
Abstract
Designing catalysts with well-defined, identical sites that achieve site-specific selectivity, and activity remains a significant challenge. In this work, we introduce a design principle of topological-single-atom catalysts (T-SACs) guided by density functional theory (DFT) and Ab initio molecular dynamics (AIMD) calculations, where metal single atoms are arranged in asymmetric configurations that electronic shield topologically misorients d orbitals, minimizing unwanted interactions between reactants and the support surface. Mn1/CeO2 catalysts, synthesized via a charge-transfer-driven approach, demonstrate superior catalytic activity and selectivity for NOx removal. A life-cycle assessment (LCA) reveals that Mn1/CeO2 significantly reduces environmental impact compared to traditional V-W-Ti catalysts. Through in-situ spectroscopic characterizations combined with DFT calculations, we elucidate detailed reaction mechanisms. This study establishes T-SACs as a promising class of catalysts, offering a systematic framework to address catalytic challenges by defining site characteristics. The concept highlights their potential for advancing selective catalytic processes and promoting sustainable technologies.
Similar content being viewed by others


Introduction
The contemporary focus in the field of catalysis is centered on the ability to achieve precise selectivity while maintaining, or even enhancing, high catalytic activity1,2,3,4. As catalytic systems and their objectives have grown more complex, the difficulty in selectively promoting desired reactions while suppressing undesired ones has significantly increased1. For instance, in the selective catalytic reduction of nitrogen oxides (NOx) using ammonia (NH3-SCR)5,6, the challenge lies in oxidizing NH3 to NH2 without further overoxidizing it to NH species. The overoxidation of NH3 can lead to the production of N2O instead of the desired N27,8, which is problematic since N2O has approximately 300 times the global warming potential (GWP) of CO2 over a 100-year period9. Similarly, when dealing with organic molecules with multiple unsaturated bonds, selectively hydrogenating one bond without affecting others is a significant hurdle10,11,12. A key strategy for controlling reaction selectivity involves designing catalysts that exhibit specific-sites tailored to form the desired products13,14,15. This often requires exploiting or modifying the steric and electronic properties of these active sites16,17. Despite significant advances18,19,20,21,22,23, the intricate interactions between catalytic sites and substrates are often discovered by chance, making them difficult to predict or systematically apply across different systems24.
Heterogeneous single-atom catalysts (SACs)25,26,27, featuring well-defined mononuclear metal sites with tunable coordination and configuration on a support material, present a promising solution to these challenges28,29,30. The isolation of metal atoms in SACs allows for the precise design of active centers guided by theoretical principles31,32,33,34. While previous studies35,36,37 have made significant progress in designing “embedded Pt” and “adsorbed Pt” catalysts by focusing on single-atom design through geometric structures or analogies to doped/substituted surface structures, these approaches have not fully achieved the precise site-specific design from the perspectives of crystal field design or orbital symmetry. In this work, we introduce a novel design principle for topological-single-atom catalysts (T-SACs) that target site-selective reactions. In these catalysts, reactants temporarily bond with topologically arranged single atoms, allowing for precise control over reaction sites. We demonstrate this concept using the NH3-SCR reaction as a model system.
To date, Mn single-atom moieties prepared through scalable methods that simultaneously achieve high activity and excellent selectivity in NH3-SCR reactions have not been reported. Through density functional theory (DFT) and Ab initio molecular dynamics (AIMD) calculations we demonstrate that the designed Mn1/CeO2 single-atom moiety effectively stabilizes adsorbates and critical intermediates, thereby enhancing reaction performance. The metal single atoms, arranged in asymmetric configurations, utilize electronic d orbital shielding and spatial hindrance to protect the support surface from direct interactions with reactants. As a proof of concept, Mn1/CeO2 were synthesized using a charge-transfer-driven approach, which is readily scalable to kilogram-scale production and is characterized by low energy consumption, minimal environmental impact, and reduced costs. Catalytic evaluation of NH3-SCR shows that Mn1/CeO2 exhibit exceptional performance compared to both manganese oxides and other benchmark catalysts. An ex-ante Life Cycle Assessment (LCA) quantifying the environmental benefits of the Mn1/CeO2 over traditional V-W-Ti catalyst shows a much lower carbon footprint in four recognized metrics. In-situ characterization and theoretical studies unravel the complete reaction pathway of NH3-SCR over Mn1/CeO2. This study shows that Mn1/CeO2 surmounts the long-standing challenge of striking a delicate equilibrium SCR activity and N2 selectivity at low temperatures, underscoring the immense potential of this new class of SCR catalysts at the forefront of next-generation NOx removal technology. Furthermore, the concept of T-SACs offers broad potential for application in the selective processing of various reactants and catalytic processes, with opportunities for further generalization using alternative supports, single atoms, and electronic structures.
Results
Definition of topological-single-atom catalysts
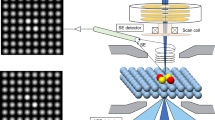
T-SACs feature metal single atoms arranged in distinctive, asymmetric configurations that, through their spatial and electronic topological structure, effectively shield the support surface from direct interaction with reactants linked to these atoms (Fig. 1). The catalytic behavior of T-SACs mirrors that of topology-defined conductive surface states, in contrast to the insulating bulk states in a topological insulator. Similar to electrons confined to moving along the surface of the material in a topological insulator, the design of T-SACs ensures that only the protruding metal single atoms actively engage in reactions, thus improving the selectivity of a designated catalytic process. This design principle relies on precise spatial arrangement enabled electronic shielding of mis-oriented d orbitals to restrict the interaction of reactive substrates with the bulk of the catalyst. Such a design effectively reduces the likelihood of side reactions that could diminish the yield of the desired product. Additionally, the symmetry breaking in the crystal field lead to a partial d electron shielding of T-SACs allows for precise control of reaction pathways, making these materials particularly advantageous for applications in site-specific catalysis, where catalyst specificity can significantly impact the purity and functionality of the end products.

a, b Conceptual framework of T-SACs: Single atoms are strategically positioned in spatially distinct and asymmetric configurations, which, within their electronic structure, act to shield the support from direct interaction with reactants attached to these metal atoms. This arrangement is complemented by the crystal field environment of topologically designed metal sites, achieving precise activation of reactants through symmetry broken. c Orbital interaction of Mn-NH2 in Mn-L4 coordination and Mn-N σ and π bonding over Mn1/CeO2. Mn 3d splitting is associated with Mn-L4 tetrahedral field in Mn1/CeO2. Colour code: Nitrogen, blue; Oxygen, red; Hydrogen, white; Cerium, yellow; Manganese, purple.
For example, low-temperature NH3-SCR reactions represent the next generation of low-energy consumption denitrification technology aimed at addressing the harm to sustainable development caused by NOx emissions5,6,38,39,40,41,42 (Fig. S1). MnOx complexes serve as effective catalysts in low temperature NH3-SCR reactions due to the ability of their Mn active sites to readily activate N-H bonds in NH38,43. However, the peroxidation of NH3 often results in the unwanted production of N2O, a by-product that reduces the selectivity for N2 (Fig. S2). The over-dissociation of NH2 on the catalyst surfaces primarily stems from an abundance of oxygen atoms, with the unsaturated oxygen atoms on the MnOx surface engaging with the hydrogen atoms in NH2 (Fig. S3). Through the constrained AIMD simulations, the dissociation process of NH2* on the MnOx surface was elucidated (Fig. S4c–f), revealing that NH2* can be easily split into NH* and OH* under the influence of surface oxygen atoms. This decomposition, particularly the formation of NH*, can contribute to the generation of the by-product N2O. It is therefore imperative to design novel Mn single-atom moieties that are fundamentally distinct from conventional MnOx, Mn-doped CeO2, MnCeOx, or MnOx/CeO2, to effectively mitigate NH3 peroxidation.
Topological design of Mn1/CeO2 catalyst for selective NOx removal
Grounded in theoretical insights, a novel Mn single-atom catalyst (Mn1/CeO2) featuring a distinctive tetrahedral coordination structure was designed to prevent adsorbed NH2* from being overwhelmed by surrounding oxygen atoms (Fig. 1b). The elevated positioning of the Mn single atoms creates a spatial buffer between NH2* and the substrate O atoms, which significantly hinders the dissociation of NH2*, a finding confirmed by AIMD simulations. Specifically, the energy barrier for NH2* dissociation on the Mn1/CeO2 surface is notably higher than 1.57 eV (Fig. S4b), presenting a stark contrast to the lower energy barriers encountered on various manganese oxide surfaces (Fig. S4, 0.63 eV for MnO, 0.61 eV for Mn2O3, 0.97 eV for Mn3O4, and 0.66 eV for MnO2). These results are further supported by climbing image nudged-elastic band (CINEB) calculations (Fig. S5), highlighting the energetic advantage of the topological Mn1/CeO2 in preventing the dissociation of NH2*.
The Walsh diagram for NH2 molecular orbitals (2a12 1b22 3a12 1b11) is shown in Fig. S6a. Following such knowledge, it is clear that electron transfer from Mn to NH2 can result in its activation; therefore, weak bonding between Mn and NH2 is demanding to suppress further oxidation. The intricate interaction between the Mn atom and adsorbed NH2* is explored through analyses of the σ and π bonds and their anti-bonding counterparts (Fig. S6b). Given the fundamental difference between MnO and Mn1/CeO2, which is the change in local coordination, crystal field theory has been employed to evaluate the Mn-N interaction. The analysis extends to the projected density of states (DOS), revealing notable spin polarization among these catalysts, dominated by spin-up states (Fig. S7), as closely examined in the orbital overview presented in Figs. 1c and S7. Specifically, MnO catalyst presents with six-coordinated Mn (Mn-L6) and leads to distorted octahedral field with spin up states, according to which several electrons fill in anti-bonding states (π*, 2eg). Accordingly, strong Mn-NH2 interaction origins from σ and π bonds (see Fig. S8a). Such picture has been confirmed by calculated Crystal Orbital Hamilton Population (COHP) profile (Fig. S7), from which an integrated bond order index (ICOBI) of 0.58 has been obtained. Clearly, to suppress NH2→NH conversion, it is highly essential to weaken Mn-N interaction, which can be achieved via the breaking of octahedral symmetry. Our strategy is to protect a part of Mn 3d orbitals by topological shielding through changing Mn-L6 to Mn-L4 as demonstrated by Mn1/CeO2. In this case, Mn 3d splits into E + T2, while E orbitals (\({\text{d}}_{{\text{x}}^{2}-{\text{y}}^{2}}\), \({\text{d}}_{{\text{z}}^{2}}\)) can hardly interact with the ligands due to the symmetry mismatch (Fig. 1c). As a result, the interaction between Mn and N is significantly reduced, as evidenced by a lower ICOBI value of 0.23, leading to a higher energy barrier for NH* formation (1.58 eV) compared to that over MnO (0.63 eV). Additionally, the Mn1/CeO2 configuration shows the weakest Mn-NH2 interaction compared to other crystal-field manganese oxides (such as ML5’s Mn3O4, see Fig. S9), fully demonstrating its superiority in effectively inhibiting the peroxidation reaction of NH2 and promoting NO removal (see Fig. S3b).
Large-scale synthesis and characterization
Given the difficulty in loading metal single atoms on a support, a charge-transfer-driven approach was devised for the synthesis of Mn1/CeO2. The cornerstone of this approach is the controlled removal of ligands from metal precursors and the careful management of their interactions with the support. For Mn1/CeO2, the key advancement involves the gradient decomposition of citric acid (CA) complexes, selectively targeting the support and single atomic elements. Initially, a mixed solution of MnCA and CeCA was prepared (Fig. 2a), ensuring homogeneity, as confirmed by the TEM and EDS images of the MnCA + CeCA precursor obtained after solvent evaporation (Figs. 2b and S10). The precursor underwent an initial annealing at 150 °C, which initiated the decomposition of CeCA into CeO2, forming CeO2 supports with (111) surfaces while avoiding the decomposition of MnCA. As depicted in Figs. 2c and S11, this process results in an amorphous layered structure corresponding to undecomposed MnCA and nanoparticles representing CeO2. Subsequently, a series of ball milling processes was conducted alongside a second annealing at 250 °C. During this stage, spontaneous charge transfer from the CA ligand to the support facilitated the kinetic migration and thermal stabilization of the Mn single atoms (further detailed in the AIMD analysis in the next section). Finally, the MnCA + CeO2 precursor underwent a final annealing to remove the remaining ligands from the Mn atoms, stabilizing them on the CeO2 lattice and resulting in the formation of Mn1/CeO2. The TEM and EDS surface scanning images in Fig. 2d and Fig. S12 confirm the complete decomposition of MnCA and CeCA, as evidenced by the presence of CeO2 nanoparticles without any amorphous precursor layers and the uniform distribution of Mn and Ce elements.

a Strategy for the preparation of Mn1/CeO2. b–d TEM images of different states of the synthesis process. e–g The difference in energy barriers (e) for the anchoring of Mn single atoms on the CeO2 surface with the use of MnCA (f) and MnCl2 (g) as metal precursors, respectively.
AIMD simulations were then employed to elucidate the critical stages in the synthesis of Mn1/CeO2 (Fig. 2e). Our theoretical analysis revealed that employing MnCA as a metal precursor (Fig. 2g) can effectively reduce the kinetic barrier for the anchoring of Mn single atoms onto CeO2 surface, reducing it from 1.41 eV to 0.89 eV, in comparation to conventional compounds like MnCl2 (Fig. 2f). The reduction in the energy barrier is primarily attributed to the weak bonding interaction between the oxygen atoms in CA and the cerium atoms in CeO2. As MnCA approaches the CeO2 surface, the spontaneous charge transfer from the CA ligand to the support simultaneously assists in the kinetic migration and thermal stabilization of the Mn single atoms. Fig. S13 illustrates that as the temperature increases, the CA component in MnCA progressively decomposes, while the single Mn atoms continue to be firmly anchored within the pockets formed by three oxygen atoms on CeO2. Moreover, stability simulations conducted in SCR reaction (Fig. S14) demonstrated that the single Mn atoms remain highly stable and do not aggregate under the harsh conditions.
The existence of Mn atoms on the surface as single atoms with a distinctive tetrahedral coordination structure was verified through multiple advanced characterization techniques. X-ray diffraction (XRD) patterns of Mn1/CeO2 showed only diffraction peaks characteristic of the cubic fluorite structure of CeO2 (Fig. 3a, b and Table S1). No lattice distortion was detected in the CeO2 support, even with the incorporation of a high Mn content exceeding 3 wt% (Table S2), ruling out the possibility of Mn doping or Mn-Ce solid solution formation. The annular dark-field scanning transmission electron microscopy (ADF-STEM) images revealed bright spots approximately 0.2 nm in diameter, which correspond to Mn atoms superimposed on the CeO2 surface (Figs. 3c and S15). This observation is in good alignment with the simulated atomic structure of Mn on the (111) surface of CeO2. The presence of single Mn atoms was further confirmed by atomic-scale electron energy loss spectroscopy (EELS) and elemental mapping of Mn1/CeO2 (Fig. 3d), where a pronounced Mn L-edge signal was observed at the corresponding position. Large-scale energy-dispersive X-ray spectroscopy (EDS) elemental mapping verified the uniform distribution of Mn, with only CeO2-related lattice structures observed (Fig. S16).

a, b Rietveld refined X-ray diffraction patterns for the CeO2, Mn1/CeO2. a. u. arbitrary units. c Atomic-resolution AC-HAADF-STEM image of Mn1/CeO2 containing single Mn atoms. d Electron energy loss spectroscopy (EELS) analysis for (c) in Mn1/CeO2. e The experimental high-resolution XANES spectrum compared with the calculated XANES data of optimized DFT-modelled structure of Mn1/CeO2. a. u. arbitrary units. f K3-weighted Mn K-edge Fourier-transformed (FT) EXAFS spectra of Mn1/CeO2 and Mn foil as reference. Colour code: Oxygen, red; Cerium, yellow; Manganese, purple.
X-ray absorption near-edge structure (XANES) spectra indicated that the Mn sites in Mn1/CeO2 are in a positive oxidation state (Fig. S17a). Fourier-transformed extended X-ray absorption fine structure (EXAFS) spectra (Figs. 3f and S17b) and wavelet-transformed EXAFS (Fig. S17c–h) confirmed the presence of Mn single-atom metal sites. Only a strong first-shell M-O peak was observed, corresponding to the Mn-O scattering pathway, with no evidence of a first-shell Mn-Mn peak. After fitting the EXAFS spectrum (Fig. S18), the specific fitting parameters, summarized in Table S3, revealed that the Mn-O coordination number and bond length closely align with the topological design. To further confirm the atomic configuration of the Mn sites, first-principles calculations were performed to explore potential models and simulate their corresponding XANES spectra. The model proposing a tetrahedral structure of a single Mn atom stabilized by four oxygen atoms on the CeO2 (111) surface showed the closest match with the experimental Mn K-edge XANES spectrum (Fig. 3e). Although there are minor discrepancies between the model and experimental data, the key criterion for validating a model is that the positions of the features in both the simulated and experimental spectra closely correspond. These results conclusively demonstrate that single Mn atoms are robustly stabilized on the top surface of the CeO2 support in a topologically tetrahedral configuration. In addition, it is worth emphasizing that the synthetic approach presented in this study offers a versatile and scalable method for preparing T-SACs beyond Mn. By preciously controlling the annealing temperature and further optimizing the process (Table S4), as determined by the thermogravimetric analysis (TGA) of the corresponding precursors (Fig. S19), we successfully synthesized catalysts with other metals supported on CeO2. The presence of these metals in single atom form was confirmed through XANES spectra, FT-EXAFS, WT-EXAFS, and EXAFS fitting (Fig. S20, Table S5 for Fe; Fig. S21, Table S6 for Ru; Fig. S22, Table S7 for Zn). Most importantly, this approach can be readily scaled up to produce quantities in the tens of kilograms, representing a significant milestone toward industrial implementation (see Fig. S23). In contrast, if the carefully planned multi-step ligand removal process and the charge transfer from the MnCA ligand to the support are bypassed, and the MnCe mixed precursor is instead subjected to a single high-temperature calcination, MnOx/CeO2 catalysts are formed. This is clearly shown by the Rietveld-refined X-ray diffraction patterns, which reveal distinct peaks corresponding to Mn2O3 and Mn3O4 phases, as seen in Fig. S24 and detailed in Table S8.
Catalytic performance of Mn1/CeO2
To demonstrate that the newly designed Mn1/CeO2 catalyst exhibits fundamentally different behavior from conventional Mn-based catalysts, we evaluated its catalytic performance in NH3 oxidation and NH3-SCR reactions. As shown in Fig. S25, we employed a systematic selection of control catalysts to ensure comprehensive and rigorous comparisons. These included Mn oxides with varying oxidation states, such as MnO2, Mn3O4, Mn2O3, and MnO, as well as several commonly used Mn-Ce-based catalysts, such as Mn-doped CeO2, MnOx/CeO2, and MnCeOx (additional synthesis and characterization details provided in Supplementary Note 1, Fig. S26–S38 and Table S9). Specifically, evidence for the proper formation of octahedral Mn sites in MnCeOx and MnOx catalysts can be found in Supplementary Note 2, Figs. S39 and S40 and Table S10. To confirm that the catalytic performance originates from the Mn active sites rather than the CeO2 support, we also synthesized pure CeO2 without incorporating the Mn precursor. Additionally, we synthesized the traditional V-W-Ti catalyst to further explore the commercial potential of Mn1/CeO2.
We first assessed the adsorption and oxidation capabilities of NH3 and NO on Mn1/CeO2. The NH3-TPD analysis (Fig. 4a) displayed desorption peaks at 300 °C, 400 °C, and 700 °C for the Mn1/CeO2, with a peak area considerably larger than that of the CeO2 samples. This indicates that the incorporation of single Mn atoms greatly enhances the catalyst’s NH3 adsorption capacity. Figure 4b shows that the onset temperature for NH3 oxidation decreased from 240 °C for CeO2 to 150 °C for Mn1/CeO2, demonstrating improved NH3 oxidation activity due to the addition of Mn single atoms. However, for MnOx/CeO2 catalysts, NH3 overoxidation was observed, with the NH3 oxidation rate exceeding 60% between 150 °C and 270 °C, leading to reduced selectivity. The NO oxidation ability of Mn1/CeO2 showed no significant enhancement (Fig. 4c), indicating that the fast-SCR pathway contributes minimally to the overall catalytic activity and selectivity of Mn1/CeO2.

a NH3-temperature programmed desorption (TPD) curve. Colour code: Nitrogen, blue; Oxygen, red; Hydrogen, white; Cerium, yellow; Manganese, purple. a. u. arbitrary units. b NH3 oxidation (reaction conditions: [NH3] = 500 ppm, [O2] = 5 vol%). c NO oxidation (reaction conditions: [NO] = 500 ppm, [O2] = 5 vol%). d–f NO conversion, N2 selectivity and N2O production for all the catalysts (reaction conditions: [NH3] = [NO] = 500 ppm, [O2] = 5 vol%). g NO conversion for all the catalysts (reaction conditions: [NH3] = [NO] = 500 ppm, [O2] = 5 vol%, [H2O] = 0 vol%, [SO2] = 100 ppm). Corresponding N2 selectivity data is presented in Fig. S41. h Effect of H2O and SO2 at 180 °C on the NH3-SCR performance of Mn1/CeO2. (Reaction conditions: [NH3] = [NO] = 50 ppm, [O2] = 5 vol%, [H2O] = 0 or 10 vol%, [SO2] = 0 or 100 ppm). i Kinetic analysis of the catalysts during the NH3-SCR reaction (further details can be found in the Methods section).
For the NH3-SCR performance evaluation, we initially conducted tests in the absence of SO2 and H2O to effectively assess the intrinsic catalytic capabilities of the Mn1/CeO2 (Fig. 4d–f). Following this, we introduced SO2 and H2O to the reaction environment to evaluate the catalysts’ resistance to poisoning and their overall stability, which are crucial for commercial applications (Fig. 4g–h). Figure 4d–e present the NO conversion and N2 selectivity as functions of temperature. Notably, the incorporation of a single Mn atom into CeO2 led to a increase in NO conversion from 4% to an impressive 99% at a low reaction temperature of 90 °C. This modification extended the effective denitrification temperature range to 90–240 °C, maintaining SCR activity above 95% while achieving consistently high N2 selectivity, remaining above 90% throughout this temperature range. This performance significantly surpasses that of all other catalysts tested. Although MnOx/CeO2 catalysts achieved NO conversion rates comparable to Mn1/CeO2, Fig. 4e indicates a marked decline in N2 selectivity for MnOx/CeO2 catalysts as temperature increased, dropping below 20% at 240 °C. Similarly, the N2 selectivity of other MnOx catalysts (MnO2, Mn3O4, Mn2O3, and MnO) also fell below 20% at 240 °C. The sharp rise in N2O production for MnOx and MnOx/CeO2 catalysts with increasing temperature, exceeding 300 ppm at 210 °C (Fig. 4f), provides direct evidence of this limitation. In contrast, Mn1/CeO2 maintained N2O concentrations below 40 ppm during NH3-SCR, highlighting their superior ability to minimize unwanted byproducts.
Given that SO2 and H2O in exhaust gases from stationary and mobile sources can lead to catalyst poisoning in practical applications, we also evaluated the catalytic performance of these catalysts in the presence of 10 vol% H2O and 100 ppm SO2 (Figs. 4g and S41). Despite a slight decrease in activity at 90 °C, Mn1/CeO2 maintained over 97% NO conversion and more than 95% N2 selectivity in the 120−240 °C range. In contrast, the performance of MnOx/CeO2, Mn-doped CeO2, and MnOx catalysts significantly deteriorated upon exposure to SO2 and H2O. For example, NO conversion for MnO, MnO2, Mn2O3, and Mn3O4 catalysts fell below 40% within the 90–240 °C range, indicating that the active sites on these MnOx catalysts were severely poisoned by SO2 and water vapor. Similarly, Mn-doped CeO2, MnOx/CeO2, and V-W-Ti catalysts exhibited poor NO conversion, with values below 60% within the 90–210 °C range. The MnCeOx series of catalysts, despite achieving relatively high activity at 180 °C in the absence of SO2 and H2O (Fig. S42a), failed to sustain their activity when exposed to SO2 and H2O, highlighting the inability of traditional MnCeOx catalysts to effectively catalyze NOx removal in the sulfur stream. In contrast, Mn1/CeO2 demonstrated remarkable stability, maintaining NO conversion above 90% and N2 selectivity exceeding 90% after 300 min of testing at 180 °C in the presence of H2O and SO2 (Fig. 4h). Overall, traditional Mn-based catalysts tend to suffer from sulfate formation, which severely inhibits catalytic activity over time. Our results demonstrate that Mn1/CeO2 can effectively survive the sulfur stream, as supported by additional experiments and discussions provided in Supplementary Note 3 and Figs. S43–S47. For instance, TG-IR-MS experiments verified that no sulfate was formed on Mn1/CeO2 after the reaction. Given that sulfate formation is a widely observed phenomenon in most SCR catalysts with moderate redox properties, we further provided a detailed explanation of how Mn1/CeO2 suppresses SO2 adsorption and oxidation, thereby preventing sulfate formation.
To further evaluate the intrinsic activity of Mn1/CeO2, MnOx/CeO2, and V-W-Ti in the NH3-SCR reaction, kinetic studies were conducted. As shown in Fig. 4i, Mn1/CeO2 exhibited a significantly lower apparent activation energy (Ea) of 27 kJ·mol−1 compared to MnOx/CeO2 (48 kJ·mol−1) and V-W-Ti (49.3 kJ·mol–1). This indicates that Mn1/CeO2 are more efficient in catalyzing the NH3-SCR reaction for NO reduction. Moreover, the design principles of T-SACs can be extended to other metals and catalytic processes, for example, enabling the selective hydrogenation of specific bonds in organic molecules with multiple unsaturation, thus achieving both high catalytic activity and product selectivity (Fig. S48 and Supplementary Note 4).
Environmental viability of T-SACs production
Early-stage lifecycle assessments (LCA) can effectively bridge the molecular and process levels to rigorously quantify the impact of different catalytic systems by accounting for emissions and waste embodied in all reaction components. These metrics help determine the degree to which novel technologies are environmentally sustainable and identify critical hotspots that warrant focused research efforts. In order to assess the environmental benefits of the Mn1/CeO2, a comprehensive cradle-to-gate LCA was performed in comparison to commercially available V-W-Ti catalysts for NH3-SCR reactions. This assessment was based on their SCR activity and selectivity data under conditions that included the presence of SO2 and H2O. Distribution of LCA impacts in the catalyst production and Sankey diagram of embodied GWP flows are shown in Figs. 5a, b and S49. The LCA results reveal that the Mn1/CeO2 possess an environmental footprint that is an order of magnitude lower than that of commercially available V-W-Ti catalysts, as illustrated in Fig. 5c.

a–b Distribution of LCA impacts in the catalyst production for commercialized V-W-Ti catalysts (a) and Mn1/CeO2 (b). c Environmental impacts comparison for two types of active catalysts. It is based on 11 sustainability indicators: acidification, global warming potential, freshwater aquatic ecotoxicity, marine aquatic ecotoxicity, terrestrial ecotoxicity, abiotic depletion potential for fossil fuels, eutrophication, human toxicity, abiotic depletion potential for elements, ozone layer depletion, and photochemical oxidation. The reported values are normalized per 1 kg of active catalyst. d Global warming potential (GWP100) of different catalysts at different temperatures.
This significant reduction in environmental impact is primarily due to the exceptional performance of the Mn1/CeO2, which not only exhibits high conversion and selectivity, but also maintain these properties at lower temperatures. Figure 5d presents the Global Warming Potential (GWP100) of the two catalysts at different reaction temperatures, corresponding to their catalytic activities shown in Fig. 4g. The conventional V-W-Ti catalyst maintains a relatively high GWP100, which gradually decreases as the reaction temperature increases. In contrast, the Mn1/CeO2 consistently exhibits a significantly lower GWP100 across the entire temperature range, reflecting a substantially reduced carbon footprint. This stable performance under varying thermal conditions highlights the potential of Mn1/CeO2 to offer a more environmentally friendly alternative in processes where temperature fluctuations are common.
Mechanistic studies
In-situ Raman and DFT calculations show that during SCR pre-treatment process (following H2 purging), axially adsorbed oxygen atoms on Mn single atoms can be readily removed to form H2O, thereby releasing the Mn sites (see Fig. S50). In-situ NAP-XPS at 240 °C during NH3-SCR reveals that introducing Mn single atoms on CeO2 increases the Ce3+/Ce4+ ratio from 11% to 26%, indicating partial reduction of Ce4+ to Ce3+ (Fig. S51a). The peak at 641.2 eV (Fig. S51b) confirms Mn single atoms as Mn2+44, stable in the SCR environment. In situ DRIFT spectra were conducted on Mn1/CeO2 T-SACs and MnOx/CeO2 catalysts to capture the NH3 activation state during the reaction. As shown in Fig. 6a, b, IR bands at ~1100–1300 cm–1, corresponding to NH3 adsorption on Lewis acid sites45,46,47, appear on both catalysts after 2 min. Additionally, an IR band at 1420–1470 cm−1, assigned to NH species according to literature45,48,49,50,51,52, is detected on MnOx/CeO2. MnOx/CeO2 samples also show an N-H stretching band at 3250 cm–153,54 (Fig. S52), consistent with DFT-calculated harmonic vibrational frequencies for NH adsorbed on MnOx (Figs. S52 and 53). The DRIFT spectra of NH3 adsorption on Mn1/CeO2 differ significantly from those on MnOx/CeO2. As adsorption progresses, bands in the range of 1550–1580 cm–1, attributed to NH2* species47,55,56, are enhanced, while NH* species are scarcely detectable. This suggests that while MnOx promotes NH3 dissociation to NH, Mn1/CeO2 favors the oxidation of NH3 to NH2 and effectively prevents further oxidation to NH. DFT calculations further support these findings, confirming the correlation between IR bands and the vibrational modes of the adsorbed species (Supplementary Note 5). This underscores the catalyst’s ability to facilitate the desired conversion of NH3 to NH2 while inhibiting unwanted side reactions from NH2 to NH.

a In situ DRIFT spectra recorded at 210 °C while exposed to NH3 for 30 min, along with vibrational peaks calculated by DFT. a. u. arbitrary units. b In situ DRIFT spectra recorded at 210 °C while exposed to NH3, along with vibrational peaks calculated by DFT. a. u. arbitrary units. c, d Free-energy diagrams (T = 500 K) of reaction pathways for the NH3 activation, NH2NO formation, NH2NO decomposition and H* removal at Mn1/CeO2 surfaces.
Comprehensive DFT calculations were further conducted to explore the catalytic mechanism in the NH3-SCR reaction. The choice of the slab model for Mn1/CeO2 was informed by XAFS spectroscopy data, as shown in Fig. S54. Comparing adsorption free energies for NO, NH3, and O2 on both the pristine CeO2 catalyst (Fig. S55) and Mn1/CeO2 (Fig. S56) illustrates that the incorporation of a single Mn atom significantly improves the binding of reactant molecules. The NH3-SCR mechanism (Figs. 6c, d and S57) begins with NH3 adsorption at the Mn single-atom site, forming NH2* with a hydrogen atom transferring to a nearby lattice O site, forming O-H* species. The activation of NH3 by Mn1/CeO2 has a reaction free energy change of 0.13 eV and a kinetic barrier of 0.34 eV, much lower than the 1.77 eV barrier on pure CeO2 (Fig. S58). The activation of NH3 by MnO2 and Mn3O4 presents kinetic barriers of 0.62 eV (Fig. S59) and 0.72 eV (Fig. S60), respectively.
The kinetic barrier for NH2* oxidation to NH* on Mn1/CeO2 is 3.3 eV (Fig. S5), much higher than for MnO2 and Mn3O4. NO can then adsorb at the Mn site, forming NH2NO* via the Langmuir–Hinshelwood mechanism with a low activation barrier of 0.41 eV (TS2, Fig. 6c). The direct isomerization of NH2NO* to NHNOH faces a high barrier of 1.79 eV (Fig. S61), making it less likely. Mn1/CeO2 facilitates the dehydrogenation of NH2NO* to NHNO*, which then decomposes into N2 and H2O with a low barrier of 0.75 eV (TS4). The formation of N2O from NHNO* on Mn1/CeO2 is less favorable, requiring 0.34 eV and facing a high barrier of 1.56 eV (Fig. S62). H* removal by lattice oxygen species is unlikely due to a high barrier of 1.13 eV (Fig. S63). Instead, H* is removed through O2 activation by an oxygen vacancy (Ovac) on Mn1/CeO257. The energy required to form Ovac on Mn1/CeO2 is lower than on pure CeO2 (2.62 eV vs. 2.93 eV, Fig. S64), suggesting Mn enhances Ovac formation on CeO2. O2 adsorbs at Ovac, forming O*, which reacts with two adjacent H atoms to produce H2O (2H* + O*, Fig. 6c). The activation energy for this pathway is 0.40 eV (TS5, Fig. 6c). DFT calculations also explored competing pathways, specifically the N-N coupling reaction via \({\text{NH}}_{4}^{+}\) and \({\text{NO}}_{2}^{-}\) (Fig. S65). The formation of NH4NO2 on the Mn1/CeO2 surface was found to be highly endothermic (+2.87 eV), making this pathway less favorable. Additionally, it is well-established that Mn-based catalysts are prone to forming ammonium nitrite and nitrate, which are common causes of catalyst deactivation due to ammonium nitrate poisoning. To experimentally validate our computational findings regarding the suppression of NH4NO2 formation by Mn1/CeO2, we conducted supplementary experiments confirming that Mn1/CeO2 does not lead to the formation of ammonium nitrate during the SCR reaction. Detailed experimental evidence and discussion are provided in Supplementary Note 3 and Figs. S43–S47. This analysis clarifies the atomic-level catalytic mechanism of Mn1/CeO2 in the SCR process.
Discussion
This work creates an innovative design concept of T-SACs featuring metal single atoms in asymmetric patterns, whose spatial and electronic configurations form a protective barrier on the support surface, thereby preventing direct interactions with reactants associated with these atoms. LCA analysis quantifying the environmental benefits of the Mn1/CeO2 over traditional V-W-Ti catalyst shows a much lower carbon footprint in several recognized metrics. In-situ analysis and DFT studies highlight the strategic topological arrangement creates steric hindrance and electronic shielding effects around the isolated Mn center, significantly reducing NH3 over-oxidation and thus enhancing N2 selectivity. The development of single-atom topological catalysts not only broadens the scope of catalytic chemistry, but also opens new avenues for designing materials with tailored properties for specific applications, ushering in a new era of catalytic science where precision and efficiency are paramount.
Methods
Synthesis of Mn1/CeO2
A typical synthesis process is as follows: a precursor solution was prepared by dissolving 8 g of glycerol and 24 g of citric acid in a mixture of 400 mL deionized water and 400 mL ethanol. This solution was stirred continuously for at least 2 h to ensure complete dissolution and homogeneity. Subsequently, 24 g cerous nitrate hexahydrate (Ce(NO3)3·6H2O) and 2 g manganese nitrate solution (50 wt% aqueous solutions of Mn(NO3)2) were added to the homogeneous solution, followed by stirring at 600 rpm for another 2 h. The mixture was then heated to 80 °C to evaporate the solvent, leading to the formation of a gel-like precursor. The resulting gel-like precursor was thoroughly ground into a fine powder, placed in a sealed container, and then subjected to calcination at 150 °C for 6 h in an oven (first annealing step, see Figs. S66 and 67). The calcined powder was subsequently ball-milled for at least 2 h to ensure uniformity. The milled sample was then annealed in a reducing atmosphere of N2/H2 (4 vol% H2 and 96 vol% N2) at 250 °C for 2 h (second annealing step), followed by multiple repetitions of the ball-milling and annealing process. Finally, the resulting solid powder was subjected to annealing in a tubular furnace at 600 °C under an N2 atmosphere for 4 h, followed by calcination in a muffle furnace at 300 °C for 3 h (third annealing step). For kilogram-scale production, the synthesis was scaled up by 2000 times through proportional scaling of both reactants and reaction vessel volume.
The synthesis of other metal T-SACs (Fe, Ru, Zn) follows the same steps, using the corresponding metal precursors and adjusting the annealing temperatures accordingly. The decomposition temperatures of the respective metal citrates were obtained from thermogravimetric analysis curves, which were used to confirm the temperature for each annealing step.
Synthesis of MnOx/CeO2
As shown in Fig. S26a, a precursor solution was prepared by dissolving 8 g of glycerol and 24 g of citric acid in a mixture of 400 mL deionized water and 400 mL ethanol. This solution was stirred continuously for at least 2 h to ensure complete dissolution and homogeneity. Subsequently, 24 g cerous nitrate hexahydrate (Ce(NO3)3·6H2O) and 2 g manganese nitrate solution (50 wt% aqueous solutions of Mn(NO3)2) were added to the homogeneous solution, followed by stirring at 600 rpm for another 2 h. The mixture was then heated to 80 °C to evaporate the solvent, forming a gel-like precursor. This gel was then annealed in a tubular furnace at 800 °C under an N2 atmosphere for 4 h, followed by calcination in a muffle furnace at 300 °C for 3 h.
Synthesis of CeO2
In the synthesis of the CeO2 catalyst, manganese nitrate was not added; the other synthesis steps remained the same as for Mn1/CeO2.
Synthesis of Mn-doped CeO2
As shown in Fig. S26b, Glucose (3.94 g) was dissolved in 160 mL of deionized water with magnetic stirring for approximately 60 min. Acrylamide (2.13 g), cerous nitrate hexahydrate (Ce(NO3)3·6H2O), and manganese nitrate solution (50 wt% aqueous solutions of Mn(NO3)2) were then added to form a transparent solution. The pH of the solution was adjusted to 10 by adding 6 mL of ammonia solution (25–28 wt%) dropwise while stirring. This addition caused the solution to transition into a stiff gel. The gel mixture was stirred for an additional 6 h and then transferred to a 200 mL Teflon-lined autoclave. The autoclave was sealed and heated in an oven at 180 °C for 72 h. After the reaction, the autoclave was allowed to cool naturally to room temperature. The resulting orange suspension and precipitate were separated by centrifugation. The suspension was washed three times with deionized water and ethanol, and then dried at 80 °C for approximately 24 h. The dried precipitate was calcined under N2 atmosphere with a flow rate of 5 mL/min in a tube furnace at 600 °C for 6 h. Finally, the calcined product was further treated in a muffle furnace at 400 °C for 4 h under an air atmosphere.
Synthesis of MnCeOx and MnCeOx-2
As shown in Fig. S26d, MnCeOx catalysts were prepared by a standard co-precipitation method from aqueous solutions of nitrates similar to that used by Gongshin et al.58. First, 4.34 g cerous nitrate hexahydrate (Ce(NO3)3·6H2O) and 0.36 g manganese nitrate solution (50 wt% aqueous solutions of Mn(NO3)2) were dissolved in water. Then, an excess amount of ammonium carbonate solution was slowly added to the mixture while stirring continuously. The resulting mixture was aged for 1 h and subsequently filtered. The obtained solid sample was dried at 120 °C in air for 12 h and then calcined at 600 °C for 6 h. For the synthesis of MnCeOx-2, the amount of manganese nitrate solution was increased to 1.432 g, and the calcination temperature was raised to 650 °C in a muffle furnace.
Synthesis of MnO, Mn3O4, Mn2O3 and MnO2
MnO was prepared by first synthesizing Mn3O4 through a hydrothermal method and then subjecting it to H2 reduction59. Initially, 0.353 g of manganese acetylacetonate was dissolved in 40 mL of absolute ethanol. The solution was then transferred into an 80 mL Teflon-lined autoclave and heated at 120 °C for 10 h. After heating, the mixture was washed three times with ethanol and once with deionized water through centrifugal separation. The resulting powder (Mn3O4) was collected after freeze-drying for 12 h. Finally, the Mn3O4 powder was thermally treated at 600 °C for 2 h in a tube furnace under a reducing gas flow (5 vol% H2 and 95 vol% argon) with a heating rate of 5 °C/min, yielding the final MnO catalyst.
As shown in Fig. S26c, MnO2, Mn3O4, and Mn2O3 catalysts were prepared using a hydrothermal synthesis method similar to that described by Leilei et al. 60. First, 0.118 g of KMnO4 and 0.6 mL ethylene glycol (EG) were dispersed in 44 mL distilled water. The mixed solution was stirred for 5 h and then transferred into a 50 mL Teflon-lined stainless-steel autoclave. The autoclave was sealed and heated at 120 °C for 10 h, then allowed to cool naturally to room temperature. The products (c-MnOOH) were collected by centrifugation, washed several times with ethanol and distilled water to remove excess reactants and byproducts, and then dried in a vacuum at 50 °C for 12 h. MnO2 were prepared by treating c-MnOOH at 400 °C for 4 h in air. Mn2O3 and Mn3O4 were obtained by calcining c-MnOOH at 700 °C for 5 h in air and argon atmosphere, respectively.
Synthesis of V-W-Ti
V2O5-WO3/TiO2 catalysts (denoted as V-W-Ti) were synthesized using the classical impregnation method. Ammonium metatungstate ((NH4)10H2W12O42·4H2O) was used as the precursor for WO3. To prepare the impregnation solution, 0.404 g of ammonium metatungstate was dissolved in 100 mL of deionized water in a beaker. In a separate beaker, 0.07 g of ammonium metavanadate (NH4VO3) and 0.151 g of oxalic acid (C2H2O4) (used as a solubilizing agent for vanadium, at twice the molar amount of vanadium) were dissolved in 100 mL of deionized water. The solutions were then combined and stirred at 60 °C for 30 min until complete dissolution (the solution should turn deep blue, with no crystalline particles remaining at the bottom). The prepared ammonium metavanadate and ammonium metatungstate solution was then gradually titrated onto 5 g of TiO2 powder using a pipette. The mixture was stirred thoroughly to ensure uniform distribution of the solution onto the TiO2 powder, followed by ultrasonic treatment for 30 min to enhance mixing. After allowing the mixture to stand for 2 h, it was dried in a conventional oven at 100 °C for at least 12 h with adequate ventilation. The dried precursor was then calcined in a muffle furnace at 600 °C for 5 h. Following the calcination, the catalyst was allowed to cool naturally to room temperature, resulting in the formation of the V-W-Ti catalyst.
NOx removal catalytic activity measurement
The self-designed and built fixed-bed reactor was used to test the performance of the catalyst for NH3-SCR reaction, including the catalytic activity and N2 selectivity of the catalyst. The reaction device is shown in Fig. S68. First, the prepared catalyst powder was pressed into tablets, sieved to 60–100 mesh, placed in a reaction tube (8 mm inner diameter), and then a thermocouple was inserted into the reaction tube to detect the reaction temperature. The gas concentration of the reaction gas was adjusted by a flow meter. The reaction gas comprised 500 ppm NO, 500 ppm NH3, 5 vol% O2, 100 ppm SO2 (when used) and 10 vol% H2O (when used), and the balance gas was N2 or Ar. The reaction space velocity (GHSV) was set to 50,000 h–1 or 100,000 h–1 according to the actual sample and experimental settings. The weight of a typical catalyst is provided in Table S11. When the reaction reached a stable state, the gas concentrations (NO, NO2, N2O, and NH3) in the inlet and outlet streams were measured using an infrared gas analyzer (GASMET FTIR DX4000). The SCR performance was evaluated across a temperature range of 90 °C to 300 °C, with NO conversion and N2 selectivity calculated using Eq. (1) and Eq. (2). NH3 and NO oxidation rates were determined using Eq. (4) and Eq. (5), respectively.
For the kinetic studies of the NH3-SCR reaction, the gas composition was consistent with that of the standard SCR reaction, and a gas hourly space velocity (GHSV) of approximately 150,000 h–1 was employed. Reaction rates (r) were normalized to the catalyst mass using Eq. (6), and activation energies (Ea) were calculated using the Arrhenius Eq. (7).
In these calculations, \({\text{F}}_{{\text{NO}}_{x}}\) represents the NOx flow rate (mol/s), \({\text{X}}_{{\text{NO}}_{x}}\) is the NOx conversion, Wcat is the catalyst mass (g), and \({[{\text{NO}}_{x}]}_{0}\) denotes the initial NOx concentration, encompassing NO, NO2, or a mixture of both.
Catalyst characterization
We characterized the crystal phase, morphology, and microstructure of the samples by XRD (Rigaku Dmax-2400, RIGAKU, Japan, Cu Kα radiation, 40 kV, 40 mA). The surface characteristics of single-atom catalysts were studied by aberration-corrected (AC) high-angle annular dark-field (HAADF) STEM and sub-angstrom-scale resolution was realized. The AC-HAADF-STEM images in this article were all obtained using a Titan Cubed Themis G2 300 spherical AC scanning transmission electron microscope at the Electron Microscopy Laboratory of Peking University. The X-ray absorption spectrum was obtained at the 1W1B-XAFS experimental station of the Beijing Synchrotron Radiation Facility. Athena software was used for data processing. NH3-TPD experiments were performed on automatic chemical adsorption analyzers (ChemBET Pulsar TPR/TPD, Quantachrome Instruments, USA and AUTOCHEM II 2920, USA).
We obtained in situ DRIFTS from an FT-IR spectrophotometer (Nicolet 6700, Thermo Fisher, US) by accumulating 32 scans with a resolution of 4 cm–1. For in situ Raman measurements, a 532 nm laser was used for excitation. We recorded the Raman spectra on a Horiba LaRAM HR Evolution (Horiba Jobin Yvon) with a resolution of 5 cm–1. Furthermore, we performed in situ NAP-XPS measurements on a SPECS NAP-XPS system, and XPS spectra were obtained using monochromatized Al Kα irradiation (1486.6 eV) generated by an Al anode (SPECS XR-50) and an excitation source power fixed at 50 W. In situ NAP-XPS measurements at pressures up to 25 mbar were possible because of a differential pumping system, which separated the electron analyzer (SPECS Phoibos NAP-150) from the reaction area.
The TG-FTIR-MS experiment was conducted using Perkin Elmer instruments: TGA 8000 thermogravimetric analyzer, FRONTIER FTIR spectrometer, and Clarus SQ8T mass spectrometer. Samples were heated from 30 °C to 900 °C at 20 °C/min under nitrogen flow (50 mL/min for balance and 20 mL/min for purge gas) in a corundum crucible. Transfer lines and interfaces were maintained at 280 °C, with an effluent gas flow of 50 mL/min. FTIR detection used a DTGS detector with 8 cm−1 resolution, and MS operated at 230 °C with an m/z range of 35–300. FTIR and MS detections were synchronized with the TGA program.
DFT methods
Plane-wave DFT calculations were performed in the Vienna Ab-initio Simulation Package (VASP)61,62,63 code in our study. With a kinetic energy cutoff of 600 eV, the projector augmented wave (PAW)64 scheme was employed to simulate the atoms’ potentials. We used Perdew-Burke-Ernzerhof (PBE)65 generalized gradient approximation functional for all calculations. A Hubbard U value (Ueff = U–J, J = 0) of 4.5 eV and 3.5 eV66,67,68 were added to the PBE functional to describe the localized electrons on the 4f orbitals of Ce and the 3d orbitals of Mn. Optimized structures were obtained by minimizing the forces on each ion using the conjugate gradient algorithm until the forces were below 0.02 eV/Å. We set the convergence criterion for the total energies of the investigated systems to 10–6 eV. LOBSTER program was employed to calculate COHP to analysis bonding states69.
For a certain reaction (A→B), the reaction free-energy ΔG [including the DFT total energy, zero-point energy, vibrational enthalpy (thermal corrections), and vibrational entropy (at T = 500 K)] is defined as follows:
We employed the CINEB method70 with a force tolerance of 0.02 eV/Å to calculate kinetic barriers. We further verified the optimized TS structures by which only having a single imaginary frequency. The kinetic barrier (Ea) was calculated as the energy difference between the TS and the IS.
We used a slab model (15.21 Å × 13.17 Å × 30.72 Å) to simulate the CeO2 (111) surface, which is a nine-layer slab containing 48 Ce and 96 O atoms, separated by a vacuum space of 21.51 Å. Furthermore, the top six atomic layers and surface species were allowed to relax, while the bottom three layers remained fixed in their bulk position. We implemented a capping strategy that involved using pseudo-hydrogen (pseudo-H) atoms71 to prevent the occurrence of dangling bonds.
The Mn1/CeO2 catalyst model was constructed by placing a Mn single atom on the CeO2 (111) surface. All possible sites were selected and optimized to obtain the most stable Mn1/CeO2 configuration based on the principle of lowest energy. The crystal plane model of MnO2 was a periodic slab model of the supercell constructed by cutting along the (211) plane on the MnO2 primitive cell. The supercell size of the simulated MnO2 (211) surface was 13.95 Å × 11.76 Å × 24 Å. Contains 72 Mn and 96 O atoms. The crystal plane model of Mn3O4 was a periodic plate model of the supercell constructed by cutting along the (101) plane on the MnO2 primitive cell. The supercell size of the simulated MnO2 (101) surface is 16.11 Å × 15.98 Å × 24 Å. Contains 48 Mn and 96 O atoms.
AIMD methods
All AIMD simulations were conducted using the VASP to investigate the dissociation process of NH2 and the formation of Mn single atoms on the CeO2 surface. The details of the AIMD calculations largely align with those described in the DFT methods section, with some adjustments made to the computational approach to reduce computational costs, as outlined below.
The wave functions were expanded with a cutoff energy of 400 eV and the Gaussian smearing with a width of 0.2 eV was employed. No spin polarization is considered to reduce the computational cost because it has almost no impact on the overall energies72,73,74,75. Two layers of CeO2 (111) facets were cleaved, incorporating a vacuum layer of 25 Å, to construct the surface structures. Additionally, one manganese salt (either MnCA or MnCl2, used for simulating the Mn single atom formation process) or NH2 (for simulating the NH2 dissociation process) was introduced into the slab model.
Firstly, the model has been fully relaxed with the energy convergence criterion of 10–4 eV and the force convergence criterion of 0.02 eV/Å, respectively. Only the Γ point has been used in the Brillouin zone since the model is large.
To simulate the formation process of single atoms and the dehydrogenation steps of NH2, as well as to calculate their activation free energy barriers, we employed the slow-growth sampling approach within the constrained molecular dynamics framework. Below, we present an illustrative example that details the simulation specifics of synthesizing Mn single atoms. This process utilizes the constrained AIMD method within our synthesis strategy.
The optimized model has been heated up from 0 K to 523 K by velocity scaling over 2615 fs every MD step and equilibrated at 523 K for 1 ps with a 1-fs time step. Finally, the constrained AIMD has been used with slow-growth method to simulate the dynamic process. The collective variable is the combination of the distance between the atom in manganese salt and CeO2. All collective variables increase with 0.0005 Å per MD step. Besides, to verify the stability of Mn-single atom on CeO2 surface, the CeO2 surface with three Mn-atoms have been built. After optimization, the model has been heated up from 0 K to 1073 K by velocity scaling over 5365 fs every MD step and equilibrated at 1073 K for 10 ps. The energy of all process has been counted every MD step and smoothed using Savitzky-Golay method per 1000 points.
LCA methods
This investigation conducts a life cycle assessment (LCA) of Mn1/CeO2 catalysts alongside commercially available V-W-Ti catalysts, utilizing 1 kg of the active catalyst as the functional unit for comparison. The LCA follows the CML baseline76 analytical framework, employing eChemstore® Intelligence software for its execution. This assessment rigorously adheres to the four distinct phases of LCA as delineated in the ISO 14040/14044 standards77. The activity and emission factor data employed in this life cycle assessment are derived from a combination of experimental findings, scholarly literature, and the ecoinvent 3.9 database. For any materials not cataloged in the ecoinvent 3.9 database, this study meticulously retraces the preparation process of these materials to the point where the raw materials are identifiable and locatable within the database.
Data availability
Relevant data supporting the key findings of this study are available within the article and the Supplementary Information file. All raw data generated during this study are available from the corresponding authors upon request.
References
Zaera, F. Designing sites in heterogeneous catalysis: are we reaching selectivities competitive with those of homogeneous catalysts? Chem. Rev. 122, 8594–8757 (2022).
Sun, G.-Q. et al. Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations. Nature 615, 67–72 (2023).
Fu, X. et al. Medium entropy alloy wavy nanowires as highly effective and selective alcohol oxidation reaction catalysts for energy-saving hydrogen production and alcohol upgrade. Ees. Catal. 2, 1285–1292 (2024).
Wang, X. et al. Discovery of promising Cu-based catalysts for selective propane dehydrogenation to propylene assisted by high-throughput computations. J. Catal. 438, 115718 (2024).
Inomata, Y. et al. Bulk tungsten-substituted vanadium oxide for low-temperature NOx removal in the presence of water. Nat. Commun. 12, 557 (2021).
Song, I. et al. Simple physical mixing of zeolite prevents sulfur deactivation of vanadia catalysts for NOx removal. Nat. Commun. 12, 901 (2021).
Chen, W., Zou, R. & Wang, X. Toward an atomic-level understanding of the catalytic mechanism of selective catalytic reduction of NOx with NH3. ACS Catal. 12, 14347–14375 (2022).
Han, L. et al. Selective catalytic reduction of NOx with NH3 by using novel catalysts state of the art and future prospects. Chem. Rev. 119, 10916–10976 (2019).
Voigt, C. et al. Nitrous oxide emissions from permafrost-affected soils. Nat. Rev. Earth Environ. 1, 420–434 (2020).
Shi, S. et al. Selective hydrogenation via precise hydrogen bond interactions on catalytic scaffolds. Nat. Commun. 14, 429 (2023).
Lan, X. & Wang, T. Highly selective catalysts for the hydrogenation of unsaturated aldehydes: a review. ACS Catal. 10, 2764–2790 (2020).
Li, C. et al. Selective hydroboration of unsaturated bonds by an easily accessible heterotopic cobalt catalyst. Nat. Commun. 12, 3813 (2021).
Frei, M. S. et al. Nanostructure of nickel-promoted indium oxide catalysts drives selectivity in CO2 hydrogenation. Nat. Commun. 12, 1960 (2021).
Vanni, M. et al. Selectivity control in palladium-catalyzed CH2Br2 hydrodebromination on carbon-based materials by nuclearity and support engineering. ACS Catal. 13, 5828–5840 (2023).
Veenstra, F. L. P., Ackerl, N., Martín, A. J. & Pérez-Ramírez, J. Laser-microstructured copper reveals selectivity patterns in the electrocatalytic reduction of CO2. Chem 6, 1707–1722 (2020).
Wan, C. et al. Amorphous nickel hydroxide shell tailors local chemical environment on platinum surface for alkaline hydrogen evolution reaction. Nat. Mater. 22, 1022–1029 (2023).
Zhao, X. & Liu, Y. Origin of selective production of hydrogen peroxide by electrochemical oxygen reduction. J. Am. Chem. Soc. 143, 9423–9428 (2021).
Davis, H. J. & Phipps, R. J. Harnessing non-covalent interactions to exert control over regioselectivity and site-selectivity in catalytic reactions. Chem. Sci. 8, 864–877 (2017).
Mahatthananchai, J., Dumas, A. M. & Bode, J. W. Catalytic selective synthesis. Angew. Chem. Int. Ed. 51, 10954–10990 (2012).
Bai, X. et al. Dynamic stability of copper single-atom catalysts under working conditions. J. Am. Chem. Soc. 144, 17140–17148 (2022).
Yu, S., Levell, Z., Jiang, Z., Zhao, X. & Liu, Y. What is the rate-limiting step of oxygen reduction reaction on Fe–N–C catalysts? J. Am. Chem. Soc. 145, 25352–25356 (2023).
Prasoon, A. et al. Site-selective chemical reactions by on-water surface sequential assembly. Nat. Commun. 14, 8313 (2023).
Pang, S. H., Schoenbaum, C. A., Schwartz, D. K. & Medlin, J. W. Directing reaction pathways by catalyst active-site selection using self-assembled monolayers. Nat. Commun. 4, 2448 (2013).
Qing, Y., Tamagaki-Asahina, H., Ionescu, S. A., Liu, M. D. & Bayley, H. Catalytic site-selective substrate processing within a tubular nanoreactor. Nat. Nanotechnol. 14, 1135–1142 (2019).
Mitchell, S. & Pérez-Ramírez, J. Single atom catalysis: a decade of stunning progress and the promise for a bright future. Nat. Commun. 11, 4302 (2020).
Qiao, B. et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 3, 634–641 (2011).
Xu, H., Cheng, D., Cao, D. & Zeng, X. C. Revisiting the universal principle for the rational design of single-atom electrocatalysts. Nat. Catal. 7, 207–218 (2024).
Zhang, S. et al. Atomically dispersed bimetallic Fe-Co electrocatalysts for green production of ammonia. Nat. Sustain. 6, 169–179 (2023).
Guo, X. et al. Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Science 344, 616–619 (2014).
Cao, L. et al. Atomically dispersed iron hydroxide anchored on Pt for preferential oxidation of CO in H2. Nature 565, 631–635 (2019).
Hai, X. et al. Geminal-atom catalysis for cross-coupling. Nature 622, 754–760 (2023).
Hai, X. et al. Scalable two-step annealing method for preparing ultra-high-density single-atom catalyst libraries. Nat. Nanotechnol. 17, 174–181 (2022).
Lang, R. et al. Single-atom catalysts based on the metal–oxide interaction. Chem. Rev. 120, 11986–12043 (2020).
Han, L. et al. A single-atom library for guided monometallic and concentration-complex multimetallic designs. Nat. Mater. 21, 681–688 (2022).
Xie, S. et al. Pt Atomic single-layer catalyst embedded in defect-enriched ceria for efficient CO oxidation. J. Am. Chem. Soc. 144, 21255–21266 (2022).
Merte, L. R. et al. CO-induced embedding of Pt adatoms in a partially reduced FeOx film on Pt (111). J. Am. Chem. Soc. 133, 10692–10695 (2011).
Yu, J. et al. Highly stable Pt/CeO2 catalyst with embedding structure toward water-gas shift reaction. J. Am. Chem. Soc. 146, 1071–1080 (2024).
Kim, C. H., Qi, G., Dahlberg, K. & Li, W. Strontium-doped perovskites rival platinum catalysts for treating NOx in simulated diesel exhaust. Science 327, 1624–1627 (2010).
Becher, J. et al. Chemical gradients in automotive Cu-SSZ-13 catalysts for NOx removal revealed by operando X-ray spectrotomography. Nat. Catal. 4, 46–53 (2021).
Shindell, D. et al. Climate, health, agricultural and economic impacts of tighter vehicle-emission standards. Nat. Clim. Chang. 1, 59–66 (2011).
Chaudhary, E. et al. Reducing the burden of anaemia in Indian women of reproductive age with clean-air targets. Nat. Sustain. 5, 939–946 (2022).
Krishna, S. H. et al. Influence of framework Al density in chabazite zeolites on copper ion mobility and reactivity during NOx selective catalytic reduction with NH3. Nat. Catal. 6, 276–285 (2023).
Liu, C., Shi, J.-W., Gao, C. & Niu, C. Manganese oxide-based catalysts for low-temperature selective catalytic reduction of NOx with NH3: A review. Appl. Catal. A-Gen. 522, 54–69 (2016).
Wang, Y. et al. Synergistic effect of N-decorated and Mn2+ doped ZnO nanofibers with enhanced photocatalytic activity. Sci. Rep. 6, 32711 (2016).
Hu, W. et al. Mechanistic investigation of NH3 oxidation over V-0.5Ce(SO4)2/Ti NH3-SCR catalyst. Catal. Commun. 112, 1–4 (2018).
Jin, R. et al. The role of cerium in the improved SO2 tolerance for NO reduction with NH3 over Mn-Ce/TiO2 catalyst at low temperature. Appl. Catal. B-Environ. 148, 582–588 (2014).
Liu, Y. et al. DRIFT studies on the selectivity promotion mechanism of Ca-modified Ce-Mn/TiO2 catalysts for low-temperature NO reduction with NH3. J. Phys. Chem. C. 116, 16582–16592 (2012).
Busca, G., Lietti, L., Ramis, G. & Berti, F. Chemical and mechanistic aspects of the selective catalytic reduction of NOx by ammonia over oxide catalysts: a review. Appl. Catal. B-Environ. 18, 1–36 (1998).
Suárez, S., Jung, S. M., Avila, P., Grange, P. & Blanco, J. Influence of NH3 and NO oxidation on the SCR reaction mechanism on copper/nickel and vanadium oxide catalysts supported on alumina and titania. Catal. Today 75, 331–338 (2002).
Slavinskaya, E. M. et al. Studies of the mechanism of ammonia oxidation into nitrous oxide over Mn-Bi-O/α-Al2O3 catalyst. J. Catal. 222, 129–142 (2004).
Peña, D. A., Uphade, B. S., Reddy, E. P. & Smirniotis, P. G. Identification of surface species on titania-supported manganese, chromium, and copper oxide low-temperature SCR catalysts. J. Phys. Chem. B 108, 9927–9936 (2004).
Liang, H., Gui, K. & Zha, X. DRIFTS study of γFe2O3 nano-catalyst for low-temperature selective catalytic reduction of NOx with NH3. Can. J. Chem. Eng. 94, 1668–1675 (2016).
Krueger, P. J. Fundamental NH2 stretching frequencies in amines and amides. Nature 194, 1077–1078 (1962).
Wilfong, W. C., Srikanth, C. S. & Chuang, S. S. C. In situ ATR and DRIFTS studies of the nature of adsorbed CO2 on tetraethylenepentamine films. ACS Appl. Mater. Inter. 6, 13617–13626 (2014).
Chen, Y. et al. Promoting effect of Nd on the reduction of NO with NH3 over CeO2 supported by activated semi-coke: an in situ DRIFTS study. Catal. Sci. Technol. 5, 2251–2259 (2015).
Qi, G. & Yang, R. T. Characterization and FTIR studies of MnOx-CeO2 catalyst for low-temperature selective catalytic reduction of NO with NH3. J. Phys. Chem. B 108, 15738–15747 (2004).
Chen, W., Wang, X. & Xu, S. Investigation of intrinsic catalytic mechanism for NO oxidation to NO2 in CeO2 used for NO removal. Chem. Eng. J. 460, 141801 (2023).
Qi, G., Yang, R. T. & Chang, R. MnOx-CeO2 mixed oxides prepared by co-precipitation for selective catalytic reduction of NO with NH3 at low temperatures. Appl. Catal. B-Environ. 51, 93–106 (2004).
Tian, H. et al. In situ electrochemical Mn(III)/Mn(IV) generation of Mn(II)O electrocatalysts for high-performance oxygen reduction. Nano-Micro Lett. 12, 161 (2020).
Lan, L., Li, Q., Gu, G., Zhang, H. & Liu, B. Hydrothermal synthesis of γ-MnOOH nanorods and their conversion to MnO2, Mn2O3, and Mn3O4 nanorods. J. Alloy. Compd. 644, 430–437 (2015).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B-Condens Matter 48, 13115–13118 (1993).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kildgaard, J. V., Hansen, H. A. & Vegge, T. DFT + U study of strain-engineered CO2 reduction on a CeO2–x (111) facet. J. Phys. Chem. C. 125, 14221–14227 (2021).
Wu, T., Deng, Q., Hansen, H. A. & Vegge, T. Mechanism of water splitting on gadolinium-doped CeO2(111): A DFT + U study. J. Phys. Chem. C. 123, 5507–5517 (2019).
Su, Y.-Q., Liu, J.-X., Filot, I. A. W., Zhang, L. & Hensen, E. J. M. Highly active and stable CH4 oxidation by substitution of Ce4+ by two Pd2+ ions in CeO2(111). ACS Catal. 8, 6552–6559 (2018).
Maintz, S., Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. LOBSTER: a tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 37, 1030–1035 (2016).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Xu, S. & Carter, E. A. CO2 Photoelectrochemical reduction catalyzed by a GaP(001) photoelectrode. ACS Catal. 11, 1233–1241 (2021).
Cheng, T., Wang, L., Merinov, B. V. & Goddard, W. A. III Explanation of dramatic pH-dependence of hydrogen binding on noble metal electrode: Greatly weakened water adsorption at high pH. J. Am. Chem. Soc. 140, 7787–7790 (2018).
Li, P., Huang, J., Hu, Y. & Chen, S. Establishment of the potential of zero charge of metals in aqueous solutions: different faces of water revealed by Ab initio molecular dynamics simulations. J. Phys. Chem. C. 125, 3972–3979 (2021).
Li, C.-Y. et al. In situ probing electrified interfacial water structures at atomically flat surfaces. Nat. Mater. 18, 697–701 (2019).
Li, P. et al. Revealing the role of double-layer microenvironments in pH-dependent oxygen reduction activity over metal-nitrogen-carbon catalysts. Nat. Commun. 14, 6936 (2023).
Guinée, J. B. Handbook on Life Cycle Assessment: Operational Guide to the ISO Standards, Vol. 7 (Springer Science & Business Media, 2002).
Standardization, I. O. F. Environmental Management: Life Cycle Assessment; Requirements and Guidelines, Vol. 14044 (ISO Geneva, Switzerland, 2006).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52332007Z) and the NRF, Prime Minister’s Office, Singapore, under the Competitive Research Program Award (NRF-CRP29-2022-0004L). The authors would like to acknowledge the support from the AI for Science (AI4S) Preferred Program at Peking University Shenzhen Graduate School, China. The DFT calculations were supported by Shanxi Supercomputing Center of China (TianHe−2), High-Performance Computing Platform of Peking University. The authors also acknowledge the Beijing Beilong Super Cloud Computing Co., Ltd for providing HPC resources that have contributed to the research results reported in this paper. We thank the Materials Processing and Analysis Center, Peking University, and the Beijing Synchrotron Radiation Facility (BSRF) at beamline 1W1B for their support in material characterization.
Author information
Authors and Affiliations
Contributions
J. Lu and R. Zou supervised the project and organized the collaboration. W. Chen and M. Bao designed the experiments and synthesized the materials. W. Chen and B. Ma conceived the reaction mechanism and carried out quantum-chemical calculations. M. Bao and L. Feng discovered the catalytic activity. X. Zhang and S. Gao conducted the comparison studies and synthetic applications. S. Xi performed the XAFS measurement and structural analysis. F. Meng and Z. Qiu performed the electron microscopy experiments and data analysis. R. Zhong advised on the experiments, methodologies and data presentation. W. Chen wrote the draft, with the assistance of X. Hai. All authors discussed the results and edited and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yaqiong Su, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, W., Bao, M., Meng, F. et al. Designer topological-single-atom catalysts with site-specific selectivity. Nat Commun 16, 574 (2025). https://doi.org/10.1038/s41467-025-55838-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-55838-6
This article is cited by
-
The precise regulation of noble metal-loaded layered double hydroxides nanocatalysts: synthesis and structure-activity relationship
Science China Chemistry (2026)
-
Sustainable DNA-polysaccharide hydrogels as recyclable bioplastics
Nature Communications (2025)
