into deionized water at 60 oC for partial swelling and dissolving of crystalline domains for 1 h. The swollen hydrogels were then fixed at their original length and immersed into 1.5 M sodium citrate solution for 3 h to acquire the regenerated samples. For the adhesion process, the surfaces of the adhered samples were firstly wetted for partial dissolving of crystalline domains for 3 s. Wet regions of two slices were then gently pressed together and immersed into 1.5 M sodium citrate solution. After 1 h of soaking, the slices were adhered together based on the reformation of crystalline domains at the interfaces.
Mechanical tests
The tensile tests were performed on the hydrogel piece with a length of 2 mm at a stretch speed of 10 mm/min along the direction parallel to the fibrils at room temperature. For cyclic tensile tests, the speed of 50 mm/min was used. The tensile stress was calculated from the applied force (F) divided by the cross-sectional area (A). The toughness was calculated as the area covered by the stress-strain curve at the fracture point.
Pure shear tests were applied to examine the fracture resistance13,58,59. The stress-strain curves under the same testing conditions were measured for both pre-notched and unnotched specimens. For the heterogeneous hydrogel, the critical strain for initiating crack propagation of core layer of pre-notched samples was denoted as εc. H and σ were the initial clamp distance (2 mm) and the stress of unnotched specimens, respectively. Thus, the fracture energy Γ was calculated as
A single-notch method was used to evaluate fatigue resistance. All the samples in fatigue tests were coated with oil to prevent dehydration-induced crack propagation. Cyclic tensile tests were performed on both pre-notched and unnotched samples with identical size and testing conditions. The strain energy density of the unnotched sample under the Nth cycle with maximum applied stretch of λmax was estimated as
The pre-notched specimens with an initial crack (c0) smaller than 1/5 of original length were cyclically stretched for ~1000 times with an ahead of measurements for a consistent crack tip. The average crack advance per cycle dc/dN after another Nth with the maximum applied stretch of λmax was observed using an optical microscope. Therefore, the applied energy release rate G in the notched samples under the Nth cycle with maximal applied stretch of λmax can be calculated as
Where k is a slowly varying function of the applied stretch and determined by
With the variation of λmax, the curve of crack extension per cycle dc/dN versus the applied energy release rate G was obtained. Thereafter, the fatigue threshold Γ0 can be evaluated by linearly extrapolating the curve of dc/dN vs. G to the intercept with the abscissa.
Measurement of crystallinities
To avoid crystallization during the freeze-drying process, samples were firstly immersed into the solution composed of 100 mL of 1.5 M sodium citrate, 500 μL of hydrochloric acid (36.5%) and 5 mL of glutaraldehyde (50 vol%) for 1 h, leading to the excessive crosslinks of PVA chains. Subsequently, excessively crosslinked samples were washed by 1.5 M sodium citrate solutions to remove the residual hydrochloric acid and glutaraldehyde. Finally, the samples were freeze-dried for 3 days and the mass before and after was weighted as m0 and m, respectively. Therefore, the water content can be calculated as
The freeze-dried samples were cut into small pieces and used for a typical DSC measurement. They were thereafter heated up from 50 to 250 °C at the rate of 20 °C/min under a N2 atmosphere with flow rate of 30 mL/min. There was a broad peak from 60 to 180 °C in the curve of heat flow corresponding to the transition temperature of water. The integration of the endothermic transition ranging from 60 to 180 °C gave the enthalpy for evaporation of the residual water per unit mass of the freeze-dried sample Hresidual. The mass of residual water mresidual was obtained as
Where \({H}_{{\mbox{water}}}^{0}=2260 J/g\) is the latent heat for water evaporation. The narrow peak ranging from 200 °C to 250 °C indicated the melting of crystalline domains. The integration of the endothermic transition ranging from 200 to 250 °C gave the enthalpy for melting crystalline domains per unit mass of the freeze-dried sample Hcrystalline. The mass of crystalline mcrystalline was calculated as
Where \({H}_{{{{\rm{crystalline}}}}}^{0}=138.6 J/g\) is enthalpy for evaporation of the fusion of 100 wt% crystalline PVA measured at the equilibrium melting point \({T}_{m}^{0}\). Therefore, the crystallinity of dry samples Xdry (without residual water) can be calculated as
And the crystallinity in swollen state Xswollen can be obtained as
Materials characterization
SEM images were obtained on a Zeiss Merlin Compact scanning electron microscope. The hydrogels were freeze-dried using a Labconco FreeZone freeze-dryer after washing. TEM images were performed on a JEM-2100F field-emission transmission electron microscope. AFM images were taken with a Bruker Dimension FastScan in tapping mode and the diameter of CNFs were measured by software NanoScope Analysis 2.0. FT-IR spectra were collected in wavenumber range of 4000-500 cm-1 on a Thermo Nicolet instrument assisted by ATR attachment. The spin-spin relaxation time (T2) was measured by a NMI20-040V-I NMR analyzer (Suzhou Niumag Corporation, China). The magnetic field strength was set as 21 MHz and the magnet temperature was set to 25 °C constantly. DMA measurement was conducted on a TA HR10 rheometer. Storage and loss modulus (G’ and G”) measurements were carried out over shear rates ranging from 0.01 to 500 s–1 at a constant temperature of 25 °C. XRD patterns were performed on the Rigaku instrument with an X ray of λ = 0.154 nm, operated at 40 kV and 200 mA, and the XRD patterns were collected in the 2θ range of 5° to 35° with a rotate speed at 2 °/min. For the XRD tests, the hydrogels were stacked to the similar thickness of 1 mm. Furthermore, the stretched HHPC-1.5 hydrogels at a certain strain were used to avoid potential beam damage from continuous X-ray irradiation. The USAXS-SAXS tests were conducted at Xeuss 3.0 UHR system (XENOCS SAS, France) with an X ray of λ = 1.5406 Å. The sample-to-detector distances were set to be 1000 mm for SAXS tests and 4550 mm for USAXS tests, permitting the collection of the scattered intensity across a range of scattering vectors from 0.01 to 0.35 Å-1 and lower vectors from 0.002 to 0.01 Å-1, respectively. All the samples were encapsulated in 1-mm path-length quartz cells in preventing from the dehydration of hydrogels. The exposure times were set at 5 min for SAXS measurements and 30 min for USAXS measurements. 1D SAXS curves were obtained by integrating two-dimensional SAXS patterns as a function of
Where q, θ and λ are the modules of scattering vector, scattering angle and wavelength of X ray, respectively. The scattering intensity were Lorentz corrected by multiplying q2 (Iq2) to track the evolution of the lamellar crystal domains.
Orientation degree was calculated through Herman’s method based on the azimuthal intensity. The Herman’s factor f was calculated as
Where <cos2θ>hkl was used to express the average orientation of hkl crystal plane which can be calculated mathematically using equation
Where θ and I(θ) are the azimuthal angle and scattering intensity, respectively.
Data availability
The data supporting the findings of this study are included in the paper and its Supplementary Information. All data are available from the corresponding author on request. Source data are provided with this paper.
References
Fu, L. et al. Cartilage-like protein hydrogels engineered via entanglement. Nature 618, 740–747 (2023).
Peng, Y. H. et al. Dynamic matrices with DNA-encoded viscoelasticity for cell and organoid culture. Nat. Nanotechnol. 18, 1463–1473 (2023).
Hou, G. et al. Self-regulated underwater phototaxis of a photoresponsive hydrogel-based phototactic vehicle. Nat. Nanotechnol. 19, 77–84 (2023).
Na, H. et al. Hydrogel-based strong and fast actuators by electroosmotic turgor pressure. Sciecne 376, 301–307 (2022).
Fusi, G., Del Giudice, D., Skarsetz, O., Di Stefano, S. & Walther, A. Autonomous soft robots empowered by chemical reaction networks. Adv. Mater. 35, 2209870 (2022).
Lang, C. et al. Nanostructured block copolymer muscles. Nat. Nanotechnol. 17, 752–758 (2022).
Zhang, M. et al. Hydrogel muscles powering reconfigurable micro-metastructures with wide-spectrum programmability. Nat. Mater. 22, 1243–1252 (2023).
Zhang, L. et al. High‐performance organohydrogel artificial muscle with compartmentalized anisotropic actuation under microdomain confinement. Adv. Mater. 35, 2202193 (2023).
Wegst, U. G. K., Bai, H., Saiz, E., Tomsia, A. P. & Ritchie, R. O. Bioinspired structural materials. Nat. Mater. 14, 23–36 (2014).
Chen, S.-M. et al. Mechanically robust bamboo node and its hierarchically fibrous structural design. Nat. Sci. Rev. 10, nwac195 (2023).
Mao, L.-B. et al. Synthetic nacre by predesigned matrix-directed mineralization. Science 354, 107–110 (2016).
Yeom, B. et al. Abiotic tooth enamel. Nature 543, 95–98 (2017).
Lin, S., Liu, J., Liu, X. & Zhao, X. Muscle-like fatigue-resistant hydrogels by mechanical training. Proc. Natl. Acad. Sci. USA. 116, 10244–10249 (2019).
Liang, X. et al. Anisotropically fatigue‐resistant hydrogels. Adv. Mater. 33, 2102011 (2021).
Feng, Y. et al. Muscle‐inspired MXene conductive hydrogels with anisotropy and low‐temperature tolerance for wearable flexible sensors and arrays. Adv. Funct. Mater. 31, 2105264 (2021).
Weldon, C. et al. Nanoscale bupivacaine formulations to enhance the duration and safety of intravenous regional anesthesia. ACS Nano 13, 18–25 (2018).
Mredha, M. T. I. et al. A facile method to fabricate anisotropic hydrogels with perfectly aligned hierarchical fibrous structures. Adv. Mater. 30, 1704937 (2018).
Liang, X. et al. Impact‐resistant hydrogels by harnessing 2D hierarchical structures. Adv. Mater. 35, 2207587 (2022).
Kim, J., Zhang, G., Shi, M. & Suo, Z. Fracture, fatigue, and friction of polymers in which entanglements greatly outnumber cross-links. Science 374, 212–216 (2021).
Creton, C. 50th anniversary perspective: Networks and gels: Soft but dynamic and tough. Macromolecules 50, 8297–8316 (2017).
Lake, G. J. & Thamos, A. G. The strength of highly elastic materials. Proc. R. Soc. London Ser. A 300, 108–119 (1967).
Hua, M. et al. Strong tough hydrogels via the synergy of freeze-casting and salting out. Nature 590, 594–599 (2021).
Cui, W. et al. Strong tough conductive hydrogels via the synergy of ion‐induced cross‐linking and salting‐out. Adv. Funct. Mater. 32, 2204823 (2022).
Xu, L., Qiao, Y. & Qiu, D. Coordinatively stiffen and toughen hydrogels with adaptable crystal‐domain cross‐linking. Adv. Mater. 35, 2209913 (2023).
Ho, S. P. et al. Structure, chemical composition and mechanical properties of human and rat cementum and its interface with root dentin. Acta Biomater. 5, 707–718 (2009).
Cuya, J. L. et al. Nanoindentation mapping of the mechanical properties of human molar tooth enamel. Arch. Oral Biol. 47, 281–291 (2002).
Liu, Z., Meyers, M. A., Zhang, Z. & Ritchie, R. O. Functional gradients and heterogeneities in biological materials: Design principles, functions, and bioinspired applications. Prog. Mater Sci. 88, 467–498 (2017).
Zhang, Z. B. et al. Scalable manufacturing of mechanical robust bioinspired ceramic–resin composites with locally tunable heterogeneous structures. Adv. Mater. 35, 2209510 (2023).
de Lima, G. G. et al. Effect of cellulose size-concentration on the structure of polyvinyl alcohol hydrogels. Carbohyd. Polym. 245, 116612 (2020).
Liu, L. et al. Dynamic nanoconfinement enabled highly stretchable and supratough polymeric materials with desirable healability and biocompatibility. Adv. Mater. 33, 2105829 (2021).
Song, P. et al. Granular nanostructure: A facile biomimetic strategy for the design of supertough polymeric materials with high ductility and strength. Adv. Mater. 29, 1704661 (2017).
Wu, S. et al. Poly(vinyl alcohol) hydrogels with broad‐range tunable mechanical properties via the Hofmeister effect. Adv. Mater. 33, 2007829 (2021).
Wang, Y. et al. Quantitative analysis of compatibility and dispersibility in nanocellulose-reinforced composites: Hansen solubility and Raman mapping. ACS Nano 15, 20148–20163 (2021).
Müller, L. A. E., Zimmermann, T., Nyström, G., Burgert, I. & Siqueira, G. Mechanical properties tailoring of 3D printed photoresponsive nanocellulose composites. Adv. Funct. Mater. 30, 2002914 (2020).
Zhang, H., Tang, N., Yu, X., Li, M. H. & Hu, J. Strong and tough physical eutectogels regulated by the spatiotemporal expression of non‐covalent interactions. Adv. Funct. Mater. 32, 2206305 (2022).
Wan, H., Wu, B., Hou, L. & Wu, P. Amphibious polymer materials with high strength and superb toughness in various aquatic and atmospheric environments. Adv. Mater. 36, 2307290 (2023).
Lee, W. J., Clancy, A. J., Kontturi, E., Bismarck, A. & Shaffer, M. S. P. Strong and stiff: High-performance cellulose nanocrystal/poly(vinyl alcohol) composite fibers. ACS Appl. Mater. Interfaces 8, 31500–31504 (2016).
Sun, Z., Jiang, Z. & Qiu, Z. Thermal, crystallization and mechanical properties of branched Poly(butylene succinate) copolymers with 1,2-decanediol being the comonomer. Polymer 213, 123197 (2021).
Wu, Y. et al. Solvent‐exchange‐assisted wet annealing: A new strategy for superstrong, tough, stretchable, and anti‐fatigue hydrogels. Adv. Mater. 35, 2210624 (2023).
Lei, Z., Gao, W., Zhu, W. & Wu, P. Anti‐fatigue and highly conductive thermocells for continuous electricity generation. Adv. Funct. Mater. 32, 2201021 (2022).
Wang, K., Li, Y., Zhang, Y., Sun, J. & Qiao, C. Preheating and high-intensity ultrasound synergistically affect the physicochemical, structural, and gelling properties of chicken wooden breast myofibrillar protein. Food Res. Int. 162, 111975 (2022).
Miyazaki, T., Hoshiko, A., Akasaka, M., Shintani, T. & Sakurai, S. SAXS studies on structural changes in a poly(vinyl alcohol) film during uniaxial stretching in water. Macromolecules 39, 2921–2929 (2006).
Ruland, W. The evaluation of the small-angle scattering of lamellar two-phase systems by means of interface distribution functions. Colloid. Polym. Sci. 255, 417–427 (1977).
Miyasaka, K. PVA-iodine complexes: Formation, structure, and properties. Adv. Polym. Sci. 108, 91 (1993).
Ricciardi, R. et al. Structural organization of poly(vinyl alcohol) hydrogels obtained by freezing and thawing techniques: A SANS study. Chem. Mater. 17, 1183–1189 (2005).
Auriemma, F. et al. Time-resolving analysis of cryotropic gelation of water/poly(vinyl alcohol) solutions via small-angle neutron scattering. J. Phys. Chem. B 112, 816–823 (2008).
Zhang, Q. et al. Stretch-induced structural evolution of poly (vinyl alcohol) film in water at different temperatures: An in-situ synchrotron radiation small- and wide-angle X-ray scattering study. Polymer 142, 233–243 (2018).
Kojio, K. et al. Simultaneous small-angle X-ray scattering/wide-angle X-ray diffraction study of the microdomain structure of polyurethane elastomers during mechanical deformation. Polym. J. 43, 692–699 (2011).
Kim, K. et al. 4D printing of hygroscopic liquid crystal elastomer actuators. Small 17, 2100910 (2021).
Duan, T., Xu, H., Tang, Y., Jin, J. & Wang, Z. Effect of epitaxial crystallization on the structural evolution of PCL/RGO nanocomposites during stretching by in-situ synchrotron radiation. Polymer 159, 106–115 (2018).
Zheng, Y., Zhang, L. & Duan, B. Anisotropic chitosan/tunicate cellulose nanocrystals hydrogel with tunable interference color and acid-responsiveness. Carbohyd. Polym. 295, 119866 (2022).
Sørensen, B. E. A revised Michel-Lévy interference colour chart based on first-principles calculations. Eur. J. Mineral. 25, 5–10 (2013).
Bai, R. et al. Fatigue fracture of tough hydrogels. Extreme Mech. Lett. 15, 91–96 (2017).
Xu, L. et al. A solvent‐exchange strategy to regulate noncovalent interactions for strong and antiswelling hydrogels. Adv. Mater. 32, 2004579 (2020).
Wen, X. et al. Flexible, multifunctional neural probe with liquid metal enabled, ultra-large tunable stiffness for deep-brain chemical sensing and agent delivery. Biosen. Bioelectron. 131, 37–45 (2019).
Mredha, M. T. I. & Jeon, I. Biomimetic anisotropic hydrogels: Advanced fabrication strategies, extraordinary functionalities, and broad applications. Prog. Mater. Sci. 124, 100870 (2022).
Guan, Q.-F. et al. A general aerosol-assisted biosynthesis of functional bulk nanocomposites. Nat. Sci. Rev. 6, 64–73 (2019).
Sun, J.-Y. et al. Highly stretchable and tough hydrogels. Nature 489, 133–136 (2012).
Rivlin, R. S. & Thomas, A. G. Rupture of rubber. I. Characteristic energy for tearing. J. Polym Sci. 10, 291–318 (1953).
Acknowledgements
This work is supported by the National Natural Science Foundation of China (grant nos. 22471052, 22171066, 21922104 (H.P.C.), and U1932213 (S.H.Y.)), the National Key Research and Development Program of China (grant nos. 2021YFA0715700 and 2018YFE0202201 (S.H.Y.)) and the Fundamental Research Funds for the Central Universities (grant nos. JZ2023YQTD0074 (H.P.C.) and JZ2021HGPA0064 (H.Q.)).
Author information
Authors and Affiliations
Contributions
H.P.C. and S.H.Y. supervised the project, conceived the idea and designed the experiments. Z.X. planned and performed the experiments, collected and analyzed the data. H.C. contributed to the structural and mechanism analyses. H.B.Y. synthesized the CNFs. X.Y. and H.Q. helped with the characterizations. Z.X., H.P.C. and S.H.Y. wrote the paper, and all authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, Z., Chen, H., Yang, HB. et al. Hierarchically aligned heterogeneous core-sheath hydrogels. Nat Commun 16, 400 (2025). https://doi.org/10.1038/s41467-024-55677-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55677-x
This article is cited by
-
Molecular clustering unlocks high-performance hydrovoltaics across temperatures from −35 °C to 80 °C
Nature Communications (2026)
-
Anisotropic hydrogel enabled by infusion of lubricating phase into macroporous robust skeleton with alignment structure
Advanced Composites and Hybrid Materials (2026)
-
Tough and tear resistant hydrogel with a sandwich mineralized structure induced by bidirectional ion migration
Nature Communications (2025)
-
Hierarchical crack-resistant, tissue-mimetic hydrogels enabled by progressive nanocrystallization of anisotropic polymer networks
Nature Communications (2025)
-
Hierarchically structuralized hydrogels with ligament-like mechanical performance
Nature Communications (2025)
